

Abstract
In modeling evolutionary genetics, it is often assumed that mutational effects are assigned according to a continuous probability distribution, and multiple distributions have been used with varying degrees of justification. For mutations with beneficial effects, the distribution currently favored is the exponential distribution, in part because it can be justified in terms of extreme value theory, since beneficial mutations should have fitnesses in the extreme right tail of the fitness distribution. While the appeal to extreme value theory seems justified, the exponential distribution is but one of three possible limiting forms for tail distributions, with the other two loosely corresponding to distributions with right-truncated tails and those with heavy tails. We describe a likelihood-ratio framework for analyzing the fitness effects of beneficial mutations, focusing on testing the null hypothesis that the distribution is exponential. We also describe how to account for missing the smallest-effect mutations, which are often difficult to identify experimentally. This technique makes it possible to apply the test to gain-of-function mutations, where the ancestral genotype is unable to grow under the selective conditions. We also describe how to pool data across experiments, since we expect few possible beneficial mutations in any particular experiment.
ADAPTATION at the molecular level involves the fixation of beneficial mutations over time through natural selection. Although there is an extensive body of theoretical work on the process of natural selection, the overall process of adaptation is not as well characterized theoretically (Orr 2005a). Because beneficial mutations are rare and often have small effects, theoreticians have little information on the raw material upon which natural selection acts. Despite this dearth of data, there has been a recent emergence of work on the theory of adaptation (Gillespie 1983, 1984, 1991; Orr 2002, 2003a, 2005b, 2006b; Rokyta et al. 2006). Building on ideas of a sequence space originally proposed by Maynard Smith (1962, 1970), Gillespie introduced the primary assumption leading to much of the current theoretical work by arguing that extreme value theory (EVT) can help circumvent our lack of information regarding the nature of benficial mutations. Gillespie (1983, 1984, 1991) posited that, given an initial genotype, one can imagine the fitnesses of mutants being drawn from some underlying probability distribution. The majority of these mutations will be neutral, deleterious, or even lethal to the organism and only a small number will be beneficial. The fitness effects assigned to these mutations can thus be assumed to reside in the extreme right tail of the underlying fitness distribution.
Assuming that the fitnesses of interest lie in the right tail of the fitness distribution allows the use of EVT to predict the characteristics of beneficial mutations. Theoretical work thus far, however, has relied on one further assumption. It has always been assumed that the underlying fitness distribution is in the Gumbel domain of attraction. If this is true, EVT shows that the limiting distribution of the tail is exponential. However, this need not be the case. In fact, EVT describes three types of limiting tail distributions (i.e., domains of attraction), and furthermore, not all distributions have even a limiting tail distribution. This choice of the Gumbel domain has been rationalized by arguing that the other two types are biologically unreasonable (Orr 2006a), and some theoretical (Orr 2006a), computational (Cowperthwaite et al. 2005), and empirical data (Kassen and Bataillon 2006) support this contention.
As the Gumbel domain of attraction is such a prominent component of the current theory of adaptation, it is necessary to provide a thorough empirical test of this assumption. Any attempt to test the Gumbel hypothesis ultimately reduces to a determination of whether or not data from the extreme right tail of a distribution appear to be exponential. Of course, in the construction of any statistical test, the appropriate alternative hypothesis must be determined. Commonly, in tests for exponentiality, the alternative selected is the gamma distribution (e.g., Kassen and Bataillon 2006), which has the attractive property that it subsumes the exponential distribution as a special case. However, since there seems to be little reason to doubt that fitnesses of genotypes possessing beneficial mutations can be considered draws from the tail of the fitness distribution, this may not be the most appropriate alternative hypothesis. In fact, the test of interest is whether the Gumbel domain is the correct domain for the unknown fitness distribution. The gamma distribution is in the Gumbel domain, so at best, testing against the gamma might provide information on whether the observed fitness values are indeed drawn from the tail.
According to EVT, there are three domains of attraction, the Gumbel, Fréchet, and Weibull domains (Figure 1). All distributions in the Gumbel domain have the exponential as the limiting distribution of their tail. This domain contains the majority of well-known distributions such as the normal, the exponential, and the gamma. The Fréchet domain contains distributions with an infinite yet heavier tail than the exponential, (e.g., the Cauchy distribution). The Weibull domain (distinct from the Weibull distribution, which in fact belongs to the Gumbel domain) contains distributions with lighter tails than exponential, which possess a finite upper bound (e.g., the uniform distribution).
Figure 1.—

An illustration of the different possible tail behaviors corresponding to the three domains of attraction under extreme value theory. The top describes a general fitness distribution of all genotypes within one mutational step of the wild type, with wild-type fitness given by Wi. We are interested in the distribution of values bigger than Wi. The bottom shows hypothetical examples of the three alternative tail distributions.
There are two standard approaches to EVT. The classical approach considers the distribution of the largest value, the second largest value, etc. This naturally leads to results on the distribution of spacings between consecutive extreme observations. These results are leveraged by Gillespie and Orr in their theory of adaptation, as the spacings can be used to calculate fitness effects. However, when considering alternative models, the distribution of the spacings is not straightforward. A more natural approach is to consider the distribution of values above a threshold, i.e., the wild type. This is often referred to as the “peaks over threshold” approach. Under this framework all three domains can be described by a single family of distributions called the generalized Pareto distribution (GPD) (Pickands 1975).
The cumulative distribution function for the GPD is given by
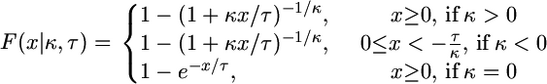 |
(1) |
and the probability density function is given by
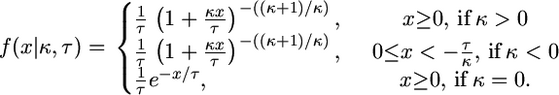 |
(2) |
This parameterization unifies all three limiting distributions in terms of the distribution of values exceeding a high threshold, where the data are transformed such that the threshold is at x = 0. A location parameter can be added to move the threshold, but it is equivalent to transforming the data relative to the threshold. The form of the GPD is determined by a scale parameter τ and shape parameter κ, commonly referred to as the tail index, which specifies the weight of the tail. Note that the exponential distribution is nested in the GPD as a special case when κ = 0, making it an ideal candidate for likelihood-ratio methods. Further, the domain of attraction is exactly determined by the shape parameter κ, the case κ = 0 corresponding exactly to the Gumbel, κ > 0 to the Fréchet, and κ < 0 to the Weibull domain of attraction (Castillo 1988). EVT assures us that these other domains are the best alternative to the Gumbel hypothesis of an exponential tail and therefore will produce the most powerful statistical tests, provided the data lie in the extreme right tail of some underlying distribution falling into one of the three domains of attraction. In terms of empirical data, we can assume that the threshold is the fitness of the wild type and that the fitness effects of beneficial mutations follow some form of the GPD.
We imagine that the methodology we present herein will prove to be most useful for microbial experimental evolution experiments, although the test is applicable to other forms of data. Particularly in viral experimental evolution, it is possible to isolate multiple beneficial mutations arising from the same ancestral genotype, as well as identify the mutations involved (e.g., Bull et al. 2000; Rokyta et al. 2005). Yet in these experiments, there is an inherent difficulty associated with testing for the domain of attraction for the fitness distribution. As we are assuming that our observations represent extreme values from the tail of a distribution, we can expect only a small number of unique beneficial mutations. However, having only a handful of beneficial mutations implies low statistical power to perform a test for domain of attraction. The usual solution to this problem is to collect more data, but as the number of beneficial mutations is small, this results in little or no improvement in statistical power, since replicating will tend to produce the same mutants. As any test with a low number of observations will be plagued by at best mediocre power, we present methods that allow for an additional increase in power through pooling data across distinct experiments. We also address the issue of missing small-effect mutations. Since identifying beneficial mutations experimentally involves selecting for them, it may prove difficult to identify those mutations with very small beneficial fitness effects. This bias toward seeing larger-effect mutations could have profound effects on the outcome of the data analysis. Under our statistical framework, accounting for this turns out to require only a simple shift of the data.
STRUCTURE OF THE DATA
When fitting a statistical distribution to data, one usually views the data as a random sample, where this is defined to be a collection of independent observations from a common probability distribution. However, a sample of observed first-step mutations and their corresponding fitnesses that arise from an adaptive evolution experiment cannot be viewed as a random sample from a probability distribution. Since evolution favors the fixation of more-fit mutants over less-fit mutants, there is a greater chance of observing a mutation with large effect over one with small effect. In fact, it is likely that mutations with effects below some threshold may not be observed at all, even after a large number of replicate experiments. In addition, another threshold exists where mutations cannot be resolved as significantly beneficial given some assay to measure fitness. Both of these aspects contribute to the possibility of some data not being observed during the course of the experiment. This is known as a censored data problem. As a solution to this, we present a way to transform the data through a shift relative to the smallest observation.
Censored data:
Consider the fitness distribution of all one-step mutations from some genotype. Let i be the rank of the wild type. Thus, from the ancestral genotype, there are a total of i − 1 beneficial mutations with selection coefficients in rank order s = s1, s2,…, si−1, which are drawn from an unknown probability density f(s) with cumulative distribution F(s). Thus, s1 is the selection coefficient for the largest-effect mutation, s2 is the selection coefficient for the second largest, etc. Note that selection coefficients are just the fitness difference relative to the wild type normalized to the wild-type fitness. All of what follows works equivalently if selection coefficients are replaced with fitness effects. Now suppose that due to the experimental protocol, it is not possible to observe all possible mutations, only the largest n of the total i − 1, with selection coefficients s1, s2,…, sn, where n < i − 1. In other words, we failed to observe the leftmost selection coefficients sn+1, sn+2,…, si−1 in the collected data set. Denote the observed selection coefficients as sn = (s1, s2,…, sn).
Using standard results from order statistics (see Rice 1995, p. 100), the distribution of sn depends on i and is given by
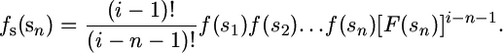 |
(3) |
If fitness effects are measured relative to the wild type and there exists the possibility that selection coefficients smaller than sn are missing from the data set, then Equation 3 represents the appropriate likelihood equation. However, because there are missing data, the true number of selection coefficients, i − 1, is unknown. Thus, to use Equation 3, an estimate for i − 1 would need to be obtained while accounting for uncertainty in this estimate. This unfortunate complication of the likelihood analysis can be avoided at the cost of 1 d.f. Instead of measuring fitness relative to the wild type, we shift the distribution relative to the smallest observed selection coefficient,
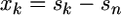 |
(4) |
for k = 1, 2,…, n − 1. Now we have a transformed data set x = (x1, x2,…, xn−1) with one less observation. The likelihood for this shifted data is
 |
Note that the probability density function for the selection coefficients shifted by sn simplifies to
 |
(5) |
Therefore,
 |
(6) |
which is the distribution of a rank-ordered random sample of n − 1 selection coefficients drawn from the density fX(x). The key observation is that the probability density function given by (6) does not depend on the unobserved selection coefficients between si−1 and sn. This result is independent of the particular distribution. Assuming that all of the unobserved mutations fall below a set threshold, shifting the probability distribution further out into the tail resolves the missing data problem and eliminates the need to know i, the rank of the wild type. This comes at the moderate cost of losing a single degree of freedom. While this may reduce power, it results in the avoidance of the potentially serious consequence of misinterpreting the data (Figure 2).
Figure 2.—

The impact of missing small-effect mutations on the type I error for the LRT. Values were simulated from a shifted exponential. The horizontal axis represents the shift, measured as a fraction of the mean fitness effect. The vertical axis represents the true type I error for the likelihood-ratio test when one fails to account for the shift. One hundred thousand replicate tests were performed for each point.
Applying (6) to the GPD yields a curious result. As has been previously noted (Castillo and Hadi 1997), the GPD shape parameter κ is stable with respect to shifts in the threshold. Thus, upon shifting the threshold, κ remains the same and only the scale τ changes. For our purposes, we are concerned only with the shape, as it determines the domain of attraction. To see this, suppose the f(s) is given by (2); then
 |
Thus, a GPD with a change in threshold will again follow the GPD with the same shape but new scale parameter. This stability property is not true in general for other distributions, such as the normal, lognormal, and gamma. Note that for κ = 0 this result simply states the well-known memoryless property for the exponential. This implies that the likelihood function for shifted data is of the same form as for the unshifted data, differing only by a factor of (n − 1)!, which cancels out in a likelihood ratio and is needed only in the case of ordered observations.
LIKELIHOOD-RATIO TEST
We propose a likelihood-ratio test (LRT) framework for testing whether an empirical sample of beneficial mutations have fitness effects that are consistent with having been drawn from a distribution in the Gumbel domain of attraction. While likelihood analysis is well established, there are several aspects to our problem that make it unique. First, if we believe that some of the possible selection coefficients were not observed, we need to change the threshold and measure fitness relative to the smallest observed fitness as described above. Second, we argue that the GPD given by (2) is a more appropriate alternative to the exponential distribution than some of the more familiar distributions. Third, because there may be only a small number of mutations that are adaptive, the power of any test may be low. To improve power we consider pooling data from many experiments. The flexibility of the likelihood approach allows a simplified adaptation of the test to this general framework. Finally, the likelihood framework extends to incorporate measurement error associated with the observed fitness effects.
Likelihood-ratio test:
After shifting the selection coefficients appropriately (see Equation 4), we can view the data X = (X1, X2,…, Xn−1) as a random sample of n − 1 observations from the GPD. The log-likelihood function under the GPD is given by
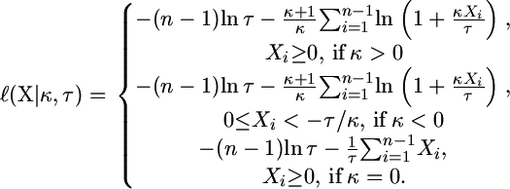 |
(7) |
The likelihood framework allows great flexibility in hypothesis testing. If we are interested in testing the null model that the data are from the exponential distribution, we can simply optimize the likelihood under the restriction of κ = 0 and then under the alternative model where κ is unrestricted. The LRT statistic is usually calculated on the log scale and with the standard formulation,
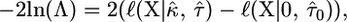 |
(8) |
where 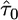 is the maximum-likelihood estimate (MLE) for τ under the exponential model, and
is the maximum-likelihood estimate (MLE) for τ under the exponential model, and 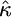 and
and 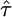 are the maximum-likelihood estimates under the full GPD.
are the maximum-likelihood estimates under the full GPD.
Although often −2 ln(Λ) asymptotically follows a 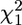 -distribution, we do not know the sample sizes for which this approximation is appropriate. Instead, the distribution of the test statistic can be generated using parametric bootstrap based specifically on the size of a particular sample. First, the MLEs of the parameters are found under the restricted model, which in our case is the scale parameter of the exponential. A data set is generated under the null model using this estimated parameter. The LRT is performed on this simulated data set and the test statistic −2 ln(Λ) is calculated. This procedure for the calculation of the test statistic is replicated to generate an empirical distribution of the test statistic from which an approximation of the P-value is obtained. The parametric bootstrap approach approximates the distribution of −2 ln(Λ) in two ways. Because the approach is based on simulation of data, there is simulation error. However, this error is controllable, as we can obtain any degree of accuracy needed by increasing the number of bootstrap replicates. The second way in which the parametric bootstrap approximates the true distribution of −2 ln(Λ) is that we simulate using the estimated parameter
-distribution, we do not know the sample sizes for which this approximation is appropriate. Instead, the distribution of the test statistic can be generated using parametric bootstrap based specifically on the size of a particular sample. First, the MLEs of the parameters are found under the restricted model, which in our case is the scale parameter of the exponential. A data set is generated under the null model using this estimated parameter. The LRT is performed on this simulated data set and the test statistic −2 ln(Λ) is calculated. This procedure for the calculation of the test statistic is replicated to generate an empirical distribution of the test statistic from which an approximation of the P-value is obtained. The parametric bootstrap approach approximates the distribution of −2 ln(Λ) in two ways. Because the approach is based on simulation of data, there is simulation error. However, this error is controllable, as we can obtain any degree of accuracy needed by increasing the number of bootstrap replicates. The second way in which the parametric bootstrap approximates the true distribution of −2 ln(Λ) is that we simulate using the estimated parameter 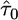 rather than the unknown true parameter τ. For small sample sizes, the low accuracy of the estimate could affect the approximation. There are ways to adjust the parametric bootstrap approach to account for this error, but these adjustments are not necessary for the problem at hand. In general, the fact that τ is a scale parameter would imply that the distribution of Xj/τ is independent of τ. Specifically, under the null model, Xj/τ follows the standard exponential distribution with mean one. Note that the likelihood of the data under every form of the GPD (Equation 2) is a function of Xj/τ, so the distribution of
rather than the unknown true parameter τ. For small sample sizes, the low accuracy of the estimate could affect the approximation. There are ways to adjust the parametric bootstrap approach to account for this error, but these adjustments are not necessary for the problem at hand. In general, the fact that τ is a scale parameter would imply that the distribution of Xj/τ is independent of τ. Specifically, under the null model, Xj/τ follows the standard exponential distribution with mean one. Note that the likelihood of the data under every form of the GPD (Equation 2) is a function of Xj/τ, so the distribution of 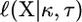 does not depend on τ. (However,
does not depend on τ. (However, 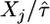 does depend on τ, but the dependence is so weak that it can be ignored, even for small sample sizes).
does depend on τ, but the dependence is so weak that it can be ignored, even for small sample sizes).
Care must be taken when applying likelihood theory to the Weibull domain of attraction (κ < 0). Here, the truncation point depends on the parameters to be estimated. In the statistical literature this is referred to as a range-dependent model. It is well known that standard asymptotic theory does not apply for range-dependent models. This issue for parameter estimation under maximum likelihood has been previously noted for the GPD (Smith 1985). However, since we are using parametric bootstrap, we do not rely on asymptotic theory. Also, note that if κ < −1, the likelihood can become infinite due to the distribution increasing the weight on the rightmost observation, and therefore the maximum-likelihood estimate does not exist. The problem can be remedied by restricting κ > −1, which excludes “reverse” tails (Figure 1) from consideration. If the true value of κ is indeed < −1, the likelihood-ratio test nearly always leads to rejection of the null model κ = 0. This restriction is conservative and has little effect on the analysis (see Power analysis).
Pooling data across experiments:
The problem of low power is inherent in any statistical test involving extreme values. The nature of extreme value theory dictates that when observing data from the extreme right tail of a distribution, we will have relatively few observations. The observation of a large sample contraindicates this assumption. This presents a problem in an experiment that relies on the observation of adaptive mutations. Not only is there an expectation of a small number of observations, the number of possible observations is actually fixed. This prevents the standard solution of increasing power through the collection of additional observations through replicate experiments. As the number of replicate experiments is increased, data collection suffers from a diminishing return; previously observed mutants will occur more often, culminating at a point where all possible mutants have been observed and no new information can be gained. Alternatively, there may be enough beneficial mutations to achieve a reasonable amount of power, but many of these mutations will be of small effect and require a prohibitive number of replicate experiments before they will be observed.
Seemingly caught in a catch-22 (Heller 1961), the only hope for increasing power is through pooling data across nonreplicate experiments with different ancestral genotypes or different environmental conditions. In this case the data can now be thought of as an array of observations, where Xj,k represents the jth fitness effect from the kth experiment. Consider a total of m experiments. The formal hypothesis test is of the form
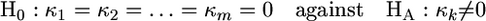 |
for at least one k. The likelihood-ratio statistic in this scenario generalizes to
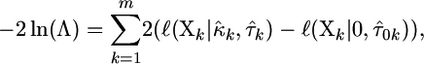 |
(9) |
where 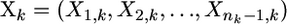 are the observed fitnesses for the kth experiment,
are the observed fitnesses for the kth experiment, 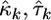 are the parameter estimates under the GPD for the kth experiment, and
are the parameter estimates under the GPD for the kth experiment, and 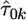 is the estimate for τ under the exponential model.
is the estimate for τ under the exponential model.
To illustrate the improvement in power that occurs by pooling nonreplicate experiments consider the following example. Suppose that one performs 10 nonreplicate experiments generated from distinct ancestral genotypes, each of which results in the observation of 10 distinct beneficial mutations. After shifting the threshold relative to the smallest observed, we have nine fitness effects for the 10 experiments. For each experiment we are required to estimate two parameters and shift relative to the smallest observation, which reduces the degrees of freedom by 3 for each replicate experiment. The effective sample size would then be 70, since 10(10 − 1 d.f. − 2 d.f.) = 70. Now consider the case of observing 73 distinct adaptive mutations in a single replicate experiment. We lose 3 d.f. from the shift of the threshold and the estimation of two model parameters, leaving us again with an effective sample size of 70. Therefore pooling 10 observations from 10 experiments is equivalent to observing 73 from a single experiment.
Incorporating measurement error:
In microbial evolution experiments, the beneficial mutations are first identified and then the fitness effect of each is estimated from the results of a separate experiment. The precision of the fitness assays associated with this second step can be a source of significant measurement error and could influence the analysis. Kassen and Bataillon (2006) appropriately account for this measurement error in their likelihood analysis. It is possible to minimize the effect of this error on the test for domain of attraction by conducting a larger number of replicate fitness assays. However, it is possible that the number of replicates required is too large or not cost effective, so that accounting for measurement error is required. In this case the likelihood equations can be easily extended to account for both normal and lognormal error. The type of error that is operating can be determined by standard methods such as a Q-Q plot against the standard normal. Here we present an efficient algorithm for estimating the appropriate parameters when measurement error cannot be ignored. Let yij be the jth replicate for the ith largest fitness effect. Suppose that f(x | θ) is the distribution for fitness effects. We use θ as the generic parameter, where θ could represent a vector of parameters, for example, the scale and shape parameter of the GPD. Let g(y | x, σ2) be the normal density with mean x and variance σ2. Let 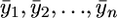 be the average of the observed fitness effects under the assumption of equal replications per mutant genotype. Let θ0 be the MLE for θ when measurement error is ignored. Let
be the average of the observed fitness effects under the assumption of equal replications per mutant genotype. Let θ0 be the MLE for θ when measurement error is ignored. Let  be the pooled variance based on the observed fitness effects. Now, the likelihood of the data is given by
be the pooled variance based on the observed fitness effects. Now, the likelihood of the data is given by
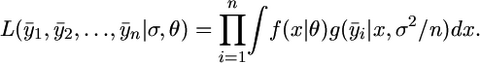 |
We can approximate 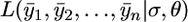 , using a Monte Carlo algorithm as follows. Since
, using a Monte Carlo algorithm as follows. Since 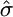 is the appropriate estimate of σ we need only calculate
is the appropriate estimate of σ we need only calculate 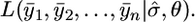 Calculate θ0, the MLE for θ when measurement error is ignored. Simulate X1,j, X2,j,…, Xn,j for j = 1,…, N, from f(x | θ0). Order the X 's so they match up with the corresponding
Calculate θ0, the MLE for θ when measurement error is ignored. Simulate X1,j, X2,j,…, Xn,j for j = 1,…, N, from f(x | θ0). Order the X 's so they match up with the corresponding 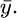 The maximum-likelihood estimate can now be obtained by maximizing the following approximation to the likelihood with respect to θ:
The maximum-likelihood estimate can now be obtained by maximizing the following approximation to the likelihood with respect to θ:
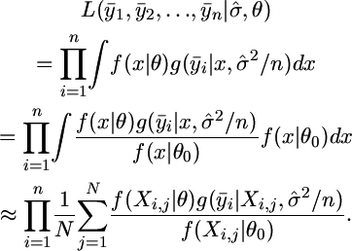 |
(10) |
To account for lognormal error, a similar algorithm can be described. The algorithm utilizes a special case of importance sampling, where samples are drawn from a fixed distribution f(x | θ0) to approximate a likelihood over a range of θ's. Importance sampling is a commonly used Monte Carlo technique in population genetics (e.g., Griffiths and Tavaré 1994, Section 7). While Kassen and Bataillon (2006) use a numerical technique to approximate the likelihood rather than importance sampling, the two methods are in fact equivalent.
To gauge how susceptible the algorithm is to Monte Carlo error, simulations were performed with N = 10,000 under the null hypothesis, where κ = 0, τ = 1, σ = 0.1, and a sample size n = 20. The algorithm provided reasonable estimates of the parameters (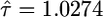 ,
, 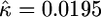 ). Note the coefficient of variation (σ/τ = 0.1) represents measurement error less than what is expected to influence the results of the test (see Figure 4). The Monte Carlo error, due to the approximation in Equation 10, for the estimates of the parameters and the log-likelihood achieved a coefficient of variation <5 × 10−5 and 4 × 10−5, respectively. In a second example, we simulated a sample of size n = 20 from the null model with κ = 0, τ = 1, and σ = 0.3. Note that the coefficient of variation in this example is much larger (σ/τ = 0.3) yet the algorithm still was able to recover reasonable estimates of the parameters (
). Note the coefficient of variation (σ/τ = 0.1) represents measurement error less than what is expected to influence the results of the test (see Figure 4). The Monte Carlo error, due to the approximation in Equation 10, for the estimates of the parameters and the log-likelihood achieved a coefficient of variation <5 × 10−5 and 4 × 10−5, respectively. In a second example, we simulated a sample of size n = 20 from the null model with κ = 0, τ = 1, and σ = 0.3. Note that the coefficient of variation in this example is much larger (σ/τ = 0.3) yet the algorithm still was able to recover reasonable estimates of the parameters (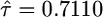 and
and 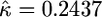 ). The Monte Carlo errors for the estimates and log-likelihood were again more than three orders of magnitude smaller than the parameters, 1 × 10−5 and 1 × 10−4, respectively. The results of these two simulations suggest that the algorithm as described is suitable for maximum-likelihood methods with N remaining computationally tractable.
). The Monte Carlo errors for the estimates and log-likelihood were again more than three orders of magnitude smaller than the parameters, 1 × 10−5 and 1 × 10−4, respectively. The results of these two simulations suggest that the algorithm as described is suitable for maximum-likelihood methods with N remaining computationally tractable.
Figure 4.—


The impact of ignoring measurement error. Data were simulated under the exponential distribution and both normal errors (A) and lognormal (B) were included in the simulations of each data set. The likelihood-ratio test was performed for the exponential against a GPD ignoring measurement error in the data. The type I error was plotted against the coefficient of variation for the distribution of measurement error.
SIMULATION RESULTS
Optimization in a multidimensional parameter space that contains a boundary restriction on the values of the parameters can prove to be difficult. To simplify the computational burden of optimizing the likelihood equations, we make use of a reparameterization of the GPD (Davison and Smith 1990; Grimshaw 1993). Let λ = −κ/τ. Note that if λ was known then the MLE for κ can be written
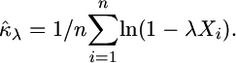 |
Now, substituting λ and 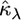 into the log-likelihood for the GPD we arrive at the reparameterization,
into the log-likelihood for the GPD we arrive at the reparameterization,
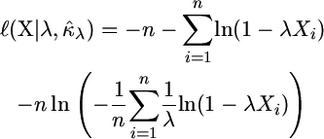 |
This form allows for single-dimensional optimization of the log-likelihood over the single parameter λ, which is more reliable and computationally efficient, although under this reparameterization it is not possible to restrict values of κ < −1. All of the calculations in this section were conducted using the freely available statistical package R (R Development Core Team 2006). Log-likelihoods were optimized with the Nelder–Mead algorithm. Implementations of the test with and without measurement error are available from the author's web site at http://www.uidaho.edu/∼joyce/lab page/computer-programs.html.
Power vs. sensitivity:
Two types of statistical analysis based on simulations are presented. We present a power analysis, summarized in Figure 3 and sensitivity analyses, summarized in Figures 2 and 4. Power analysis determines how many data are needed to distinguish between the null model and alternatives. Estimating the power of a statistical test requires simulating many data sets under various alternatives to the null model. In contrast, sensitivity analysis involves simulating data under the null model, where the structure of the data is included in the simulations. In one set of simulations, we add measurement error to each observation and in another we shift each data point by a percentage of the mean, equivalent to censoring the small selection coefficients. In essence, we simulate data under the null hypothesis κ = 0 and then transform this simulated data set to make it appear more like data that might arise in an experiment.
Figure 3.—

The power of the GPD likelihood-ratio test. The null hypothesis is the exponential distribution corresponding to κ = 0 and the type I error was set at α = 0.05. Power was calculated for the test for sample sizes of n = 10, 20, 30, 50, and 100. Critical values of the test statistic were estimated by 10 million simulations. One million replicate tests were performed for each point and power was taken as the percentage of tests that correctly rejected the null hypothesis.
In the language of statistical inference, power analysis is concerned with avoiding type II errors. A type II error occurs when one accepts the null model when an alternative is more appropriate. Power is one minus the probability of a type II error. Sensitivity analysis is concerned with evaluating how much the probability of a type I error is inflated when the structure of the data is ignored. A type I error occurs if we incorrectly reject the null model. If the null model is an appropriate description of the data, but the data include measurement error or exclude small-effect mutations, then failing to account for these effects will inflate the type I error rate.
Power analysis:
The critical value of the test statistic was calculated for sample sizes considered typical of a single experiment or obtainable by pooling experiments, n = 10, 20, and 30. For comparison, larger data sets of size 50 and 100 were also simulated. This calculation was performed using 10 million replicate simulations. For each replicate, a data set of size n was simulated according to the null hypothesis where κ = 0 and τ = 1. The LRT statistics from all such replicates generate an empirical distribution of the test statistic that allows for an approximation of the critical value for a given type I error rate α. Simulations were then performed to estimate the power of the test against a GPD alternative at varying values of the shape parameter, −1 ≤ κ ≤ 1.
Note in Figure 3 that for a sample of size 10, κ = 0 is virtually indistinguishable from κ > 0. For 0 < κ < 0.8, the percentage of simulated data sets that reject the Gumbel domain is ∼0.05, exactly the type I error rate (α = 0.05) of the test when κ = 0. When κ = 1 the GPD reduces to the distribution with similar tail properties as the Cauchy distribution, well known for having a heavy tail. When κ = 1, the tail of the distribution is so heavy that both the mean and the variance of the distribution are infinite. However, a sample of size 10 drawn from the GPD with κ = 1 will produce samples that are distinguishable from the exponential distribution only 20% of the time. Power increases appreciably for a sample of size 20, but a sample of size 50 is required to get reasonable power. However, if we consider κ < 0 then we see that even a sample size as low as 10 has reasonable power. The case of κ = −1 corresponds to the uniform distribution, and 60% of all samples of size 10 drawn from the uniform distribution are distinguishable from the exponential under the GPD likelihood-ratio test.
Sensitivity analysis:
Ignoring the fact that small-effect mutations are missing from the data has a major impact on the data analysis. We outline above a simple adjustment to account for this missing data. Figure 2 shows the implications associated with failing to make this adjustment. The type I error increases dramatically as the shift size increases. The effect on the type I error rate is less pronounced when using the GPD alternative over the gamma.
The test is fairly insensitive to ignoring measurement error under both lognormal and normal error structures (Figure 4). A coefficient of variation as high as 20% has virtually no effect on the probability of a type I error. We did not assess the effect of measurement error on power. The measurement error in the fitness assay can be reduced through replication.
CONCLUSION
The distribution of beneficial fitness effects for new mutations remains the primary unknown in emerging theories of adaptation. Thus far, theoretical work has relied heavily on the assumption of an exponential tail for fitness distributions (i.e., the assumption of a Gumbel-type fitness distribution). This assumption has arisen in studies of clonal interference (e.g., Gerrish and Lenski 1998; Rozen et al. 2002; Kim and Orr 2005) and in the development of the mutational landscape model of adaptation (e.g., Gillespie 1983, 1984, 1991; Orr 2002, 2003b; Rokyta et al. 2006). Justification for this assumption is provided by an appeal to EVT, and we have presented a framework for testing the Gumbel hypothesis firmly grounded in EVT. Furthermore, we have described methodology for overcoming the difficulties inherent in testing this assumption. As beneficial mutations are assumed to be rare, the power of our statistical test may be low due to small sample sizes. Therefore, to improve power, it may become necessary to pool data across experiments involving different starting genotypes or under different selective conditions. However, as indicated in Figure 3, the power to distinguish between the Gumbel domain of attraction and the truncated alternatives within the Weibull domain is quite high, even for small sample sizes. Small-effect mutations may be difficult to detect experimentally, and thus we have described a method for shifting data to remove them from consideration. Small-effect mutations can be problematic in two ways. In experiments such as that described by Rokyta et al. (2005), which rely on mutations fixing in experimental populations, selection will favor large-effect mutations, and the number of replicate experiments necessary to identify small-effect mutations may be prohibitively large. In experiments involving the identification of beneficial mutations either through screening random mutations or mutations found to be beneficial under different conditions from those used to measure fitness (e.g., Sanjuán et al. 2004; Kassen and Bataillon 2006), it may prove difficult to distinguish small-effect mutations from neutral or slightly deleterious mutations due to measurement error. In both cases, shifting the data eliminates the issue at the cost of only a single degree of freedom.
We envision this test as being most useful for experimental microbial evolution studies, because these systems allow the isolation of a number of beneficial mutations from a single ancestral background and, through sequencing, the identification and verification of those mutations. Previous studies identifying beneficial mutations have been quite labor intensive, involving either long fixation times (Rokyta et al. 2005) or screening a large number of mutations (Sanjuán et al. 2004; Kassen and Bataillon 2006). However, the framework we have described will allow testing of beneficial mutations identified through gain-of-function experiments (Ferris et al. 2007). In these types of experiments, microbes are exposed to conditions under which the ancestral genotype cannot grow. For example, Bull et al. (2000) isolated mutants of the phage φX174 capable of growing at 45°, a temperature at which wild type fails to grow. The same type of experiment can be done by isolating, for example, antibiotic resistance mutations in bacteria or host range mutations in a virus. The difficulty with these experiments is that the wild type has a fitness near or at zero, and there may be many mutations that confer a slight advantage but not enough for measurable growth (i.e., colony or plaque formation). Since the ancestral genotype cannot grow, it might appear that this type of data violates an underlying assumption used to justify EVT—that the wild type is already well adapted to the current environment and thus its fitness along with the fitnesses of all one-step beneficial mutations are in the tail of the fitness distribution. However, we are concerned only with whether or not beneficial mutations are in the tail of the fitness distribution, not whether the ancestral genotype is in the tail. If we think of creating an ordered list containing the fitness of each one-step mutant, listed from largest to smallest, the beneficial mutations that formed plaques or colonies would be at the top of the list. If the beneficial mutations represent a small subset among all possible mutations, then they meet the criteria of being in the extreme right tail of the fitness distribution. For example, if an experiment is replicated 20 times and the same five mutations are observed four times each, then this would suggest that there are only a small number of beneficial mutations. By establishing ahead of time that the number of adaptive changes is relatively small, and by shifting fitnesses appropriately, one can confidently use the tests described here for beneficial mutations observed through gain-of-function experiments. It is important to note we are not limited in shifting relative to the genotype with the smallest observed fitness and can in fact shift relative to any observation deemed far enough out in the tail to warrant the use of EVT. Thus, not only does the methodology described provide the appropriate test for the type of tail distribution, but also it allows experimentalists to use simple but powerful gain-of-function techniques for isolating beneficial mutations, greatly facilitating the characterization of the distribution of beneficial fitness effects.
Acknowledgments
The authors thank Jim Bull for numerous discussions and encouragement on this project. This work was supported by a grant from the National Institutes of Health (NIH), R01GM076040 to P. Joyce and H. A. Wichman. C. J. Beisel was supported in part by NIH P20 RR16448 and a grant from the National Science Foundation, DEB-0515738 to P. Joyce; D. R. Rokyta was supported in part by NIH P20 RR16454. Analytical resources were provided by NIH P20 RR16448 and NIH P20 RR16454.
References
- Bull, J. J., M. R. Badgett and H. A. Wichman, 2000. Big-benefit mutations in a bacteriophage inhibited with heat. Mol. Biol. Evol. 17: 942–950. [DOI] [PubMed] [Google Scholar]
- Castillo, E., 1988. Extreme Value Theory in Engineering. Academic Press, New York/London/San Diego.
- Castillo, E., and A. S. Hadi, 1997. Fitting the generalized Pareto distribution to data. J. Am. Stat. Assoc. 92: 1609–1620. [Google Scholar]
- Cowperthwaite, M. C., J. J. Bull and L. Ancel Myers, 2005. Distributions of beneficial fitness effects. Genetics 170: 1449–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davison, A. C., and R. L. Smith, 1990. Models for exceedances over high thresholds. J. R. Stat. Soc. Ser. B 52: 393–442. [Google Scholar]
- Ferris, M. T., P. Joyce and C. L. Burch, 2007. High frequency of mutations that expand the host range of an RNA virus. Genetics 176: 1013–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerrish, P. J., and R. E. Lenski, 1998. The fate of competing beneficial mutations in an asexual population. Genetica 102/103: 127–144. [PubMed] [Google Scholar]
- Gillespie, J. H., 1983. A simple stochastic gene substitution model. Theor. Popul. Biol. 23: 202–215. [DOI] [PubMed] [Google Scholar]
- Gillespie, J. H., 1984. Molecular evolution over the mutational landscape. Evolution 38: 1116–1129. [DOI] [PubMed] [Google Scholar]
- Gillespie, J. H., 1991. The Causes of Molecular Evolution. Oxford University Press, New York.
- Griffiths, R. C., and S. Tavaré, 1994. Simulating probability distributions in the coalescent. Theor. Popul. Biol. 46: 307–319. [Google Scholar]
- Grimshaw, S. D., 1993. Computing maximum likelihood estimates for the generalized pareto distribution. Technometrics 35: 185–191. [Google Scholar]
- Heller, J., 1961. Catch-22. Simon & Schuster, New York.
- Kassen, R., and T. Bataillon, 2006. Distribution of fitness effects among beneficial mutations before selection in experimental populations of bacteria. Nat. Genet. 38: 484–488. [DOI] [PubMed] [Google Scholar]
- Kim, Y., and H. A. Orr, 2005. Adaptation in sexuals vs. asexuals: clonal interference and the Fisher–Muller model. Genetics 171: 1377–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard Smith, J., 1962. The limitations of molecular evolution, pp. 252–256 in The Scientist Speculates: An Anthology of Partly-Baked Ideas, edited by I. J. Good. Basic Books, New York.
- Maynard Smith, J., 1970. Natural selection and the concept of a protein space. Nature 225: 563–564. [DOI] [PubMed] [Google Scholar]
- Orr, H. A., 2002. The population genetics of adaptation: the adaptation of DNA sequences. Evolution 56: 1317–1330. [DOI] [PubMed] [Google Scholar]
- Orr, H. A., 2003. a The distribution of fitness effects among beneficial mutations. Genetics 163: 1519–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr, H. A., 2003. b A minimum on the mean number of steps taken in adaptive walks. J. Theor. Biol. 220: 241–247. [DOI] [PubMed] [Google Scholar]
- Orr, H. A., 2005. a The genetic theory of adaptation: a brief history. Nat. Rev. Genet. 6: 119–127. [DOI] [PubMed] [Google Scholar]
- Orr, H. A., 2005. b The probability of parallel evolution. Evolution 59: 216–220. [PubMed] [Google Scholar]
- Orr, H. A., 2006. a The distribution of beneficial fitness effects among beneficial mutations in Fisher's geometric model of adaptation. J. Theor. Biol. 238: 279–285. [DOI] [PubMed] [Google Scholar]
- Orr, H. A., 2006. b The population genetics of adaptation on correlated fitness landscapes: the block model. Evolution 60: 1113–1124. [PubMed] [Google Scholar]
- Pickands, III, J., 1975. Statistical inference using extreme order statistics. Ann. Stat. 3: 119–131. [Google Scholar]
- R Development Core Team, 2006. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna.
- Rice, J. A., 1995. Mathematical Statistics and Data Analysis. Duxbury Press, Belmont, CA.
- Rokyta, D. R., P. Joyce, S. B. Caudle and H. A. Wichman, 2005. An empirical test of the mutational landscape model of adaptation using a single-stranded DNA virus. Nat. Genet. 37: 441–444. [DOI] [PubMed] [Google Scholar]
- Rokyta, D. R., C. J. Beisel and P. Joyce, 2006. Properties of adaptive walks on uncorrelated landscapes under strong selection and weak mutation. J. Theor. Biol. 243: 114–120. [DOI] [PubMed] [Google Scholar]
- Rozen, D. E., J. A. G. M. de Visser and P. J. Gerrish, 2002. Fitness effects of fixed beneficial mutations in microbial populations. Curr. Biol. 12: 1040–1045. [DOI] [PubMed] [Google Scholar]
- Sanjuán, R., A. Moya and S. E. Elena, 2004. The distribution of fitness effects caused by single-nucleotide substitutions in an rna virus. Proc. Natl. Acad. Sci. USA 101: 8395–8401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, R. L., 1985. Maximum likelihood estimation in a class of nonregular cases. Biometrika 72: 67–90. [Google Scholar]