

Summary
Meiosis is a specialized cell division that halves the genome complement, producing haploid gametes/spores from diploid cells. Proper separation of homologous chromosomes at the first meiotic division requires the production of physical connections (chiasmata) between homologs through recombinational exchange of chromosome arms after sister-chromatid cohesion is established but before chromosome segregation takes place. The events of meiotic prophase must thus occur in a strict temporal order, but the molecular controls coordinating these events have not been well elucidated. Here we demonstrate that the budding yeast cyclin-dependent kinase Cdc28 directly regulates the formation of the DNA double-strand breaks that initiate recombination by phosphorylating the Mer2/Rec107 protein and thereby modulating interactions of Mer2 with other proteins required for break formation. We propose that this function of Cdc28 helps to coordinate the events of meiotic prophase with each other and with progression through prophase.
Keywords: Meiosis, recombination, Mer2, Spo11, double strand breaks, cell cycle regulation
Introduction
During the first meiotic division, homologous maternal and paternal chromosomes are segregated. In most organisms, homologs must be physically connected to ensure their proper alignment on the meiosis I spindle (reviewed in Page and Hawley, 2003; Petronczki et al., 2003). Physical connections are achieved by exchange of chromosome arms through chiasma formation, while sister chromatid cohesion is maintained distal to the exchange. Failure to establish chiasmata causes missegregation at meiosis I and results in meiotic cell death or aneuploid gametes. Although the cues triggering meiotic entry differ between organisms, the orderly progression of events leading to chiasma formation and many of the proteins involved are widely conserved (reviewed in Gerton and Hawley, 2005; Zickler and Kleckner, 1999).
Each chiasma is an ensemble of local DNA exchange (a crossover recombination product) plus higher order chromosome structure changes at the site of recombination (exchange of chromosome axes and local separation of sister chromatids) (Jones, 1987; Page and Hawley, 2003; Zickler and Kleckner, 1999). To build this ensemble, different processes must occur in the correct order: premeiotic DNA replication and establishment of sister chromatid cohesion must precede initiation of recombination and, in turn, these events must precede dissolution of arm cohesion and chromosome segregation (Page and Hawley, 2003; Petronczki et al., 2003). Thus, several processes must be temporally coordinated with each other and with progression through meiotic prophase. The molecular basis for this coordination is not well understood.
One candidate for coordinating events of meiotic prophase is the cell-cycle regulator, cyclin-dependent kinase (CDK; in budding yeast, Cdc28). Indeed, Cdc28 is required for exit from the pachytene stage (Shuster and Byers, 1989) and, along with its B-type cyclin activators, Clb5 and Clb6, for premeiotic DNA replication, analogous to its role in promoting S phase during vegetative growth (Benjamin et al., 2003; Stuart and Wittenberg, 1998). Although the relevant substrates of Cdc28 remain to be identified, these findings demonstrate that CDK does regulate at least some of the processes required for chiasma formation.
It has proven more difficult to establish whether CDK also directly promotes meiotic recombination. In the absence of Clb5 and Clb6, the double-strand breaks (DSBs) that initiate recombination are not formed (Smith et al., 2001). Because several conditions that block premeiotic S phase also block DSB formation (Borde et al., 2000; Simchen et al., 1976), it appeared that DSB formation might be indirectly promoted by CDK through control of DNA replication (reviewed in Baudat and Keeney, 2001; Smith et al., 2001). However, other studies in budding and fission yeasts suggest that recombination is not absolutely dependent on premeiotic DNA replication (e.g., Hochwagen et al., 2005; Murakami and Nurse, 2001), raising the alternative possibility that recombination is more directly dependent on CDK activity.
Here, we demonstrate that CDK directly promotes DSB formation and we identify a phosphorylation target important in this process. DSBs are formed by the conserved Spo11 protein plus accessory factors that in budding yeast include Mer2/Rec107 (hereafter Mer2) (Keeney, 2001). Phosphorylation of Mer2 by cyclin-CDK complexes modulates interactions of Mer2 with other DSB proteins and is critical for DSB formation. These results suggest that regulation of DSB formation by CDK is part of the mechanism that coordinates recombination with progression through meiotic prophase.
Results
Mer2 is chromatin-associated and is phosphorylated during meiosis
MER2 was isolated in two screens for genes involved in meiotic recombination (Engebrecht et al., 1990; Malone et al., 1991). Null mer2 mutants do not make DSBs, leading to spore inviability. The predicted 35.5 kDa protein has no obvious motifs aside from a region of heptad repeats (Rockmill et al., 1995). MER2 homologs are present in database sequences from six more Saccharomyces species and two other ascomycetes, with 25–90% amino acid sequence identity (Supplemental Figure 1A and 1B).
An intriguing feature of MER2 is its meiosis-specific regulation: its transcript has an intron with a non-canonical 5′ splice site that is efficiently spliced only during meiosis under the control of Mer1, a meiosis-specific RNA-binding protein (Engebrecht et al., 1991; Nandabalan and Roeder, 1995). Each putative MER2 ortholog has an intron within the same poorly conserved region (Supplemental Figure 1A). Splicing regulation may be conserved, because the five homologs most closely related to S. cerevisiae have the same non-canonical splice site (Supplemental Figure 1C) and because Mer1 homologs are also present (data not shown). Conservation of sequence and (apparently) regulation suggests that Mer2 has a similar role in meiosis in these organisms.
To characterize S. cerevisiae Mer2, we epitope-tagged the protein at the C-terminus with multiple copies of the myc epitope. MER2myc complemented a mer2 null mutant and supported nearly normal levels of meiotic recombination (see Experimental Procedures). Given its role in DSB formation, Mer2 is expected to localize to meiotic chromosomes. Nuclear spreads were double-stained for Mer2myc and Zip1, a component of the synaptonemal complex (SC) (Sym et al., 1993). The Zip1 pattern indicates the stage in meiosis (Figures 1A–1C). In leptonema (prior to SC formation), Mer2myc was on chromatin in many foci (Figure 1A), which places Mer2 on chromosomes prior to or at the time DSBs are formed. Staining was brighter and patchy in zygonema (during SC formation) (Figure 1B) and remained on chromosomes but became weaker in pachynema (full-length SC), i.e., past the period when DSBs form (Figure 1C). In contrast to sudden loss of Rec102 and Rec104 from chromosomes at mid-pachynema (Kee et al., 2004), Mer2myc persisted, similar to Ski8 and Spo11 (Arora et al., 2004; Prieler et al., 2005). Mer2myc staining only partially overlapped with Zip1 (Figure 1B and 1C), suggesting that much of Mer2myc localized to chromatin loops rather than chromosome axes, similar to Rec102, Rec104, and Ski8 (Arora et al., 2004; Kee et al., 2004). Bulk differential extraction confirmed association of Mer2 with meiotic chromatin (described below in Figure 4) and revealed that chromatin association did not require any of the other DSB proteins tested (Spo11, Ski8, Mei4, Rec114, Rec102, Rec104, Xrs2, Mre11, data not shown).
Figure 1. Localization of Mer2 to premeiotic and meiotic chromosomes.
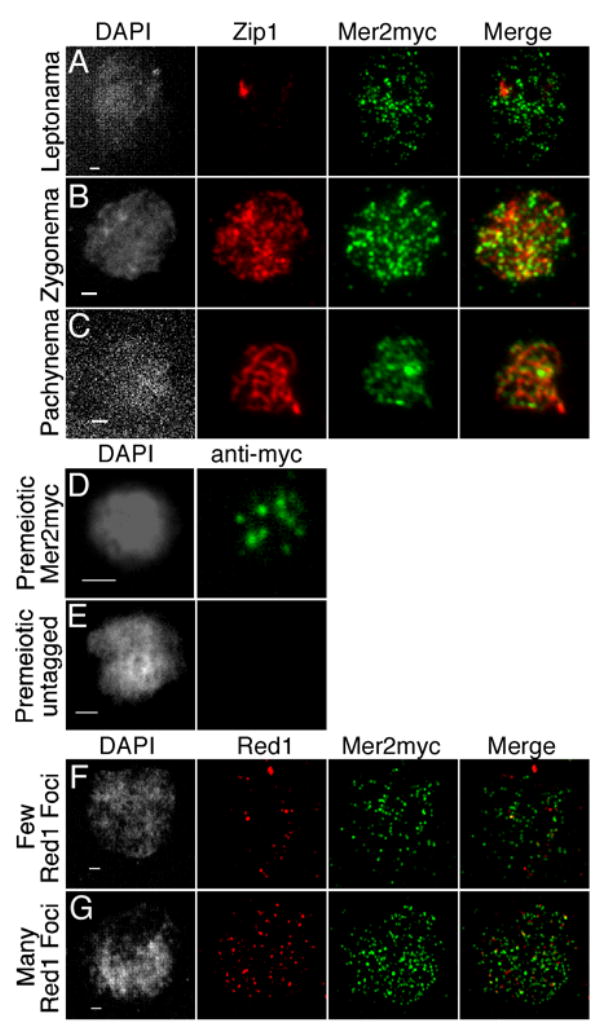
(A–C) Meiotic prophase. Nuclear spreads from meiotic MER2myc cultures were stained with DAPI (grayscale), anti-myc antibodies (green), and anti-Zip1 antibodies (red). Equivalent exposures of representative nuclei at the indicated stages are presented.
(D, E) Premeiotic nuclei. Spreads from MER2myc (D) and untagged MER2 (E) strains were stained with DAPI (grayscale) and anti-myc (green). Equivalent exposures are presented.
(F, G) Early meiotic nuclei. Spreads from a MER2myc strain were prepared two hr after transfer to sporulation medium, and stained with DAPI (grayscale), anti-myc (green), and anti-Red1 (red). All scale bars represent 1 μm.
Figure 4. Physical and functional characterization of Mer2 phosphorylation.
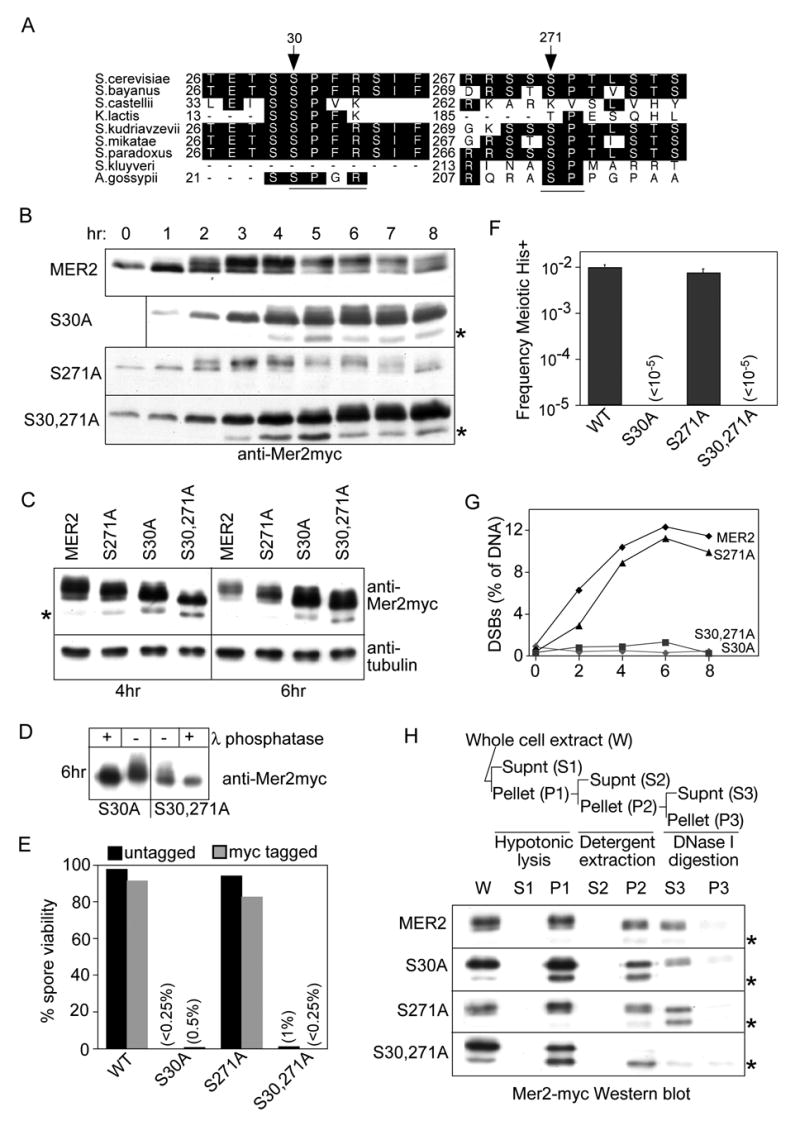
(A) Two Cdc28 consensus sites in Mer2 (underlined). Black boxes indicate residues identical to those in S. cerevisiae. N-terminal sequences were not available for S. kluyveri.
(B, C) Western blot analysis of meiotic time courses of wild-type and Cdc28-consensus-site mutants (MER2myc, mer2(S30A)myc, mer2(S271A)myc, and mer2(S30,271A)myc). In panel C, samples from 4 hr and 6 hr time points are compared side by side; half as much extract was loaded for the mer2 mutants. Blots were stripped and reprobed for tubulin as a loading control.
(D) Residual phosphorylation of Mer2(S30A) and Mer2(S30,271A). Myc-tagged protein was immunoprecipitated from denaturing whole-cell extracts prepared at 6 hr in meiosis, and left untreated or treated with lambda phosphatase.
(E) Spore viabilities (100 tetrads per strain).
(F) Intragenic meiotic recombination at the his4LEU2 hotspot in return-to-growth assays (untagged MER2 alleles). Each value is the mean ± s.d. for three independent cultures.
(G) DSBs at the YCR048w hotspot, measured by Southern blotting of genomic DNA from rad50S strains (untagged MER2 alleles).
(H) Association of Mer2myc with meiotic chromatin. The subcellular fractionation assay is outlined at the top. Extracts from cells at 4 hr in meiosis were fractionated and analyzed by western blotting. Lanes contain equal cell equivalents. Asterisks denote a Mer2myc proteolytic product.
Because Mer2 was already abundant on leptotene chromosomes, we also examined earlier times. Mer2myc was present in extracts from premeiotic cells immediately upon transfer to sporulation medium and from cycling haploid and diploid cells (Figure 2A), consistent with small amounts of spliced message detected in non-meiotic cells (Engebrecht et al., 1991). Mer2myc formed a few distinct foci (7.3 ± 5.1, mean ± SD, n=107) on chromosomes from premeiotic cells (Figure 1D). Foci were also found on nuclei from haploid cells cultured identically to the premeiotic cells, and on nuclei from vegetatively growing haploid and diploid cells (data not shown). No foci were detected on spreads from an untagged diploid control (Figure 1E). Thus, Mer2 forms chromatin-associated complexes in non-meiotic cells. The function of these complexes is not known.
Figure 2. Phosphorylation of Mer2 in meiotic prophase, independent of DSB formation.
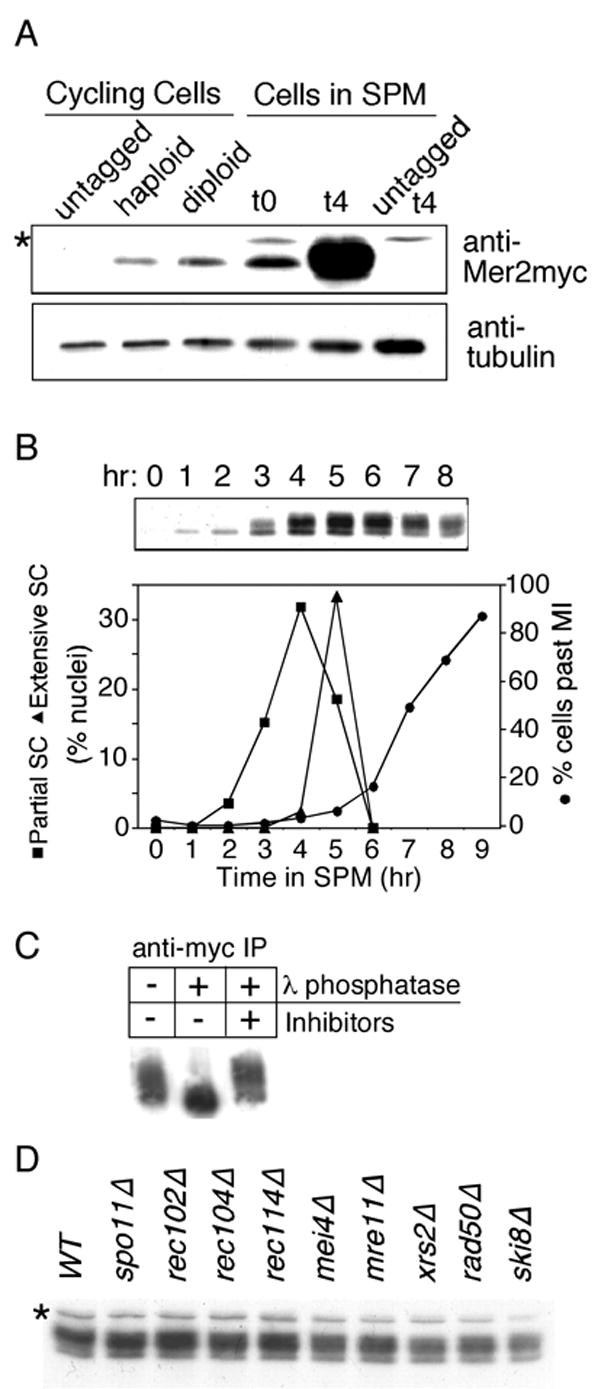
(A) Mer2myc expression. Denaturing whole-cell extracts were prepared from untagged diploid cells or MER2myc haploid (MATa) or diploid cells in log phase culture (cycling cells) or at the indicated times after transfer to sporulation medium (cells in SPM). Extracts were analyzed by SDS-PAGE and western blotting with anti-myc antibody. The asterisks in (A) and (D) indicate a non-specific band frequently seen with cells subjected to the standard meiotic pre-growth regimen. The blot was stripped and reprobed with anti-tubulin as a loading control.
(B) Time course of Mer2myc expression in meiosis. Denaturing extracts were prepared at the indicated times after transfer to sporulation medium and analyzed by anti-myc western blotting. A lighter exposure is shown than the blot in (A) to reveal multiple shifted forms. Meiotic timing of the same culture was determined by measuring SC formation and nuclear division.
(C) Mer2myc phosphorylation. Denaturing whole-cell extract was prepared at 4 hr in meiosis, then Mer2myc was immunoprecipitated and treated with lambda phosphatase alone or in the presence of inhibitors.
(D) Mer2myc phosphorylation in DSB-defective mutants. Denaturing extracts from the indicated mutants at 4 hr in meiosis were analyzed by anti-myc western blotting.
Nuclear spreads were also prepared from cells early in meiosis (2 hr after transfer to sporulation medium) and double-stained for Mer2myc and Red1. Red1 is a meiosis-specific protein associated with chromosomes prior to SC formation and can be used to define early meiotic stages (Smith and Roeder, 1997). In nuclei with a few Red1 foci, the number of Mer2myc foci had increased substantially from premeiotic levels (Figure 1F; 75 ± 54 foci per spread, n=7). Nuclei with more Red1 immunostaining structures showed even more foci, reaching 150 or more per nucleus (Figure 1G; 166 ± 53 foci per spread, n=8). Thus, progression into premeiotic S phase is associated with formation of numerous Mer2 foci. Aside from the Rad50-Mre11-Xrs2 complex, none of the other DSB proteins can be detected on chromosomes so abundantly this early (Arora et al., 2004; Kee et al., 2004; Prieler et al., 2005, and our unpublished results).
In pre- and non-meiotic cells, Mer2myc migrated on SDS-PAGE as a single band at ~54 kDa, larger than its predicted size of ~43 kDa. Similar aberrant mobilities of tagged proteins were observed previously (Kee et al., 2004). After transfer to sporulation medium, Mer2myc levels increased and the mobility changed so that multiple forms were seen (Figures 2A and 2B). Slower migrating species first appeared between ~2–3 hr, at or before the beginning of SC formation, and the protein persisted past the first meiotic division (Figure 2B). Phosphatase treatment converted slow-migrating to rapidly migrating forms, revealing that Mer2 is a phosphoprotein (Figure 2C). Mer2myc phosphorylation was similar to wild type in DSB-defective mutants (Figure 2D). Hence, phosphorylation is not a consequence of DSB formation.
Mer2 phosphorylation requires CDK activity in vivo
Cdc28-Clb5/Clb6 activity accumulates during premeiotic S phase, increases through prophase and peaks at about MI (Stuart and Wittenberg, 1998). We tested whether this kinase is required for Mer2 phosphorylation. First, we used a mutant version of Cdc28 (Cdc28-as1) that can be inactivated in vivo by the inhibitor 1-NM-PP1 (Bishop et al., 2000). Addition of 0.5 μM 1-NM-PP1 to the sporulation medium allows DNA replication but arrests cells prior to the first meiotic division, whereas more complete inhibition with 5 μM 1-NM-PP1 prevents both S phase and the first division (Benjamin et al., 2003). Mer2myc was phosphorylated in a cdc28-as1 mutant in the absence of 1-NM-PP1 (Figure 3A). In the presence of 0.5 μM 1-NM-PP1, meiotic induction of Mer2myc occurred, but phosphorylation was delayed and reduced (Figure 3A). Phosphorylation was reduced even further when the culture was exposed to 5 μM 1-NM-PP1 (Figure 3A). We also reduced CDK activity by deleting CLB5 and CLB6. Mer2myc levels increased as cells entered meiosis but the protein mobility did not change, indicating that the protein was not phosphorylated normally (Figure 3B).
Figure 3. Direct phosphorylation of Mer2 by Cdc28-Clb5/Clb6.
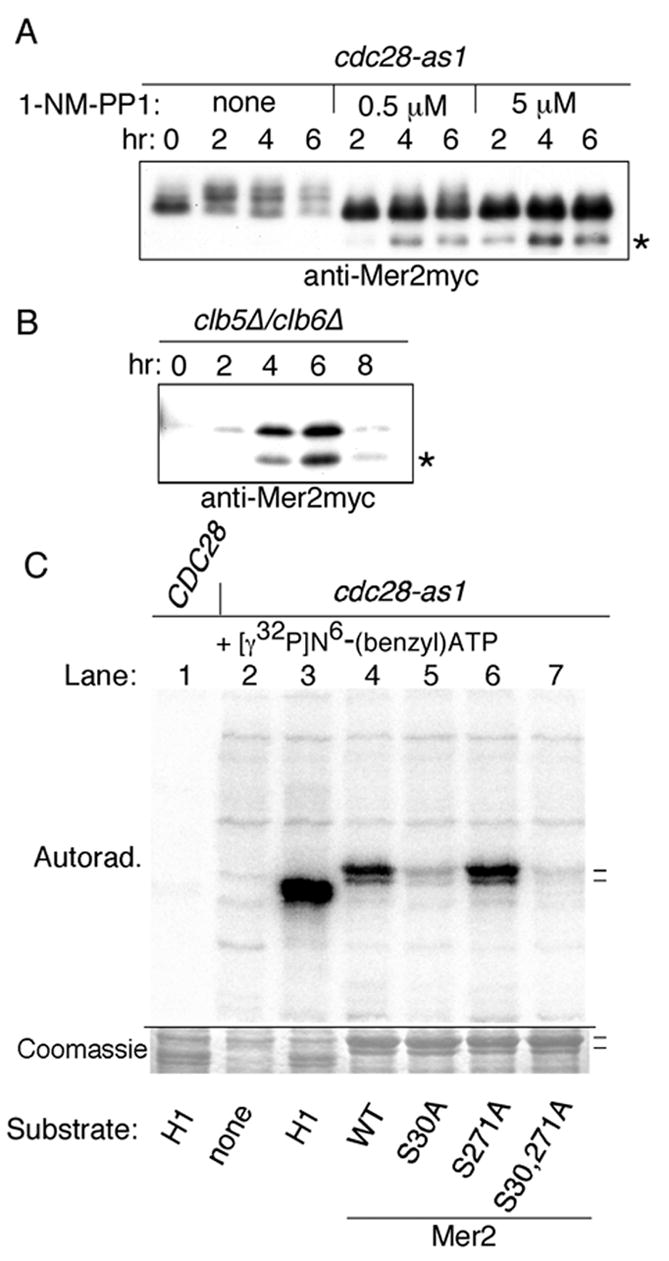
(A) Western blot of whole-cell extracts from cdc28-as1 meiotic time courses. The culture was divided and either left untreated or treated starting at 0 hr with 0.5 or 5 μM 1-NM-PP1. The asterisk denotes a Mer2myc proteolytic product. Half as much material was loaded for the drug-treated samples.
(B) Western blot of whole-cell extracts from a clb5δ clb6 δ strain.
(C) Direct phosphorylation of Mer2 by Cdc28 in vitro. Kinase assays with [γ32P] N6-(benzyl)ATP were performed with whole-cell extract prepared at 3 hr in meiosis from CDC28 (lane 1) and cdc28-as1 cultures (lanes 2–7). Extracts were incubated without added substrate (lane 2), with histone H1 (lanes 1 and 3), or with the indicated recombinant Mer2 proteins purified from E. coli. The upper panel is an autoradiograph; the lower panel shows the relevant portion of the same gel stained with Coomassie Blue. Dashes indicate positions of full-length Mer2 and a proteolytic fragment present in the recombinant protein preparation.
Steady-state Mer2myc levels were elevated when Cdc28as1 activity was decreased (Figure 3A). The reason for this effect is not known, but analysis of nonphosphorylatable mer2 mutants suggests that this is a consequence of blocking Mer2 phosphorylation (see below). In addition, a faster migrating form of Mer2myc accumulated when CDK activity was reduced (asterisks in Figures 3A and 3B). This species is presumably an N-terminally truncated proteolytic product (the epitope tag is on the C terminus), although it is not known whether proteolysis occurred in vivo or during extraction. Based on analysis of mer2 mutants, susceptibility to cleavage also appears to be a specific consequence of blocking Mer2 phosphorylation (see below).
CDK-cyclin complexes phosphorylate Mer2 directly
These findings demonstrate that phosphorylation of Mer2 requires CDK activity. To determine whether this requirement is direct, we tested for Mer2 phosphorylation by CDK in vitro. Whole-cell extracts were prepared from CDC28 and cdc28-as1 strains at 3 hr in meiosis and incubated with [γ32P] N6-(benzyl)ATP with or without purified recombinant Mer2 protein (Figure 3C). The source of radioactive label is a bulky ATP analog that is used by Cdc28-as1 protein 130-fold more efficiently than ATP but is used very inefficiently by wild-type Cdc28 (Ubersax et al., 2003). Under these conditions, proteins in the extract (Figure 3C, lane 2) as well as added histone H1 (Figure 3C, lane 3) were labeled by Cdc28-as1 but not by wild-type Cdc28 (Figure 3C, lane 1). The cdc28-as1 extract also labeled Mer2 (Figure 3C, lane 4). Because Cdc28-as1 is the only kinase in the extract capable of using N6-(benzyl)ATP efficiently, we conclude that Mer2 can be directly phosphorylated by CDK-cyclin complexes.
Identification of CDK target sites on Mer2
The consensus CDK target is the sequence T/S-P-x-K/R where x is any amino acid, although CDK targets can also be phosphorylated at a minimal site, S/T-P (Nigg, 1993; Ubersax et al., 2003). Mer2 contains an optimal consensus site (SPFR) at Ser-30 and a minimal site (SP) at Ser-271 (Figure 4A). Both sites are well conserved despite low overall conservation (Figure 4A).
Mutating Ser-30 to alanine decreased the electrophoretic mobility shift and delayed the appearance of shifted species until after the normal time of DSB formation (Figures 4B and 4C). Mutating Ser-271 also altered the mobility pattern, but less drastically, and with kinetics similar to wild type (Figures 4B and 4C). Mutating both residues led to greater loss and delay of shifted species (Figures 4B and 4C). Phosphatase treatment increased the mobility of both Mer2(S30A)myc and Mer2(S30,271A)myc (Figure 4D), indicating that both are still phosphorylated. Similar to global inhibition of CDK, mutation of Ser-30 increased steady-state Mer2 levels, particularly at late time points (Figure 4C) and increased the proteolytic product (asterisks in Figures 4B and 4C). Thus, Ser-30 and Ser-271 are both required for normal Mer2 phosphorylation in vivo, with mutation of Ser-30 having a more profound effect. A simple interpretation is that these residues are both phosphorylated by Cdc28-Clb5/Clb6, although these are not the only sites that can be phosphorylated.
To confirm that Ser-30 and Ser-271 are direct targets of CDK, recombinant mutant proteins were tested as substrates in cdc28-as1 extracts with [γ32P] N6-(benzyl)ATP. Mer2(S271A) was efficiently phosphorylated, but phosphorylation of Mer2(S30A) and Mer2(S30,271A) was reduced ~5-fold and ~20-fold, respectively, compared to wild-type Mer2 (Figure 3C). We conclude that Cdc28 directly phosphorylates Mer2 on Ser-30. Because Mer2(S30,271A) phosphorylation was decreased even more, it appears that Ser-271 is also phosphorylated.
CDK targets on Mer2 are required for normal DSB formation
To determine the significance of Mer2 phosphorylation, phenotypes of phosphorylation mutants were analyzed. In mer2(S30A), spore viability was greatly reduced and intragenic recombination and DSB formation were not detectable over background (Figures 4E–4G). These phenotypes are indistinguishable from a mer2 null, revealing that Ser-30 is essential for meiotic DSB formation. The mer2(S30,271A) mutant was similar (Figures 4E–4G).
In contrast, Ser-271 phosphorylation is less important for recombination, because spore viability in untagged mer2(S271A) was only slightly reduced compared to wild type (94% vs. 98%; Figure 4E), DSB formation was also slightly reduced (96% of wild type at 6 hr in meiosis; Figure 4G), and intragenic recombination was similar to wild type (Figure 4F). Interestingly, combining the mer2(S271A) mutation with the myc tag caused a more severe defect than either change alone. Addition of the epitope tag to otherwise wild-type Mer2 gave a small but significant reduction in spore viability from 98% to 91% (p<0.01, chi-square test), indicating that the tag slightly reduces Mer2 activity in vivo (Figure 4E). However, when mer2(S271A) was tagged, spore viability was further reduced (83%, Figure 4E), suggesting that mer2(S271A) is not fully functional and that its mild defect is exacerbated by the tag. Similar effects from combining point mutations with epitope tags were observed for SPO11 (Diaz et al., 2002).
Phosphorylation is not required for association of Mer2 with meiotic chromatin
To determine if Mer2 phosphorylation is required for proper localization of the protein, we used a previously described differential extraction assay to follow the subcellular distribution of wild-type and mutant Mer2myc (Figure 4H) (Kee et al., 2004). Meiotic cells were hypotonically lysed to generate whole-cell extract (W) which was separated by centrifugation into a soluble cytoplasmic fraction (S1) and a pellet (P1) containing nuclei and other insoluble material. The pellet was extracted with nonionic detergent to release nucleoplasmic proteins, then separated by centrifugation again into a soluble fraction (S2) and a pellet containing chromosomes and other insoluble material (P2). Mer2myc was insoluble after detergent extraction (P2) and was quantitatively solubilized by DNase I (S3), indicating that Mer2myc is chromatin-associated (Figure 4H), consistent with the immunocytological analysis above.
Both single mutant proteins were quantitatively chromatin associated, with no detectable increase in the non-chromatin bound fractions (S1 and S2) compared to wild type (Figure 4H). However, both proteins were more susceptible to degradation by endogenous proteases during extraction, showing increases of the N-terminally truncated species (Figure 4H). Mer2(S30A)myc was more sensitive, because the truncated form was apparent at the first step (P1), whereas the truncated form did not become abundant for Mer2(S271A)myc until the DNase digestion (S3). The final yield of Mer2(S30A)myc was also reduced. Both the increased truncation product and decreased overall yield were even more pronounced for the double mutant Mer2(S30,271A)myc (Figure 4H). Thus, protease sensitivity correlated with the extent of the phosphorylation defect. Even so, this protein was also chromatin-associated as judged by the quantitative recovery in the first pellet (P1). Immunocytological analysis confirmed that the mutant proteins still associated with meiotic chromosomes (data not shown). These results reveal that CDK-dependent phosphorylation is not required to target Mer2 to chromatin, consistent with the finding that Mer2 was associated with chromosomes of premeiotic cells, i.e., when it was not yet detectably phosphorylated (Figure 1D). Moreover, the DSB defect of mer2(S30A) cannot be attributed to improper localization.
Protein degradation in this assay probably reflects activation of cellular proteases during extraction and is thus likely to be non-physiological (Kee et al., 2004). This result may indicate that the degradation described above (Figure 3) also occurred ex vivo. More importantly, however, the increased degradation suggests that the mutations alter Mer2 conformation and/or association with other factors such that the mutant proteins are more accessible to proteases.
CDK target site mutations perturb Mer2 interaction with other DSB proteins
Mer2 interacts physically with itself and with other DSB proteins (Arora et al., 2004). To determine if the DSB defect of the phosphorylation site mutants can be attributed to defects in Mer2 association with its known partners, we analyzed a subset of two-hybrid interactions that produced the highest β-galactosidase signals with wild-type proteins: Mer2 with itself and Mer2 with Rec114, Mei4 and Xrs2 (Figure 5A) (Arora et al., 2004). These interactions can be detected when the two-hybrid reporter strain is grown vegetatively, with the exception of the Mer2-Rec114 interaction, which is only detected when the reporter strain is induced to enter meiosis (Arora et al., 2004). Expression levels for two-hybrid fusion proteins were indistinguishable for wild type and mutant Mer2 constructs (data not shown). All of the mutant Mer2 proteins retained the capacity to interact with wild-type Mer2 and with Mei4, albeit with reduced signal for Mer2(S30A) and Mer2(S30,271A) (Figures 5B and 5C). In contrast, when each mutant was assayed for interaction with itself, all three showed severe defects, although they continued to yield β-galactosidase expression significantly above background (Figure 5D). Interactions with Rec114 and Xrs2 were even more substantially compromised for some or all of the mutants. For both Mer2(S30A) and Mer2(S30,271A), signals were similar to background controls (Figures 5E and 5F). For Mer2(S271A), the interaction with Rec114 was unaffected, but the interaction with Xrs2 was decreased to nearly the same extent as for the other two mutants, although still above background (Figure 5F).
Figure 5. Effects of phosphorylation site mutations on Mer2 interactions with other DSB proteins.
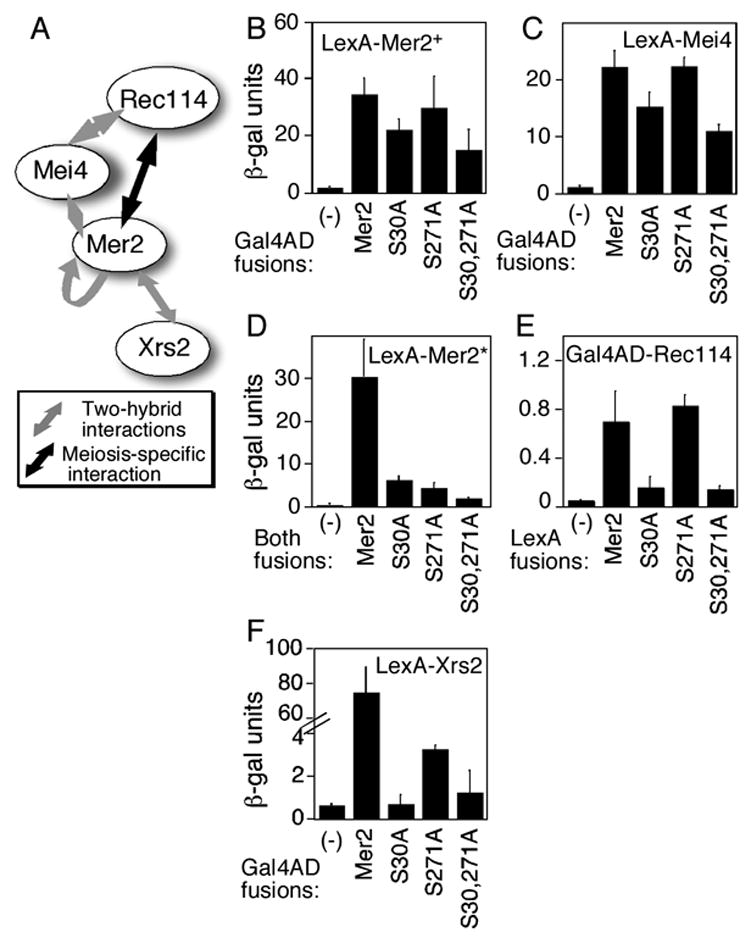
(A) Schematic of the Mer2 interactions analyzed.
(B–F) Yeast two-hybrid analysis of wild-type and mutant Mer2 proteins. β–galactosidase (β–gal) reporter expression (mean ± s.d. for three independent cultures) is given for LexA and Gal4 activating domain (AD) fusions in the indicated combinations. In (D), “Mer2*” indicates that LexA was fused to the same version of Mer2 (wild type or mutant) as Gal4AD. Minus signs denote empty vector controls.
Discussion
Chiasma formation requires temporal coordination of the initiation of meiotic recombination with progression through prophase, but the molecular events ensuring this coordination are not well understood. We provide evidence that Mer2 is phosphorylated by Cdc28-Clb5/Clb6 complexes early in meiosis and that this phosphorylation promotes DSB formation by fostering physical interactions between Mer2 and other proteins. These results provide a mechanistic link between DSB formation and the activity levels of specific cyclin-Cdc28 complexes. We propose that this link is one facet of the mechanism for coordinating DSB formation with progression through meiotic prophase. Although it is certainly likely that many other processes also contribute to controlling the timing of DSB formation (discussed further below), these studies are the first to establish a direct connection between initiation of meiotic recombination and a key regulator of cell cycle progression.
CDK-dependent phosphorylation patterns of Mer2 in meiosis
These studies reveal complex relationships between Mer2 phosphorylation patterns and biochemical properties of the protein (Figure 6A). Mutation of Ser-30 substantially reduced Mer2 phosphorylation in vivo and in vitro, eliminated DSB formation, eliminated or reduced protein-protein interactions with itself, Mei4, Rec114 and Xrs2, and increased the sensitivity of Mer2 to protease. It is likely that some or all of these defects are sufficient to explain the DSB defect when Ser-30 is mutated. It is also likely that these defects are caused by the inability to phosphorylate Ser-30, although we cannot rule out a phosphorylation-independent requirement for a serine at this position. (Clb5 and Clb6 are expressed in vegetative cells, so two-hybrid fusion proteins are likely to be phosphorylated during at least some portion of the cell cycle.) An attempt to make a phosphomimetic mutant by substituting Ser-30 with aspartate yielded a severely crippled protein: mer2(S30D) supported only 3.5% of normal recombination levels in return-to-growth assays (data not shown).
Figure 6. CDK-dependent regulation of meiotic DSB formation.
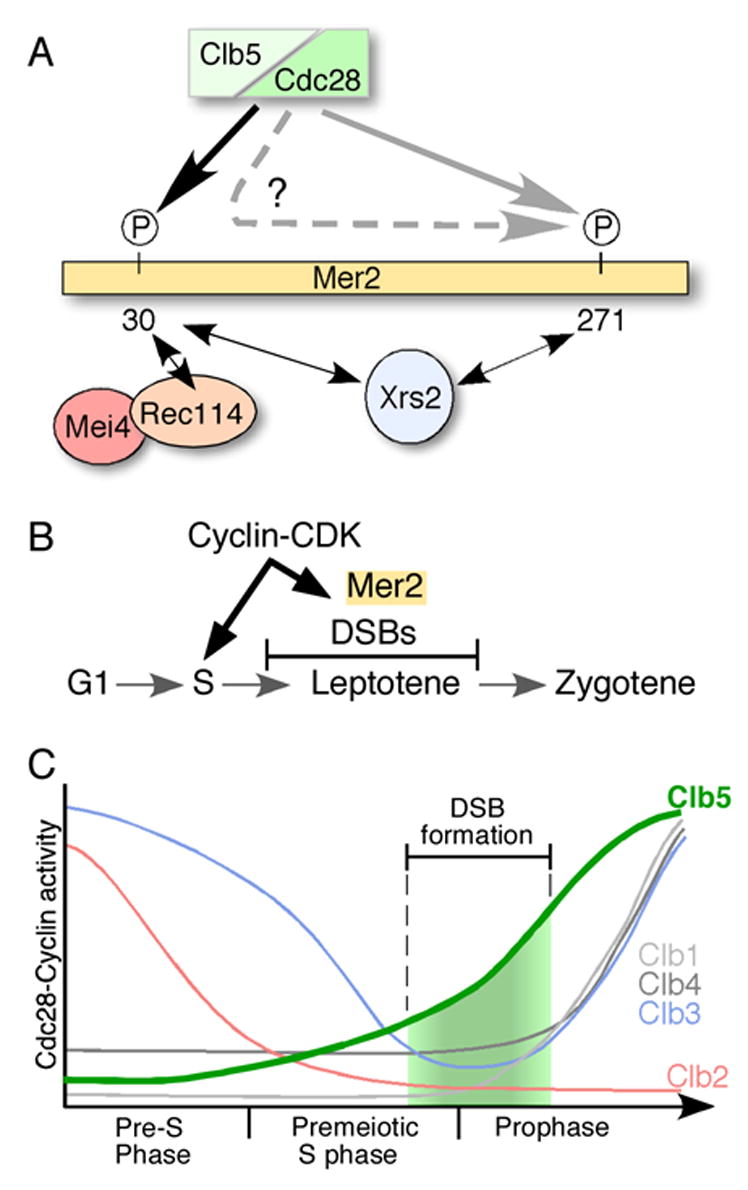
(A) Model for establishment of Mer2 phosphorylation patterns and interactions with other DSB proteins. See text for details.
(B) CDK directly promotes both premeiotic DNA replication and DSB formation.
(C) Fluctuation of B-type cyclin-Cdc28 kinase activities (based on Grandin and Reed, 1993; Stuart and Wittenberg, 1998). DSB formation is restricted to a window (shaded green) at the beginning of meiotic prophase. In this window, Clb5-Cdc28 activity is at intermediate levels but other cyclin-CDK activities are relatively low. We propose that a threshold of Clb5-Cdc28 activity is required to establish conditions permissive for DSB formation, and that this contributes to the appropriate timing of recombination initiation. Although DSBs form after DNA replication is completed locally, asynchrony of replication of different parts of the genome may result in formation of DSBs in early replicating regions before later regions have been replicated. The time (horizontal) axis is not drawn to scale.
Ser-271 is also likely to be phosphorylated because Mer2 with both serines mutated showed less phosphorylation than Mer2(S30A), both in vivo and in vitro. Ser-271 is important for homotypic interactions and interaction with Xrs2, but mutation of this residue alone had only modest effects on phosphorylation patterns, sensitivity to protease, and, most importantly, recombination levels. The phenotype of mer2(S271A) suggests either that these interactions are not essential for DSB formation or that the residual interaction ability of the mutant Mer2 protein is sufficient.
Maximal phosphorylation of wild-type Mer2 (based on electrophoretic mobility shift) occurred prior to pachynema (Figure 2B), whereas residual phosphorylation of Mer2(S30A) was delayed 1–2 hr and that of Mer2(S30,271A) occurred even later (Figure 4B). The residual phosphorylation may be due to other kinases (although their activity must be Clb5/Clb6-dependent) but we note that the context of both serine residues is serine/threonine-rich (Figure 4A). Perhaps Cdc28 can inefficiently phosphorylate these residues when the principal target serine is mutated. The delay in phosphorylation may reflect a need for higher levels of Clb5/Clb6 and/or other cyclins (see Figure 6C).
We suggest the following scenario to account for our findings (Figure 6A), although we stress that other interpretations are possible. We propose that phosphorylation of Ser-30 and Ser-271 by CDK occurs in concert, with Ser-271 phosphorylation at least partially dependent on phosphorylated Ser-30 (PSer-30). Mutation of Ser-30 reduces and delays Ser-271 phosphorylation, but Ser-271 eventually becomes phosphorylated because higher Clb5/Clb6 levels overcome the need for PSer-30 and/or because phosphorylation of other residues near Ala-30 promotes Ser-271 phosphorylation. Both PSer-30 and PSer-271 stabilize multiple protein-protein interactions. It is attractive to think that at least one interaction involves direct binding of a partner to PSer-30. Moreover, we suggest that the protease sensitivity of the mutants reflects altered Mer2 conformation and/or lack of binding partner(s). Consistent with this idea, the size of the Mer2 fragment was consistent with cleavage in the vicinity of residue 30, and Mer2 protease sensitivity in the chromatin fractionation assay appeared to increase specifically in rec114 and mei4 mutants (data not shown).
Predictions of this working model remain to be tested, but this scenario provides a framework for understanding the results presented here. Elucidating these details is an important challenge for understanding the function of Mer2 phosphorylation.
Relationship of DSB formation to premeiotic DNA replication
Although it is clear that recombination initiates after local replication of DNA (Borde et al., 2000), just how these events are related is not well understood (reviewed in Baudat and Keeney, 2001). DSBs do not form when bulk DNA replication is prevented by hydroxyurea treatment, leading to the hypothesis that there is a strict dependence of DSB formation on replication (Borde et al., 2000; Simchen et al., 1976). However, interpretation of these results is complicated because hydroxyurea also blocks induction of early meiotic genes, including SPO11 (Lamb and Mitchell, 2001). Perhaps more compelling was the observation that replication and DSB formation are both eliminated by mutation of CLB5 and CLB6 without effects on early meiotic gene induction (Smith et al., 2001; Stuart and Wittenberg, 1998). Because cyclin-CDK activity directly regulates DNA replication in vegetative cells (reviewed in Bell and Dutta, 2002), these findings appeared to be consistent with the hypothesis that DSB formation is dependent on DNA replication. However, our results show that this conclusion cannot be drawn, because DSB formation itself is also directly dependent on CDK activity (Figure 6B). Other recent studies challenge the idea of a strict dependence of recombination initiation on replication. When meiotic expression of the replication initiation factor Cdc6 was eliminated, DNA replication was confined to cycling cells but meiotic DSB formation occurred nonetheless (Hochwagen et al., 2005). In Schizosaccharomyces pombe, reducing levels of core replication proteins prevented completion of premeiotic DNA replication but not formation of DSBs (Murakami and Nurse, 2001) and elimination of replication checkpoint genes allowed recombination to proceed even in the presence of hydroxyurea (which does not block early gene expression in this organism) (Ogino and Masai, 2006; Tonami et al., 2005).
Nevertheless, even if replication is not a strict prerequisite for DSB formation, there remains compelling evidence that the two processes are coupled. Most telling are experiments in which delaying replication of the left arm of chromosome III delayed DSB formation by the same margin locally without affecting timing on the right arm (Borde et al., 2000; Murakami et al., 2003). One possibility is that active replication forks are sensed by a regulatory checkpoint mechanism that prevents premature DSB formation, analogous to S-phase checkpoints that couple mitosis with DNA replication (Hochwagen and Amon, 2006; Hochwagen et al., 2005). Another possibility is that replication fork passage (or associated processes) promotes installation of chromosomal features (e.g., chromatin or higher order chromosome structures) that constrain subsequent DSB formation. DSB-independent effects of spo11 mutations on replication timing (Cha et al., 2000) are consistent with this hypothesis. The early localization of Mer2 to chromosomes may also play a role (Figure 1).
Temporal regulation of DSB formation and the role of CDK
DSB formation is restricted to a narrow window of time around the beginning of meiotic prophase (Figure 6C) (Padmore et al., 1991), and this temporal pattern is important both for chiasma formation and to prevent production of potentially toxic DNA lesions at inappropriate times. The mechanism for establishing the beginning of this window is not well understood, but meiosis-specific control of the expression of Spo11 and other DSB proteins (reviewed in Kassir et al., 2003) as well as possible mechanistic links to replication (see above) probably contribute. Our results reveal another potential facet of early temporal regulation by showing that Mer2 phosphorylation absolutely requires Cdc28-Clb5/Clb6. Clb5-dependent activity is low for most of premeiotic S phase and then steadily rises, peaking prior to the first meiotic division (Stuart and Wittenberg, 1998). (Cdc28-Clb6 activity has not been analyzed but may be similar.) DSB formation occurs when these cyclin-CDK activities are at intermediate levels (Figure 6C). We therefore propose that the onset of DSB production is controlled in part by a requirement for a threshold level of Cdc28-Clb5/Clb6 activity. Because DSB timing is controlled locally rather than globally in the nucleus (Borde et al., 2000), we envision this CDK threshold as one of several factors that establish conditions permissive for Spo11 activity rather than as a master trigger of the time of DSB formation.
Interestingly, Mer2 was not phosphorylated at early times or in the absence of Clb5/Clb6 even though other B-type cyclin-CDK complexes are active (Grandin and Reed, 1993; Stuart and Wittenberg, 1998) (Figure 6C). Clearly, Mer2 is not a substrate of other CDK complexes under physiological conditions and, indeed, it appears that DSB formation coincides with a period of very low levels of these other kinases. This pattern raises the possibility that other cyclin-CDK complexes might antagonize DSB formation, perhaps through inhibition of another process necessary for DSB formation and/or through competition between cyclins for available Cdc28, which does not fluctuate in meiosis (Stuart and Wittenberg, 1998). Such antagonism could also contribute to proper timing.
Less clear is what sets the late boundary and restricts DSB formation to leptonema, because Spo11 and other DSB proteins remain associated with chromosomes until later stages of prophase (Arora et al., 2004; Kee et al., 2004; Prieler et al., 2005, this study). Cdc28-Clb5 activity continues to increase, so attenuation of this activity is not what causes the end of DSB production (Figure 6C). One possible factor could be inhibition of DSB formation (independent of Mer2 phosphorylation status) by cyclin-CDK complexes that accumulate later. It has been noted (Keeney, 2001) that DSB formation appears to continue longer than normal under conditions that attenuate or eliminate later CDK activities, e.g., in an ndt80 mutant (Xu et al., 1995) (in which Clb1, Clb3, Clb4 are not induced (Chu and Herskowitz, 1998; Hepworth et al., 1998)) or in a conditional cdc28 mutant at a nonpermissive temperature (Shuster and Byers, 1989).
Of course, other mechanisms unrelated to CDK regulation certainly contribute to proper DSB timing. Deciphering these molecular pathways remains an important challenge, but the demonstration of a direct role for CDK and the identification of Mer2 as one critical target have provided new insight into this central aspect of meiotic chromosome dynamics.
Experimental Procedures
Yeast strains and culture methods
Strains are isogenic diploid derivatives of SK1 (Kane and Roth, 1974) and are listed in Supplemental Table 1. The mer2δ::kanMX4 allele was amplified from a Research Genetics knockout strain. Strains expressing Mer2 tagged with 5 myc repeats were used for all experiments except those in Figure 1, in which Mer2 was tagged with 8 myc repeats. Similar immunocytological results were obtained with a MER2myc5 strain (data not shown). MER2myc8::URA3 was generated using the tag-URA3-tag strategy previously described (Kee et al., 2004). A MER2myc5 derivative of a MER2myc8::URA3 strain was generated by recombination to lose the URA3 cassette. To generate subsequent MER2myc5::URA3 constructs, MER2myc5 plus ~500 bp upstream and downstream was PCR-amplified from genomic DNA and cloned into pRS314 (Sikorski and Hieter, 1989). Desired mutations were introduced by the QuickChange system (Stratagene), verified by sequencing, then subcloned into pRS306 (URA3 integrating vector). Untagged versions of the mer2 mutants were created similarly, except that the starting plasmid contained untagged MER2 amplified from SK1 genomic DNA and cloned directly into pRS306. Tagged and untagged MER2 alleles were integrated at mer2δ::kanMX4 by linearization of vectors with EcoRI and yeast transformation by electroporation. Correct targeting was confirmed by Southern blotting of genomic DNA, and retention of the desired point mutations was confirmed by diagnostic restriction patterns of PCR-amplified genomic DNA. MER2myc5 complemented a mer2 null mutant well, but not completely as indicated by a slight decrease in spore viability (Figure 4E) and by an increase in the frequency of polycomplexes observed in meiotic chromosome spreads (data not shown). The clb5δ::kanMX4 and clb6δ::TRP1 alleles (Stuart and Wittenberg, 1998) were obtained from strains provided by C. Wittenberg (Scripps Research Institute) and the cdc28as1 allele from a strain (Benjamin et al., 2003) provided by K. Benjamin and I. Herskowitz (University of California, San Francisco).
Meiotic cultures were prepared as decribed (Kee and Keeney, 2002). Briefly, cultures were grown in liquid YPA (1% yeast extract, 2% Bacto Peptone, and 1% potassium acetate) for 13.5 hr at 30°C, followed by harvesting and resuspension in SPM (0.3% potassium acetate and 0.02% raffinose) preequilibrated at 30°C. Cells were cultured in SPM at 30°C and samples taken at the times indicated.
Meiotic recombination and two-hybrid assays
Spore viabilities were determined by dissection of four-spored asci produced in liquid SPM. Meiotic intragenic recombination was measured 8 hr after transfer to SPM using his4LEU2 heteroalleles as described (Diaz et al., 2002). Premeiotic (t=0 hr) recombinant frequencies were subtracted. Physical analysis of DSBs by Southern blot was performed at the YCR048w locus as described (Kee and Keeney, 2002).
Two-hybrid assays were performed using methods and fusion constructs previously described (Arora et al., 2004). Mutant versions of the intronless cMER2 fusions were created using the QuickChange method and confirmed by diagnostic restriction digest. Some known Mer2 interactions depend on the N- or C-terminal orientation of the fusion constructs (Arora et al., 2004). Combinations of orientations used here were as follows: LexA-cMer2/cMer2-Gal4AD; LexA-Mei4/cMer2-Gal4AD; Gal4AD-Rec114/LexA-cMer2, Xrs2-LexA/cMer2-Gal4AD. One unit of β-galactosidase hydrolyzes 1 μmol of o-nitrophenyl-β-D-galactopyranoside per min per OD600.
Cytological methods
Surface spread meiotic chromosomes were prepared as described (Kee et al., 2004). Staining was performed as described (Gasior et al., 1998) with the following primary antibodies: mouse monoclonal anti-myc (1:500, Covance), guinea pig polyclonal anti-GST-Zip1 (1:1000, this laboratory), and rabbit polyclonal Red1 (1:100, G.S. Roeder, Yale University). Secondary antibodies (Molecular Probes) were used at a 1:500 dilution: goat anti-mouse Alexa-488, goat anti-guinea pig Alexa-546, and donkey anti-rabbit Alexa-594. The anti-myc antibody yielded no detectable chromatin labeling in either meiotic or nonmeiotic cells (Figure 1E and data not shown). Indirect immunofluorescence images were captured as described (Kee and Keeney, 2002).
Protein analysis
Denaturing whole cell extracts were prepared from cell suspensions in 20% trichloroacetic acid by agitation with glass beads. Precipitated proteins were solubilized in SDS-PAGE sample buffer and appropriate dilutions were analyzed by SDS-PAGE and western blotting. Immunoprecipitation (mouse monoclonal anti-myc, 1:150), phosphatase treatment, and the subcellular fractionation protocol were as described (Kee et al., 2004). Antibodies for western blotting were mouse monoclonal anti-myc (1:1000), rat monoclonal anti-tubulin (1:500; Harlan Sera-Lab, Ltd.), goat anti-mouse IgG conjugated to horseradish peroxidase (1:10,000; Jackson Labs), and donkey anti-rat IgG conjugated to horseradish peroxidase (1:1000; Accurate Chemicals and Scientific Corp., NJ). Blots of anti-myc immunoprecipitations were probed with mouse monoclonal anti-myc conjugated to horseradish peroxidase (1:1000, Covance).
In vitro kinase assay
Recombinant Mer2 protein was expressed in E. coli as a fusion with the yeast SUMO homolog Smt3 essentially as described by the manufacturer for the Champion pET SUMO expression system (Invitrogen). His6-Smt3-Mer2 fusion protein was purified on Ni-NTA agarose (Qiagen), and cleaved with recombinant SUMO protease Ulp1 to remove the His6-Smt3. Details are available upon request. [γ32P] N6-(benzyl)ATP was a generous gift of S. LaRochelle and R. Fisher (MSKCC), prepared as described (Larochelle et al., 2005). Whole cell extract was prepared from CDC28 and cdc28as1 strains by glass bead lysis in 25 mM HEPES-NaOH pH 7.4, 10 mM NaCl, 2 mM MgCl2, 0.1% Triton X-100, 1 mM EDTA, 10 mM NaF plus protease inhibitors. Kinase reactions were performed essentially as described (Larochelle et al., 2005). Briefly, 50 μg cell extract was preincubated 30 min at 30°C with ATP and an ATP regenerating system prior to addition of [γ32P] N6-(benzyl)ATP and exogenous substrate (either 2 μg histone H1 or 20 μg Mer2) and further incubation 10 minutes at 30°C. The preincubation step substantially reduces background labeling of proteins in the extract (Larochelle et al., 2005). Reactions were stopped with SDS sample buffer and separated by SDS-PAGE. Radioactivity was quantified on a Fuji BAS-2500 phosphorimager system.
Supplementary Material
Acknowledgments
We are particularly grateful to Stéphane Larochelle and Rob Fisher for providing advice and the [γ32P] N6-(benzyl)ATP for in vitro kinase assays. We also thank Kelly Yule and Katya Bojilova for help in early stages of the project, Rob Diaz for recombinant Mer2 expression constructs and purification protocols, and Kirsten Benjamin, Shirleen Roeder, and Curt Wittenberg for providing strains or antibodies. We thank members of the laboratory for helpful discussions, especially Charan Arora, Matt Neale, and Jing Pan. This work was supported in part by NIH grant R01 GM58673 (to S.K.). K.H. was supported in part by a Frank Lappin Horsfall, Jr. Fellowship and an Association for Women in Science Predoctoral Fellowship. S.K. is a Leukemia and Lymphoma Society Scholar.
References
- Arora C, Kee K, Maleki S, Keeney S. Antiviral protein Ski8 is a direct partner of Spo11 in meiotic DNA break formation, independent of its cytoplasmic role in RNA metabolism. Mol Cell. 2004;13:549–559. doi: 10.1016/s1097-2765(04)00063-2. [DOI] [PubMed] [Google Scholar]
- Baudat F, Keeney S. Meiotic recombination: Making and breaking go hand in hand. Curr Biol. 2001;11:R45–R48. doi: 10.1016/s0960-9822(01)00013-6. [DOI] [PubMed] [Google Scholar]
- Bell SP, Dutta A. DNA replication in eukaryotic cells. Annu Rev Biochem. 2002;71:333–374. doi: 10.1146/annurev.biochem.71.110601.135425. [DOI] [PubMed] [Google Scholar]
- Benjamin KR, Zhang C, Shokat KM, Herskowitz I. Control of landmark events in meiosis by the CDK Cdc28 and the meiosis-specific kinase Ime2. Genes Dev. 2003;17:1524–1539. doi: 10.1101/gad.1101503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop AC, Ubersax JA, Petsch DT, Matheos DP, Gray NS, Blethrow J, Shimizu E, Tsien JZ, Schultz PG, Rose MD, et al. A chemical switch for inhibitor-sensitive alleles of any protein kinase. Nature. 2000;407:395–401. doi: 10.1038/35030148. [DOI] [PubMed] [Google Scholar]
- Borde V, Goldman ASH, Lichten M. Direct coupling between meiotic DNA replication and recombination initiation. Science. 2000;290:806–809. doi: 10.1126/science.290.5492.806. [DOI] [PubMed] [Google Scholar]
- Cha RS, Weiner BM, Keeney S, Dekker J, Kleckner N. Progression of meiotic DNA replication is modulated by interchromosomal interaction proteins, negatively by Spo11p and positively by Rec8p. Genes Dev. 2000;14:493–503. [PMC free article] [PubMed] [Google Scholar]
- Chu S, Herskowitz I. Gametogenesis in yeast is regulated by a transcriptional cascade dependent on Ndt80. Mol Cell. 1998;1:685–696. doi: 10.1016/s1097-2765(00)80068-4. [DOI] [PubMed] [Google Scholar]
- Diaz RL, Alcid AD, Berger JM, Keeney S. Identification of residues in yeast Spo11p critical for meiotic DNA double-strand break formation. Mol Cell Biol. 2002;22:1106–1115. doi: 10.1128/MCB.22.4.1106-1115.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engebrecht J, Hirsch J, Roeder GS. Meiotic gene conversion and crossing over: Their relationship to each other and to chromosome synapsis and segregation. Cell. 1990;62:927–937. doi: 10.1016/0092-8674(90)90267-i. [DOI] [PubMed] [Google Scholar]
- Engebrecht JA, Voelkel-Meiman K, Roeder GS. Meiosis-specific RNA splicing in yeast. Cell. 1991;66:1257–1268. doi: 10.1016/0092-8674(91)90047-3. [DOI] [PubMed] [Google Scholar]
- Gasior SL, Wong AK, Kora Y, Shinohara A, Bishop DK. Rad52 associates with RPA and functions with Rad55 and Rad57 to assemble meiotic recombination complexes. Genes Dev. 1998;12:2208–2221. doi: 10.1101/gad.12.14.2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerton JL, Hawley RS. Homologous chromosome interactions in meiosis: Diversity amidst conservation. Nat Rev Genet. 2005;6:477–487. doi: 10.1038/nrg1614. [DOI] [PubMed] [Google Scholar]
- Grandin N, Reed SI. Differential function and expression of Saccharomyces cerevisiae B-type cyclins in mitosis and meiosis. Mol Cell Biol. 1993;13:2113–2125. doi: 10.1128/mcb.13.4.2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hepworth SR, Friesen H, Segall J. Ndt80 and the meiotic recombination checkpoint regulate expression of middle sporulation-specific genes in Saccharomyces cerevisiae. Mol Cell Biol. 1998;18:5750–5761. doi: 10.1128/mcb.18.10.5750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochwagen A, Tham WH, Brar GA, Amon A. The FK506 binding protein Fpr3 counteracts protein phosphatase 1 to maintain meiotic recombination checkpoint activity. Cell. 2005;122:861–873. doi: 10.1016/j.cell.2005.07.010. [DOI] [PubMed] [Google Scholar]
- Hochwagen A, Amon A. Checking your breaks: surveillance mechanisms of meiotic recombination. Curr Biol. 2006;16:R217–228. doi: 10.1016/j.cub.2006.03.009. [DOI] [PubMed] [Google Scholar]
- Jones GH. Chiasmata. In: Moens PB, editor. Meiosis. New York: Academic Press; 1987. pp. 213–244. [Google Scholar]
- Kane S, Roth R. Carbohydrate metabolism during ascospore development in yeast. J Bacteriol. 1974;118:8–14. doi: 10.1128/jb.118.1.8-14.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassir Y, Adir N, Boger-Nadjar E, Raviv NG, Rubin-Bejerano I, Sagee S, Shenhar G. Transcriptional regulation of meiosis in budding yeast. Int Rev Cytol. 2003;224:111–171. doi: 10.1016/s0074-7696(05)24004-4. [DOI] [PubMed] [Google Scholar]
- Kee K, Keeney S. Functional interactions between SPO11 and REC102 during initiation of meiotic recombination in Saccharomyces cerevisiae. Genetics. 2002;160:111–122. doi: 10.1093/genetics/160.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kee K, Protacio RU, Arora C, Keeney S. Spatial organization and dynamics of the association of Rec102 and Rec104 with meiotic chromosomes. EMBO J. 2004;23:1815–1824. doi: 10.1038/sj.emboj.7600184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeney S. Mechanism and control of meiotic recombination initiation. Curr Top Dev Biol. 2001;52:1–53. doi: 10.1016/s0070-2153(01)52008-6. [DOI] [PubMed] [Google Scholar]
- Lamb TM, Mitchell AP. Coupling of Saccharomyces cerevisiae early meiotic gene expression to DNA replication depends upon Rpd3 and Sin3. Genetics. 2001;157:545–556. doi: 10.1093/genetics/157.2.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larochelle S, Batliner J, Gamble MJ, Barboza NM, Kraybill BC, Blethrow JD, Shokat KM, Fisher RP. Dichotomous but stringent substrate selection by the dual-function Cdk7 complex revealed by chemical genetics. Nat Struct Mol Biol. 2005 doi: 10.1038/nsmb1028. [DOI] [PubMed] [Google Scholar]
- Malone RE, Bullard S, Hermiston M, Rieger R, Cool M, Galbraith A. Isolation of mutants defective in early steps of meiotic recombination in the yeast Saccharomyces cerevisiae. Genetics. 1991;128:79–88. doi: 10.1093/genetics/128.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami H, Nurse P. Regulation of premeiotic S phase and recombination-related double-strand DNA breaks during meiosis in fission yeast. Nat Genet. 2001;28:290–293. doi: 10.1038/90142. [DOI] [PubMed] [Google Scholar]
- Murakami H, Borde V, Shibata T, Lichten M, Ohta K. Correlation between premeiotic DNA replication and chromatin transition at yeast recombination initiation sites. Nucleic Acids Res. 2003;31:4085–4090. doi: 10.1093/nar/gkg441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandabalan K, Roeder GS. Binding of a cell-type-specific RNA splicing factor to its target regulatory sequence. Mol Cell Biol. 1995;15:1953–1960. doi: 10.1128/mcb.15.4.1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigg EA. Cellular substrates of p34(Cdc2) and its companion cyclin-dependent kinases. Trends Cell Biol. 1993;3:296–301. doi: 10.1016/0962-8924(93)90011-o. [DOI] [PubMed] [Google Scholar]
- Ogino K, Masai H. Rad3-Cds1 mediates coupling of initiation of meiotic recombination with DNA replication. Mei4-dependent transcription as a potential target of meiotic checkpoint. J Biol Chem. 2006;281:1338–1344. doi: 10.1074/jbc.M505767200. [DOI] [PubMed] [Google Scholar]
- Padmore R, Cao L, Kleckner N. Temporal comparison of recombination and synaptonemal complex formation during meiosis in S. cerevisiae. Cell. 1991;66:1239–1256. doi: 10.1016/0092-8674(91)90046-2. [DOI] [PubMed] [Google Scholar]
- Page SL, Hawley RS. Chromosome choreography: The meiotic ballet. Science. 2003;301:785–789. doi: 10.1126/science.1086605. [DOI] [PubMed] [Google Scholar]
- Petronczki M, Siomos MF, Nasmyth K. Un menage a quatre: The molecular biology of chromosome segregation in meiosis. Cell. 2003;112:423–440. doi: 10.1016/s0092-8674(03)00083-7. [DOI] [PubMed] [Google Scholar]
- Prieler S, Penkner A, Borde V, Klein F. The control of Spo11’s interaction with meiotic recombination hotspots. Genes Dev. 2005;19:255–269. doi: 10.1101/gad.321105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockmill B, Engebrecht JA, Scherthan H, Loidl J, Roeder GS. The yeast MER2 gene is required for chromosome synapsis and the initiation of meiotic recombination. Genetics. 1995;141:49–59. doi: 10.1093/genetics/141.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuster EO, Byers B. Pachytene arrest and other meiotic effects of the start mutations in Saccharomyces cerevisiae. Genetics. 1989;123:29–43. doi: 10.1093/genetics/123.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simchen G, Idar D, Kassir Y. Recombination and hydroxyurea inhibition of DNA synthesis in yeast meiosis. Mol Gen Genet. 1976;144:21–27. doi: 10.1007/BF00277299. [DOI] [PubMed] [Google Scholar]
- Smith AV, Roeder GS. The yeast Red1 protein localizes to the cores of meiotic chromosomes. J Cell Biol. 1997;136:957–967. doi: 10.1083/jcb.136.5.957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KN, Penkner A, Ohta K, Klein F, Nicolas A. B-type cyclins CLB5 and CLB6 control the initiation of recombination and synaptonemal complex formation in yeast meiosis. Curr Biol. 2001;11:88–97. doi: 10.1016/s0960-9822(01)00026-4. [DOI] [PubMed] [Google Scholar]
- Stuart D, Wittenberg C. CLB5 and CLB6 are required for premeiotic DNA replication and activation of the meiotic S/M checkpoint. Genes Dev. 1998;12:2698–2710. doi: 10.1101/gad.12.17.2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sym M, Engebrecht JA, Roeder GS. Zip1 is a synaptonemal complex protein required for meiotic chromosome synapsis. Cell. 1993;72:365–378. doi: 10.1016/0092-8674(93)90114-6. [DOI] [PubMed] [Google Scholar]
- Tonami Y, Murakami H, Shirahige K, Nakanishi M. A checkpoint control linking meiotic s phase and recombination initiation in fission yeast. Proc Natl Acad Sci USA. 2005;102:5797–5801. doi: 10.1073/pnas.0407236102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubersax JA, Woodbury EL, Quang PN, Paraz M, Blethrow JD, Shah K, Shokat KM, Morgan DO. Targets of the cyclin-dependent kinase Cdk1. Nature. 2003;425:859–864. doi: 10.1038/nature02062. [DOI] [PubMed] [Google Scholar]
- Xu L, Ajimura M, Padmore R, Klein C, Kleckner N. NDT80, a meiosis-specific gene required for exit from pachytene in Saccharomyces cerevisiae. Mol Cell Biol. 1995;15:6572–6581. doi: 10.1128/mcb.15.12.6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zickler D, Kleckner N. Meiotic chromosomes: Integrating structure and function. Annu Rev Genet. 1999;33:603–754. doi: 10.1146/annurev.genet.33.1.603. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.