

Abstract
Research to understand and remediate coastal pollution is moving toward a multitiered approach in which traditional enumeration of fecal indicators is accompanied by molecular analysis of a variety of targets. Technology that rapidly detects multiple microbial contaminants would benefit from such an approach. The Luminex® 100™ system is a suspension array that assays multiple analytes rapidly in a single well of a microtiter plate. The ability of the system to simultaneously detect multiple fecal indicating bacteria in environmental samples was tested. Primer/probe sets were designed to simultaneously detect the following fecal indicators: the Bacteroides fragilis group, Enterococcus spp., Escherichia coli & Shigella spp., B. distasonis, and Ent. faecalis. Specificity and sensitivity of the Luminex probes was tested against laboratory cultures. In addition, sequencing, culture plate testing, and specificity testing with environmental isolates were steps taken to validate the function of the assay with environmental samples. Luminex response to cultures and to environmental samples was consistent with sequencing results, suggesting that the technology has the potential to simultaneously detect multiple targets for coastal water quality applications, particularly as progress is made to efficiently extract DNA from water and sediment matrices.
Keywords: Bacteroides, beaches, fecal indicating bacteria, Luminex, recreational water quality
1. Introduction
Microbial contamination of coastal waters is deleterious to the health of humans, marine mammals, and ecosystems with corresponding negative feedback on coastal economies (Dwight et al., 2005). Fecal indicator concentrations have been correlated to the risk of human gastrointestinal (GI) illness when subjects were exposed to waters impacted by point sources of human fecal pollution (Cabelli et al., 1979; Dufour, 1984; Wade et al., 2003). Therefore, fecal indicators have been used to manage human contact with contaminated waters and seafood products. Standard methods for managing water quality include culture-based techniques to assess the concentration of enterococci for marine waters, E. coli or enterococci for freshwater, and total or fecal coliform for shellfish harvesting waters (EPA, 2003; FDA/ISSC, 2003).
Culture methods, however, have a variety of drawbacks. A primary concern is the time (>18 h) between sample collection and result reporting. This lag time creates a risk that humans will be exposed to contaminated water. Alternatively, the lag time creates a risk of false positive reporting (posting that waters are contaminated when clean) because quickly fluctuating conditions (Boehm et al., 2002) give rise to indicator concentrations that are poorly correlated between the sampling and the reporting day (Kim et al., 2004; Whitman and Nevers, 2003). In addition, standard culture methods do not give species level-identification, although this might provide a better indication of fecal contamination (Bonilla et al., 2006). For example, certain species of Enterococcus appear to be strongly associated with fecal pollution (Blanch et al., 2003), but the media used with membrane filtration methods culture a variety of enterococci and some species other than members of the genus Enterococcus (Ferguson et al., 2005).
There is growing consensus that alternate indicators are needed (Griffin et al., 2001). Traditional indicators such as Enterococcus spp. and E. coli may persist or grow in sediment and sand environments (Alm et al., 2006; Anderson et al., 2005; Desmarais et al., 2002; Ferguson et al., 2005; Lee et al., 2006; Whitman et al., 2003; Whitman and Nevers, 2003), thereby creating a source of indicators to the nearshore waters. Regrowth violates a fundamental axiom of indicator theory (National Research Council, 2004). Bacteroides spp. have been suggested as promising alternatives to E. coli and enterococci analysis (Allsop and Stickler, 1985; Bernhard and Field, 2000a; Bernhard and Field, 2000b; Fiksdal et al., 1985; Kreader, 1995), and these anaerobic gut microbes may provide a useful source tracking tool (Noble et al., 2006). Molecular methods primarily have been used for evaluation of this indicator because culturing requires maintenance of anaerobic conditions. Regardless of the indicator, momentum is growing for inclusion of sand or sediment in analysis of recreational water quality (Clean Beaches Council, 2005).
Overall, no single indicator or pathogen is likely to monitor all exposure routes adequately (National Research Council, 2004). Therefore, a suite of indicators may provide a better approach than single-species analysis (Harwood et al., 2005). Rapid detection of multiple species could yield a “fingerprint” of water quality that would be a useful addition to fecal indicator enumeration. Such a multitiered approach (Boehm et al., 2003; Noble et al., 2006) could yield more information about the source of contamination, the potential health risk, and the best strategy for remediation. This approach could benefit environmental research, epidemiological studies, and routine water quality analysis.
Investigators have increasingly turned toward molecular technologies to meet the need for rapid, multiplexed, species-level detection. For example, microarrays have the ability to simultaneously assay hundreds of specific probes, but suspension arrays can offer more flexibility, cost effectiveness, statistical power, and faster hybridization kinetics than planar arrays (Dunbar, 2006). In this study, the Luminex® 100™ suspension array system was adapted to detect several fecal indicators important to water quality analysis.
The Luminex system has primarily been used for clinical applications (Dunbar et al., 2003; Dunbar, 2006), although some environmental applications have been reported (Ellison and Burton, 2005; Spiro et al., 2000; Spiro and Lowe, 2002). Any molecule or chemical group that can be recognized by reactive or complementary functional groups can be immobilized to the surface of the microspheres. As a result, Luminex can detect a variety of compounds, including proteins or nucleic acids. For DNA hybridization assays, DNA is amplified with a biotin-labeled primer, and the amplicons are hybridized to capture probes bound to microspheres. With Luminex xMAP® technology, the microspheres (“beads”) have varying ratios of red and infrared fluorophores, giving a unique spectral address to each set of beads. The beads are coupled to a reporter molecule (streptavidin R-phycoerythin) to generate fluorescence. Microfluidics control the flow of the microspheres though the path of two lasers. The red laser (636 nm) identifies the spectral address of the color-coded beads and the green laser (532 nm) registers whether or not the probe has captured a target. There are 100 microsphere colors available, allowing detection of many targets in a single sample well (Diaz and Fell, 2005; Diaz et al., 2006). Hybridization time is approximately one hour and each well of a 96-well microtiter plate is assayed in approximately 0.47 s; thus the Luminex system has the potential to provide rapid, high-throughput detection of multiple targets for environmental samples.
In this study, we investigated the potential of the Luminex® 100™ system to be adapted to water quality applications. Luminex probes were designed to detect DNA from the following fecal indicating bacteria: the Bacteroides fragilis group, Escherichia coli & Shigella spp., Enterococcus spp., Bacteroides distasonis, and Enterococcus faecalis. The Luminex assay was first tested with laboratory cultures and then with river water, seawater, beach sand, and beach water spiked with raw sewage. Steps taken to verify Luminex results obtained from environmental samples included culturing, amplicon sequencing, specificity testing of selective media, and specificity testing of Luminex probes with environmental isolates. Strategies and obstacles to transferring clinical molecular methodologies such as Luminex to environmental settings are discussed.
2. Materials and methods
2.1. Sample collection
Samples were collected in sterile containers from the following sites in Miami, Florida: the Miami River, Wagner Creek, Bear Cut, and Hobie Beach. The Miami River and Wagner Creek (a tributary of the Miami River) are urbanized, tidally influenced river sites, located downstream from flood control gates. Bear Cut is a manmade channel allowing tidal exchange between Biscayne Bay and the Atlantic Ocean. Hobie Beach is a limited circulation seawater site located within Biscayne Bay (Shibata et al., 2004). Surface water was collected from docks except for Hobie Beach samples, which were collected at knee-deep depth. In addition, raw sewage water was collected from the primary settling tank of a local wastewater treatment plant. Sand was collected from Hobie Beach above the high water mark (“dry sand”) or from the surf zone (“wet sand”). All samples were transported on ice and kept cool until processed within 6 hours of collection.
2.2. Bacterial enumeration
Water samples (including raw sewage) were filtered onto 0.45-μm, 47-mm, cellulose nitrate membrane filters (Whatman) and rinsed with 20 ml phosphate buffered saline (PBS), according to standard membrane filtration protocols (EPA, 2002). Typically, five to seven sample volumes were used in order to obtain one or more plates that were “countable” (4–100 colonies).
For sand, bacteria were extracted by hand-shaking 2 g of sand in 80 ml PBS for 2 min and filtering the water though a 30-μm, 47-mm, nylon net filter (Millipore Corp.) using two, 10 ml rinses to remove all the sand from the container. This process was repeated until the volume of water needed for culture analysis was generated. The resultant water was hand mixed and processed by membrane filtration methods, as described below. Sand aliquots were dried overnight at 110°C and the calculated water content was used to normalize cell densities to g dry weight of sand.
Membrane filtration methods were used to enumerate E. coli (mTEC medium, EPA method 1103.1), enterococci (mEI medium, EPA method 1600), fecal coliform, (mFC medium, Standard Method 9222D), total coliform (M-Endo-LES medium, Standard Method 9222B), and Bacteroides (Bacteroides Bile Esculin Agar -- BBE or Bacteroides Vulgatus Selective Agar -- BVSA). Both BBE and BVSA (Anaerobe Systems, Morgan Hill, CA) are selective and differential media for isolation and presumptive identification of members of the Bacteroides fragilis group. These plates were incubated at 35 °C for 22–48 h using the Gas Pak EZ Anaerobe Container System with GasPak indicators (BD, Franklin Lakes, NJ). Concentrations of fecal indicating bacteria were calculated in terms of colony forming units (CFU) per 100 ml of water or CFU per g dry sand.
2.3. DNA extraction
For DNA analysis, water samples were filtered onto 47-mm, 0.2-μm, hydrophilic polyethersulfone membrane filters (Supor-200, Pall Inc.). Filters were aseptically cut and placed into tubes provided by DNA extraction kits. The Luminex response to DNA extracted from E. coli culture or river water with several commercially available kits was compared. Tested kits included the QBiogene FAST DNA kit (bacteria, plant, and animal protocols), QBiogene FastDNA for Soil (QBiogene, Irvine, CA), DNeasy (Qiagen, Valencia, CA), EpiCenter MasterPure, and EpiCenter SoilMaster (Epicenter, Madison, WI). DNA was typically extracted from filters using FastDNA kits (plant protocol or the kit for soil) (QBiogene, Irvine, CA), according to the manufacture’s instructions. RNase A was added (20 μl of 20 mg/ml stock) following a 10 min vortex. For some extractions with the plant protocol, 25 mg of polyvinylpyrrolidone (PVP-360; Sigma, St. Louis, MO) was added to aid removal of phenolics and alkaloids associated with humic substances.
DNA was extracted from beach sand using the QBiogene FastDNA kit for Soil. Sand was placed directly into the tubes provided or was processed as for culture analysis with subsequent extraction of DNA from the filters, as described for water. Bacterial cultures and samples of raw sewage were pelleted and extracted via spin kit (Promega Wizard) according to the instruction of the manufacturer for gram positive bacteria.
Bacterial enumeration data were used to estimate the number of cells of a particular type that were filtered for DNA extraction. The value was calculated by multiplying the volume of water (or mass of sand) processed through a filter by the concentration of bacteria in the water (or sand), as determined by enumeration on a selective agar medium. This method did not account for DNA contributed by viable-but-nonculturable (VBNC) cells; nonetheless, the value should overestimate the number of cells available for molecular detection because of low DNA extraction efficiencies (see discussion).
2.4. PCR primers and conditions
PCR primers were designed to target rRNA genes of a variety of fecal indicating bacteria based on sequences available in GenBank. Sequences were aligned with the MegAlign program (DNASTAR, Inc.), and conserved areas were analyzed for possible locations of group-specific primers. Primers (Table 1) were inspected with Oligo software (Molecular Biology Insights) for secondary structure and the likelihood of dimer formation. Primers were 5′-biotin-labeled (Integrated DNA Technologies) so that the strand hybridized to the Luminex probe would carry a fluorescent label after coupling to a phycoerythin-streptavidin conjugate.
Table 1.
Description of primers designed to amplify regions of the 16S or 23S rRNA gene of various fecal indicating bacteria
| Primer Name | Primer Description | Sequence 5′ → 3′ | Reference | Primer pair; amplicon size (bp) |
|---|---|---|---|---|
| E135 | Enterobacteriaceae group forward | TGA TGG AGG GGG ATA ACT AC | this work | E135/Univer800; ~ 700 bp |
| Bacterfor | Bacteroides genus forward | AAG GTC CCC CAC ATT GG | this work | Bacterfor/Unirev800; ~ 500 bp |
| EN393* | Enterococcus genus forward | AAG TCT GAC CGA GCA ACG | this work | EN393/EN650; 252 bp |
| SCST748F | Enterococcus genus forward | AGA AAT TCC AAA CGA ACT TG | Haugland et al. 2005 | SCST748F/ENC854R; 92 bp |
| Unirev800* | Bacterial universal reverse | GAC TAC CAG GGT ATC TAA TCC T | this work | |
| EN650 | Enterococcus genus reverse | ACC CTC CCC GGT TGA GCC | this work | |
| ENC854R* | Enterococcus genus reverse | CAG TGC TCT ACC TCC ATC ATT | Haugland et al. 2005 |
biotinylated
Singleplex reactions were used during initial testing, and multiplex reactions were carried out in the final format of the assay. Singleplex reactions typically contained 1X Optimized Dynazyme buffer (containing 1.5 mM MgCl2), 0.2 mM dNTP, 20 pmol of each primer, 2 U Dynazyme EXT DNA polymerase (New England Biolabs, Ipswich, Ma), 1 μl bacterial genomic DNA of concentration ranging from 500 fg to 1 ng, and nuclease-free water for a final volume of 50 μl. Samples were PCR amplified on a PTC-100 thermocycler (MJ Research) with the following conditions: 94°C for 5 min; 30 cycles of 94°C for 1 min, 55°C for 1 min, 72°C for 1 min; a final 8 min extension at 72°C. Multiplex reactions typically contained 25 μl of HotStarTaq MasterMix (Qiagen Corp., Valencia, CA), 80 pmol of Unirev800, 40 pmol each of the other primers used, 3 μl bacterial genomic DNA of concentration ranging from 500 fg to 1 ng, and nuclease-free water for a final volume of 50 μl. Reactions were PCR amplified on a PTC-100 thermocycler (MJ Research) with the following conditions: 94°C for 10 min; 40 cycles of 94°C for 1 min, 55°C for 1 min, 72°C for 1 min; a final 8 min extension at 72°C. The resulting amplicons were about 400 bp to 700 bp in length (Table 1).
2.5. Probe design
Six group- and species-specific Luminex probes were designed to hybridize within the amplicons generated by PCR (Table 1). Probes (Table 2) targeted the following: anaerobic gut microbes belonging to the Bacteroides fragilis group (Bfra3), the fecal indicating bacterium E. coli and bacteria of the genus Shigella (Colinsitu2), the fecal indicating bacteria Enterococcus spp. (Entero23S), the anaerobic bacterium B. distasonis (Bdis3), and the fecal indicating bacterium Ent. faecalis (Efaec2). Probes had Tm’s higher than 55°C, GC contents around 50%, minimum secondary structure and primer dimer potential, and lengths between 20–25 base pairs. Basepair differences conferring specificity were located near the center of the probe (Diaz and Fell, 2004). Probes were manufactured with a C-12 modifier on the 5′ end (Integrated DNA Technologies) to reduce steric hindrance from microspheres.
Table 2.
Description of probes designed to hybridize to regions of the 16S or 23S gene of various fecal indicating bacteria and within the amplicons described in Table 1
| Probe name | Probe Target | Probe Sequence 5′ → 3′ | Reference; modification |
|---|---|---|---|
| Bfra3 | Bacteroides fragilis group16S; bp 586–607 of B. fragilis AB05106 sequence | CAG TTG TGA AAG TTT GCG GCT C | Franks et al. 1998; 3 bp to 5′ end |
| Entero23S | Enterococcus genus 23S; bp 846–874 of E. faecalis AJ295306 sequence | TGG TTC TCT CCG AAA TAG CTT TAG GGC TA | Haugland et al 2005; none |
| Colinsitu2 | E. coli 16S; bp 623–646 of E. coli X80723 sequence | CTG ATA CTG GCA AGC TTG AGT CTC | Regnault et al. 2000; none |
| Bdis3 | Bacteroides distasonis 16S; bp 711–723 of B. distasonis M86695 sequence | ATA TCA CGC AGA ACC CCG ATT G | This work |
| Efaec2 | Enterococcus faecalis 16S; bp 491–494 of E. faecalis AF515223 sequence | GTC AGG GGA CGT TCA GTT ACT AAC | This work |
2.6. Coupling of probes to beads
Each probe was assigned a particular bead “color.” Probes were covalently coupled to 5.6-μm, polystyrene, carboxylated beads (Miraibio, Alameda, CA) by a carbodiimide coupling method (Diaz and Fell, 2004; http://www.miraibio.com) with slight modifications. The uncoupled bead stock was vortexed and sonicated for 20 s and 200 μl (5 × 106 beads) was transferred to a low binding 1.5 ml microcentrifuge tube (USA Scientific, Ocala, FL). The beads were pelleted by centrifugation at 8,000 × g for 3 min and the supernatant was removed. The beads were resuspended in 25 μl of 0.1 M MES (2[N-Morpholino]ethanesulfonic acid), pH 4.5 with vortex and sonication (20 s each). Probe (0.2 nmol) was added and the solution was vortexed. A 1 μg/μl solution of EDC (1-Ethyl-3-[3-dimethylaminopropyl]carbodiimide Hydrochloride) (Pierce, Rockford, IL) was freshly made, 2.5 μl was added to the bead solution and vortexed, and the EDC solution was incubated in the dark, at room temperature for 30 min. A fresh aliquot of EDC solution was made, and the EDC addition and incubation steps were repeated. The microspheres were washed (vortexed, spun for 2 min at 8,000 × g, and supernatant discarded) with 0.5 ml of 0.02% Tween 20 followed by a wash with 0.5 ml of 0.1% SDS, 20% Sarkosyl. The beads were resuspended in 100 μl of TE (Tris-EDTA) buffer (10mM Tris-HCl; 1mM EDTA, pH 8). The probe-coupled beads were stored in the dark at 4°C until use.
2.7. Hybridization of amplicons to Luminex probes
DNA amplified via PCR was hybridized to the probe-coupled beads. During initial development, probes were tested in singleplex bead format (only one bead “color” per well) with amplicons derived from either singleplex or multiplex PCR. Successful probes were next tested in multiplex bead format with amplicons derived from a multiplex PCR reaction. A typical multiplex assay utilized beads coupled to the following probes: Colinsitu2, Bdis3, Bfra3, and Efaec2.
Probe-coupled beads were vortexed and sonicated (20 s each) prior to addition of 1.5× TMAC (tetramethylammonium chloride) Hybridization Buffer (4.5 M TMAC; 0.15% Sarkosyl (N-Lauroylsarcosine), 75 mM Tris-HCl, pH 8.0; 6 mM EDTA, pH 8.0) for a final concentration of 150 beads/μl, or approximately 5000 beads of each type per reaction well. Bead solution (33 μl) and biotinylated amplicons (17 μl for environmental samples, 5 μl for cultures) were placed into the wells of a 96-well microtiter plate. If needed, 1× TE (pH 8.0) was added to achieve a final reaction volume of 50 μl and the reaction was mixed by pipetting.
Reactions were covered to prevent evaporation and denatured in the dark at 95°C for 10 min. Hybridization was initially carried out at 55°C for 15 min, but conditions were later relaxed to 53°C for 60 min to increase signal strength for environmental samples. After hybridization, solutions were pelleted by centrifugation at 2,500 rpm for 3 min, the supernatant was carefully removed to eliminate unbound PCR products, and the pellet was incubated at the hybridization temperature for 5 min. The hybridized amplicons were fluorescently labeled by resuspending the pellet in 75 μl of 1× TMAC solution containing a freshly made 1:250 dilution of streptavidin-R-phycoerythin conjugate (final concentration 4 ng/μl) and incubated in the dark at the hybridization temperature for a further 5 min. To wash unbound label, solutions were pelleted again by centrifugation at 2,500 rpm for 3 min, the supernatant was removed, and the reactions were re-suspended in 75 μl of 1× TMAC solution and incubated at the hybridization temperature for 5 min.
Reactions were placed into the Luminex unit for bead enumeration and quantification of phycoerythin fluorescence by the Luminex 100 analyzer. For each probe, 100 microspheres were analyzed, representing 100 replicate measurements (Diaz and Fell, 2005). The median fluorescence of the 100 measurements (defined here as F) was calculated by the Luminex digital signal processor and proprietary software (v. 1.7). In addition to the 100 microsphere measurements made for each sample, samples were processed in duplicate (including the amplification step).
All reported median fluorescence intensity measurements (MFI) were background corrected (F-Fo), where Fo was the background signal determined from the fluorescence measurement of a blank sample (TE buffer only). A positive Luminex response has been defined as a signal twice the background, after the background has been subtracted (Diaz and Fell, 2005). This description was parameterized here as the corrected median fluorescent intensity (cMFI), where cMFI = (F-Fo)/Fo. Therefore, a positive Luminex response was defined as cMFI>2. For sensitivity experiments, the cMFI value was reported to facilitate visual interpretation of results. Otherwise, values of MFI were reported to show the intensity of fluorescence for comparison to other work (Dunbar et al., 2003).
In addition to a blank, each assay incorporated a positive and negative control. The positive control consisted of known amounts of DNA extracted from cultured targets. The negative control was the PCR blank (all PCR reagents minus DNA template). The fluorescence from the TE blank was consistently higher than the fluorescence from the negative control.
2.8. Culture testing and sensitivity analysis of the Luminex assay
Initial assessment of assay design, cross-reactivity studies, and sensitivity analysis were performed with DNA from cultured organisms. Bacterial cultures were obtained from the American Type Culture Collection (ATCC) or from CultiLoops (Remel). Aerobic Gram-negative bacteria were grown on Nutrient Broth (DIFCO 233000). Enterococcus spp. were grown on LB (Luria-Bertani) Broth (BBL 244620) or Brain Heart Infusion Broth (BBL 211059). Anaerobic bacteria were grown on pre-reduced Chopped Meat Carbohydrate Broth (BBL 297307), except for P. ruminicola, which was grown on pre-reduced Chopped Meat Glucose Broth (BBL 297305).
DNA was isolated from laboratory cultures with a standard spin kit (Promega Wizard, Madison, WI), except for Shigella flexneri in which the DNA was obtained directly from the ATCC. Positive control DNA included E. coli ATCC 25922, S. flexneri ATCC 29903, Ent. faecalis ATCC 29212, B. distasonis ATCC 8503, B. vulgatus ATCC 8482, and B. thetaiotaomicron ATCC 29741. Negative controls included DNA isolated from Enterobacter aerogenes ATCC 13048, Klebsiella pneumoniae ATCC 35657, Enterococcus hirae ATCC 8043, Prevotella ruminicola ATCC 19189, and Pseudomonas aeruginosa ATCC 27853. Environmental isolates also were used to test the specificity of the Luminex assay (see last section of Methods).
For sensitivity analysis, the concentration of DNA extracted from E. coli, Ent. faecalis, B. distasonis, and B. vulgatus was determined by spectrophotometer (Nanodrop Technologies, Wilmington, Delaware). The concentration of genomic DNA from each organism was standardized, serially diluted, and PCR amplified prior to Luminex analysis. Luminex assays were performed at 55°C using 5 μl of a 50 μl PCR reaction. Genome equivalents (analogous to cells) were calculated based on an average base pair molecular weight of 660 and the following genome sizes: E. coli K12-MG1655, 4.64 mb (www.tigr.org); Ent. faecalis V583, 3.2 mb (www.tigr.org); B. distasonis ATCC 8503, 4.28 mb (www.genome.washington.edu/UWGC/); B. vulgatus ATCC 8482, 5.17 mb (www.genome.washington.edu/UWGC).
2.9. Verification of Luminex by DNA sequencing of environmental amplicons
Environmental sequence libraries were constructed to validate Luminex results obtained from environmental samples. DNA was extracted from river and beach water and the 16S rRNA gene was amplified with the primer sets Bacterfor/Unirev800 or EN393/Unirev800 to enrich for sequences related to Bacteroides or Enterococcus, respectively (Table 3). Amplification reactions contained 25 μl of HotStarTaq MasterMix (Qiagen Inc., Valencia, CA), 80 pmol of each primer, 3 μl genomic DNA, and nuclease-free water for a final volume of 50 μl. Reactions were PCR amplified on a PTC-100 thermocycler (MJ Research) with the following conditions: 94°C for 10 min; 40 cycles of 94°C for 1 min, 55°C for 1 min, 72°C for 1 min; a final 8 min extension at 72°C. The amplicons were cloned into competent bacterial cells using the TOPO TA cloning kit (Invitrogen Corp., Carlsbad, CA). Single colonies that contained the insert were grown overnight and the plasmid was purified with the Wizard SV 96 Plasmid Purification System (Promega, Madison, WI). Sequencing reactions were set-up as follows: 1/16 dilution of BigDye 3.1 mix, 1.075× buffer, 0.32 pmol of M13 primer, 6 μl plasmid DNA, and nuclease-free water for a final volume of 10 μl. Reactions were sequenced in both directions on an ABI 3730 capillary sequencer (Applied Biosystems). Sequences were aligned with appropriate sequences in GenBank and with Luminex probes using Sequencher 4.5 (Gene Codes, Ann Arbor, MI) and Vector NTI 9.1 (Invitrogen, Carlsbad, CA). Phylogenetic trees were constructed with PAUP* 4.0 (Sinauer Associates, Inc.) and with Vector NTI 9.1.
Table 3.
Performance of various selective agar media to grow the intended organism from filtered river or beach samples. The per cent match is the ratio of target colonies (identified by BLAST analysis) to the total number of colonies successfully sequenced for that medium
| medium | target | % match (target:total) river | % match (target:total) beacha |
|---|---|---|---|
| MEI | enterococci | 63% (41/65) | 92% (13/14) |
| MTEC | E. coli | 73% (45/62) | 0% (0/14) |
| BBE or BVSA | Bacteroides spp. | 6% (2/35) | 39% (26/67) |
water or sand
2.10. Verification of Luminex by sequencing and colony growth
Culture methods are a logical initial approach to corroborate molecular methods such as Luminex; however, the culture methods themselves require verification for the comparison to be valid. Agar specificity was checked in order to validate the comparison to Luminex data. To verify agar specificity, colonies picked from agar plates of mEI, mTEC, BBE, and BVSA were sequenced. A single colony in sterile water or an aliquot of culture grown overnight from a single colony was used as the PCR template by subjecting the colony solution (25 μl) to 10 min of heat stress at 95°C to release DNA. An aliquot of this (1 to 5 μl) was PCR amplified with the Unifor/Unirev800 primer set (Table 1) using the protocol described above for singleplex reactions. Amplicons were purified with the QIAquick PCR Purification Kit (Qiagen, Valencia, CA) or ExoSAP-IT (USB, Cleveland, OH). Sequencing reactions were performed as described above except that 0.32 pmol of Unifor or Unirev800 primer and 2 μl (~50 ng) of purified amplicon were used. The sequences were submitted for BLAST analysis (http://www.ncbi.nlm.nih.gov) for identification.
Although BBE and BVSA culture plates were incubated anaerobically, samples were typically held 4 – 6 h prior to processing, samples were aerated during membrane filtration, and plates were not sealed in anaerobic conditions until all samples had been processed in order to conserve the number of catalyst packs used. An experiment was performed with B. fragilis culture to investigate the possible effect of sample processing on anaerobic cells. The number of colonies resulting from an aliquot of B. fragilis culture spread directly onto agar plates was compared to the number of colonies grown from the same amount of B. fragilis culture added to 0.2 μm-filtered river water, held for 4 h, and then processed by membrane filtration.
2.11. Luminex specificity testing with DNA sequenced from environmental isolates
Sequencing was used to verify the identity of colonies grown on selective agar media (see above). In turn, the DNA obtained from the environmental isolates was used to test the specificity of Luminex. An aliquot of the colony suspension (5 μl) was amplified by multiplex PCR and subjected to Luminex analysis. Presence and absence of Luminex probe response was compared to the results of the sequencing analysis.
3. Results
3.1 Testing of Luminex primers and probes
Sets of primers and probes were developed to adapt Luminex to detect several groups and species of fecal indicating bacteria. Earliest iterations of the assay used only the single Unifor/Unirev800 primer set in an attempt to avoid multiplex PCR. However, the sensitivity of this approach with environmental samples was unsatisfactory probably because excess nontarget amplicons were generated. Therefore, a multiplex PCR with group-specific primers was designed. Group-specific forward primers increased target amplification while retention of the Unirev800 primer minimized reagent expense and optimization needed upon incorporation of additional targets. The combination of primers (Table 1) and probes (Table 2) targeted the following fecal indicating bacteria: the Bacteroides fragilis group (probe Bfra3), B. distasonis (probe Bdis3), E. coli & Shigella spp. (probe Colinsitu2), Enterococcus spp. (probe Entero23S), and Ent. faecalis (probe Efaec2).
Specificity testing with DNA isolated from laboratory cultures indicated that the Luminex probes Colinsitu2, Bdis3, Bfra3, and Efaec2 functioned as designed (Fig. 1). DNA from E. coli and S. flexneri produced a positive response from the Colinsitu2 probe; DNA from B. distasonis produced a positive response from the Bdis3 probe; DNA from B. vulgatus, B. fragilis and B. thetaiotaomicron produced a positive response from the Bfra3 probe; and Ent. faecalis DNA produced a positive response from Efaec2 probe. No significant cross-hybridization between probes was observed and no response was observed for the negative controls Enterobacter aerogenes, K. pneumoniae, and Enterococcus hirae (Fig. 1) or the negative controls P. ruminicola and Ps. aeruginosa (data not shown).
Fig. 1.
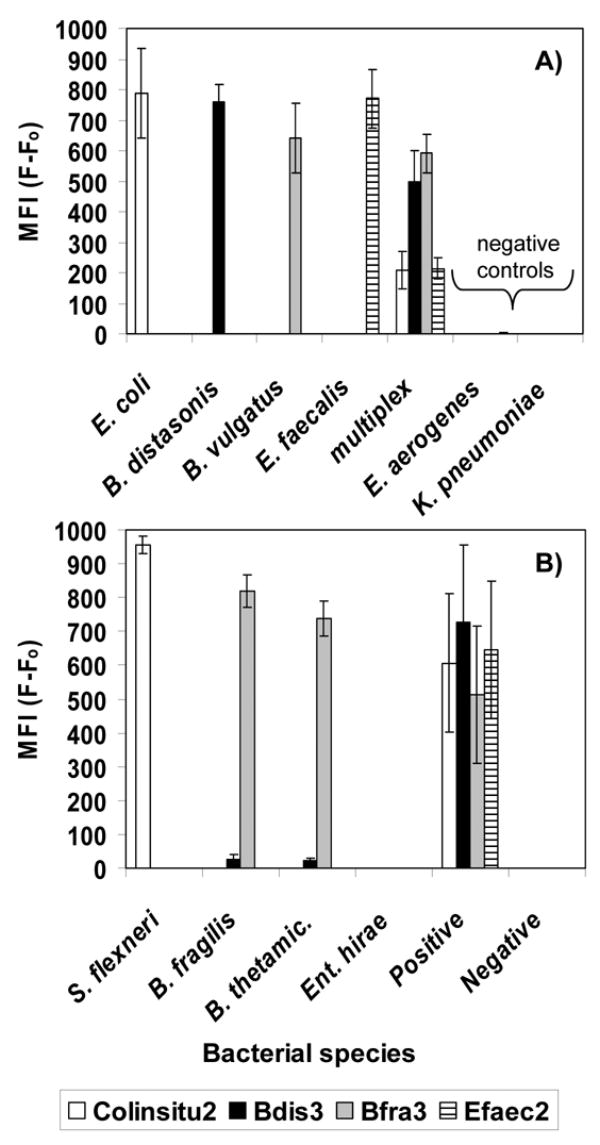
Median fluorescence intensity (MFI) of Luminex probes hybridized against DNA from a variety of bacterial cultures. Results are shown for beads in multiplex format for A) DNA amplified by singleplex or multiplex PCR and hybridized at 55 °C and B) DNA amplified by multiplex PCR containing a single DNA template or a pool of template “positive” and hybridized at 53 °C. “thetamic.” is thetaiotaomicron; “Positive” was a mixture of E. coli, B. distasonis, B. vulgatus, and Ent. Faecalis DNA; “Negative” was a mixture of Enterobacter aerogenes and K. pneumoniae DNA.
The multiplex ability of the Luminex system was successfully demonstrated in several ways. First, amplicons generated in singleplex reactions were hybridized against probes in multiplexed bead format (Fig. 1 A). Next, amplicons generated in multiplex PCR reactions, but that contained only the DNA from a single target, were hybridized against probes in multiplexed bead format (Fig. 1B). Finally, amplicons generated in multiplex PCR reactions, which contained a pool of target DNA, were hybridized against multiplexed probes (Fig. 1A “multiplex”; Fig. 1B “positive”).
3.2. Sensitivity testing with genomic DNA from cultures
The sensitivity of the Luminex assay was estimated by analysis of serially diluted amplicons for DNA extracted from bacterial cultures. For the probes Colinsitu2, Bdis3, Bfra3, and Efaec2, target sequences were detected with as little as 500 fg of genomic DNA as template in the PCR amplification reactions, as determined by cMFI values greater than two (Fig. 2). This amount of genomic DNA corresponded to 97 genome equivalents for E. coli, 137 genome equivalents for Ent. faecalis., 94 genome equivalents for B. distasonis, and 89 genome equivalents for B. vulgatus.
Fig. 2.
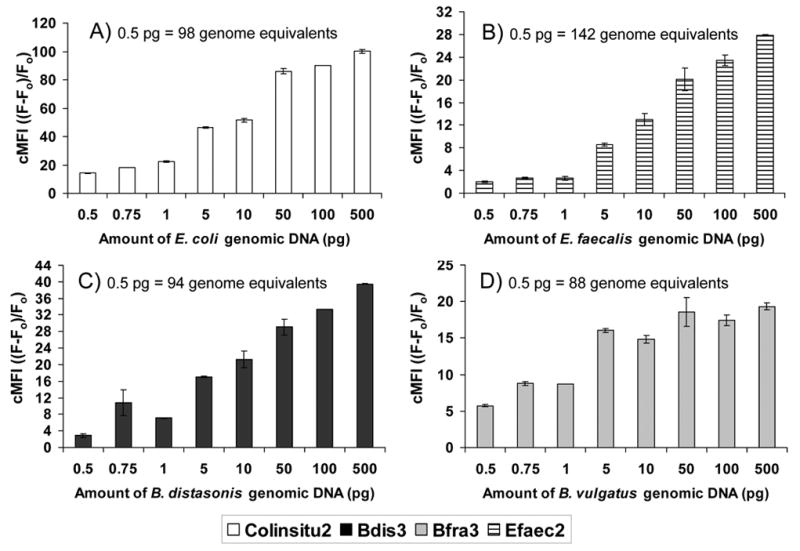
Corrected fluorescence intensity (cMFI) of Luminex probes versus concentration of genomic DNA used in the PCR reaction for sensitivity analysis of A) E. coli B) Ent. faecalis C) B. distasonis and D) B. vulgatus. Column height depicts the mean and bars the range for replicate analysis, which included separate PCR and detection reactions. A positive response is a cMFI value >2.
3.3 Comparison of DNA extraction protocols
Obtaining adequate amounts of clean DNA is a challenging, but critical, step for any molecular technique used with environmental samples. The relative performance of several commercial kits was evaluated by comparing the Luminex response to DNA extracted from E. coli culture or from river water. Evaluation of extraction protocols with the metric of DNA yield was of limited value because yield was not a good predictor of amplification success or Luminex response.
Overall, the Luminex response to DNA extracted from cultures was similar for all the kits and protocols tested (see methods). Specifically, the Luminex response to E. coli did not differ significantly for DNA extracted by the DNeasy kit or by the FastDNA kit using either the animal, bacteria, or plant protocol (data not shown). The Luminex response to river water for the probes Colinsitu2, Bfra3, and Bdis3 did not differ significantly for total DNA extracted by the FastDNA for Soil kit versus the FastDNA kit using the plant protocol. Although the soil kit tended to yield higher mean fluorescence readings than the plant protocol, the range of values was high. The per cent relative range (range/median*100) of the Luminex response (MFI) for replicate samples varied from 0.4–14% for the plant protocol (n=6) but 25–290% for the soil protocol (n=6). Both protocols include substances to neutralize humic substances (Guy et al., 2003; Rivera et al., 2003) commonly found in sub-tropical and tropical aquatic environments that can inhibit downstream DNA applications (Wilson, 1997). RNase A addition with the FastDNA kit appeared to be beneficial, as it slightly incrased the Colinsitu2 probe response to DNA from filtered E. coli (data not shown).
3.4. Luminex response with environmental samples and comparison to culture counts
The Luminex assay worked as designed based on laboratory studies (Fig. 1, Fig. 2); therefore, efforts turned toward environmental samples. Samples of river water, beach water, and beach sand (wet and dry) were tested. Based on culture results, the river site offered the highest number of targets available for DNA extraction (Fig. 3). Results are reported as the number of cells filtered for DNA extraction (see methods) to avoid assumptions regarding the extraction efficiency achieved for each sample. Correspondingly, the Luminex response was strongest for the river samples, and the response was often below detection for the beach samples (Fig. 4).
Fig. 3.
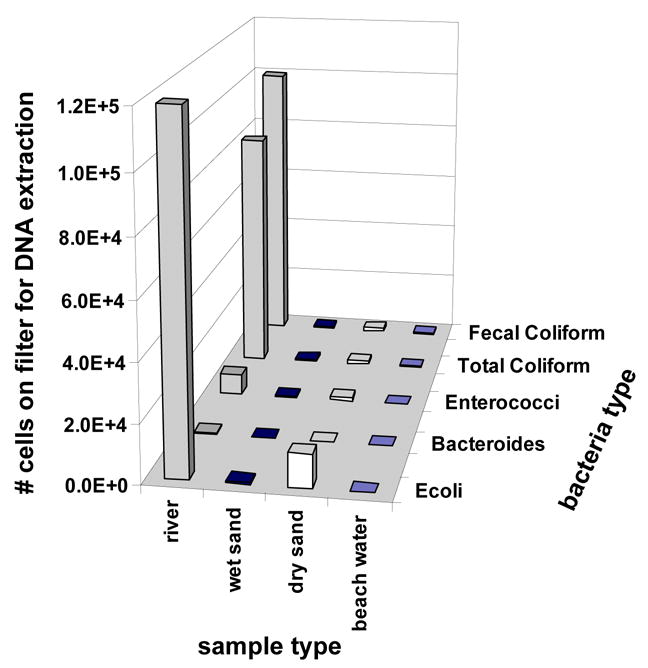
The average number of cells of various bacterial types filtered for DNA extraction for river water (n=15), wet sand (n=6), dry sand (n=6), and beach water (n=11) for samples collected between February 2004 and April 2005. Values of %CV ranged from 52% for Bacteroides spp. in creek water to 240% for E. coli in river water.
Fig. 4.
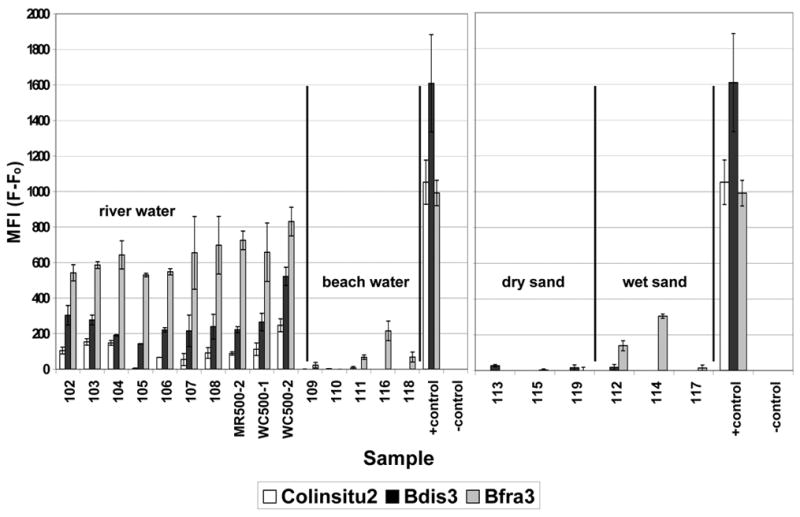
Luminex response to DNA extracted from various types of environmental samples. The positive controls are E. coli, B. distasonis, and B. vulgatus; the negative is a no template control. Background fluorescence (Fo) from TE controls containing no PCR product was removed to yield values of median fluorescence intensity (MFI). Column height depicts the mean and bars the range for replicate analysis, which included separate multiplex PCR and detection reactions.
The highest MFI values for environmental samples resulted from the Bacteroides probes (Fig. 4), despite similar sensitivity observed for all probes with laboratory samples (Fig. 2) and the relatively low numbers of colonies obtained on BBE or BVSA plates (Fig. 3). In contrast to Bacteroides, the sensitivity obtained for the Ent. faecalis Luminex assay in laboratory samples (Fig. 2) was not observed with environmental samples. Sensitivity was greatly enhanced by switching from the primer/probe set targeting the 16S rRNA gene of Ent. faecalis to a Luminex assay for Enterococcus (Fig. 5), which targeted the 23S rRNA gene of Enterococcus spp. (Haugland et al., 2005). Using this assay, sensitivity of 4 genome equivalents was achieved for Ent. faecalis culture (Fig. 5 A), and detection was achieved for samples of raw sewage and for Hobie Beach seawater (Fig. 5B).
Fig. 5.
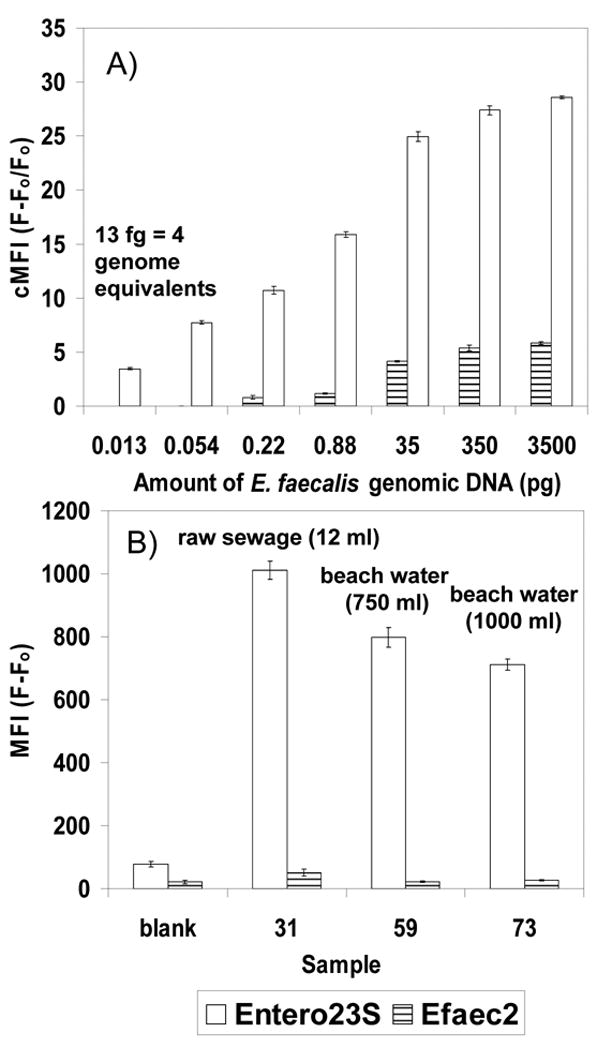
A) Sensitivity of the Luminex assay designed to target the 23S rRNA gene region of Enterococcus (Entero23S) versus the assay designed for the 16S rRNA gene region of Ent. faecalis (Efaec2) for a serial dilution of Ent. faecalis DNA. Results are reported as cMFI to aid interpretation of the detection limit; positive is cMFI >2. B) Comparison of probe sensitivity for samples of raw sewage (#31) and for seawater collected from Hobie Beach (#59, #73). Column height depicts the mean and bars the range for 2 separate detection reactions from a single multiplex PCR. Results are reported as MFI (F-Fo) to show the intensity of the fluorescence obtained.
3.5. Luminex verification through DNA sequencing of environmental amplicons
Probes were designed in silico; however, the molecular microbial diversity of coastal waters is poorly documented. Therefore, environmental sequence libraries were constructed to better understand the sequence environment in which the probes functioned. Despite sensitive and specific detection with laboratory cultures (Fig. 1, Fig. 2), sequence analysis of cloned amplicons generated from EN393/EN650 and EN393/Unirev800 indicated that PCR reactions with environmental samples were not specific enough for the probe to function adequately (data not shown). Therefore, the Luminex probe for enterococci was switched to a 23S target, and sensitivity was significantly increased (Fig. 5).
Sequence analysis of Bacterfor/Unirev800 amplicons verified the presence of target organisms in DNA amplified from environmental samples. However, samples also revealed rich molecular diversity. Many of the obtained sequences were closely related to sequences available in GenBank, but few were exact matches. Amplicons generated from a river sample produced a variety of sequences related to uncultured members of the phylum Bacteroidetes (Fig 6), mostly obtained from environmental water samples (Bernhard et al., 2005). Despite the molecular diversity, the probes appeared to be specific. The only sequence that contained a match to a Luminex Bacteroides spp. probe (sequence G7; Bfra3 probe) was most closely related to an uncultured bacterium obtained from the human gut (496/498 bp identical) (Fig. 6).
Fig. 6.
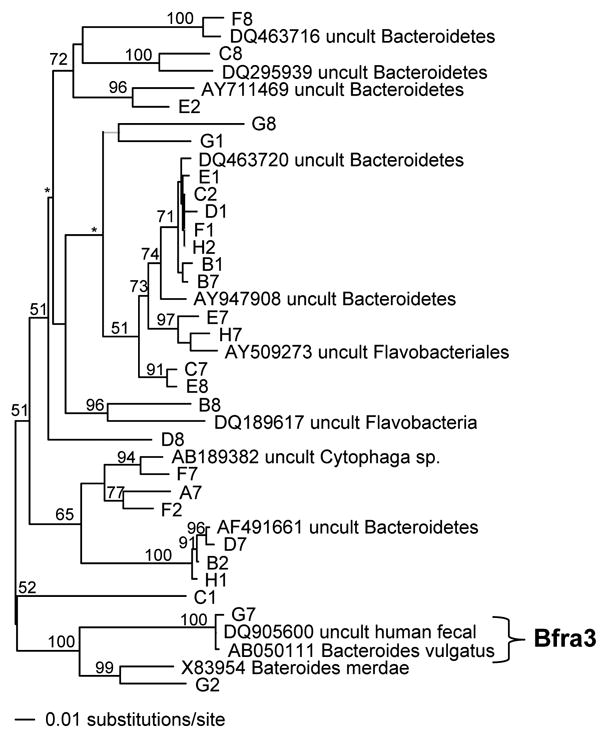
Phylogram of PAUP* neighbor joining tree analysis of 16S rRNA gene sequences (498 positions) obtained by Bacterfor/Unirev 800 amplification of river water followed by shot-gun cloning. The sequence labeled G7 contained a perfect match to the Bfra3 probe. Bootstrap values are shown on internal nodes for values greater than 50, as space allowed; “*” denotes visible nodes with bootstrap values <50.
3.6. Verification of plate specificity used for comparison to Luminex
Agar specificity was tested to validate the comparison of Luminex molecular probing to plate counts. DNA was sequenced from colonies picked from selective agar media. Plate performance varied depending on the type of water used (Table 3). Overall, sequence analysis of the environmental isolates highlighted the difficulty of comparing molecular to culture-based methods.
For colonies picked from mEI agar selective for enterococci, sequences related to the genus Enterococcus were obtained from 68% of the colonies grown from river, beach, or sand samples with remaining colonies yielding sequences related to Streptococcus spp. The mEI agar performed better with beach samples than with river samples; 92% of the sequences from beach water or sand were related to Enterococcus spp. in comparison to only 63% for river samples. In contrast, mTEC isolation of E. coli was better for river samples, with 73% of colonies from river samples yielding sequences related to E. coli versus 0% for beach water or sand (Table 3).
The BBE and BVSA agar was not highly specific when used with environmental samples. Only 27% of the sequences obtained from BBE or BVSA plates were related to species of Bacteroides. Agar performance was worse for the river samples (6%) than for beach water or sand (39%) (Table 3). Common nontarget sequences were closely related to Aeromonas spp. and Cetobacterium somerae, particularly from the river sites (Fig. 7). The sequences that were related to Bacteroides spp. (Fig 7) were most closely related to uncultured bacterial clones isolated from feces (for example, >97% identity to ATCC# AY684430 for ~600 bp of sequence), rather than named members of the genus Bacteroides. In addition, five sequences were obtained from colonies grown on BVSA plates from samples of raw sewage or seawater spiked with 10% raw sewage. For these colonies presumptively identified as Bacteroides spp. (black with black halo), 3/5 colonies yielded sequences related to species of Bacteroides. One sequence was related to Klebsiella spp. and the other appeared to be a mix of Enterobacter and Bacteroides, despite the use of streak plating for isolation.
Fig. 7.
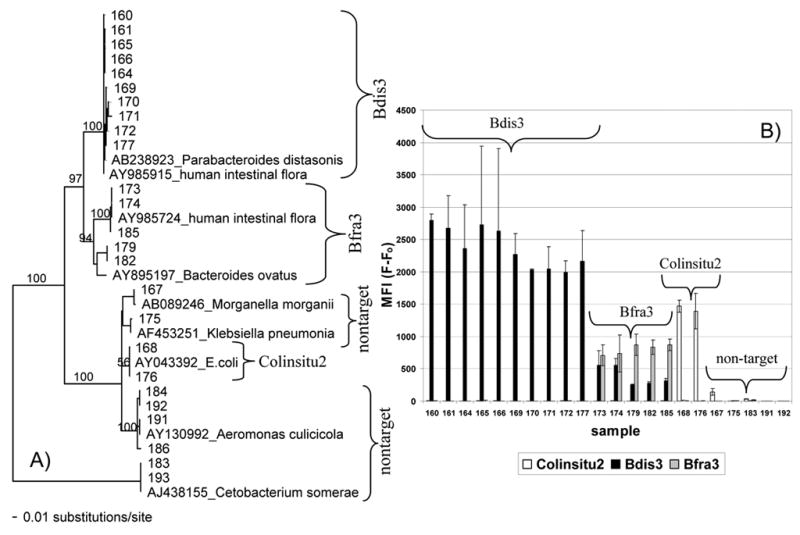
A) Phylogram of PAUP* neighbor joining tree analysis of 16S rRNA sequences (444 positions) obtained by Unifor/Unirev800 amplification of bacterial colonies isolated from BBE or BVSA media used to isolate Bacteroides spp. from river and beach water. Bootstrap values are shown on internal nodes for values greater than 50, as space allowed. B) Luminex response for samples included in the phylogram, except for samples 184, 186, and 193, which were not tested. Column height depicts the mean and bars the range for replicate analysis, which included separate multiplex PCR and detection reactions. Groups labeled with the probe names Colinsitu2, Bfra3, and Bdis3 (see Table 2) contained sequence that matched the probe within 0–1 bp. The group labeled Bfra3 also contained sequences with 2 bp mismatches to the Bdis3 probe.
Although culture plates were incubated anaerobically, membrane filtration was performed aerobically with typical sample hold times of 4 – 6 h. A brief test suggested that the sample processing that occurred prior to anaerobic incubations may have caused an underestimate of the Bacteroides spp. available in these experiments. An aliquot of B. fragilis culture spread directly onto agar yielded 334 CFU. In contrast, the same volume added to 0.2 μm-filtered river water and held for 4 h prior to aerobic membrane filtration resulted in 2.4 CFU after 24 hours of incubation.
3.7. Luminex verification through specificity testing with environmental isolates
DNA from environmental isolates that had been characterized by sequencing was used to test the specificity of the Luminex assay. Luminex results were consistent with colony sequences for colonies picked from BBE and BVSA plates and streaked for isolation (Fig. 7). The Bdis3 probe showed the strongest response, with high signal strength only seen for organisms containing a maximum of 1 bp variation within the region of the probe. Response to the Bfra3 probe was only observed for organisms containing the sequence that exactly matched the probe. These organisms also produced a response from the Bdis3 probe. Alignment of the sequences showed a 2 bp variation in the region of the Bdis3 probe. A similar pattern of Bfra3 and Bdis3 response (data not shown) was seen for high numbers of B. fragilis loaded onto membrane filters (≥6 ×105 cells), perhaps from low levels of contamination or intraspecies variability, particularly with a relatively low hybridization temperature of 53° C. However, no response to Bdis3 was observed for lower numbers of B. fragilis (≤6×104 cells). Cross-hybridization with high concentrations of target also has been observed in other Luminex assays (Dunbar et al., 2003).
Luminex probes did not appear to cross-hybridize with nontarget species isolated from BBE or BVSA plates (Fig. 7). Although sequencing sometimes identified black with black halo colonies to be Aeromonas spp. or Cetobacterium somerae rather than Bacteroides spp., the DNA from these colonies did not yield false positive results via Luminex (Fig. 7). In addition, Luminex analysis revealed that colonies isolated from BBE or BVSA agar often were not pure. The Luminex results (not shown) were consistent with electrophoresis gels showing multiple banding patterns. In addition, repeated streak-plating confirmed contamination by loss of black-haloed colonies with only a white contaminant surviving (usually Enterobacteriaceae). For example, the Colinsitu2 probe responded to colonies picked from BBE or BVSA plates, and analysis showed that these colonies were indeed E. coli-like organisms containing a sequence that exactly matched the probe (Fig. 7). For colonies grown on Bacteroides media, Luminex analysis or gel electrophoresis better revealed colony impurities than did sequencing.
4. Discussion
The underlying hypothesis of this project was that sensitive and rapid molecular technologies designed for clinical applications could be transferred to environmental water quality research and monitoring. There are a number of promising technologies on the horizon to meet the needs of biodetection (Deisingh and Thompson, 2004), and many emerging technologies are being tried in the arena of recreational water quality (Noble and Weisberg, 2005; Tallon et al., 2005). Technology validation in seawater is complicated by the wealth of molecular diversity that is little documented to date. Although Luminex and other molecular technologies are a promising means to improve analysis of coastal water quality, environmental samples offer critical challenges, some of which are discussed below. Challenges to molecular technologies include choice of appropriate targets, the rich molecular diversity inherent in environmental samples, and difficulties in delivering adequate amounts of clean DNA to the detector system.
4.1. Verification of Luminex performance with environmental samples
The Luminex® 100™ system has been used to rapidly detect a variety of proteins and nucleic acids, primarily in clinical applications (Dunbar et al., 2003; Dunbar, 2006). This study adapted the Luminex system to detect several common fecal indicating bacteria (Table 2; Fig. 1, Fig. 2) to assess whether this system might be useful to multitiered analyses of coastal water quality (Noble et al., 2006).
Members of the genus Bacteroides were most easily detected by Luminex in environmental samples (Fig 4), but verification of results was complicated by difficulties with culturing techniques. Several steps were undertaken to assess the performance of the Luminex assay with environmental samples. Verification procedures included the following: A) cloning and sequencing of environmental amplicons (Fig. 6); B) testing of agar specificity and growth conditions to validate the comparison between Luminex molecular probing and culture results (Table 3; Fig. 7); C) specificity testing of the Luminex probes against characterized DNA obtained from environmental isolates (Fig. 7). Overall, sequence analysis indicated that the Luminex primer/probe sets hybridized as designed and detected targets of interest in environmental samples.
Sequence analysis also highlighted the difficulty of comparing molecular to culture-based methods. DNA sequence data and target identification via selective media were sometimes poorly matched (Table 3). In the case of Bacteroides spp., Luminex appeared to be more sensitive and specific than culture techniques. For example, DNA sequencing suggested that Aeromonas spp. and Cetobacterium somerae from environmental samples sometimes yielded false positive results when grown on BBE or BVSA agar and that isolated Bacteroides colonies were often contaminated with other organisms. In contrast, Luminex probes designed for Bacteroides spp. were selective and did not cross-react with the DNA of these contaminants (Fig. 7), and Luminex probes were able to reveal colony contamination (data not shown). In addition, the data suggested that the sample processing used here (> 4 h hold times and aeration during membrane filtration) may have caused cell counts from BBE or BVSA culturing (Fig. 3) to be an underestimate relative to the available molecular signature (Fig. 4). DNA from damaged, viable-but-nonculturable (VBNC), and dead cells can be detected with molecular methods (Lipp et al., 2003; Lleo et al., 2003; Noble et al., 2006); therefore, Luminex can overcome the obstacle of oxygen sensitivity because live cells are not necessary for analysis. Overall, these results suggest that molecular analysis offers a convenient way to detect gut anaerobes such as Bacteroides spp. and imply that culture methods should not be the only means by which molecular technologies are validated.
4.1. The challenge of choosing appropriate targets
The use of fecal indicating bacteria to manage coastal water quality has come under increasing scrutiny (Griffin et al., 2001). As the search for the ideal indicator continues, a convenient attribute of the Luminex technology is the ability to readily modify the probes incorporated into the assay. Rather than a single indicator, a combination of targets may provide a water quality fingerprint useful to scientists and managers, particularly for devising remediation strategies. In this case, technologies that offer multi-target detection, such as Luminex, will be desired. Appropriate targets may include alternate indicators and source tracking markers (Bernhard et al., 2003; Carson et al., 2003; Noble et al., 2003; Scott et al., 2005) or detection of pathogens or markers of pathogenicity (Blanch et al., 2003; Fuhrman et al., 2005; Garcia-Aljaro et al., 2004; Shetab et al., 1998). Ultimately, epidemiologic studies are needed to establish the correlation between health effects and a given determinant of microbiological water quality (National Research Council, 2004).
4.3. The challenge to overcome environmental molecular diversity
Rich molecular diversity is a fundamental difference between environmental and clinical samples. Fecal contaminants in surface waters are rare in comparison to the indigenous microbial population; therefore, methods of sample concentration based on size exclusion produce samples containing large amounts of nontarget organisms. Initial assay development attempted to use a single amplification with universal primers, analogous to what has been done in DNA hybridization assays for harmful algae (Scorzetti and Fell, unpublished data; Goodwin et al., 2005). For the bacterial indicators used here, this approach did not prove successful (data not shown), presumably because samples predominately contained nontarget organisms that prevented adequate amplification of the intended targets and thus decreased the sensitivity of the assay. A multiplex PCR with group-specific forward primers (Table 1) increased the efficiency of target over non-target amplification. Switching from the 16S rRNA target to the 23S rRNA target for Enterococcus also appeared to reduce the background of nontarget amplicons, allowing detection of Enterococcus spp. with environmental samples (Fig. 5 A, B).
Investigations are on-going to assess the benefit of even more specific priming. For example, use of the Bacterfor/Unirev800 primer set generates amplicons to which both Bfra3 and Bids3 probes can hybridize. This permits cross-hybridization and competition for fluorescent beads if DNA is not pure or if hybridization temperatures are too low (Fig. 7), but generation of more specific amplicons would remove this possibility. Although multiplex PCR requires that reactions be optimized for each set of targets, a variety of strategies exist to optimize multiplex PCR reactions (Elnifro et al., 2000; Schoske et al., 2003). In addition, programs to support the design of multiplex PCR reactions (Rachlin et al., 2005) have become available.
Even under the most specific and stringent DNA amplification and hybridization conditions, environmental molecular diversity makes it difficult to prove correspondence of detected and intended targets. For example, the environmental sequence diversity observed in amplicons (Fig. 6) and media (Fig. 7) designed to select Bacteroides species implied that environmental samples harbored sequences identical and closely related to those found in feces, but did not prove that those sequences originated from feces. Although Bacteroides spp. are considered to be of animal origin (O’Sullivan et al., 2002), the diversity of sequences present in environmental waters and sediments is poorly catalogued. The sites sampled here are known to have inputs from bird, dog, and probably human fecal sources. The majority of the sequences were closely related to, but not exact matches to sequences available in GenBank. These Bacteroides-like sequences may or may not have originated recently from animal guts. Unfortunately, a simple survey of “naturally occurring” sequences has little power to resolve this issue because even “pristine” sites will contain animal fecal inputs.
Protein analysis may provide an approach to increase sensitivity of the assay by reducing the background of nontarget organisms amplified during PCR of environmental samples. Fundamentally, bacterial species differ from one another because of variability at the DNA sequence level. This sequence variability confers differences at the protein level which further translate into differences at the physiological level. Identification of these unique proteins can lead to the development of taxon-specific diagnostic targets. Databases are available that catalog proteins thought to be unique to certain organisms. One such database is CUPID (Core and Unique Protein Identification System), which can provide a list of proteins specific to genus, species, or strain (Mazumder et al., 2005). The gene sequences of these proteins could be mined for primer and probe sequences to be used in the Luminex detection assay.
4.3. The need to improve nucleic acid extraction
The ability to efficiently extract nucleic acids from environmental samples constrains the utility of any molecular technology for water quality research and management, including the one described here. Sample concentration is necessary to achieve detection of fecal indicators and human pathogens in coastal water samples, despite the sensitivity offered by molecular methods (Fig. 2; Fig. 5). Low extraction efficiencies and PCR inhibition (Wilson, 1997) create a gap between the sensitivity achieved with diluted genomic DNA and what can be achieved with environmental samples (Fig. 4). Variable and low DNA extraction efficiencies from soil have been observed with standard spin kits (0–43% recovery) (Mumy and Findlay, 2004). We have observed DNA recoveries of 0–2% and 0–31% for filtered Ent. faecalis processed by the FastDNA spin kit and the crude lysate method of Haugland et al. (2005), respectively (Goodwin et al. unpublished results). Simply increasing the sample amount does not compensate for inefficient DNA extraction because concentration of PCR inhibitors results in diminishing returns -- increasing the level of concentration often reduces the likelihood of achieving detection. Progress in the area of upstream sample processing is needed to realize the full potential of emerging technologies (Noble and Weisberg, 2005), particularly as molecular assays attempt to move away from indicators to rare targets such as human pathogens.
4.4. Summary
The Luminex system provides high throughput, multiplex detection that allows a matrix of targets to be identified in samples (Fig 4). A benefit of Luminex is that it returns numeric results, unlike a series of electrophoresis gels, which is convenient for statistical analysis. In addition, probe hybridization adds specificity in comparison to PCR alone. The system is versatile in that the user can easily add or subtract probes (bead sets) for specific studies. The entire procedure, including water filtration and nucleic acid extraction can be performed in a matter of hours; therefore, Luminex assays have the potential to meet the needs of water quality research. If DNA extraction and cataloguing of environmental sequence diversity can be improved, suspension arrays could provide a useful tool to multitiered analysis (Boehm et al., 2003; Noble et al., 2006) and epidemiologic studies of recreational water and sand quality.
Acknowledgments
We are indebted to Mara Diaz for providing Luminex training. We thank Claudia Garcia for her work with field sampling, cultures, DNA extraction, and sequencing. Lisa Matragrano helped with sequencing of sewage and sewage-spiked samples. We acknowledge the help of Anjali Sardeshmukh and the Recreational Microbes team at the University of Miami Center of Excellence for Oceans and Human Health with field sampling and sample processing. Financial support is gratefully acknowledged from the NOAA Center of Excellence for Oceans and Human Health at the Hollings Marine Laboratory, the Cooperative Institute of Estuarine and Environmental Technology (CICEET), and funding provided in part by the NSF-NIEHS Oceans and Human Health Center Program (NSF 0432368 and NIEHS P50 ES12736). This research was carried out in part under the auspices of the Cooperative Institute for Marine and Atmospheric Studies (CIMAS), a joint institute of the University of Miami and the National Oceanic and Atmospheric Administration, cooperative agreement #NA17RJ1226.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allsop K, Stickler DJ. An assessment of Bacteroides fragilis group organisms as indicators of human faecal pollution. Journal of Applied Bacteriology. 1985;58:95–99. doi: 10.1111/j.1365-2672.1985.tb01433.x. [DOI] [PubMed] [Google Scholar]
- Alm EW, Burke J, Hagan E. Persistence and potential growth of the fecal indicator bacteria, Escherichia coli in shoreline sand at Lake Huron. J Great Lakes Res. 2006;32:401–405. [Google Scholar]
- Anderson KL, Whitlock JE, Harwood VJ. Persistence and differential survival of fecal indicator bacteria in subtropical waters and sediments. Applied and Environmental Microbiology. 2005;71:3041–3048. doi: 10.1128/AEM.71.6.3041-3048.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernhard AE, Field KG. Identification of nonpoint sources of fecal pollution in coastal waters by using host-specific 16S ribosomal DNA genetic markers from fecal anaerobes. Applied and Environmental Microbiology. 2000a;66:1587–1594. doi: 10.1128/aem.66.4.1587-1594.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernhard AE, Colbert D, McManus J, Field KG. Microbial community dynamics based on 16S rRNA gene profiles in a Pacific Northwest estuary and its tributaries. FEMS Microbiology Ecology. 2005;52:115–128. doi: 10.1016/j.femsec.2004.10.016. [DOI] [PubMed] [Google Scholar]
- Bernhard AE, Field KG. A PCR assay to discriminate human and ruminant feces on the basis of host differences in Bacteroides-Prevotella genes encoding 16S rRNA. Applied and Environmental Microbiology. 2000b;66:4571–4574. doi: 10.1128/aem.66.10.4571-4574.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernhard AE, Goyard T, Simonich MT, Field KG. Application of a rapid method for identifying fecal pollution sources in a multi-use estuary. Water Research. 2003;37:909–913. doi: 10.1016/s0043-1354(02)00384-6. [DOI] [PubMed] [Google Scholar]
- Blanch AR, Caplin JL, Iversen A, Kuhn I, Manero A, Taylor HD, Vilanova X. Comparison of enterococcal populations related to urban and hospital wastewater in various climatic and geographic European regions. Journal of Applied Microbiology. 2003;94:994–1002. doi: 10.1046/j.1365-2672.2003.01919.x. [DOI] [PubMed] [Google Scholar]
- Boehm AB, Furhman JA, Mrse RD, Grant SB. Tiered approach for identification of a human fecal pollution source at a recreational beach: Case study at Avalon Bay, Catalina Island, CA. Environmental Science and Technology. 2003;37:673–680. doi: 10.1021/es025934x. [DOI] [PubMed] [Google Scholar]
- Boehm AB, Grant SB, Kim JH, Mowbray SLO, Mcgee Cd, Clark CD, Foley DM, Wellman DE. Decadal and shorter period variability of surf zone water quality at Huntington Beach, California. Environmental Science and Technology. 2002;36:3885–3892. doi: 10.1021/es020524u. [DOI] [PubMed] [Google Scholar]
- Bonilla TD, Nowosielski K, Esiobu N, McCorquodale DS, Rogerson A. Species assemblages of Enterococcus indicate potential sources of fecal bacteria at a south Florida recreational beach. Marine Pollution Bulletin. 2006;52:807–810. doi: 10.1016/j.marpolbul.2006.03.004. [DOI] [PubMed] [Google Scholar]
- Cabelli VJ, Dufour AP, Levin MA, McCabe J, Haberman PW. Relationship of microbial indicators to health effects at bathing beaches. American Journal of Public Health. 1979;69:690–696. doi: 10.2105/ajph.69.7.690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson CA, Shear BL, Ellersieck MR, Schnell JD. Comparison of ribotyping and repetitive extragenic palindromic-PCR for identification of fecal Escherichia coli from humans and animals. Applied and Environmental Microbiology. 2003;69:1836–1839. doi: 10.1128/AEM.69.3.1836-1839.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clean Beaches Council. 2005 State of the Beach Report: Bacteria and Sand, A National Call to Action. 2005 http://www.cleanbeaches.org/ewebeditpro/items/O31F6450.pdf.
- Deisingh AK, Thompson M. Biosensors for the detection of bacteria. Canadian Journal of Microbiology. 2004;50:69–77. doi: 10.1139/w03-095. [DOI] [PubMed] [Google Scholar]
- Desmarais TR, Solo-Gabriele HM, Palmer CJ. Influence of soil on fecal indicator organisms in a tidally influenced subtropical environment. Applied and Environmental Microbiology. 2002;68:1165–1172. doi: 10.1128/AEM.68.3.1165-1172.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz MR, Fell JW. High through-put detection of pathogenic yeasts in the genus Trichosporon. Journal of Clinical Microbiology. 2004;42:3696–3706. doi: 10.1128/JCM.42.8.3696-3706.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz MR, Fell JW. Use of a suspension array for rapid identification of the varieties and genotypes of the Cryptococcus neoformans species complex. Journal of Clinical Microbiology. 2005;43:3662–3672. doi: 10.1128/JCM.43.8.3662-3672.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz MR, Boekhout T, Theelen B, Bovers M, Cabanes FJ, Fell JW. Microcoding and flow cytometry as a high-throughput fungal identification system for Malassezia species. Journal of Medical Microbiology. 2006;55:1197–1209. doi: 10.1099/jmm.0.46630-0. [DOI] [PubMed] [Google Scholar]
- Dufour AP. Bacterial indicators of recreational water quality. Can J Public Health. 1984;75:49–56. [PubMed] [Google Scholar]
- Dunbar SA, Vander Zee CA, Oliver KG, Karem KL, Jacoboson JW. Quantitative, multiplexed detection of bacterial pathogens: DNA and protein application of the Luminex LabMAP™ system. Journal of Microbiological Methods. 2003;53:245–252. doi: 10.1016/s0167-7012(03)00028-9. [DOI] [PubMed] [Google Scholar]
- Dunbar SA. Applications of LuminexR xMAP™ technology for rapid, high-throughput multiplexed nucleic acid detection. Clinica Chimica Acta. 2006;363:71–82. doi: 10.1016/j.cccn.2005.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwight RH, Fernandez LM, Baker DB, Semenza JC, Olson BH. Estimating the economic burden from illnesses associated with recreational coastal water pollution--a case study in Orange County, California. Journal of Environmental Management. 2005;76:95–103. doi: 10.1016/j.jenvman.2004.11.017. [DOI] [PubMed] [Google Scholar]
- Ellison CK, Burton RS. Application of bead array technology to community dynamics of marine phytoplankton. Marine Ecology Progress Series. 2005;288:75–85. [Google Scholar]
- Elnifro EM, Ashshi AM, Cooper RJ, Klapper PE. Multiplex PCR: optimization and application in diagnostic virology. Clinical Microbiology Reviews. 2000;13:559–570. doi: 10.1128/cmr.13.4.559-570.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EPA. EPA-821-R-02-022. United States Environmental Protection Agency; 2002. Method 1600: Enterococci in water by membrane filtration using membrane-Enterococcus indoxyl-B-D-glucoside agar (mEI) [Google Scholar]
- EPA. Guidelines establishing test procedures for the analysis of pollutants; Analytical methods for biological pollutants in ambient water; Final Rule. Federal Register V68, No. 139 40 CFR Part 136. 2003:43272–43283. [Google Scholar]
- FDA/ISSC. National Shellfish Sanitation Program Guide for the Control of Molluscan Shellfish 2003. US Food and Drug Administration & the Interstate Shellfish Sanitation Conference.2003. [Google Scholar]
- Ferguson DM, Moore DF, Getrich MA, Zhowandai MH. Enumeration and speciation of enterococci found in marine and intertidal sediments and coastal water in southern California. Journal of Applied Microbiology. 2005;99:598–608. doi: 10.1111/j.1365-2672.2005.02660.x. [DOI] [PubMed] [Google Scholar]
- Fiksdal L, Maki JS, LaCroix SJ, Staley JT. Survival and detection of Bacteroides spp., prospective indicator bacteria. Applied and Environmental Microbiology. 1985;49:148–150. doi: 10.1128/aem.49.1.148-150.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrman JA, Liang X, Noble RT. Rapid detection of enteroviruses in small volumes of natural waters by real-time quantitative reverse transcriptase PCR. Applied and Environmental Microbiology. 2005;71:4523–4530. doi: 10.1128/AEM.71.8.4523-4530.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Aljaro C, Muniesa M, Jofre J, Blanch AR. Prevalence of the stx2 gene in coliform populations from aquatic environments. Applied and Environmental Microbiology. 2004;70:3535–3540. doi: 10.1128/AEM.70.6.3535-3540.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin KD, Cotton SA, Scorzetti G, Fell JW. A DNA hybridization assay to identify toxic dinoflagellates in coastal waters: detection of Karenia brevis in the Rookery Bay National Estuarine Research Reserve. Harmful Algae. 2005;4:411–422. [Google Scholar]
- Griffin DW, Lipp EK, McLaughlin MR, Rose JB. Marine recreation and public health microbiology: quest for the ideal indicator. BioScience. 2001;51:817–825. [Google Scholar]
- Harwood VJ, Levine AD, Scott TM, Chivukula V, Lukasik J, Farrah SR, Rose JB. Validity of the indicator organism paradigm for pathogen reduction in reclaimed water and public health protection. Applied and Environmental Microbiology. 2005;71:3163–3170. doi: 10.1128/AEM.71.6.3163-3170.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haugland RA, Siefring SC, Wymer LJ, Brenner KP, Dufour AP. Comparison of Enterococcus measurements in freshwater at two recreational beaches by quantitative polymerase chain reaction and membrane filter culture analysis. Water Research. 2005;39:559–568. doi: 10.1016/j.watres.2004.11.011. [DOI] [PubMed] [Google Scholar]
- Kim JH, Grant SB, Mcgee Cd, Sanders BF, Largier JL. Locating sources of surf zone pollution: a mass budget analysis of fecal indicator bacteria at Huntington Beach, California. Environmental Science and Technology. 2004;38:2626–2636. doi: 10.1021/es034831r. [DOI] [PubMed] [Google Scholar]
- Kreader CA. Design and evaluation of Bacteroides DNA probes for the specific detection of human fecal pollution. Applied and Environmental Microbiology. 1995;61:1171–1179. doi: 10.1128/aem.61.4.1171-1179.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CM, Lin TY, Lin CC, Kohbodi GA, Bhatt A, Lee R, Jay JA. Persistence of fecal indicator bacteria in Santa Monica Bay beach sediments. Water Research. 2006;40:2593–2602. doi: 10.1016/j.watres.2006.04.032. [DOI] [PubMed] [Google Scholar]
- Lipp EK, Rivera ING, Gil AI, Espeland EM, Choopun N, Louis VR, Russek-Cohen E, Huq A, Colwell RR. Direct detection of Vibrio cholerae and ctxA in Peruvian coastal water and plankton by PCR. Applied and Environmental Microbiology. 2003;69:3676–3680. doi: 10.1128/AEM.69.6.3676-3680.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lleo MdM, Bonato B, Signoretto C, Canepari P. Vancomycin resistance is maintained in enterococci in the viable but nonculturable state and after division is resumed. Antimicrobial Agents and Chemotherapy. 2003;47:1154–1156. doi: 10.1128/AAC.47.3.1154-1156.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazumder R, Natale DA, Murthy S, Thiagaranjan R, Wu CH. Computational identification of strain-, species-, and genus-specific proteins. BMC Bioinformatics. 2005;6:279–287. doi: 10.1186/1471-2105-6-279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumy KL, Findlay RH. Convenient determination of DNA extraction efficiency using an external DNA recovery standard and quantitative-competitive PCR. Journal of Microbiological Methods. 2004;57:259–268. doi: 10.1016/j.mimet.2004.01.013. [DOI] [PubMed] [Google Scholar]
- National Research Council. Committee on Indicators for Waterborne Pathogens. National Academy of Sciences; Washington, DC: 2004. Indicators for Waterborne Pathogens. [Google Scholar]
- Noble RT, Weisberg SB. A review of technologies being developed for rapid detection of bacteria in recreational waters. Journal of Water and Health. 2005;3:381–392. doi: 10.2166/wh.2005.051. [DOI] [PubMed] [Google Scholar]
- Noble RT, Griffith JF, Blackwood AD, Fuhrman JA, Gregory JB, Hernandez X, Liang X, Bera AA, Schiff K. Multitiered approach using quantitative PCR to track sources of fecal pollution affecting Santa Monica Bay, California. Applied and Environmental Microbiology. 2006;72:1604–1612. doi: 10.1128/AEM.72.2.1604-1612.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble RT, Weisberg SB, Leecaster MK, McGee CD, Ritter K, Walker KO, Vainik PM. Comparison of beach bacterial water quality indicator measurement methods. Environmental Monitoring and Assessment. 2003;81:301–312. [PubMed] [Google Scholar]
- O’Sullivan LA, Weightman AJ, Fry JC. New degenerate Cytophaga-Flexibacter-Bacteroides-specific 16S ribosomal DNA-targeted oligonucleotide probes reveal high bacterial diversity in river taff epilithon. Applied and Environmental Microbiology. 2002;68:201–210. doi: 10.1128/AEM.68.1.201-210.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rachlin J, Ding C, Cantor C, Kasif S. MuPlex: multi-objective multiplex PCR assay design. Nucleic Acids Research. 2005;33:W544–W547. doi: 10.1093/nar/gki377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoske R, Vallone PM, Ruitberg CM, Butler JM. Multiplex PCR design strategy used for simultaneous amplification of 10 Y chromosome short tandem repeat (STR) loci. Analytical and Bioanalytical Chemistry. 2003;375:333–343. doi: 10.1007/s00216-002-1683-2. [DOI] [PubMed] [Google Scholar]
- Scott TM, Jenkins TM, Lukasik J, Rose JB. Potential use of a host associated molecular marker in Enterococcus faecium as an index of human fecal pollution. Environmental Science and Technology. 2005;39:283–287. [PubMed] [Google Scholar]
- Shetab R, Cohen SH, Prindiville T, Tang YJ, Cantrell M, Rahmani D, Silva J., Jr Detection of Bacteroides fragilis enterotoxin gene by PCR. Journal of Clinical Microbiology. 1998;36:1729–1732. doi: 10.1128/jcm.36.6.1729-1732.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata T, Solo-Gabriele HM, Fleming LE, Elmir S. Monitoring marine recreational water quality using multiple microbial indicators in an urban tropical environment. Water Research. 2004;38:3119–3131. doi: 10.1016/j.watres.2004.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiro A, Lowe M. Quantitation of DNA sequences in environmental PCR products by a multiplexed, bead-based method. Applied and Environmental Microbiology. 2002;68:1010–1013. doi: 10.1128/AEM.68.2.1010-1013.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiro A, Lowe M, Brown D. A bead-based method for multiplexed identification and quantitation of DNA sequences using flow cytometry. Applied and Environmental Microbiology. 2000;66:4258–4265. doi: 10.1128/aem.66.10.4258-4265.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallon P, Magajna B, Lofranco C, Leung KT. Microbial indicators of faecal contaminatin in water: a current perspective. Water, Air, and Soil Pollution. 2005;166:139–166. [Google Scholar]
- Wade TJ, Pai N, Eisenberg JNS, Colford JM., Jr Do U.S. Environmental Protection Agency water quality guidelines for recreational waters prevent gastrointestinal illness? A systematic review and meta-analysis. Environmental Health Perspectives. 2003;111:1102–1109. doi: 10.1289/ehp.6241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitman RL, Nevers MB. Foreshore sand as a source of Escherichia coli in nearshore water of a Lake Michigan beach. Applied and Environmental Microbiology. 2003;69:5555–5562. doi: 10.1128/AEM.69.9.5555-5562.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitman RL, Shively DA, Pawlik H, Nevers MB, Byappanahalli MN. Occurrence of Escherichia coli and enterococci in Cladophora (Chlorophyta) in nearshore water and beach sand of Lake Michigan. Applied and Environmental Microbiology. 2003;69:4714–4719. doi: 10.1128/AEM.69.8.4714-4719.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson IG. Inhibition and facilitation of nucleic acid amplification. Applied and Environmental Microbiology. 1997;63:3741–3751. doi: 10.1128/aem.63.10.3741-3751.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]