

Summary
Collapsed DNA replication forks must be reactivated through origin-independent reloading of the replication machinery (replisome) to ensure complete duplication of cellular genomes. In E. coli, the PriA-dependent pathway is the major replication restart mechanism and requires primosome proteins PriA, PriB, and DnaT for replisome reloading. However, the molecular mechanisms that regulate origin-independent replisome loading are not fully understood. Here, we demonstrate that assembly of primosome protein complexes represents a key regulatory mechanism, as inherently weak PriA-PriB and PriB-DnaT interactions are strongly stimulated by single-stranded DNA. Furthermore, the binding site on PriB for single-stranded DNA partially overlaps the binding sites for PriA and DnaT, suggesting a dynamic primosome assembly process in which single-stranded DNA is handed off from one primosome protein to another as a repaired replication fork is reactivated. This model helps explain how origin-independent initiation of DNA replication is restricted to repaired replication forks, preventing over-replication of the genome.
Introduction
Cells must grow and divide in the face of numerous pressures that challenge genome integrity. Chemical damage to the genome arising from the environment and from cellular metabolism creates barriers that impede cellular processes that replicate and transcribe the information contained in DNA. As a counter to these stresses, cells dedicate several metabolic pathways to monitoring their genomes and making repairs when needed. DNA replication is particularly sensitive to DNA damage, as advancing replisomes can derail upon encountering the vast arrays of DNA lesions that are possible in cells, including single-stranded nicks, gaps, double-stranded breaks, and modified bases (Cox, 2001; Cox et al., 2000). To ensure complete and faithful replication of the genome, these lesions must be repaired and DNA replication must resume.
In Escherichia coli, DNA replication forks that form at the origin of replication, oriC, frequently encounter DNA damage that can result in their inactivation (Cox et al., 2000). Since origin-dependent replisome loading is carefully regulated and sequence-specific, cells require a distinct set of machinery to reload replisomes at non-origin sequences where replication forks have been disrupted. The proteins responsible for this activity are collectively known as the replication restart primosome and include PriA, PriB, PriC, DnaT, and Rep proteins. Genetic lines of investigation in E. coli led to the “multiple replication restart pathways” model, stemming from observations that null mutations in priA, priB, priC, and dnaT genes do not all display identical cellular phenotypes (McCool et al., 2004; Sandler et al., 1999). Two distinct replisome loading systems have been biochemically defined, one requiring PriA, PriB, and DnaT, and the other requiring only PriC (Heller and Marians, 2005). In vivo, pathways involving PriC are thought to require activities of either PriA helicase or Rep helicase (Sandler, 2000; Sandler et al., 2001). The existence of multiple replication restart pathways likely underscores the importance of reactivating replication forks that have diverse structures resulting from various genome maintenance processes (Heller and Marians, 2006).
A major challenge in understanding replication restart pathways has been delineating the sequence of events needed to reload a replisome at a repaired DNA replication fork. PriA binds DNA in a structure-specific manner and unwinds duplex DNA in the 3’ to 5’ direction, making it a likely candidate for the initiator of the PriA-PriB-DnaT-dependent replication restart pathway at repaired replication forks and D-loops (Lee and Marians, 1987; McGlynn et al., 1997). In the case of replication restart from a forked DNA structure, it is thought that PriA can unwind the lagging strand arm if one is present to produce the single-stranded DNA (ssDNA) that will ultimately be used as a loading site for the replicative helicase, DnaB. PriB stabilizes PriA on the DNA (Ng and Marians, 1996a), stimulates its helicase activity (Cadman et al., 2005), and is thought to help recruit DnaT (Liu et al., 1996). While ssDNA binding by PriB plays an important role in its ability to stimulate PriA helicase (Cadman et al., 2005), the mechanisms by which PriB contributes to primosome assembly have not been clearly defined. The role of DnaT in primosome assembly is even more mysterious. Based on poor viability of dnaT null E. coli cells, DnaT is clearly a critical component of replication restart, but at present there is no specific function attributed to it (McCool et al., 2004). Once DnaB is loaded onto the lagging strand template, priming by DnaG occurs and recruitment of DNA polymerase III holoenzyme results in complete reactivation of the replication fork, allowing DNA synthesis to resume.
Although the proteins involved in replisome reloading and the DNA substrates on which they operate are known, the molecular underpinnings of origin-independent replisome reloading and regulation remain elusive. In particular, how the components of the primosome coordinate their activities in a regulated manner to prevent unnecessary and deleterious over-replication of the genome is unknown. We sought to address this question by examining the individual protein:protein and protein:nucleic acid complexes that form among members of the PriA-PriB-DnaT replication restart primosome. Here, we report detection of direct, physical interactions between individual components of the PriA-PriB-DnaT primosome and demonstrate that these interactions are greatly stimulated by DNA. Furthermore, we have identified binding sites for PriA, ssDNA, and DnaT on the surface of PriB and show that the spatial relation of these binding sites has important ramifications for regulation of primosome protein assembly into a multi-protein complex. Our results provide a mechanistic explanation for primosome activity at the level of assembly of individual nucleoprotein complexes and offer important insights into the regulation of early stages of replisome reloading in replication restart pathways.
Results
Recently reported crystal structures of E. coli PriB revealed structural similarity between PriB and ssDNA-binding proteins (SSBs) and offered important insights into the function of PriB in DNA replication restart (Liu et al., 2004; Lopper et al., 2004; Shioi et al., 2005) (Figure 1). Indeed, as shown by mutational analysis in an earlier report from our laboratory and by others through co-crystallization studies with ssDNA, the classic ligand-binding surface of PriB’s oligonucleotide/oligosaccharide-binding (OB) folds mediates interactions with ssDNA, akin to SSB (Huang et al., 2006; Lopper et al., 2004; Raghunathan et al., 2000). One notable difference between PriB and SSB is their oligomeric state: PriB forms homodimers whereas SSB forms homotetramers. A consequence of this difference is exposure of a surface of PriB that is used for higher order assembly in SSB (Figure 1). As PriB is one component of a multi-protein assembly, we hypothesized that this surface serves as a binding platform for other primosome proteins such as PriA and DnaT. This hypothesis led us to investigate which surfaces of PriB mediate its interactions with other primosome components. We generated a panel of 16 single and double alanine-substitution mutants of priB that systematically alter the surface structure of each portion of the PriB homodimer. PriB variants were tested for their ability to interact individually with PriA, DnaT, and ssDNA in vitro, and to support replication restart in vitro and in vivo.
Figure 1. Model for PriB function based on structural similarity to SSB.
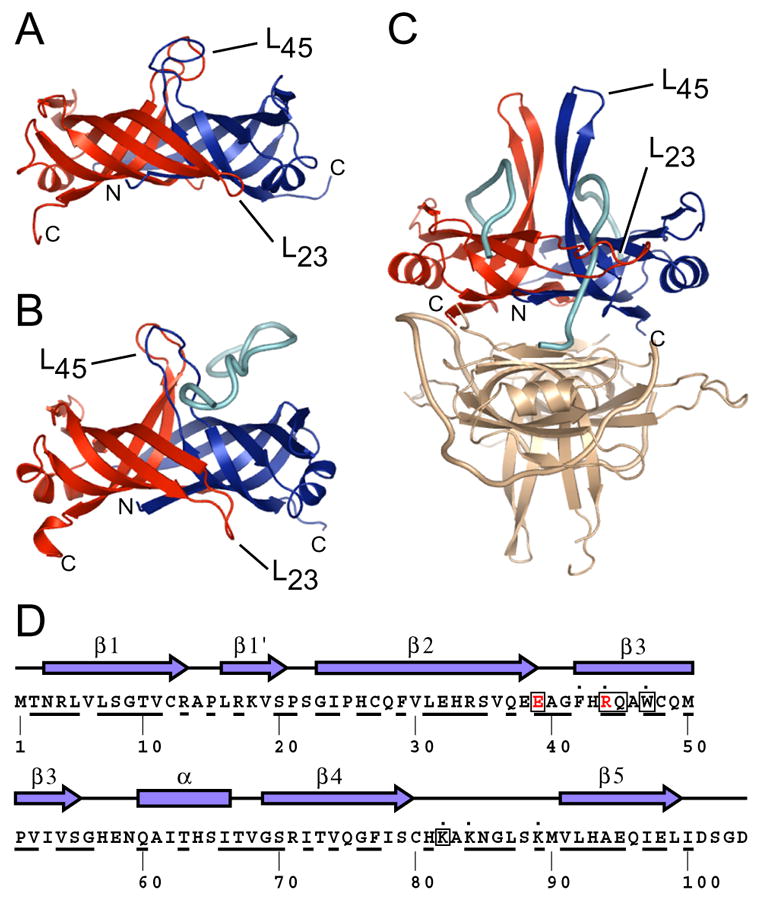
Ribbon diagrams of crystal structures of (A) apo PriB (Protein Databank accession code 1V1Q) (Liu et al., 2004), (B) PriB:ssDNA (Protein Databank accession code 2CCZ) (Huang et al., 2006), and (C) SSB:ssDNA (Protein Databank accession code 1EYG) (Raghunathan et al., 2000) were rendered with Pymol (DeLano, 2002) and are colored as follows: chain A (PriB and SSB), red; chain B (PriB and SSB), blue; ssDNA, cyan. One homodimer of the SSB:ssDNA homotetramer is colored beige. The amino- and carboxy-termini of chains A and B for PriB and SSB are indicated, as are the L23 and L45 loops. (D) The secondary structural elements of PriB are shown above the primary sequence and residues that are highly conserved among sequenced priB homologs are underlined. PriB residues are indicated that are important for interactions with PriA (red), DnaT (boxed), and ssDNA (dot).
PriB physically interacts with the helicase domain of PriA
We first determined if direct, physical interactions exist between PriB and PriA or DnaT. While it has been assumed that PriA, PriB, and DnaT associate with one another, interactions have not been examined in the absence of DNA. Therefore, we employed a fluorescence-based assay to probe for direct, physical interactions between purified PriA, PriB, and DnaT proteins and to ascertain the role of DNA in mediating complex formation among primosome proteins.
We labeled PriB with fluorescein isothiocyanate (FITC) and measured the fluorescence anisotropy of FITC-PriB in the presence and absence of PriA. If the two proteins associate, a PriA-dependent increase in FITC-PriB fluorescence anisotropy should be observed due to the greater mass of the PriA:FITC-PriB complex. However, only a modest increase in fluorescence anisotropy was observed, even in the presence of 500-fold molar excess PriA relative to FITC-PriB (Figure 2A), indicating that PriA and FITC-PriB interact only weakly under these conditions. Therefore, we tested whether association of PriA and PriB requires DNA by including PhiX174 virion ssDNA in the binding assay. PhiX174 virion ssDNA contains a primosome assembly site (PAS) sequence and PriA and PriB were originally isolated based on their requirement for replication of this bacteriophage (Schekman et al., 1975; Wickner and Hurwitz, 1974). Upon addition of PhiX174 ssDNA to mixtures of PriA and FITC-PriB, we observed a PriA-dependent increase in fluorescence anisotropy that saturates at approximately 500 nM PriA and has an apparent dissociation constant (Kd,app) of 46.2 +/− 3.2 nM (Figure 2A). This is consistent with ternary complex formation among PriA, FITC-PriB, and PhiX174 ssDNA. It is significant that the Kd,app of PriA:FITC-PriB:ssDNA is at least several orders of magnitude lower than that of PriA:FITC-PriB, for which a Kd,app could not be determined, suggesting that DNA is a major regulatory determinant for the PriA-PriB interaction.
Figure 2. PriB physically interacts with the helicase domain of PriA.
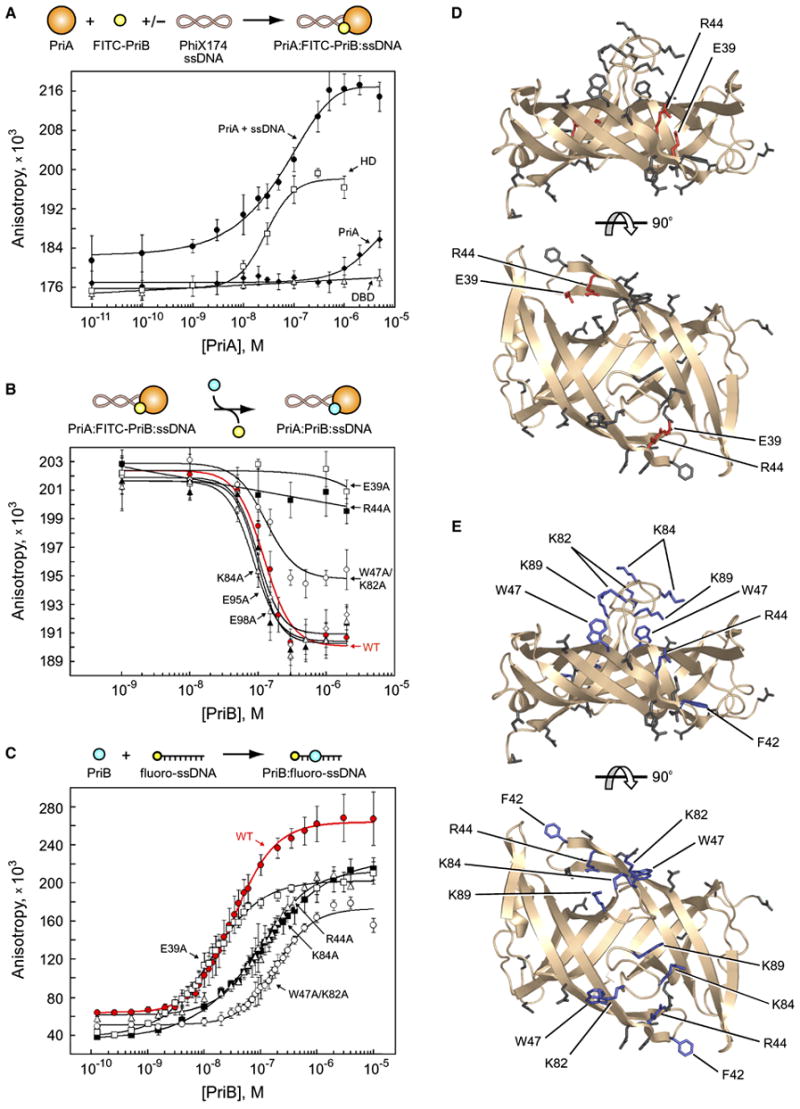
(A) Intact PriA (closed diamonds), PriA DBD (open triangles), PriA HD (open squares), and intact PriA in the presence of PhiX174 virion ssDNA (closed circles) were serially diluted and incubated with FITC-PriB. (B) Wild type PriB (red closed circles), E39A (open squares), W47A/K82A (open circles), R44A (closed squares), E95A (open diamonds), E98A (closed triangles), and K84A (open triangles) were serially diluted and incubated with PriA, FITC-PriB, and PhiX174 virion ssDNA. (C) Wild type PriB (red closed circles), E39A (open squares), R44A (closed squares), K84A (open triangles), and K89A (open circles) were serially diluted and incubated with fluorescein-labeled ssDNA oligonucleotide. Data are reported in triplicate and error bars are one standard deviation of the mean. For the sake of clarity, not all variants that were tested are shown. (D) Orthogonal views of ribbon diagrams of PriB show residues that are important for the PriA:PriB interface of the PriA:PriB:DNA ternary complex (red). Residues that were mutated but have no effect on the PriA:PriB:DNA ternary complex are shown in gray. The top view is the same as in Figure 1. (E) Orthogonal views of ribbon diagrams of PriB show residues that are important for ssDNA binding (blue). Residues that were mutated but have no effect on ssDNA binding are shown in gray.
While these results support the hypothesis that PriA and PriB physically interact in a DNA-dependent manner, we could not determine if the observed interaction involves direct contact between PriA and PriB. To determine if PriA and FITC-PriB form a protein complex on DNA, we tested the ability of unlabeled PriB to compete with FITC-PriB for association with PriA and DNA. If PriA and FITC-PriB independently bind to the DNA then addition of unlabeled PriB would further increase the fluorescence anisotropy since it would bind to the abundance of ssDNA within the large PhiX174 virion ssDNA molecule. However, if PriA and FITC-PriB form a specific complex with the DNA then addition of unlabeled PriB would decrease the fluorescence anisotropy since it would compete with FITC-PriB for binding within the ternary complex. We found that when unlabeled PriB is titrated into a mixture of PriA, FITC-PriB, and PhiX174 virion ssDNA, the fluorescence anisotropy decreases in direct proportion to the concentration of unlabeled PriB that is added (Figure 2B), indicating that the PriA:FITC-PriB:DNA interaction is specific.
To investigate the structural basis for DNA stimulation of the PriA:PriB interaction, we tested the ability of individual PriA domains to interact with FITC-PriB in the absence of DNA. PriA comprises two structurally separable functional domains, an amino-terminal DNA-binding domain (DBD, amino acids 1–198), and a carboxy-terminal helicase domain (HD, amino acids 199–732) (Chen et al., 2004; Tanaka et al., 2002). We mixed purified DBD with FITC-PriB and observed a negligible increase in fluorescence anisotropy, even in the presence of 500-fold molar excess DBD relative to FITC-PriB (Figure 2A), indicating that the DBD and FITC-PriB do not interact under these conditions. However, we observed robust binding of HD to FITC-PriB (Figure 2A). This interaction has a Kd,app of 25.3 +/− 3.4 nM and is similar to the Kd,app of intact PriA for FITC-PriB in the presence of DNA. These results indicate that the binding site on PriA for PriB lies within the HD and suggest that DNA binding by intact PriA might serve to make the PriB binding site accessible.
To test the hypothesis that DNA binding induces a conformational change in PriA that exposes the PriB binding site, we employed limited proteolysis experiments to probe for large-scale structural differences between apo and DNA-bound PriA. PriA contains a hypersensitive trypsin cleavage site that defines the boundary between the DBD and the HD (Chen et al., 2004). If DNA binding alters the spatial orientation of the DBD relative to the HD, we predicted that there will be differences in the accessibility of the hypersensitive trypsin cleavage site in apo PriA compared to the DNA-bound form. We found that trypsin cleavage of PriA into the DBD and HD is greatly stimulated in the presence of DNA, indicating that the region of PriA that connects the DBD and HD is more accessible to trypsin when PriA is bound to DNA (Supplementary Figure 1). These results support the hypothesis that DNA binding by PriA induces a conformational change that could regulate primosome assembly by exposing the PriB binding site.
Identification of PriB’s binding site for PriA
The ability of unlabeled PriB to compete for FITC-PriB within the PriA:FITC-PriB:DNA ternary complex provided an opportunity to identify residues of PriB that are important for the interaction by testing the ability of PriB variants to compete with FITC-PriB within the ternary complex. Based on our initial hypothesis, we predicted that mutation of residues on the surface of PriB opposite the L45 loops would result in PriB variants that would compete poorly with FITC-PriB (Figure 1). Furthermore, as DNA is essential for robust interaction between PriA and PriB, we expected that PriB variants with defects in ssDNA binding would also compete poorly with FITC-PriB in this assay.
Contrary to our initial expectation, none of the PriB variants with alterations in residues opposite the L45 loops shows a defect in its ability to compete with FITC-PriB (Figure 2B and Table 1). Of the 17 PriB variants tested (16 mutants plus wild type PriB), only two are defective in their ability to compete with FITC-PriB: E39A and R44A (Figure 2B and Table 1). We were unable to determine Kd,app values for these variants as both competed poorly even at 200-fold molar excess of competitor relative to FITC-PriB. A third PriB variant, W47A/K82A, also shows a difference in its ability to compete with FITC-PriB relative to wild type PriB competitor. This variant is in a class apart from the E39A and R44A variants and is discussed further below. Residues E39 and R44 lie on the surface of a shallow pocket between PriB monomers near the L23 loops (Figure 2D and Supplementary Figure 2) and are well-conserved among sequenced bacterial priB homologs (Figure 1D).
Table 1.
Apparent dissociation constants for interactions involving PriB.
| PriB variant | Kd,app, nM PriA:PriB:DNA | Kd,app, nM PriB:ssDNA | Kd,app, nM PriB:ssDNA:DnaT | Relative affinity PriB:DnaT |
|---|---|---|---|---|
| wild type | 120.0 +/− 16.2 | 34.6 +/− 7.7 | 355.7 +/− 31.0 | ++++ |
| E39A | ≫ 2000a | 14.7 +/− 5.1 | ≫ 1000a | +++a |
| R44A | ≫ 2000a | 87.9 +/− 5.2a | > 1000a | +++a |
| F42A | 105.8 +/− 34.3 | 72.8 +/− 11.5a | > 1000a | ++++ |
| W47A | 147.7 +/− 19.7 | 112.3 +/− 7.2a | > 1000a | ++a |
| K82A | 73.4 +/− 18.2 | 218.2 +/− 9.4a | ≫ 1000a | +++a |
| W47A/K82A | 105.3 +/− 9.6 | 426.6 +/− 52.3a | ≫ 1000a | +a |
| K84A | 74.3 +/− 11.7 | 96.0 +/− 25.6a | > 1000a | ++++ |
| K89A | 85.3 +/− 17.0 | 194.9 +/− 8.9a | ≫ 1000a | ++++ |
| Q45A | 112.7 +/− 17.5 | 35.7 +/− 5.9 | 352.2 +/− 54.4 | ++++++a |
| H43A | 95.0 +/− 20.4 | 21.5 +/− 3.1 | 303.1 +/− 7.7 | ++++ |
| E32A | 109.8 +/− 10.1 | 36.7 +/− 10.8 | 483.0 +/− 94.8 | +++++ |
| E38A | 105.6 +/− 10.4 | 19.2 +/− 1.7 | 254.2 +/− 37.8 | ++++ |
| E95A | 84.2 +/− 12.5 | 20.5 +/− 3.1 | 327.2 +/− 38.9 | ++++ |
| E98A | 84.0 +/− 5.6 | 23.8 +/− 7.3 | 300.2 +/− 32.1 | ++++ |
| D101A | 118.4 +/− 13.8 | 20.1 +/− 2.4 | 384.2 +/− 109.9 | ++++ |
| D104A | 102.7 +/− 22.2 | 29.1 +/− 5.7 | 635.2 +/− 72.9 | ++++ |
Kd,app values are the mean value derived from three independent experiments and associated uncertainties are one standard deviation of the mean. In the case that a Kd,app could not be determined, a lower limit is indicated with either a “>” symbol or a “≫” symbol, indicating that the actual Kd,app is likely to be greater than, or substantially greater than, the reported value, respectively. For the PriA:PriB:DNA interaction, affinities reported are based on the competition experiments shown in Figure 2B. For the PriB:DnaT interaction, affinities reported are relative to wild type PriB.
Apparent dissociation constants that are significantly different from wild type.
While PriB residues E39 and R44 lie at the dimerization interface, mutation to alanine does not result in a misfolded PriB. Indeed, the E39A variant crystallizes under the same conditions used to crystallize wild type PriB, implying that it is properly folded. We solved the crystal structure of the E39A variant to 2.25 Å resolution and found that it is virtually identical to the wild type PriB crystal structure with a root mean square deviation (RMSD) of 0.8 Å across all polypeptide backbone atoms (Supplementary Figure 2 and Supplementary Table 2). Furthermore, both the E39A and R44A PriB variants migrate through a size exclusion column similarly to wild type PriB (data not shown), suggesting that these alterations do not compromise dimerization. While PriB residues E39 and R44 clearly are important for formation of the PriA:PriB:DNA ternary complex, it remained to be determined what effect mutation of these residues has on PriB’s ssDNA binding activity, as this activity could be critical for PriB’s involvement in the PriA:PriB:DNA ternary complex.
Identification of PriB’s binding site for ssDNA
We performed equilibrium binding studies in which the fluorescence anisotropy of a fluorescein-labeled ssDNA oligonucleotide was measured in the presence and absence of each PriB variant. As expected, our identification of PriB residues that are important for ssDNA binding is consistent with our earlier (but more limited) report and with a recently published co-crystal structure of PriB and ssDNA (Huang et al., 2006; Lopper et al., 2004). The ssDNA binding site on PriB lies along the L45 loops and adjacent beta strands and involves contributions from basic residues such as K82, K84, and K89, and aromatic residue W47 (Figures 1B, 2C, 2E and Table 1). Mutation of residues on the surface of PriB opposite the L45 loops does not disrupt ssDNA binding, and even confers a slight enhancement in some cases (Figures 2C, 2E and Table 1). This is likely due to reducing charge repulsion forces between ssDNA and the sidechains of the acidic residues. The R44A variant shows only a modest defect in ssDNA binding with a Kd,app of 87.9 +/− 5.2 nM, compared to a Kd,app of 34.6 +/− 7.7 nM for wild type PriB. This level of ssDNA binding is similar to two other PriB variants, F42A and K84A, whose Kd,app values for ssDNA are 72.8 +/− 11.5 nM and 96.0 +/− 25.6 nM, respectively. Neither F42A nor K84A is defective in its ability to compete with FITC-PriB in the PriA:FITC-PriB:DNA ternary complex, suggesting that this level of defect in ssDNA binding is not sufficient to explain the failure of the R44A variant to compete with FITC-PriB. Furthermore, the E39A variant binds ssDNA with slightly higher affinity than wild type PriB with a Kd,app of 14.7 +/− 5.1 nM. Therefore, we conclude that residues E39 and R44 most likely make direct contacts between PriA and PriB in the PriA:PriB:DNA ternary complex.
In addition to the E39A and R44A variants, the W47A/K82A double mutant competes with FITC-PriB in a manner that is different from wild type PriB. In this case, however, the double mutant still competes with FITC-PriB, and does so with the same Kd,app as wild type PriB competitor, but it does not decrease the fluorescence anisotropy to the same degree as wild type PriB at high concentrations of competitor (Figure 2B and Table 1). We conclude that these residues are not crucial for the PriA:PriB interface of the PriA:PriB:DNA ternary complex, but that the subtle difference in behavior of this variant compared to wild type PriB is likely due to its severely weakened ssDNA binding activity (Kd,app = 426.6 +/− 52.3 nM). All other PriB variants with defects in ssDNA binding activity, with the exception of R44A, compete at wild type levels with FITC-PriB in the PriA:FITC-PriB:DNA ternary complex (Figure 2B and Table 1). This indicates that defects in PriB’s ssDNA binding activity can be overcome in the context of a PriA:PriB:DNA ternary complex, likely as a result of the increased number of total contacts made possible by crosstalk among three components.
PriB and DnaT physically interact
After ruling out the surface of PriB opposite the L45 loops as a protein interaction site for PriA, we sought to determine if this region is important for interactions between PriB and DnaT. To probe for a direct, physical interaction between PriB and DnaT, we labeled DnaT with FITC and measured the fluoresence anisotropy of FITC-DnaT in the presence and absence of wild type PriB. While we observed an increase in fluorescence anisotropy of FITC-DnaT following addition of PriB, the interaction requires high concentrations of PriB and is not saturable, precluding Kd,app determination (Figure 3A). Thus, while a direct, physical interaction exists between PriB and DnaT, it is clearly not robust in the absence of additional factors.
Figure 3. PriB and DnaT physically interact.
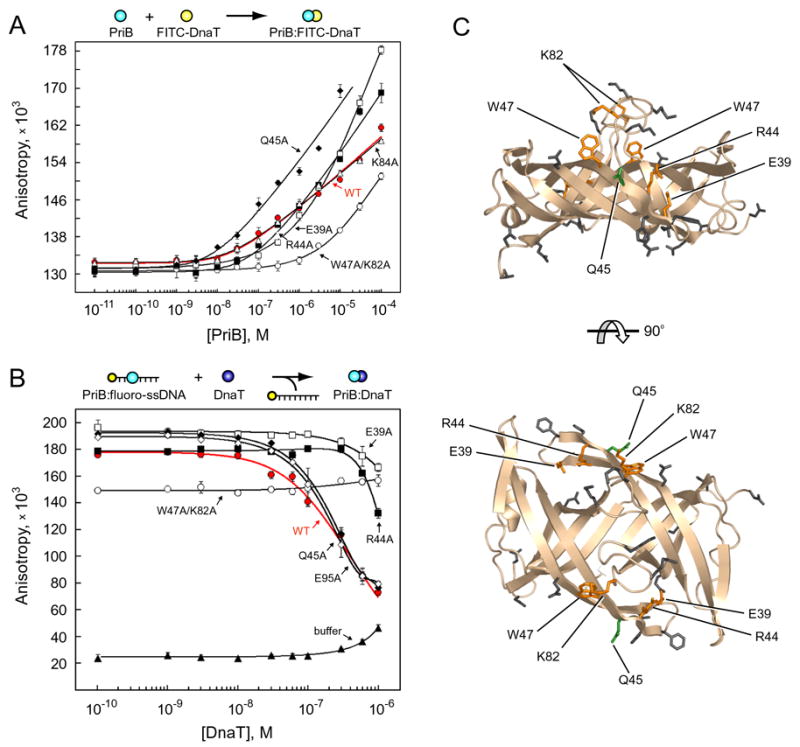
(A) Wild type PriB (red closed circles), W47A/K82A (open circles), E39A (open squares), R44A (closed squares), K84A (open triangles), and Q45A (closed diamonds) were serially diluted and incubated with FITC-DnaT. (B) Wild type PriB (35 nM, red closed circles), W47A/K82A (427 nM, open circles), E39A (15 nM, open squares), R44A (88 nM, closed squares), Q45A (36 nM, closed diamonds), E95A (21 nM, open diamonds), or buffer alone (closed triangles) were incubated with fluorescein-labeled ssDNA oligonucleotide and indicated concentrations of DnaT. Data are reported in triplicate and error bars are one standard deviation of the mean. For the sake of clarity, not all variants that were tested are shown. (C) Orthogonal views of ribbon diagrams of PriB show residues for which mutation to alanine results in defects in DnaT binding (orange). Residue Q45 (green) causes an increase in DnaT binding when mutated to alanine. Residues that were mutated but have no effect on DnaT binding are shown in gray. The top view is the same as in Figure 1.
Identification of PriB’s binding site for DnaT
We analyzed our panel of PriB variants to identify residues that are important for the PriB:FITC-DnaT interaction. Based on our initial hypothesis, we expected that mutation of residues on the surface of PriB opposite the L45 loops would result in PriB variants that would interact weakly with FITC-DnaT (Figure 1). Contrary to our initial expectation, none of the PriB variants with alterations in these residues shows a defect in its ability to interact with FITC-DnaT (Figures 3A, 3C, and Table 1). However, we identified five PriB residues that are important for FITC-DnaT binding. The PriB variant that shows the greatest defect in binding FITC-DnaT is the W47A/K82A variant, requiring concentrations at least two orders of magnitude greater than wild type PriB to reach an equivalent fluorescence anisotropy (Figures 3A, 3C, and Table 1). PriB variants E39A and R44A also show significantly reduced fluorescence anisotropy relative to wild type PriB at concentrations below 1 μM and 300 nM, respectively (Figures 3A, 3C, and Table 1). We identified one gain-of-function PriB variant: Q45A. This variant has enhanced binding to FITC-DnaT, perhaps by as much as two orders of magnitude (Figures 3A, 3C, and Table 1). Residue Q45 lies on the surface of PriB in beta strand 3 and is well conserved among sequenced priB homologs (Figure 1D).
DnaT and ssDNA compete for binding to PriB
Following identification of PriB surface residues important for the PriA:PriB:DNA interaction, the PriB:ssDNA interaction, and the PriB:DnaT interaction, it became clear that significant overlap exists among PriB binding sites for PriA, ssDNA, and DnaT (Figure 4). One prediction that emerges from this observation is that components of replication restart nucleoprotein complexes, such as ssDNA and DnaT, will compete with one another for access to binding sites on PriB. To test this prediction, we determined the effect of adding DnaT to PriB:fluoro-ssDNA complexes. If DnaT and ssDNA compete with one another for binding to PriB, addition of DnaT to PriB:fluoro-ssDNA would result in a decrease in fluorescence anisotropy as fluoro-ssDNA is released from PriB. When DnaT was added to PriB:fluoro-ssDNA complexes, we observed a decrease in fluorescence anisotropy that is proportional to the concentration of DnaT added (Figure 3B). While we did not observe complete inhibition of PriB’s ssDNA binding activity, likely due to DnaT’s ssDNA binding activity at high DnaT concentrations, we were able to derive a Kd,app of 355.7 +/− 31.0 nM for the PriB:fluoro-ssDNA:DnaT interaction (Table 1). It is notable that at these concentrations PriB and DnaT interact only weakly in the absence of ssDNA (Figure 3A). Clearly, DnaT interacts preferentially with a PriB:ssDNA complex compared to apo PriB.
Figure 4. PriA, DnaT, and ssDNA binding sites on PriB partially overlap.
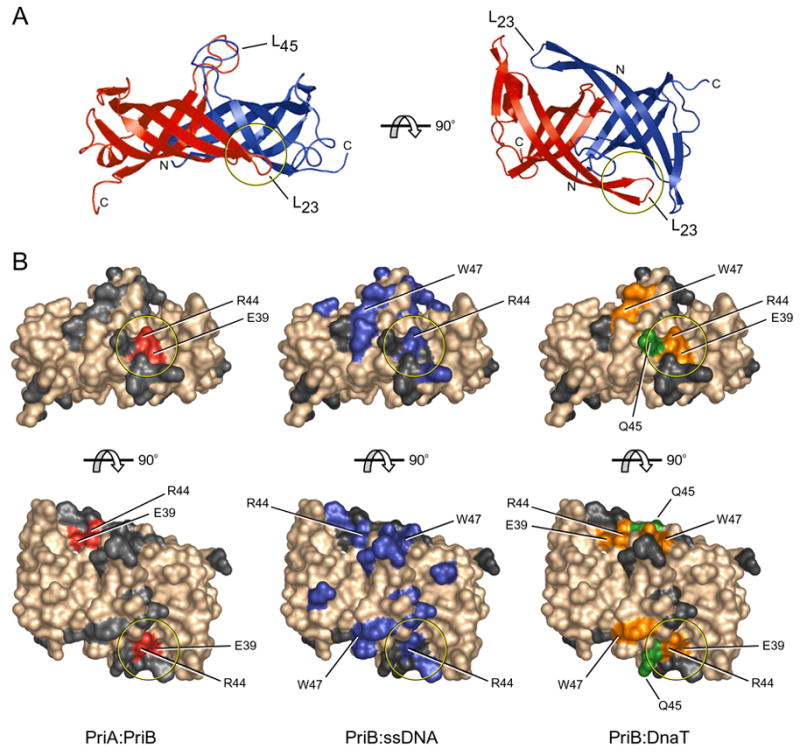
(A) Orthogonal views of ribbon diagrams of the crystal structure of apo PriB are colored and labeled as in Figure 1. (B) Surface renderings of PriB are colored according to the specific interaction in which each residue is involved. Residues colored red (left) are important for the PriA:PriB interface of the PriA:PriB:DNA ternary complex. Residues colored blue (middle) are important for ssDNA binding. Residues colored orange and green (right) are important for DnaT binding as described in Figure 3. Residues colored gray are not involved in interactions with PriA, DnaT, or ssDNA. The top row of surface renderings matches the orientation of PriB at left in (A) and the bottom row matches the orientation of PriB at right in (A). The circle in each rendering highlights important regions of overlap in the binding sites for PriA, DnaT, and ssDNA.
We tested PriB variants for their ability to interact with DnaT in the presence of fluorescein-labeled ssDNA. Not surprisingly, all of the PriB variants that bind FITC-DnaT at wild type levels in the absence of ssDNA show wild type levels of competition between fluoro-ssDNA and DnaT in this experiment, with the exception of the Q45A variant (Figure 3B and Table 1). However, mutations that impair the ability of PriB to bind ssDNA decrease the ability of DnaT to interact with the PriB:fluoro-ssDNA complex, even if the mutated residues are not directly involved in contacting DnaT. For example, PriB variant F42A shows essentially wild type levels of FITC-DnaT binding in the absence of ssDNA (Table 1), suggesting that this residue is not important for the PriB:DnaT interaction. However, DnaT is severely impaired in its ability to compete with fluoro-ssDNA in the context of a PriB:F42A:fluoro-ssDNA complex. The Kd,app for the PriB:F42A:fluoro-ssDNA:DnaT interaction is greater than 1 μM (Table 1). The same relationship exists for residues K84 and K89, and their Kd,app values in the context of a PriB:fluoro-ssDNA:DnaT complex are also greater than 1 μM (Table 1). These results underscore the importance of ssDNA binding by PriB for the PriB:DnaT interaction.
As expected, all of the PriB variants that are defective for FITC-DnaT binding in the absence of ssDNA give rise to a decreased ability of DnaT to interact with the PriB:ssDNA complex. These residues include E39, R44, W47, and K82 and PriB variants that harbor mutations of these residues have Kd,app values greater than 1 μM for the PriB:fluoro-ssDNA:DnaT interaction (Figure 3B and Table 1). Clearly, these residues play important roles in the PriB:DnaT interaction and likely represent direct points of contact between PriB and DnaT.
Interactions involving PriB promote PriA:PriB:DnaT ternary complex formation and DnaB loading
To determine the importance of individual PriB interactions with PriA, DnaT, and ssDNA for primosome assembly, we tested the ability of PriB variants to support assembly of PriA:PriB:DnaT ternary complexes on DNA. We incubated PriA, PriB variants, and FITC-DnaT with PhiX174 virion ssDNA and measured the fluorescence anisotropy of the resulting complexes. As expected, we observed an increase in fluorescence anisotropy when PriA was incubated with DNA, wild type PriB, and FITC-DnaT (Figure 5A). In the absence of PriB we observed a small increase in fluorescence anisotropy at high concentrations of PriA, consistent with previous reports indicating that PriA and DnaT can form a complex on DNA at high protein concentrations (Liu et al., 1996). When PriB variants E39A and R44A were individually incubated with PriA, FITC-DnaT, and DNA, we observed a small increase in fluorescence anisotropy at levels similar to reactions in which PriB is omitted (Figure 5A). This indicates that PriB variants E39A and R44A are defective in facilitating assembly of PriA:PriB:DnaT ternary complexes on DNA, presumably due to disruption of PriB’s individual interactions within the complex.
Figure 5. PriA:PriB:DnaT ternary complex formation and DnaB loading.
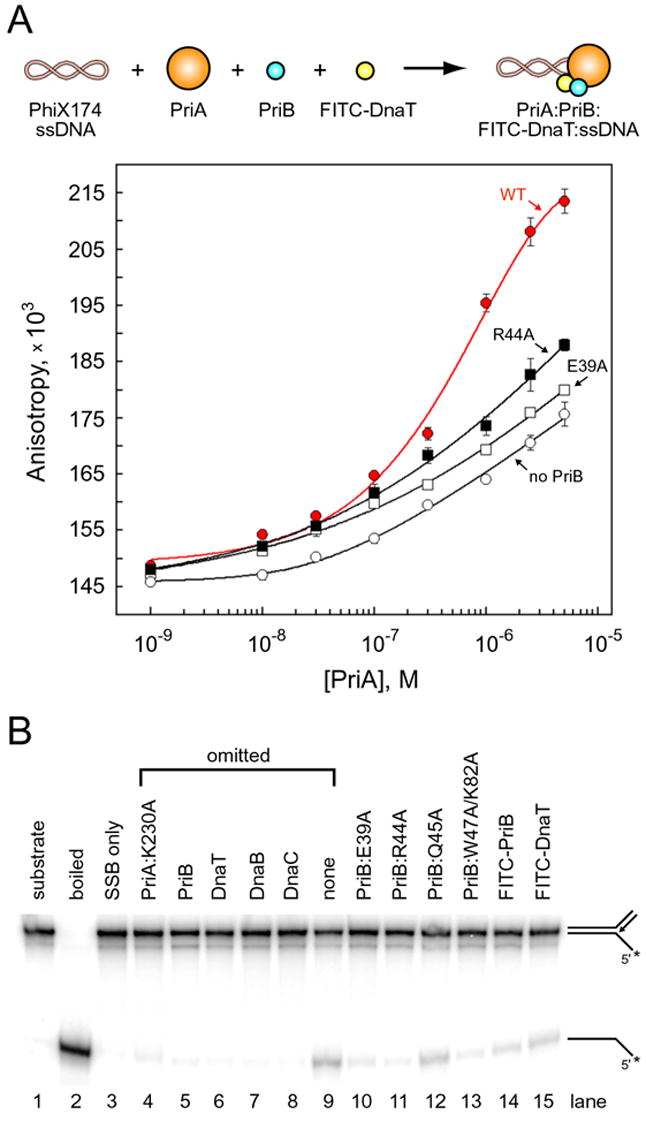
(A) PriA was serially diluted and incubated with PhiX174 virion ssDNA, FITC-DnaT, and either wild type PriB (red, closed circles), E39A (open squares), R44A (closed squares), or no PriB (open circles). Data are reported in triplicate and error bars are one standard deviation of the mean. (B) Lane 1, intact substrate (a fork with 60 bp of duplex DNA, a 38 nucleotide lagging strand arm, and a 38 bp duplex DNA leading strand arm); lane 2, boiled substrate; lane 3, substrate incubated with SSB only. The indicated proteins were omitted from reaction mixtures (lanes 4–9). PriB and DnaT variants were substituted for wild type PriB and DnaT, respectively (lanes 10–15). The PriA:K230A variant was used because it lacks ATPase and helicase activity, therefore any observed DNA unwinding results from DnaB activity. The positions of substrate and product are shown to the right of the gel and the position of the 32P label is indicated with an asterix.
Given that disruption of individual interactions involving PriB is detrimental for PriA:PriB:DnaT assembly on PhiX174 ssDNA, we predicted that the deleterious effects of perturbing PriB’s binding sites would translate to defects in replication restart from DNA structures reminiscent of repaired replication forks. To test this prediction, we examined the ability of PriB variants to support DnaB loading onto a synthetic fork substrate in the presence of PriA, DnaT, and DnaC. We found that DnaB loading and unwinding of a synthetic fork substrate is dependent upon the activities of PriA, PriB, DnaT, DnaB, and DnaC in concordance with a previous report (Heller and Marians, 2005) (Figure 5B). PriB variants E39A, R44A, and W47A/K82A support 9%, 3%, and 7% of wild type PriB activity, respectively, indicating that PriB residues mutated in these variants are important for PriB’s role in facilitating DnaB loading on forked DNA. PriB variant Q45A supports 58% of wild type PriB activity, indicating that the Q45A mutation has only a slight affect on DnaB loading. FITC-PriB and FITC-DnaT promote 27% and 45% of wild type PriB and DnaT activity, respectively, indicating that FITC-labeling does not abolish their ability to facilitate DnaB loading.
Interactions involving PriB are important for replication restart in vivo
To examine the physiological importance of PriB’s interactions with PriA, ssDNA, and DnaT in replication restart, we tested priB variants for their ability to complement a priB-null phenotype in E. coli. In E. coli strains encoding wild type priA, a priB deletion mutant, priB302, does not produce a readily detectable phenotype, likely because alternate PriA-PriC pathways are functional (Sandler et al., 1999). However, when a priB302 deletion is combined with an allele encoding an ATPase- and helicase-deficient PriA, priA300, cells experience a 10-fold increase in SOS induction compared to wild type cells and approximately 11% of the cells are filamented (Supplementary Table 3). These results are consistent with a higher frequency of replication defects in this genetic background compared to wild type E. coli.
We transformed plasmids encoding individual PriB variants into the priA300,priB302 E. coli strain as well as isogenic control strains encoding wild type priA and priB genes and measured the levels of SOS induction and the percentage of filamented cells. We found that a plasmid encoding PriB variant E39A failed to fully complement the priB302 deletion when combined with priA300. This strain has a 7-fold increase in SOS induction compared to wild type cells and 6% of the cells are filamented (Supplementary Table 3). These results indicate that PriB residue E39, shown here to be important for PriB’s interactions with PriA and DnaT and for DnaB loading in vitro, is also important for replication restart in vivo. A plasmid encoding PriB variant R44A failed to yield viable transformants, suggesting that this priB mutation might negatively regulate replication restart (Supplementary Table 3). We have shown that PriB residue R44 is important for interactions with PriA, DnaT, and ssDNA, as well as DnaB loading in vitro, and lethality of the R44A mutation when supplied in trans in E. coli indicates that residue R44 is important in vivo as well. None of the variant priB genes produced a mutant phenotype in E. coli strains encoding wild type priB, indicating that the mutations are not dominant in this genetic background.
Discussion
In this study we have demonstrated direct, physical interactions between individual components of the PriA-PriB-DnaT replication restart primosome and mapped interaction sites on PriB for its binding partners. Furthermore, we have shown that these binding sites are important for PriB function in replication restart in vitro and in vivo. The discovery of partially overlapping binding sites on PriB for PriA, ssDNA, and DnaT (Figure 4) leads us to propose a hand-off mechanism for primosome assembly in which factors trade binding partners as additional components are recruited to a repaired replication fork. This type of hand-off mechanism is an emerging theme among pathways in which multi-protein assemblies interact with and manipulate nucleic acids (Stauffer and Chazin, 2004).
Taken together, our observations lead to a model in which PriA binds to a repaired DNA replication fork or D-loop and undergoes a conformational change, exposing the PriB binding site on the PriA HD. PriB binds to the PriA HD and to ssDNA either pre-existing at the fork or created by the helicase activity of PriA to form a PriA:PriB:DNA ternary complex. Recruitment of DnaT to the PriA:PriB:DNA nucleoprotein complex results in release of ssDNA by PriB, creating a site onto which the replicative helicase can be loaded (Figure 6). This type of hand-off of ssDNA from PriA to PriB, and ultimately to DnaB, provides an attractive model to account for the sequential nature of replisome reloading while affording many potential points for regulation.
Figure 6. Model for primosome assembly.
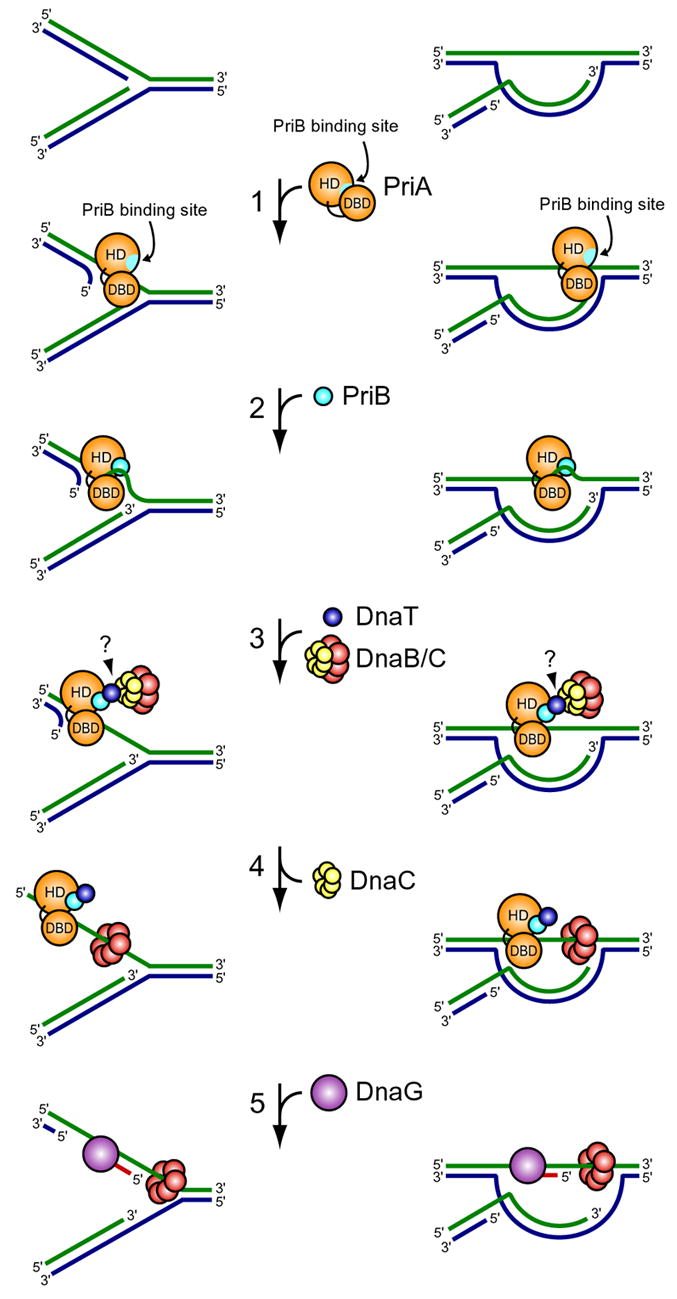
(1) PriA binds a repaired DNA replication fork or D-loop and undergoes a conformational change, exposing the PriB binding site on the PriA HD. (2) PriB binds the PriA:DNA nucleoprotein complex, making contacts with the PriA HD via a shallow pocket at the PriB dimer interface near the L23 loops and contacting ssDNA along the L45 loops of PriB’s OB folds. The PriA:PriB:DNA ternary interaction stabilizes PriA on the DNA and enhances its helicase activity, facilitating unwinding of the nascent lagging strand if one is present (Cadman et al., 2005; Ng and Marians, 1996a). (3) DnaT is recruited to the PriA:PriB:DNA ternary complex and binds PriB through contacts in a shallow pocket at the PriB dimer interface near the L23 loops and through contacts with residues bound by ssDNA. This interaction causes release of ssDNA by PriB. The DnaB/C complex is recruited to the primosome, perhaps through direct contacts with DnaT. (4) DnaB is loaded from the DnaB/C complex onto ssDNA on the lagging strand template. (5) Recruitment of DnaG allows RNA primer synthesis from which the polymerase III holoenzyme can synthesize a nascent lagging strand.
While the first stage of primosome assembly, binding of PriA to a repaired replication fork, is regulated at the level of structure-specific DNA binding by PriA (McGlynn et al., 1997; Nurse et al., 1999), the second stage of primosome assembly, recruitment of PriB, also seems to be highly regulated as PriA and PriB are likely to interact only in the context of a nucleoprotein complex. Our results suggest that the mechanism underpinning the DNA-dependence of the PriA:PriB interaction is occlusion of the PriB binding site on the PriA HD by the DBD. This inhibition is likely abrogated by structural rearrangements induced in PriA upon DNA binding that lead to exposure of the PriB binding site on the HD. While future studies will investigate the nature of the PriB binding site on the PriA HD, we identified two residues on the surface of PriB that play critical roles in the PriA interaction: E39 and R44. These residues lie adjacent to one another at the PriB dimer interface within a shallow pocket near the L23 loops and constitute an unusual binding surface for protein:protein interactions mediated by OB folds.
We have also demonstrated a direct, physical interaction between PriB and DnaT. As with the PriA:PriB interaction, the PriB:DnaT interaction is inherently weak in the absence of ssDNA and this likely represents a third stage of regulation of primosome assembly. Our experiments revealed five PriB residues that are important for the PriB:DnaT interaction in the absence of ssDNA. Curiously, two of these residues, E39 and R44, are also important for the PriA:PriB interaction. Since PriB is a homodimer, it is possible that it simultaneously binds PriA and DnaT in an asymmetric manner. This is consistent with earlier investigations of primosome assembly on DNA containing a PhiX174 PAS sequence (Liu et al., 1996). However, it could also be that competition exists between PriA and DnaT for binding to PriB. While previous studies determined that the composition of the primosome is preserved from assembly through replication of PhiX174 DNA (Ng and Marians, 1996b), competition could cause contacts between PriA, PriB, and DnaT to rearrange during primosome assembly without leading to dissociation of the entire complex.
In addition to demonstrating a direct, physical interaction between PriB and DnaT, our results clearly show that ssDNA binding by PriB stimulates the PriB:DnaT interaction, providing a mechanism whereby recruitment of DnaT to the primosome could be restricted to PriA:PriB:DNA nucleoprotein complexes. There are several mechanisms that can account for this stimulation. First, ssDNA binding by PriB might allosterically alter PriB’s conformation and enhance the binding site for DnaT. Analysis of the PriB:ssDNA co-crystal structure reveals a conformational change in the DnaT binding site within the L23 loop of the PriB:ssDNA complex relative to apo-PriB (Huang et al., 2006) (Figure 1). While this change in conformation of the L23 loop might be a crystallographic artifact, the possibility remains that ssDNA binding by PriB results in a greater degree of flexibility in the L23 loops, allowing them to adopt a conformation that is more amenable to binding DnaT. A second possible explanation for the observed increase in affinity of DnaT for the PriB:ssDNA complex compared to apo-PriB is that ssDNA provides binding sites for DnaT in addition to those contributed by PriB. Thus, ssDNA could initially tether the weakly interacting domains of PriB and DnaT together. Consistent with this interpretation, our results reveal a weak ssDNA binding activity for DnaT (Figure 3B) that could help target DnaT to a PriB:ssDNA complex.
If a ternary complex forms between PriB, ssDNA, and DnaT, how can we account for the decrease in affinity of PriB for ssDNA upon DnaT binding? According to our model, association of DnaT with the PriB:ssDNA complex occurs in at least two steps. In the first step, DnaT is recruited to the primosome through interactions with PriB residues E39 and R44, and through interactions with the ssDNA bound by PriB. Since PriB residue R44 is also involved in making contacts with ssDNA, this first stage of DnaT binding results in a small decrease in PriB’s affinity for ssDNA as DnaT competes with ssDNA for binding to PriB residue R44. We propose that at this stage, PriB residues W47 and K82, shown in this report to be important for DnaT binding, are occluded through interactions with ssDNA and are not available for interacting with DnaT. In the second step, a rearrangement occurs in which DnaT interacts with PriB residues W47 and K82, thereby strengthening its interaction with PriB. Since PriB residues W47 and K82 are involved in binding ssDNA, this transition results in a further decrease in PriB’s affinity for ssDNA and contributes to the release of ssDNA from PriB upon DnaT binding. Thus, we speculate that DnaT acts as a molecular wedge to strip ssDNA from the surface of PriB.
An important question that remains is what is the immediate fate of the ssDNA released by PriB upon DnaT binding? By extrapolating within the framework of a hand-off mechanism for primosome-mediated replisome reloading, it is tempting to speculate that the function of DnaT might be to bind to the PriA:PriB:DNA ternary complex and induce PriB to hand off its ssDNA to the DnaB/C complex. If the DnaB/C complex physically associates with DnaT, it would be in a prime position to receive the ssDNA released by PriB. While the mechanism whereby the DnaB/C complex is targeted to the primosome is unknown at present, future studies will address whether DnaB/C is directly recruited to a repaired DNA replication fork through a physical interaction with DnaT.
In conclusion, we have identified specific protein-protein and protein-nucleic acid interactions among components of the E. coli PriA-PriB-DnaT replication restart primosome. While individual interactions between primosome proteins are inherently weak, they are greatly enhanced by DNA. This provides a possible means of restricting assembly of primosome protein complexes to appropriate regions of the genome. Our findings also provide mechanistic insight into the dynamics of primosome protein assembly that suggests that ssDNA is handed off from one component of the primosome to another as a repaired replication fork is reactivated. These results offer a unique view on origin-independent replisome reloading as a dynamic and highly regulated process.
Experimental Procedures
Fluorescence anisotropy
Fluorescence anisotropy was performed at 25°C with a Beacon 2000 fluorescence polarization system (Invitrogen). For the PriA, PriA DBD, and PriA HD interactions with FITC-PriB, serial dilutions of PriA variants were incubated with 10 nM FITC-PriB in the presence or absence of 4 nM PhiX174 virion ssDNA molecules. Final dilutions were made into 20 mM Tris·HCl pH 8, 15% (v/v) glycerol, 50 mM NaCl, 1 mM MgCl2, 1 mM β-mercaptoethanol, 0.1 g·L−1 bovine serum albumin (BSA). For PriA:FITC-PriB:DNA competition experiments, serial dilutions of PriB variants were incubated with 50 nM PriA, 10 nM FITC-PriB, and 4 nM PhiX174 virion ssDNA. Final dilutions were made into 20 mM Tris·HCl pH 8, 15% (v/v) glycerol, 50 mM NaCl, 1 mM MgCl2, 1 mM β-mercaptoethanol, 0.1 g·L−1 BSA. For the PriB:fluoro-ssDNA interaction, serial dilutions of PriB variants were incubated with 1 nM fluorescein-labeled ssDNA oligonucleotide (30-mer). Final dilutions were made into 20 mM Tris·HCl pH 8, 4% (v/v) glycerol, 50 mM NaCl, 1 mM MgCl2, 1 mM β-mercaptoethanol, 0.1 g·L-1 BSA. For the PriB:FITC-DnaT interaction, serial dilutions of PriB variants were incubated with 10 nM FITC-DnaT. Final dilutions were made into 20 mM Tris·HCl pH 8, 15% (v/v) glycerol, 150 mM NaCl, 1 mM β-mercaptoethanol, 0.1 g·L−1 BSA. For the PriB:fluoro-ssDNA:DnaT interaction, serial dilutions of DnaT were incubated with mixtures of PriB variants and 1 nM fluorescein-labeled ssDNA oligonucleotide (18-mer). The concentration of each PriB variant is the Kd,app as reported in Table 1. Final dilutions were made into 20 mM Tris·HCl pH 8, 4% (v/v) glycerol, 50 mM NaCl, 1 mM β-mercaptoethanol, 0.1 g·L−1 BSA. For the DNA:PriA:PriB:FITC-DnaT interaction, serial dilutions of PriA were incubated with 4 nM PhiX174 virion ssDNA molecules, 250 nM PriB variants, and 10 nM FITC-DnaT. Final dilutions were made into 20 mM Tris·HCl pH 8, 15% (v/v) glycerol, 50 mM NaCl, 1 mM MgCl2, 1 mM β-mercaptoethanol, 0.1 g·L-1 BSA. All Kd,app values were calculated by converting fluorescence anisotropy data to fraction fluorescein-labeled component bound as a function of titrated component concentration. Data are reported in triplicate and associated uncertainties are one standard deviation of the mean.
Supplementary Material
Acknowledgments
This work was supported by a grant from the Shaw Foundation for Medical Research and an American Cancer Society Research Scholar Award to JLK, National Institutes of Health grant GM073495 to ML, and National Institutes of Health grant AI059027 to SJS and RB. X-ray diffraction data were collected at the Advanced Photon Source Beamline 14 BM-C. DnaB and DnaC proteins were generous gifts from Ken Marians. We are grateful to James Berger and members of the Keck lab for critical review of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Cadman CJ, Lopper M, Moon PB, Keck JL, McGlynn P. PriB stimulates PriA helicase via an interaction with single-stranded DNA. J Biol Chem. 2005 doi: 10.1074/jbc.M508521200. [DOI] [PubMed] [Google Scholar]
- Chen HW, North SH, Nakai H. Properties of the PriA helicase domain and its role in binding PriA to specific DNA structures. J Biol Chem. 2004 doi: 10.1074/jbc.M404769200. [DOI] [PubMed] [Google Scholar]
- Cox MM. Recombinational DNA repair of damaged replication forks in Escherichia coli: questions. Annu Rev Genet. 2001;35:53–82. doi: 10.1146/annurev.genet.35.102401.090016. [DOI] [PubMed] [Google Scholar]
- Cox MM, Goodman MF, Kreuzer KN, Sherratt DJ, Sandler SJ, Marians KJ. The importance of repairing stalled replication forks. Nature. 2000;404:37–41. doi: 10.1038/35003501. [DOI] [PubMed] [Google Scholar]
- DeLano WL. The PyMOL Molecular Graphics System. San Carlos, CA, USA: DeLano Scientific; 2002. [Google Scholar]
- Heller RC, Marians KJ. The Disposition of Nascent Strands at Stalled Replication Forks Dictates the Pathway of Replisome Loading during Restart. Mol Cell. 2005;17:733–743. doi: 10.1016/j.molcel.2005.01.019. [DOI] [PubMed] [Google Scholar]
- Heller RC, Marians KJ. Replisome assembly and the direct restart of stalled replication forks. Nat Rev Mol Cell Biol. 2006;7:932–943. doi: 10.1038/nrm2058. [DOI] [PubMed] [Google Scholar]
- Huang CY, Hsu CH, Sun YJ, Wu HN, Hsiao CD. Complexed crystal structure of replication restart primosome protein PriB reveals a novel single-stranded DNA-binding mode. Nucleic Acids Res. 2006;34:3878–3886. doi: 10.1093/nar/gkl536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MS, Marians KJ. Escherichia coli replication factor Y, a component of the primosome, can act as a DNA helicase. Proc Natl Acad Sci U S A. 1987;84:8345–8349. doi: 10.1073/pnas.84.23.8345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Nurse P, Marians KJ. The ordered assembly of the phiX174-type primosome. III. PriB facilitates complex formation between PriA and DnaT. J Biol Chem. 1996;271:15656–15661. doi: 10.1074/jbc.271.26.15656. [DOI] [PubMed] [Google Scholar]
- Liu JH, Chang TW, Huang CY, Chen SU, Wu HN, Chang MC, Hsiao CD. Crystal structure of PriB-a primosomal DNA replication protein of Escherichia coli. J Biol Chem. 2004 doi: 10.1074/jbc.M406773200. [DOI] [PubMed] [Google Scholar]
- Lopper M, Holton JM, Keck JL. Crystal Structure of PriB, a Component of the Escherichia coli Replication Restart Primosome. Structure (Camb) 2004;12:1967–1975. doi: 10.1016/j.str.2004.09.004. [DOI] [PubMed] [Google Scholar]
- McCool JD, Ford CC, Sandler SJ. A dnaT mutant with phenotypes similar to those of a priA2::kan mutant in Escherichia coli K-12. Genetics. 2004;167:569–578. doi: 10.1534/genetics.103.025296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGlynn P, Al-Deib AA, Liu J, Marians KJ, Lloyd RG. The DNA replication protein PriA and the recombination protein RecG bind D-loops. J Mol Biol. 1997;270:212–221. doi: 10.1006/jmbi.1997.1120. [DOI] [PubMed] [Google Scholar]
- Ng JY, Marians KJ. The ordered assembly of the phiX174-type primosome. I. Isolation and identification of intermediate protein-DNA complexes. J Biol Chem. 1996a;271:15642–15648. doi: 10.1074/jbc.271.26.15642. [DOI] [PubMed] [Google Scholar]
- Ng JY, Marians KJ. The ordered assembly of the phiX174-type primosome. II. Preservation of primosome composition from assembly through replication. J Biol Chem. 1996b;271:15649–15655. doi: 10.1074/jbc.271.26.15649. [DOI] [PubMed] [Google Scholar]
- Nurse P, Liu J, Marians KJ. Two modes of PriA binding to DNA. J Biol Chem. 1999;274:25026–25032. doi: 10.1074/jbc.274.35.25026. [DOI] [PubMed] [Google Scholar]
- Raghunathan S, Kozlov AG, Lohman TM, Waksman G. Structure of the DNA binding domain of E. coli SSB bound to ssDNA. Nat Struct Biol. 2000;7:648–652. doi: 10.1038/77943. [DOI] [PubMed] [Google Scholar]
- Sandler SJ. Multiple genetic pathways for restarting DNA replication forks in Escherichia coli K-12. Genetics. 2000;155:487–497. doi: 10.1093/genetics/155.2.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandler SJ, Marians KJ, Zavitz KH, Coutu J, Parent MA, Clark AJ. dnaC mutations suppress defects in DNA replication- and recombination-associated functions in priB and priC double mutants in Escherichia coli K-12. Mol Microbiol. 1999;34:91–101. doi: 10.1046/j.1365-2958.1999.01576.x. [DOI] [PubMed] [Google Scholar]
- Sandler SJ, McCool JD, Do TT, Johansen RU. PriA mutations that affect PriA-PriC function during replication restart. Mol Microbiol. 2001;41:697–704. doi: 10.1046/j.1365-2958.2001.02547.x. [DOI] [PubMed] [Google Scholar]
- Schekman R, Weiner JH, Weiner A, Kornberg A. Ten proteins required for conversion of phiX174 single-stranded DNA to duplex form in vitro. Resolution and reconstitution. J Biol Chem. 1975;250:5859–5865. [PubMed] [Google Scholar]
- Shioi S, Ose T, Maenaka K, Shiroishi M, Abe Y, Kohda D, Katayama T, Ueda T. Crystal structure of a biologically functional form of PriB from Escherichia coli reveals a potential single-stranded DNA-binding site. Biochem Biophys Res Commun. 2005;326:766–776. doi: 10.1016/j.bbrc.2004.11.104. [DOI] [PubMed] [Google Scholar]
- Stauffer ME, Chazin WJ. Structural mechanisms of DNA replication, repair, and recombination. J Biol Chem. 2004;279:30915–30918. doi: 10.1074/jbc.R400015200. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Mizukoshi T, Taniyama C, Kohda D, Arai K, Masai H. DNA Binding of PriA Protein Requires Cooperation of the N-terminal D-loop/Arrested-fork Binding and C-terminal Helicase Domains. J Biol Chem. 2002;277:38062–38071. doi: 10.1074/jbc.M204397200. [DOI] [PubMed] [Google Scholar]
- Wickner S, Hurwitz J. Conversion of phiX174 viral DNA to double-stranded form by purified Escherichia coli proteins. Proc Natl Acad Sci USA. 1974;71:4120–4124. doi: 10.1073/pnas.71.10.4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.