

Abstract
Selective DNA pooling (SDP) is a cost-effective means for an initial scan for linkage between marker and quantitative trait loci (QTL) in suitable populations. The method is based on scoring marker allele frequencies in DNA pools from the tails of the population trait distribution. Various analytical approaches have been proposed for QTL detection using data on multiple families with SDP analysis. This article presents a new experimental procedure, fractioned-pool design (FPD), aimed to increase the reliability of SDP mapping results, by “fractioning” the tails of the population distribution into independent subpools. FPD is a conceptual and structural modification of SDP that allows for the first time the use of permutation tests for QTL detection rather than relying on presumed asymptotic distributions of the test statistics. For situations of family and cross mapping design we propose a spectrum of new tools for QTL mapping in FPD that were previously possible only with individual genotyping. These include: joint analysis of multiple families and multiple markers across a chromosome, even when the marker loci are only partly shared among families; detection of families segregating (heterozygous) for the QTL; estimation of confidence intervals for the QTL position; and analysis of multiple-linked QTL. These new advantages are of special importance for pooling analysis with SNP chips. Combining SNP microarray analysis with DNA pooling can dramatically reduce the cost of screening large numbers of SNPs on large samples, making chip technology readily applicable for genomewide association mapping in humans and farm animals. This extension, however, will require additional, nontrivial, development of FPD analytical tools.
ACHIEVING reasonable statistical power of designs for detecting marker–quantitative trait loci (QTL) linkage for QTL of small effect is difficult and requires large mapping populations, with consequent high cost of marker genotyping. Similar situations also arise in association studies based on linkage disequilibrium (LD). A cost-effective solution to reduce costs associated with genotyping large mapping populations is to replace individual genotyping by DNA analysis in pools of individuals coming from the high and the low tails of the mapping population distribution. This concept, referred to as “tail analysis” (Hillel et al. 1990; Dunnington et al. 1992; Plotsky et al. 1993), “bulked segregant analysis” (Giovannoni et al. 1991; Michelmore et al. 1991), or “selective DNA pooling (SDP)” (Darvasi and Soller 1994), was proposed for QTL analysis and for testing of linkage between markers and a major gene. Darvasi and Soller (1994) provided a detailed quantitative analysis of this procedure, based on comparing marker allele frequency (which can be obtained by densitometry) in the pooled DNA samples; a number of authors have proposed useful corrections to obtain reliable estimates of SNP allele frequencies in pools (Visscher and Le Hellard 2003; Zou and Zhao 2004, 2005; Craig et al. 2005). The SDP procedure can readily be extended to situations, such as half-sib or full-sib designs, where the mapping population consists of several families. It was applied for genome scanning for QTL affecting milk production traits using microsatellite markers (Lipkin et al. 1998; Mosig et al. 2001).
Various approaches have been proposed for obtaining QTL position and its confidence interval with SDP (Dekkers 2000; Carleos et al. 2003; Brohede et al. 2005; Johnson 2005). Among the problems with such analyses are varying proportion of family founders heterozygous at both the QTL and the markers; heterogeneity of the families with respect to QTL effects; different information content of different marker loci; allele sharing between the founder sires and dams of the families; varying proportion of shared marker loci among families, laboratories, and populations; effects of population admixture; variation of PCR efficiency for marker alleles; and the use of asymptotic, difficult-to-justify approximations of test-statistic distributions. Wang et al. (2007) provide least-squares and maximum-likelihood generalizations of Dekkers (2000) and address a number of the shortcomings of existing methodology. Recently, DNA pooling analyses using SNP markers have also been employed in some human mapping studies based on populationwide association tests or involving comparison of pools of healthy and affected individuals (Sham et al. 2002; Butcher et al. 2004; Schnack et al. 2004; Brohede et al. 2005; Tamiya et al. 2005). These SNP-based association tests are also subject to many of the statistical limitations listed above. When analyses are based on individual selective genotyping, analytical solutions are available for most of these problems (Lander and Botstein 1989; Darvasi and Soller 1992; Ronin et al. 1998). This is not the case when the analyses are based on SDP. Thus, despite many publications supporting pooling analysis, concerns remain about the reliability of the marker–QTL associations obtained in this way.
A “fractioned-pool” approach, in which the tails of the population distribution are randomly allocated among a number of independent subpools, has been considered by a few authors, with the objective of obtaining an empirical standard error for estimates of marker allele frequencies in pools (e.g., Sham et al. 2002), or for optimization of pool number/pool size, from the viewpoint of amplification fidelity (Brohede et al. 2005). In the present article, the fractioned-pool concept is extended to provide a complete analytical system for QTL linkage mapping analysis by selective DNA pooling, termed fractioned-pool design (FPD) (Figure 1). The FPD removes many of the above statistical limitations. The FPD analysis is not limited by an assumption of normal distribution of the trait. However, the tails of trait distribution (corresponding to high and low trait values) must contain a sufficient number of individuals to achieve a reasonably high detection power.
Figure 1.—

Constructing multiple subpools. Trait distribution in each family is divided into three parts: individuals with high or low trait values that make up the high and low tails and individuals with intermediate trait values. At each tail, individuals are grouped randomly into subpools. NT characterizes the number of individuals with corresponding trait values in a family. L1, L2, L3, and L4 are low-tail subpools; H1, H2, H3, and H4 are high-tail subpools.
For the first time in selective DNA pooling, the FPD allows QTL detection based on permutation tests rather than on assumed asymptotic distributions of test statistics and estimation of confidence intervals for QTL position and effect based on jackknife or bootstrap resampling techniques. It also allows estimating the test statistic more accurately than in the case of a single pool per tail. The proposed method is illustrated using Monte Carlo simulations. Successful validation of the FPD for genomewide studies of quantitative variation opens a new perspective for highly reliable and cost-efficient large-scale QTL analysis, unattainable by standard SDP analytical procedures.
STANDARD SELECTIVE DNA POOLING APPROACH TO QTL MAPPING
The experimental material for QTL mapping based on SDP consists of individuals selected from the tails of the mapping population trait distributions. The procedures considered here are suitable for mapping populations composed of full- or half-sib families or multiple families within F2 or BC populations. The simulated examples employed to illustrate the proposed methodology correspond to multiple half-sib daughter families (e.g., a population based on artificial insemination as found in dairy cattle). Each family consists of the progeny of a different sire and is represented by some given number of daughters per tail selected out of all phenotyped daughters of that family.
Assume that a sire is heterozygous at a QTL affecting trait value, and designate as a positive sire QTL allele the sire QTL allele increasing trait value and as a negative sire QTL allele the sire QTL allele decreasing trait value. Then the frequency of the positive sire QTL allele will be higher in the group of daughters having high trait value and lower in the group of daughters having low trait value; the opposite will be true for the negative sire QTL allele. Through hitchhiking effects, this difference in the frequency of the positive and negative sire QTL alleles in groups with high and low trait values produces a parallel difference in the frequency of sire marker alleles at marker loci heterozygous in the sires that are in coupling linkage to these heterozygous QTL. Analyzing sire marker-allele frequency differences at several marker loci enables the position of the QTL on the chromosome to be estimated.
It is convenient to denote the two pools as high (H) and low (L), respectively, and the two sire alleles at the linked marker locus m (m = 1, …, M) as alleles Am and Bm, respectively. Using this notation, we define the statistic Dm as a characteristic of sire allele divergence in the two tails,
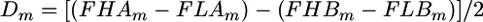 |
(1) |
(Lipkin et al. 1998), where FHAm is the frequency of allele Am in the high pool, and FHBm, FLAm, and FLBm are defined accordingly. When there are only two alleles at the marker locus as in the case of SNP markers, FAm and FBm are in perfect negative correlation, and hence only one of the alleles need be included in estimating Dm. However, when there are multiple alleles at the marker locus as in the case of microsatellite markers, FHAm and FHBm are not perfectly correlated, and hence both contain independent information on Dm. In this case, the accuracy of estimation of D is improved by averaging estimates from both alleles as shown in (1). The estimate from allele Bm is given a minus sign in (1) because changes due to a linked QTL in allele Bm are in opposite direction to those in allele Am, as noted above (see Lipkin et al. 1998 for details).
To illustrate how the QTL substitution effect influences the expected value of D-statistics, consider a single-QTL case for the half-sib design. Let QTL q be diallelic with sire QTL genotype A(q)B(q) and equal frequencies of alleles A(q) and B(q) in the dam population. In this situation, the proportions of QTL genotypes in the progeny are 25% A(q)A(q), 50% A(q)B(q), and 25% B(q)B(q). Let the targeted quantitative trait be normally distributed with residual variance σ2 and mean value dependent on QTL genotype: μ − d for B(q)B(q), μ for A(q)B(q), and μ + d for A(q)A(q). For 10% cutoff tails of trait distribution and allele substitution effect of QTL d/σ = 0.3, 0.2, and 0.15, the expected value of D(q) (defined analogously to Dm) will be 0.26, 0.17, and 0.14, respectively. Assume further that marker locus m is triallelic with alleles Am, Bm, and Cm; the sire's haplotypes are AmA(q) and BmB(q); allele frequencies in the dam population are 0.25 for Am, 0.25 for Bm, and 0.5 for Cm; and marker and QTL alleles in the dam population are in linkage equilibrium. Then, if marker m is coincident with QTL q [i.e., marker allele Am is inherited from the sire only with A(q) and Bm only with B(q)], the expectation of Dm should be half of D(q) (i.e., 0.13, 0.085, and 0.07 for d/σ = 0.3, 0.2, and 0.15, respectively).
For detecting the chromosomes with QTL effects, one can consider for every marker m the statistic 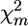 taken over all F families heterozygous for the marker m,
taken over all F families heterozygous for the marker m,
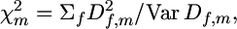 |
(2) |
where Var Df,m is the sampling variance of Df,m for the f family at the m marker. When the selected trait is not affected by the tested chromosome (H0 hypothesis), 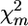 is presumed to follow a χ2-distribution with d.f. = F (number of families), enabling a χ2-test for the presence of a QTL linked to the marker (Weller et al. 1990).
is presumed to follow a χ2-distribution with d.f. = F (number of families), enabling a χ2-test for the presence of a QTL linked to the marker (Weller et al. 1990).
THE ANALYTICAL SYSTEM OF FPD
By joint analysis of these sire marker-allele frequency differences, Dm, at several marker loci, one can estimate the chromosomal position of the detected QTL. For one or several families heterozygous for the same QTL, fitting a function of chromosomal positions for observed Dm values at the polymorphic marker loci can be used for estimation of the QTL position (similar to the procedures described by Kearsey 1998 and Ronin et al. 1999).
Single-QTL model:
For a single-QTL situation, the expectation of statistic Dm is proportional to (1 − 2rm), where rm is the recombination rate between the marker m and the QTL q. In (1) the sign of statistic Dm depends on which of the two sire marker alleles was designated Am and which was designated Bm. In what follows we assume that marker haplotypes of sire are known and marker alleles from one haplotype are designated by Am and from another by Bm, m = 1, …, M, where M is the number of marker loci included in the haplotype (note that FPD methods also apply in the case of unknown phases; see Unknown marker linkage phase in the sire below). Value rm depends on location of marker m and unknown location (x(q)) of the putative QTL on the chromosome. Hence the expectation of Dm can be represented as
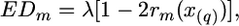 |
(3) |
where λ is the (expected) value (henceforth “λ-value”) of D for a marker that coincides with the QTL, and rm(x(q)) is the recombination rate between the marker and the QTL and will be zero for a marker located at x(q). Assuming absence of interference, rm can be calculated using the Haldane model, rm(y) = 0.5(1 − exp{−0.02y}), where y is the map distance in centimorgans between xm and the unknown coordinate x(q) of the QTL (Figure 2).
Figure 2.—

One QTL on the chromosome. Expectation of the statistic D for markers situated at various locations on the chromosome. Value ED is calculated by formula (3) (using the Haldane model of recombination). Height of the graph at the QTL position x0 = x(q) is a characteristic of the QTL effect on markers in this family (family λ-value).
The information on all markers scored for the same chromosome can be combined to derive the unknown coefficients λ and x(q). These parameters can be estimated (analogously to Wang et al. 2007) using a standard least-squares approach (by minimizing the following criterion):
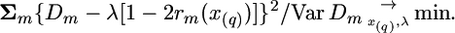 |
(4) |
The sampling variance of Dm (Var Dm) can be calculated by ways reviewed in Sham et al. (2002) and Brohede et al. (2005). Employment of expression (3) by using criterion (4) can be represented in terms of a standard linear model,
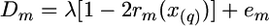 |
(Wang et al. 2007), or in matrix notations, D = Xλ + e. Here values em are residuals, including both sampling and technical errors, with variance equal to Var Dm; D, X, and e are vectors of Dm, [1 − 2rm(x(q))] and em correspondingly, m = 1, …, M, and M is the number of markers. The test statistic, calculated at given putative QTL position, can then be written as χ2 = Σm{Dm − λ [1 − 2rm(x(q))]}2/Var Dm. However, because the correlations between values of Dm for linked markers are not taken into account in (4), the statistical quality (sampling variance) of the estimates obtained by this criterion is not optimal. We therefore use a more general optimization criterion that does take correlations into account.
Let em in the linear model be correlated with correlations defined by matrix G. Then, using a generalized least-squares approach, parameters can be estimated by minimizing the following criterion (for simplicity of designation, we write it in matrix form):
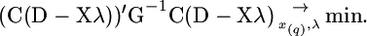 |
(5) |
Here C is the diagonal matrix of (Var Dm)−0.5. For a given x(q) putative position of the QTL q parameter λ minimizing criterion (5) is equal to (X′C′G−1CX)−1X′C′G−1CD. Coefficients of matrix G can be calculated using correlation coefficients defined under the hypothesis of no QTL in the chromosome.
For example, if sire alleles at markers m1 and m2 are not presented in the dam population and there are no technical errors, then the correlation coefficient looks like ρ = Corr( ) = 1 − 2r, where r is the recombination rate between markers m1 and m2. The estimated λ-value can serve as a test statistic combining the information from multiple markers along the chromosome. In our simulations correlations were obtained analytically using only recombination distance between markers and frequencies of the two sire alleles in dam population: ρ = Corr(
) = 1 − 2r, where r is the recombination rate between markers m1 and m2. The estimated λ-value can serve as a test statistic combining the information from multiple markers along the chromosome. In our simulations correlations were obtained analytically using only recombination distance between markers and frequencies of the two sire alleles in dam population: ρ = Corr(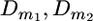 ) = (1 − 2r)Var D0/Var D, where Var D0 and Var D are analytical estimations of variances of the D-value in the cases of zero and nonzero frequencies of sire alleles in the dam population. Alternatively, correlations among Dm values can be estimated using the maximum-likelihood method (Wang et al. 2007).
) = (1 − 2r)Var D0/Var D, where Var D0 and Var D are analytical estimations of variances of the D-value in the cases of zero and nonzero frequencies of sire alleles in the dam population. Alternatively, correlations among Dm values can be estimated using the maximum-likelihood method (Wang et al. 2007).
In the same manner it is possible to combine the information from several families with respect to a given chromosome, assuming that all sires that are heterozygous at a QTL on that chromosome are heterozygous at one and the same QTL with respect to location (x(q)), although the size of the QTL effect may vary among sires. Thus, for the one-QTL assumption and multiple families and letting λf represent the λ-value for the f-sire Equation 3 will be modified as
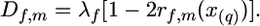 |
(3a) |
Correspondingly, the estimation criterion will be
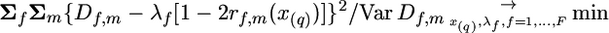 |
(4a) |
or, taking into account the correlation between values of D for linked markers,
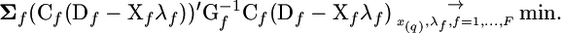 |
(5a) |
Using this expression, the unknown parameters can be obtained in the following way. At each of the chromosomal positions x = x(i) taken consecutively with some step (e.g., 1 cM), values λf, f = 1, …, F, can be found analytically. For every family, the r value in (3a) is calculated using recombination distance between location of marker m and current location x(i). Then, the position minimizing the criterion can be taken as the best position x(q).
After fitting the model (3a), by using criteria (4a) or (5a), the statistic Σ(λf)2 can serve to conduct an overall permutation test (see below), instead of using the asymptotic χ2-properties of statistic (2). If we assume one QTL in the chromosome common to all QTL-heterozygous sires, then λf will represent the expected value of the test statistic at the marker locus coinciding with (or closest to) the QTL. All other segregating markers for this sire f will display a decreasing function of the distance between the marker and the QTL. Hence, an immanent property of our approach (similar to the model of Kearsey 1998 or Ronin et al. 1999) is that for single QTL, λf represents the approximation of D at the presumed position x0 coinciding with the QTL. Thus, λf “absorbs” the information of all markers of the sire, and statistic Σ(λf)2 does this cumulatively across sires, by fitting one and only one QTL position, due to the assumption of one shared QTL.
QTL detection based on FPD permutation tests:
Employment of the FPD allows new types of tests for QTL detection, based on permutation of subpools, as an analog of permutations of individual trait or genotype scores in selective genotyping analysis. These tests do not depend on assumptions as to asymptotic distribution of the test statistics and provide a spectrum of useful analytical options. In particular, these tests can be employed for detecting chromosomes with QTL effects, discriminating between sires homozygous and heterozygous for the detected QTL, and comparing and contrasting hypotheses about one-, two-, or more QTL per chromosome. The simplest of the proposed permutation tests is based on random reshuffling of the individual subpools between tails of the trait distribution. This process is repeated many times, and each time the test statistics are recalculated. In general terms, the proportion of permuted test statistics that are greater than the observed test statistic is the type I error of the test (Doerge and Churchill 1996). If H0 {no QTL effect} is correct for a particular marker, such a permutation will not have an appreciable effect on the level of the test statistics. Thus, in most cases the observed test statistic will lie well within the range of permuted statistics. If the H1 alternative is correct, reshuffling will destroy the marker–trait (i.e., marker–tail) connection. This will be manifested as a strong reduction of the test statistics in the majority of permutation runs. Thus, the observed test statistic in this case will exceed all but a small fraction of the permuted statistics. The test can be applied to any of the possible test statistics: 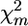 from Equation 2, estimated λ from Equations 4 or 5, or Σ(λf)2 from (4a) or (5a).
from Equation 2, estimated λ from Equations 4 or 5, or Σ(λf)2 from (4a) or (5a).
The total number of different reshuffling configurations per family, Rf, is a function of the number of subpools per tail. In the case of the same number of subpools for the high and low tails, S,
 |
In the case of an unequal number of pools per tail,
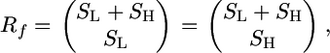 |
where SL is the number of low-trait subpools and SH is the number of high-trait subpools. Thus, for S ina the range 4–8 pools per tail, Rf varies from 35 to 6435. Clearly, the total number of configurations with multiple families is a product of corresponding numbers for families R = ∏fRf. Even for a minimal S = 4, a design with five families will give R = 355 ≈ 5.2 × 107 combinations. The number of combinations is important, because the lowest possible P-value in permutation is equal to 1/R.
Detecting chromosomes with QTL effects:
Tests based on χm2:
The significance of QTL effect for marker m in several families can be estimated as the proportion of random permutation runs of pool configurations, having test statistic value 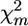 (Equation 2) ≥
(Equation 2) ≥ 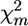 obtained on initial nonreshuffled data. To set significance levels when a number of markers are considered on the same chromosome, it is necessary to correct for multiple comparisons, e.g., by controlling the false discovery rate (FDR) (Benjamini and Hochberg 1995) or the proportion of false positives (PFP) (Fernando et al. 2004).
obtained on initial nonreshuffled data. To set significance levels when a number of markers are considered on the same chromosome, it is necessary to correct for multiple comparisons, e.g., by controlling the false discovery rate (FDR) (Benjamini and Hochberg 1995) or the proportion of false positives (PFP) (Fernando et al. 2004).
Alternatively, a chromosomewise test can be proposed analogous to the approaches applied in standard interval mapping under individual genotyping. In that case, for each set of k marker intervals, interval analysis is conducted and the maximum (across intervals) LOD value (max LODk) or the maximum F-test (max Fk) for regression-based models is calculated. Then, the significance of the putative QTL effect of the tested chromosome is estimated as the proportion of permutation runs (i.e., samples corresponding to H0 obtained by random reshuffling of the trait scores relative to the multilocus marker genotypes), where max LODk/Fk was equal to or higher than the max LOD/Fk value calculated for the nonreshuffled data (Doerge and Churchill 1996). Applying this approach to the FPD analysis, instead of max LOD we can employ max χ2 = maxm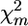 calculated for the nonreshuffled and reshuffled configurations of subpools, where maxm
calculated for the nonreshuffled and reshuffled configurations of subpools, where maxm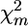 is the value for the marker for which χ2 is at a maximum. Note that in the case of max χ2-statistics, the fitted model does not include any parameters characterizing QTL effect and position, since it is based on single-marker analysis. In contrast, the max LOD/Fk test is preceded by building a genetic model that depends on unknown parameters and obtaining maximum-likelihood (least squares, in the case of the regression model) estimates of the parameters.
is the value for the marker for which χ2 is at a maximum. Note that in the case of max χ2-statistics, the fitted model does not include any parameters characterizing QTL effect and position, since it is based on single-marker analysis. In contrast, the max LOD/Fk test is preceded by building a genetic model that depends on unknown parameters and obtaining maximum-likelihood (least squares, in the case of the regression model) estimates of the parameters.
Significance of the putative QTL effect of the tested chromosome can also be estimated by the P-value of the highest significant marker on the chromosome (taking into account the problem of multiple comparisons). Individual P-values for marker m can be calculated by a permutation test (using test statistic 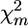 ) or a χ2-test (Weller et al. 1990). Using the FDR approach (Benjamini and Hochberg 1995) to control for multiple comparisons, we denote corresponding significance thresholds by
) or a χ2-test (Weller et al. 1990). Using the FDR approach (Benjamini and Hochberg 1995) to control for multiple comparisons, we denote corresponding significance thresholds by 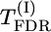 for the permutation test and
for the permutation test and 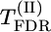 for the χ2-test, respectively.
for the χ2-test, respectively.
Permutation test based on λf:
The permutation test based on 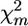 takes into account all markers on a chromosome, but information contained in the relative locations of the markers is ignored. In standard individual genotyping schemes, single-marker analysis and interval analysis are close with respect to QTL detection power at moderate to high marker density. However, at low marker density, interval analysis is more powerful. This is due to the fact that loss of power caused by QTL–marker recombination can be estimated as ∼r/2 and ∼r2/4, for single-marker analysis and interval analysis, respectively.
takes into account all markers on a chromosome, but information contained in the relative locations of the markers is ignored. In standard individual genotyping schemes, single-marker analysis and interval analysis are close with respect to QTL detection power at moderate to high marker density. However, at low marker density, interval analysis is more powerful. This is due to the fact that loss of power caused by QTL–marker recombination can be estimated as ∼r/2 and ∼r2/4, for single-marker analysis and interval analysis, respectively.
It was found that in FPD, as in standard QTL mapping analysis based on individual genotyping, hypothesis testing is more efficient and flexible, if conducted on the basis of fitting a mapping model aimed at QTL detection or at discriminating between more complex situations (such as single or multiple QTL on a chromosome, mode of QTL action and interaction, and linkage vs. pleiotropy as sources of genetic correlation). In this context, by including marker positions, models (3a), (4a), and (5a) presented above allow extracting the information about QTL presence and location on the tested chromosome through joint analysis of linked markers. As shown by simulation (Table 1), power of the max χ2-test is less than that of the Σ(λf)2 test. Presumably, this is due to the fact that the max χ2-test does not utilize all of the information potentially contributing to QTL detection power. Thus, for a single-family analysis, the estimated λ-value (from Equation 4 or 5) would be the preferred statistic for the permutation test.
TABLE 1.
Effect of number of markers (M) under the FPD on the confidence interval (C.I.) of QTL location, comparisonwise error rate (P-value), and statistical power, according to the test for significance and standardized allele substitution effect at the QTL (d/σ), using simulated data
| C.I.
|
P-value
|
Power
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| d/σ | M | Δ | SD | Σ(λf)2 | max|λ| | max-χ2 | 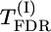 |
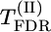 |
Σ(λf)2 (%) | max-χ2 (%) |
| 0.2 | 25 | 4.1 | 3.1 | 0.003 | 0.053 | 0.007 | 0.015 | 0.074 | 99 | 56 |
| 13 | 5.1 | 3.3 | 0.002 | 0.030 | 0.008 | 0.018 | 0.076 | 99 | 59 | |
| 7 | 6.4 | 3.6 | 0.004 | 0.049 | 0.006 | 0.016 | 0.061 | 98 | 64 | |
| 0.15 | 25 | 4.9 | 5.2 | 0.008 | 0.104 | 0.056 | 0.067 | 0.250 | 92 | 27 |
| 13 | 6.3 | 5.2 | 0.021 | 0.071 | 0.126 | 0.112 | 0.260 | 90 | 24 | |
| 7 | 7.8 | 5.9 | 0.021 | 0.098 | 0.130 | 0.101 | 0.299 | 89 | 28 | |
Tests of significance: Σ(λf)2, max|λ|, max-χ2, 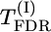 , and
, and 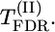 See text for details. Power was calculated at P-value = 5%. Values Δ and SD characterize the center and size of the confidence interval obtained in jackknife iterations (see text). Parameters of the simulations: chromosome length 120 cM. A single QTL was situated in position 40 cM. Number of families, F = 10 (5 families, sire heterozygous at the QTL; 5 families, sire homozygous at the QTL); number of daughters per family, N = 2000; proportion of the population selected to each tail, 0.10; number of subpools per tail, S = 4. Values are the mean based on 10 simulation data sets; for every data set, 500 permutations and 100 jackknife iterations were made.
See text for details. Power was calculated at P-value = 5%. Values Δ and SD characterize the center and size of the confidence interval obtained in jackknife iterations (see text). Parameters of the simulations: chromosome length 120 cM. A single QTL was situated in position 40 cM. Number of families, F = 10 (5 families, sire heterozygous at the QTL; 5 families, sire homozygous at the QTL); number of daughters per family, N = 2000; proportion of the population selected to each tail, 0.10; number of subpools per tail, S = 4. Values are the mean based on 10 simulation data sets; for every data set, 500 permutations and 100 jackknife iterations were made.
For multiple-family analysis statistics, Σ(λf)2 and maximum λf across all families (maxf |λf|), with family-specific least-squares estimates of λ-values being derived from (3a) and (4a) (or 5a), can serve to conduct the overall experimentwise permutation test across families and markers of the analyzed chromosome. In the FPD methodology, each marker is represented in (4a) [or in (5a)] by its position relative to the unknown location of the putative QTL, rather than by its name. Consequently, there is no need for full coincidence of polymorphic marker loci among the families. In principle, the system will work even with zero overlapping of polymorphic marker loci among families. This is an important advantage of the proposed methodology over the standard SDP methodology (Darvasi and Soller 1997), in which the test statistics is calculated for each marker locus across families polymorphic for the marker, and it is not possible to compensate for markers at which the sire is homozygous by including information from neighboring heterozygous markers.
Detecting sires heterozygous at the QTL:
For analysis of a single family, f, within a multiple-family analysis, the estimated value of |λf| or 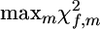 can be used as a test statistic for the permutation test. The significance of a sire f is then determined as the proportion of permutations of the runs made over all families, where the statistic of QTL effect |λf| was greater than that for nonreshuffled data. Sires of families where the test statistics (|λf| or
can be used as a test statistic for the permutation test. The significance of a sire f is then determined as the proportion of permutations of the runs made over all families, where the statistic of QTL effect |λf| was greater than that for nonreshuffled data. Sires of families where the test statistics (|λf| or 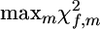 ) are not significant can be taken to be homozygous at the QTL. On this basis, sires can be subdivided into two groups, QTL homozygous and QTL heterozygous.
) are not significant can be taken to be homozygous at the QTL. On this basis, sires can be subdivided into two groups, QTL homozygous and QTL heterozygous.
Estimating the confidence interval of QTL position: bootstrap/jackknife analysis:
One of the major parameters characterizing the detected QTL is the accuracy of the estimated parameters, especially of QTL position, as given by its standard error or confidence interval. The most common way to evaluate confidence interval of QTL position within the framework of individual or selective genotyping is by using resampling procedures such as bootstrap or jackknife (Ronin et al. 1998). The 95% confidence interval of QTL location can then be taken as the narrowest interval that includes 95% of the resampling-based estimates of QTL position. Alternatively, the confidence interval of QTL location can be characterized by mean value  , standard error (SE), and standard deviation (SD) of the resampling-based estimates. The proposed FPD methodology, for the first time, allows resampling procedures to be applied for DNA pooling analysis. As in the individual genotyping application of these procedures, multiple samples are generated from the initial data set by sampling subpools within tails with return (bootstrap analysis) or without return (jackknife analysis). Each such sample is treated using the same model that was applied to the total sample, and the variation of the derived parameters among the samples is employed to get a SD for each estimated parameter and (if needed) a SE for its mean value. The only difference in application of these procedures in FPD is that pools are resampled instead of individuals.
, standard error (SE), and standard deviation (SD) of the resampling-based estimates. The proposed FPD methodology, for the first time, allows resampling procedures to be applied for DNA pooling analysis. As in the individual genotyping application of these procedures, multiple samples are generated from the initial data set by sampling subpools within tails with return (bootstrap analysis) or without return (jackknife analysis). Each such sample is treated using the same model that was applied to the total sample, and the variation of the derived parameters among the samples is employed to get a SD for each estimated parameter and (if needed) a SE for its mean value. The only difference in application of these procedures in FPD is that pools are resampled instead of individuals.
With new chip-based technologies of SNP analysis, a high number of densely spaced polymorphic markers may become available for FPD or interval-mapping analysis. In this case, the resampling procedure may be modified to include simultaneous resampling of markers within chromosomes and subpools within tails so that different jackknife or bootstrap runs may include not fully coinciding sets of markers for a given family.
Simulation data:
To illustrate the proposed methodology we simulated situations corresponding to multiple half-sib daughter families (a population based on artificial insemination, e.g., dairy cattle). Each family consists of the progeny of a different sire, with each sire family being represented by a certain number (10% of the total) of daughters per tail selected out of all phenotyped daughters of that family. In our simulations we used a normally distributed trait with constant variance σ2 and mean value depending on QTL genotype. Each of QTL q was assumed additive and diallelic with alleles A(q) and B(q). Frequencies of alleles A(q) and B(q) in dams were set to 0.50. Frequencies of marker alleles in the dams were 0.25 Am, 0.25 Bm, and 0.25 Cm, where Am and Bm are sire alleles and Cm represents all other alleles. Am and A(q) are alleles of one of the haplotypes of the sire for all m = 1, …, M, q = 1, …, Q; Bm and B(q) are alleles of the other haplotype of the sire; all loci are from one chromosome. Positions of loci (markers and simulated QTL) on the chromosome are defined by recombination distance from the most proximal locus. In the same way we define position(s) for putative QTL. Recombination events in the sire gamete were simulated as independent for different parts of the chromosome (recombination rate between loci was calculated using distance on the linkage map and the Haldane model). Linkage equilibrium among all alleles (markers and QTL) was assumed in the dams.
Each progeny genotype was simulated by independently generating a haplotype inherited from the sire and a haplotype inherited from a dam. The haplotype inherited from the dam was simulated by randomly choosing alleles for each locus proportionally to their frequencies in the dams. The haplotype inherited from the sire was simulated as follows: The allele in the most proximal locus was chosen randomly from one of the two sire alleles (with probability 0.5). This allele determined the starting sire haplotype. The allele in every subsequent locus on the chromosome was chosen with probability 1 − r from the same haplotype as in the previous locus and with probability r from the alternative haplotype, where r is the recombination rate between these two consecutive loci. The trait value for each simulated individual in the progeny was set equal to the mean trait value for the inherited QTL genotype plus a normally distributed random value with mean zero and variance σ2. In the single-QTL case, mean trait value was defined as μ − d(q), μ, and μ + d(q) for genotypes B(q)B(q), A(q)B(q), and A(q)A(q), correspondingly. Value d(q) was not necessarily the same for all families. In the case of two QTL (q = 1, 2), trait mean value was μ − d(1) − d(2), μ − d(2), μ + d(1) − d(2), μ − d(1), μ, μ + d(1), μ − d(1) + d(2), μ + d(2), and μ + d(1) + d(2) for genotypes B(1)B(1)B(2)B(2), A(1)B(1)B(2)B(2), A(1)A(1)B(2)B(2), B(1)B(1)A(2)B(2), A(1)B(1)A(2)B(2), A(1)A(1)A(2)B(2), B(1)B(1)A(2)A(2), A(1)B(1)A(2)A(2), and A(1)A(1)A(2)A(2), respectively. In the simulations, QTL-genotype frequencies in the tails of trait distribution for a given tail cutoff depend on the proportion d/σ = 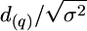 , rather than on the μ-value and σ2. In our simulations we used μ = 0 and σ2 = 1.
, rather than on the μ-value and σ2. In our simulations we used μ = 0 and σ2 = 1.
Subdivision of the individuals in the tails of the trait distribution into subpools was random. The number of individuals in each subpool was equal if the number of individuals in the tail was divisible by the number of subpools; otherwise it could differ by one individual. Simulated technical error standard deviation associated with estimation of marker allele frequencies in a pool was set at 0.02 (absolute value). For analysis of the simulated data, the marker haplotypes of the sires were assumed known.
Example of QTL analysis by FPD:
The scheme of QTL analysis by FPD for the case of a single QTL per chromosome is illustrated using a simulated example with six half-sib families, three segregating for sire alleles at the simulated QTL (i.e., the sires of the families are heterozygous at the simulated QTL) and three not segregating for the sire alleles at the simulated QTL. Results are shown in Figure 3.
Figure 3.—

QTL analysis of multiple families with some nonshared markers. Six families with 2000 daughters each were simulated (three families with sire heterozygous for a single QTL situated at position 40 cM with allele substitution effect d/σ = 0.3 and three families with sire homozygous at the QTL). Chromosome length was 120 cM with 6–10 markers per family; a proportion 0.10 of all daughters was selected to each tail in each family. Individuals in both tails were randomly subdivided into four sub-pools. (a) D-value across the markers for each family (solid and open squares, triangles, and diamonds represent D in families with QTL-heterozygous and -homozygous sires correspondingly); (b) the results of jackknife resampling analysis (90% confidence intervals of λ-values for each family are shown by vertical lines, estimated in 500 jackknifes). The experimentwise P-value in a permutation test based on Σ(λf)2 was 0.012 (in 1000 permutations). The corresponding experimentwise permutation test P-values per family were 0.018, 0.012, 0.023, 0.483, 0.344, and 0.428 Estimated QTL position on all six families or on three families with a significant (P-value <0.05) λ-value was 43.9 cM (SD = 2.8) and 43.6 (SD = 2.6) cM, respectively. Estimated power for P-value = 0.05 was 99%.
Various numbers of markers were employed in the different families (with some regions being represented by neighboring but not coinciding marker loci), illustrating the ability of the FPD analytical system to deal with cases when markers are not shared among families. To simulate such a situation, we initially generated for each family a high excess of markers with identical chromosome positions. Then, the majority of markers for each family were declared “homozygous,” and only a small proportion of markers were randomly selected to be “heterozygous.” A QTL with standardized allele substitution effect d/σ = 0.3 was simulated at location 40 cM on the chromosome of 120 cM length. There were 2000 daughters per family; a proportion 0.10 of total daughters (i.e., 200 daughters) was selected for each tail, and there were four subpools per tail. The overall permutation test conducted after fitting the estimation model (5a) gave significance P = 0.009 (in 1000 permutations). P-values per family were respectively 0.029, 0.029, 0.029, 0.94, 0.69, and 0.74 (based on permutation tests within families, where only 35 possible different permutations exist for the 4 + 4 subpool configurations). Corresponding P-values for the families obtained in an experimentwise permutation test were 0.018, 0.012, 0.023, 0.483, 0.344, and 0.428 (1000 random permutations). QTL positions estimated using all six families or only the three families with significant effect (P-value <0.05) were 43.9 cM with standard deviation of estimated position among runs (SD = 2.8) and 43.6 (SD = 2.6), respectively (based on 500 jackknifes). On the basis of the jackknife procedure, QTL detection power for the entire set of families was estimated as follows. Threshold values of the test statistics Σ(λf)2 were obtained from the permutation test for significance levels 5 and 1%. QTL “detection power” was then estimated as the proportion of jackknife runs where the test statistics exceeded the threshold value at the chosen significance level. Calculated in this way, estimated powers for P-values = 0.05 and 0.01 were 99 and 82%, respectively.
Comparing the quality of mapping for different numbers of markers:
A few more examples with single-QTL chromosomes were simulated with 10 sire families (5 with sire heterozygous and 5 with sire homozygous at the QTL), with two standardized allele substitution effects at the QTL (0.2 and 0.15) situated at position x(q) = 40 cM, and with three marker densities (9, 13, and 25 evenly spaced markers per 120-cM chromosome) (Table 1). Population size, proportion selected to the tails, and number of subpools per tail were as in Figure 1. Table 1 presents the results for the six parameter combinations, with 10 independent Monte Carlo data sets simulated for each combination; for every simulated data set 500 permutations of subpools and 100 jackknife iterations were made. For each of the 10 simulated data sets we calculated the standard deviation of the difference between estimated QTL position 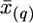 and the simulated one x(q) = 40 cM among the 100 jackknife iterations. The mean of these standard deviations across all 10 data sets, denoted SD, characterizes the size of the confidence interval of estimated QTL position. In addition, for each data set we calculated the difference between the mean of estimated QTL position based on the 100 iterations and the simulated position. The mean square of these differences, denoted Δ, characterizes the shift of the center of the confidence interval relative to the true value. Table 1 shows that increasing the number of markers reduces Δ more efficiently than SD. As one would expect, SD (and hence the size of the confidence interval) is higher in the case of d/σ = 0.15 compared to d/σ = 0.2 (5.4 vs. 3.3).
and the simulated one x(q) = 40 cM among the 100 jackknife iterations. The mean of these standard deviations across all 10 data sets, denoted SD, characterizes the size of the confidence interval of estimated QTL position. In addition, for each data set we calculated the difference between the mean of estimated QTL position based on the 100 iterations and the simulated position. The mean square of these differences, denoted Δ, characterizes the shift of the center of the confidence interval relative to the true value. Table 1 shows that increasing the number of markers reduces Δ more efficiently than SD. As one would expect, SD (and hence the size of the confidence interval) is higher in the case of d/σ = 0.15 compared to d/σ = 0.2 (5.4 vs. 3.3).
Table 1 also allows a comparison of different methods of testing the significance of QTL effect. Among the model-free tests based on  , max-χ2,
, max-χ2, 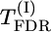 , and
, and 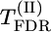 , the best results seem to be provided by the permutation test for max-χ2 statistics (for d/σ = 0.2) and by the
, the best results seem to be provided by the permutation test for max-χ2 statistics (for d/σ = 0.2) and by the 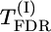 test also based on permutations (for d/σ = 0.15). According to the presented results, the
test also based on permutations (for d/σ = 0.15). According to the presented results, the 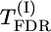 test based on permutations gave a much higher level of significance than the
test based on permutations gave a much higher level of significance than the 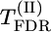 test based on χ2-asymptotic approximation (P-values were lower by an order of magnitude). The model-based test using the Σ(λf)2 statistic instead of max-χ2 resulted in a further severalfold decrease in P-values (see Table 1). In accordance with the ranking of the test statistics for P-values, Σ(λf)2 also proved to be superior with respect to detection power (i.e., resulting in the lowest proportion of false-negative declarations in the case of the given fixed P-value = 0.05). Estimated power of the test based on Σ(λf)2 was very high (∼0.9 for d/σ = 0.15 and ≥0.98 for d/σ = 0.20). When d/σ = 0.15, estimated power of this test increased slightly with increasing number of markers M. Estimated power of the test based on max-χ2 was also higher for d/σ = 0.20 than for d/σ = 0.15. Nevertheless, unlike Σ(λf)2, power for this test did not increase with increasing M; indeed, what may even be an opposite tendency was observed for d/σ = 0.20). This observation can be explained as follows: With increasing M, the probability that in permutation runs, the
test based on χ2-asymptotic approximation (P-values were lower by an order of magnitude). The model-based test using the Σ(λf)2 statistic instead of max-χ2 resulted in a further severalfold decrease in P-values (see Table 1). In accordance with the ranking of the test statistics for P-values, Σ(λf)2 also proved to be superior with respect to detection power (i.e., resulting in the lowest proportion of false-negative declarations in the case of the given fixed P-value = 0.05). Estimated power of the test based on Σ(λf)2 was very high (∼0.9 for d/σ = 0.15 and ≥0.98 for d/σ = 0.20). When d/σ = 0.15, estimated power of this test increased slightly with increasing number of markers M. Estimated power of the test based on max-χ2 was also higher for d/σ = 0.20 than for d/σ = 0.15. Nevertheless, unlike Σ(λf)2, power for this test did not increase with increasing M; indeed, what may even be an opposite tendency was observed for d/σ = 0.20). This observation can be explained as follows: With increasing M, the probability that in permutation runs, the 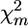 value for one of the markers will be higher than maxm
value for one of the markers will be higher than maxm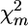 in initial pool configuration also increases. Conversely, increasing M also can increase the power of this test if the additional markers belong to the vicinity of the QTL (not shown).
in initial pool configuration also increases. Conversely, increasing M also can increase the power of this test if the additional markers belong to the vicinity of the QTL (not shown).
Multiple linked QTL analysis—two or more QTL on the chromosome:
In the case of two or more QTL per chromosome, expected D at the marker locus is defined by the expected frequencies of sire alleles in the high and low pools at the closest situated QTL and by recombination rates between marker and QTL. Let K be the number of QTL in the chromosome and denominate the QTL according to their locations [i.e., x(1) < x(2) < … < x(K)]. The expectation of D for a marker at location x can then be written in the form
 |
(6) |
where
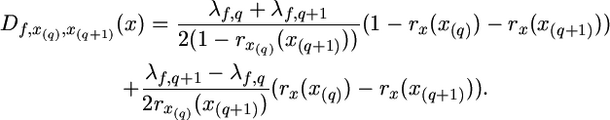 |
Here λf,q is the characteristic of the qth QTL in family f, and x(q) is the location of this QTL. Value rx(x(q)) is the recombination rate between the marker loci situated in positions x and x(q). The origin of Equation 6 is similar to Equation 3 (for details see also Wang et al. 2007): Let λf,1, …, λf,K be expectations for D-values of markers coinciding with corresponding QTL. Assuming absence of interference we can consider the expectation of D-values separately for each interval between QTL. For the two end intervals x < x(1) and x > x(K) Equation 6 has the same form as Equation 3. For other intervals the absolute value of the expectation of D is reduced by corresponding double recombination (double recombination is not a factor for the end intervals). The estimation criterion for the regression method takes the following form:
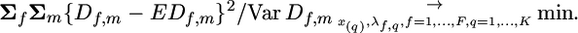 |
(7) |
Fitting the model by using criteria (7) can be expressed in terms of the linear model
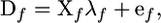 |
where λf is a vector of λf,1, …, λf,K and coefficients of matrix Xf are equal to corresponding multipliers in Equation 6. Taking into account the correlation between values of D for linked markers and using the generalized least-squares approach, the estimation criterion takes the form
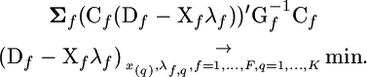 |
(8) |
Here matrices G and C are like in Equation 5a. For given putative QTL positions, vector λf of parameters λf,1, …, λf,K minimizing criterion (8) can be calculated as
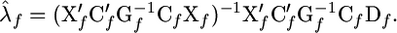 |
Even in the case of only two QTL on the chromosome, various situations can exist. These include heterozygosity of different sires for one, two, or none of the QTL and the linkage phase between the QTL (coupling vs. repulsion) in the sires that are heterozygous for both QTL. Thus, in addition to the foregoing tests of significance, the situation with linked QTL calls for comparisons of H2 vs. H1 (two-QTL vs. single-QTL hypotheses) for the entire data set as well as for each family. However, in this article we demonstrate only the potential of the FPD system to analyze linked QTL, leaving the detailed analysis of various scenarios for a future publication.
The example, presented in Figure 4, is based on one simulated data set of 10 families. Each sire was simulated heterozygous for two linked QTL (half of the sires in coupling phase and half in repulsion phase) with allele substitution effects d/σ = 0.3 at locations 30 and 80 cM on a chromosome of length 120 cM with 13 evenly spaced markers (at positions 0, 10, 20, …, 120 cM). Population size, proportion selected to the tails, and number of subpools per tail were as in Figure 1. After fitting a two-QTL model and using FPD analysis to detect the two QTL, the estimated QTL positions were within 2 cM from the simulated positions. Standard errors in 500 jackknifes were 1.7 and 0.8 for QTL 1 and QTL 2, respectively. The high quality of the analysis is due to the high allele-substitution effects in the two QTL and the relatively large map distance between them. More diverse sires with respect to their QTL structure (heterozygous at one, two, or none of the QTL) are also treatable with relative ease within the framework of the two-QTL FPD model.
Figure 4.—

Analysis with multiple-linked QTL. Simulated were 10 families heterozygous for two linked QTL, 5 in coupling and 5 in repulsion phase. Thirteen markers were evenly spaced on a chromosome of length 120 cM. QTL 1 and QTL 2 were simulated in positions 30 and 80 cM, respectively. The allele substitution effect at both QTL in all 10 families was d/σ = 0.3. Alleles at QTL 1 and QTL 2 that came from dams were simulated as independent cases. The number of daughters per family was 2000; the proportion of total population selected to each tail was 0.10. (a) D-values for all families and markers. Points corresponding to a given family are connected by a line. (b) λ-Values and their standard errors in 500 jackknifes for every family. Clear separation is observed between the first five sires (QTL in coupling phase) and the last five sires (QTL in repulsion phase). (c) Simulated (solid circle) and estimated (open circle) positions of QTL. The curve encloses the area where the position of QTL was estimated in ≥90% of 500 jackknifes {included points with integer coordinates (x, y) such that in ≥5 jackknifes, estimated QTL positions belonged in the interval (x ± 0.5, y ± 0.5 cM).
General scheme of FPD QTL analysis:
To conclude the analytical section, we present here a general scheme of the proposed system of FPD QTL analysis (Figure 5). The suggested integrative algorithm includes: (A) fitting the mapping model, (B) an overall test of significance (using λf-value-based models for conducting permutation tests), (C) detecting nonsignificant (QTL-homozygous) sires, (D) removing the homozygous sires and repeating the tests, (E) estimating QTL detection power, and (F) conducting jackknife analysis to evaluate the confidence interval for the estimated position of detected QTL. This scheme can be further extended to take into account the possibilities of multiple-linked-QTL analysis, including: fitting multiple-linked-QTL models; comparing multiple-linked and single-QTL models (testing H0 vs. H1 and H2 and H1 vs. H2); detection of sires heterozygous for zero, one, or multiple-linked QTL; and estimating the confidence intervals of the chromosomal positions of the detected QTL.
Figure 5.—

The general scheme of QTL analysis by the FPD method.
Unknown marker linkage phase in the sire:
In the case of unknown marker–QTL linkage phase (sire marker haplotypes), the algebraic sign of the statistic Dm is not uniquely defined. For markers with unknown phase these signs (plus or minus) can be found through optimization of criteria (4), (5), (4a), (5a), (7), or (8) (with the minimum now taken over all possible combinations of signs). To make optimization in this case more effective, some heuristics can be used. For a single-QTL model where marker phase in the sire is not known, it is reasonable to allocate the same sign (say, plus) to the D-values for all markers. For the model with two QTL on the chromosome, it is reasonable to consider D-values changing sign no more than once, e.g., positive for the first m markers and negative for the others (if the two QTL in the sire are in repulsive phases). Optimization of the signs of D-values can result in an increase in the false positive declaration rate. Indeed, it can convert some families with noisy fluctuating D-values around zero to have D-values of one sign. This can greatly increase |λ| and, hence, falsely cause a QTL-homozygous family to be declared heterozygous. Therefore, external information about linkage phases of the maker loci reduces the proportion of false positive families.
Choosing the number of subpools:
The multiple-pool approach was previously proposed as a means of improving the quality of allele frequency estimates (Sham et al. 2002; Brohede et al. 2005). Within this framework, the problem of “optimal size” of pools was primarily considered from the aspect of amplification fidelity (Brohede et al. 2005) and as a way to obtain an adequate estimate of variation of marker allele frequencies Var Df,m (e.g., Sham et al. 2002). In the present study, the number of pools affects the number of possible different permutations and jackknifes and hence affects P-values and power of the analysis.
To demonstrate the dependence of analysis quality on the number of subpools per tail, a series of simulation experiments were conducted. Situations with one, three, and five families were simulated. The proportion of individuals taken to the tails was 0.10 as in the previous simulations. The individuals in the tails were then randomly subdivided into four, six, or eight subpools of equal size. The family sizes were 960 and 1920. As above a chromosome of 120 cM length with 13 evenly spaced markers was assumed, and the QTL was simulated in position 40 cM with allele substitution effects d/σ = 0.3, 0.2, and 0.15. For each parameter combination, 10 Monte Carlo data sets were simulated; for every set 1000 permutations and 100 jackknife iterations were made (with exactly one pool per tail per family being excluded in each jackknife run). The results are summarized in Table 2.
TABLE 2.
Effect of number of subpools per tail (S) under the FPD on characteristics Δ and SD of the confidence interval for QTL location, comparisonwise error rate (P-value), and statistical power, according to number of families (F), number of daughters per family (N), and standardized allele substitution effect at the QTL (d/σ), using simulated data
| Δ
|
SD
|
P-value
|
Power at P = 0.05
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| F | N | d/σ | S = 4 | S = 6 | S = 8 | S = 4 | S = 6 | S = 8 | S = 4 | S = 6 | S = 8 | S = 4 (%) | S = 6 (%) | S = 8 (%) |
| 1 | 1920 | 0.3 | 10.4 | 10.1 | 10.3 | 4.7 | 4.0 | 3.5 | 0.056 | 0.007 | 0.005 | — | 79 | 89 |
| 0.2 | 14.1 | 14.0 | 13.7 | 14.6 | 12.0 | 10.7 | 0.083 | 0.043 | 0.028 | — | 32 | 56 | ||
| 0.15 | 11.0 | 10.3 | 11.1 | 11.6 | 8.8 | 6.7 | 0.156 | 0.135 | 0.110 | — | — | — | ||
| 3 | 960 | 0.3 | 4.7 | 3.8 | 3.5 | 5.4 | 4.9 | 3.2 | 0.003 | 0.003 | 0.002 | 59 | 89 | 94 |
| 0.2 | 10.3 | 12.1 | 12.0 | 11.4 | 7.1 | 6.9 | 0.030 | 0.021 | 0.023 | 30 | 52 | 52 | ||
| 0.15 | 14.6 | 14.7 | 14.9 | 19.2 | 15.7 | 13.2 | 0.195 | 0.203 | 0.208 | — | — | — | ||
| 3 | 1920 | 0.3 | 2.9 | 3.1 | 3.2 | 1.9 | 1.5 | 1.2 | 0.001 | 0.001 | 0.001 | 94 | 99 | 99 |
| 0.2 | 5.7 | 5.2 | 5.4 | 4.4 | 3.4 | 3.0 | 0.003 | 0.002 | 0.003 | 56 | 82 | 92 | ||
| 0.15 | 10.7 | 10.1 | 10.1 | 6.9 | 5.6 | 5.1 | 0.028 | 0.024 | 0.011 | 46 | 72 | 76 | ||
| 5 | 960 | 0.3 | 2.7 | 2.9 | 2.9 | 2.6 | 1.8 | 1.6 | 0.001 | 0.001 | 0.001 | 87 | 99 | 99 |
| 0.2 | 5.7 | 5.8 | 5.4 | 5.3 | 4.3 | 3.3 | 0.013 | 0.009 | 0.006 | 44 | 63 | 71 | ||
| 0.15 | 14.3 | 15.1 | 14.9 | 12.0 | 9.6 | 8.4 | 0.081 | 0.070 | 0.067 | — | — | — | ||
P-values and power were calculated using the permutation test based on Σ(Af)2 (see text). Power was calculated for the threshold of the statistics corresponding to P-value = 0.05 (shown only for situations where the observed experimentwise P-value did not exceed 0.05). Characteristics Δ and SD of the confidence interval for QTL location were obtained from the jackknife iterations. Parameters of the simulations: chromosome length 120 cM. A single QTL was situated at position 40 cM. Number of markers M = 13. Proportion of population selected to each tail, 0.10. One subpool per tail was excluded in each jackknife. Values represent mean of 10 simulation data sets; for every data set 1000 permutations of subpools and 100 jackknife iterations were made to estimate P-value, power, Δ, and SD.
It was found that a higher number of subpools does not reduce the standard error of estimated QTL location, if the percentage of excluded pools is the same in each jackknife iteration (not shown). However, if in each jackknife iteration exactly one pool per tail is excluded, SD and confidence intervals became smaller with a higher number of subpools (Table 2) but less robust (i.e., sampling variance of the confidence interval center and its size are higher), because different runs are more dependent. This can explain why value Δ does not always decrease with increasing number of subpools S. In contrast, P-values decreased asymptotically with the number of subpools until some limit determined by QTL allele substitution effect, number and proportion of QTL-polymorphic families, number of daughters per family, proportion of daughters taken to each tail, number and positions of markers on the chromosome, and technical error of densitometric estimation of pool frequencies. Results summarized in Table 2 demonstrate the variation of P-value and power of the analysis that can be achieved in different situations. As expected, better results were obtained in situations with a greater number of families, a greater number of progeny per family, and a greater allele substitution effect d/σ of QTL. The unexpected smaller Δ and SD for the one-family situation in the case of d/σ = 0.15 (compared to d/σ = 0.2) can be explained by a shortcoming of criterion (5a): In the case of absence of or very small QTL effect, the difference in the criterion values for different x(q) is very small; and the smallest value tends to be observed for x(q) close to the average marker position (60 cM in our situation). In other words, under H0, the estimated position is not uniformly distributed along the chromosome (not shown). Note that the lowest possible P-value in permutation is equal to 1/R, where R is the number of different permutations. If we are “satisfied” with P-values ≥α, then no more than 5/α different permutations are needed. Hence, in the case of only one family we need ∼S = log4R + 1.5 = log4(5/α) + 1.5 subpools. For the experimentwise permutation test in F similar families we need S = log4(R)1/F + 1.5 = 1/F log4(5/α) + 1.5 subpools per tail, per family. Thus, from the point of view of maximizing the number of different permutations, it is more effective to analyze more families than to make more subpools per family. The relative cost of additional families, subpools, markers, and desired QTL detection power and mapping accuracy defines a cost-effective strategy for the initial genome scan for QTL by FPD. Clearly, the above aspects of amplification fidelity and estimation of variation of marker allele frequencies considered by Brohede et al. (2005), Sham et al. (2002), and other authors should also be an important part of designing FPD experiments.
Correlations between D-values and quality of the analysis:
Taking into account correlations between D-values for linked markers, i.e., using a generalized least-squares method (Equations 5, 5a, and 8), will probably not increase the QTL detecting power and accuracy of the QTL position estimates in the majority of practical situations. When substitution effects, number of daughters per family, and number of families are small, the sampling variance of Dm is high relative to its expected value. Taking the correlations into account will increase the sampling variance and reduce the expected value for each marker (Montgomery and Peck 1992). This makes the analysis less robust. The least-squares optimization criterion, when H0 is true, follows a χ2-distribution with degrees of freedom equal to the number of terms in the sum. Parameters minimizing this criterion also maximize the likelihood function, but the difference between the criterion values for different putative QTL positions is small (not shown). Nevertheless, by taking the correlations into account, we reduce the confidence interval and discrepancy between the estimated and simulated QTL positions (data not shown).
DISCUSSION AND PROSPECTS
Genomewide scans for the detection of marker–QTL linkage or linkage disequilibrium for QTL of small effect require large mapping populations and hence involve a high cost of marker genotyping. Even more challenging are the requirements of population size from the viewpoint of QTL mapping accuracy. In family-based analysis, the confidence intervals for the estimated QTL chromosomal position are of tens of centimorgans even for QTL of moderate effects (Darvasi and Soller 1997; Ronin et al. 2003). A cost-effective solution is to replace individual genotyping by DNA analysis in pools using individuals from the tails of the trait distribution (Hillel et al. 1990; Darvasi and Soller 1994) or alternative phenotypic groups in the case of discontinuous variation (Giovannoni et al. 1991; Michelmore et al. 1991). To increase the fidelity of pooling analysis, Dekkers (2000) proposed a method of joint treatment of multiple markers by scanning a chromosome with a sliding window (see also Johnson 2005 for further developments in LD QTL analysis).
Although the idea of using a multiple-pool design has been discussed previously (Sham et al. 2002; Brohede et al. 2005), the objectives of those studies were to improve the quality of the allele-frequency estimates and corresponding variances. In addition to these uses, the proposed FPD system utilizes the multiple-pool design to provide a wide spectrum of new analytical options that were previously possible only with individual genotyping. These new options are of special importance in the light of accumulating evidence on reliability of pooling analysis with SNP chips. Combining SNP microarray analysis with DNA pooling can reduce dramatically the cost of screening large numbers of SNPs on large samples, making chip technology applicable for genomewide association mapping in humans and farm animals (Butcher et al. 2004; Brohede et al. 2005; Craig et al. 2005). The FPD analysis relaxes some of the previous limitations of the pooling analysis by utilizing the information provided by multiple subpools within tails. This allows a flexible analytical system in QTL detection based on resampling procedures (permutations, bootstraps, and jackknifes), rather than on asymptotic assumptions (Sham et al. 2002; Carleos et al. 2003), enabling evaluation of the confidence interval of QTL position and discriminating between different hypotheses of trait genetic architecture.
Allowing for resampling analysis via the FPD does come at a cost of requiring multiple subpools per tail. In the situations when multiple traits are analyzed, individuals need to be separated into subpools in the tails of trait distribution for every trait. In these situations the number of subpools may be close to the number of individuals in the mapping population (if traits are not strongly correlated), thereby reducing the advantage of the pooling method. Another disadvantage is that this method only partially utilizes haplotype information compared to individual selective genotyping. However, a partial solution to this problem could be provided by using multivariate tails of the multidimensional trait distribution rather than trait-specific tails (Ronin et al. 1998).
The proposed methodology allows joint analysis of multiple families and multiple markers across a chromosome, even if the markers are only partly shared (or even not shared at all) among families. Resampling procedures permit confidence intervals to be constructed for family-specific λ-values. These intervals allow identification of families for which the founder sire was homozygous at the QTL. The FPD analysis permits extension to cases of two or more QTL on the same chromosome. All this provides cost-effective options for sequential family- and region-specific increase of marker density to improve the QTL mapping resolution and accuracy and to reduce type I (false positive) and type II (false negative) errors. Of special interest is the extension of pooling methodology to genome expression analysis (Alba et al. 2004; Kendziorski et al. 2005). The cautious optimism of pooling RNA expressed by these authors can be considered as justifying the extension of the FPD to RNA analysis.
The major advantage of population-based rather than family-based mapping is in its potential for fine and ultra-fine mapping due to accumulation of historical recombination events. Recent findings on the existence of linkage disequilibrium block and estimates of the sizes of these blocks establish a basis for LD (association) mapping. Still, for loci with small to moderate effects on the target traits one of the major limiting factors is the size of the effect and not the degree of recombination (diversity of haplotypes). Consequently, very large sample sizes are required making pooling analysis extremely attractive. Therefore, we plan to extend the fractionated pooling design to LD-based QTL analysis.
Acknowledgments
We thank J. Dekkers for very constructive criticism and helpful suggestions. This research was supported in part by grant QLK5-CT-2001-02379 (BovMAS project) under the European Union FP5 program and by a Ph.D. fellowship from the University of Haifa to Z. F.
References
- Alba, R., Z. J. Fei, P. Payton, Y. Liu, S. L. Moore et al., 2004. ESTs, cDNA microarrays, and gene expression profiling: tools for dissecting plant physiology and development. Plant J. 39: 697–714. [DOI] [PubMed] [Google Scholar]
- Benjamini, Y., and Y. Hochberg, 1995. Controlling the false discovery rate - a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 57: 289–300. [Google Scholar]
- Brohede, J., R. Dunne, J. D. Mckay and G. N. Hannan, 2005. PPC: an algorithm for accurate estimation of SNP allele frequencies in small equimolar pools of DNA using data from high density microarrays. Nucleic Acids Res. 33: e142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butcher, L. M., E. Meaburn, L. Liu, C. Fernandes, L. Hill et al., 2004. Genotyping pooled DNA on microarrays: a systematic genome screen of thousands of SNPs in large samples to detect QTLs for complex traits. Behav. Genet. 34: 549–555. [DOI] [PubMed] [Google Scholar]
- Carleos, C., J. A. Baro, J. Canon and N. Corral, 2003. Asymptotic variances of QTL estimators with selective DNA pooling. J. Hered. 94: 175–179. [DOI] [PubMed] [Google Scholar]
- Craig, D. W., M. J. Huentelman, D. Hu-Lince, V. L. Zismann, M. C. Kruer et al., 2005. Identification of disease causing loci using an array-based genotyping approach on pooled DNA. BMC Genomics 6: 138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darvasi, A., and M. Soller, 1992. Selective genotyping for determination of linkage between a marker locus and a quantitative trait locus. Theor. Appl. Genet. 85: 353–359. [DOI] [PubMed] [Google Scholar]
- Darvasi, A., and M. Soller, 1994. Selective DNA pooling for determination of linkage between a molecular marker and a quantitative trait locus. Genetics 138: 1365–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darvasi, A., and M. Soller, 1997. A simple method to calculate resolving power and confidence interval of QTL map location. Behav. Genet. 27: 125–132. [DOI] [PubMed] [Google Scholar]
- Dekkers, J. C. M., 2000. Quantitative trait locus mapping based on selective DNA pooling. Anim. Breed. Genet. 117: 1–16. [Google Scholar]
- Doerge, R. W., and G. A. Churchill, 1996. Permutation tests for multiple loci affecting a quantitative character. Genetics 142: 285–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunnington, E. A., A. Haberfeld, L. C. Stallard, P. B. Siegel and J. Hillel, 1992. Deoxyribonucleic-acid fingerprint bands linked to loci coding for quantitative traits in chickens. Poult. Sci. 71: 1251–1258. [DOI] [PubMed] [Google Scholar]
- Fernando, R. L., D. Nettleton, B. R. Southey, J. C. Dekkers, M. F. Rothschild et al., 2004. Controlling the proportion of false positives in multiple dependent tests. Genetics 166: 611–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giovannoni, J. J., R. A. Wing, M. W. Ganal and S. D. Tanksley, 1991. Isolation of molecular markers from specific chromosomal intervals using DNA pools from existing mapping populations. Nucleic Acids Res. 19: 6553–6558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillel, J., R. Avner, C. Baxter-Jones, E. A. Dunnington, A. Cahaner et al., 1990. DNA fingerprints from blood mixes in chickens and turkeys. Anim. Biotechnol. 2: 201–204. [Google Scholar]
- Johnson, T., 2005. Multipoint linkage disequilibrium mapping using multilocus allele frequency data. Ann. Hum. Genet. 69: 474–497. [DOI] [PubMed] [Google Scholar]
- Kearsey, M. J., 1998. The principles of QTL analysis (a minimal mathematics approach). J. Exp. Bot. 49: 1619–1623. [Google Scholar]
- Kendziorski, C., R. A. Irizarry, K. S. Chen, J. D. Haag and M. N. Gould, 2005. On the utility of pooling biological samples in microarray experiments. Proc. Natl. Acad. Sci. USA 102: 4252–4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander, E. S., and D. Botstein, 1989. Mapping Mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics 121: 185–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipkin, E., M. O. Mosig, A. Darvasi, E. Ezra, A. Shalom et al., 1998. Quantitative trait locus mapping in dairy cattle by means of selective milk DNA pooling using dinucleotide microsatellite markers: analysis of milk protein percentage. Genetics 149: 1557–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelmore, R. W., I. Paran and R. V. Kesseli, 1991. Identification of markers linked to disease-resistance genes by bulked segregant analysis: a rapid method to detect markers in specific genomic regions by using segregating populations. Proc. Natl. Acad. Sci. USA 88: 9828–9832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery, D. C., and E. A. PECK, 1992. Introduction to Linear Regression Analysis, Ed. 2. John Wiley & Sons, New York.
- Mosig, M. O., E. Lipkin, G. Khutoreskaya, E. Tchourzyna, M. Soller et al., 2001. A whole genome scan for quantitative trait loci affecting milk protein percentage in Israeli-Holstein cattle, by means of selective milk DNA pooling in a daughter design, using an adjusted false discovery rate criterion. Genetics 157: 1683–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotsky, Y., A. Cahaner, A. Haberfeld, U. Lavi, S. J. Lamont et al., 1993. DNA fingerprint bands applied to linkage analysis with quantitative trait loci in chickens. Anim. Genet. 24: 105–110. [DOI] [PubMed] [Google Scholar]
- Ronin, Y., A. Korol, M. Shtemberg, E. Nevo and M. Soller, 2003. High-resolution mapping of quantitative trait loci by selective recombinant genotyping. Genetics 164: 1657–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronin, Y. I., A. B. Korol and J. I. Weller, 1998. Selective genotyping to detect quantitative trait loci affecting multiple traits: interval mapping analysis. Theor. Appl. Genet. 97: 1169–1178. [Google Scholar]
- Ronin, Y. I., A. B. Korol and E. Nevo, 1999. Single- and multiple-trait mapping analysis of linked quantitative trait loci: some asymptotic analytical approximations. Genetics 151: 387–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnack, H. G., S. C. Bakker, R. Van't Slot, B. M. Groot, R. J. Sinke et al., 2004. Accurate determination of microsatellite allele frequencies in pooled DNA samples. Eur. J. Hum. Genet. 12: 925–934. [DOI] [PubMed] [Google Scholar]
- Sham, P., J. S. Bader, I. Craig, M. O'Donovan and M. Owen, 2002. DNA pooling: a tool for large-scale association studies. Nat. Rev. Genet. 3: 862–871. [DOI] [PubMed] [Google Scholar]
- Tamiya, G., M. Shinya, T. Imanishi, T. Ikuta, S. Makino et al., 2005. Whole genome association study of rheumatoid arthritis using 27 039 microsatellites. Hum. Mol. Genet. 14: 2305–2321. [DOI] [PubMed] [Google Scholar]
- Visscher, P. M., and S. Le Hellard, 2003. Simple method to analyze SNP-based association studies using DNA pools. Genet. Epidemiol. 24: 291–296. [DOI] [PubMed] [Google Scholar]
- Wang, J., K. J. Koehler and J. C. M. Dekkers, 2007. Interval mapping of quantitative trait loci with selective DNA pooling data. Genet. Sel. Evol. (in press). [DOI] [PMC free article] [PubMed]
- Weller, J. I., Y. Kashi and M. Soller, 1990. Power of daughter and granddaughter designs for determining linkage between marker loci and quantitative trait loci in dairy-cattle. J. Dairy Sci. 73: 2525–2537. [DOI] [PubMed] [Google Scholar]
- Zou, G. H., and H. Y. Zhao, 2004. The impacts of errors in individual genotyping and DNA pooling on association studies. Genet. Epidemiol. 26: 1–10. [DOI] [PubMed] [Google Scholar]
- Zou, G. H., and H. Y. Zhao, 2005. Family-based association tests for different family structures using pooled DNA. Ann. Hum. Genet. 69: 429–442. [DOI] [PMC free article] [PubMed] [Google Scholar]