

Abstract
The mammalian APOBEC3 proteins are cytidine deaminases that function as inhibitors of retrovirus replication and retrotransposon mobility. An issue that has remained controversial is whether the editing of deoxycytidine residues to deoxyuridine is necessary and sufficient for this inhibition or whether APOBEC3 proteins also exert a second, distinct inhibitory mechanism. Here, we present an analysis of the ability of mutants of APOBEC3G and APOBEC3B, both of which contain two consensus cytidine deaminase active sites, to inhibit the replication of Human Immunodeficiency Virus. Our data confirm that APOBEC3G only contains a single, carboxy-terminal active site but, surprisingly, reveal that both cytidine deaminase consensus sequences in APOBEC3B are enzymatically active. Enzymatically inactive mutant forms of APOBEC3G and APOBEC3B were found to retain the ability to inhibit the infectivity of HIV-1 virions produced in their presence by ~4 fold and ~8 fold, respectively. While this inhibition was significantly less than the level seen with wild-type forms of A3G or A3B, these data, nevertheless argue that the inhibition of HIV-1 by APOBEC3 proteins is at least partly independent of DNA editing.
Keywords: APOBEC3G, DNA editing, intrinsic immunity, HIV-1
Introduction
The mammalian APOBEC3 protein family has emerged as a key mediator of intrinsic immunity to retroviruses and retrotransposons (reviewed by Cullen, 2006). This activity was first observed in the case of human APOBEC3G (A3G), which was identified as the key cellular factor blocking the replication of human immunodeficiency virus type 1 (HIV-1) variants lacking a functional vif gene (HIV-1Δ Vif) (Sheehy et al., 2002). In contrast, in cells infected with wild-type HIV-1, the viral Vif protein directly binds to A3G and induces its degradation via the proteasome (Conticello et al., 2003; Marin et al., 2003; Sheehy et al. 2003; Stopak et al., 2003; Yu et al., 2003; Mehle et al., 2004). Two other human APOBEC3 proteins, A3F and A3B, are also potent inhibitors of HIV-1Δ Vif. Like A3G, A3F is expressed ubiquitously, including in the CD4+ T cells and macrophages normally infected by HIV-1, and A3F is also subject to degradation by Vif (Bishop et al., 2004; Liddament et al., 2004; Wiegand et al., 2004; Zheng et al., 2004; Liu et al., 2005). In contrast, A3B is not detectably expressed in CD4+ T cells and macrophages and, presumably as a result, the HIV-1 Vif protein has not evolved the ability to neutralize A3B function (Bishop et al., 2004; Yu et al., 2004a; Doehle et al., 2005).
In addition to A3G, A3F and A3B the human APOBEC3 family also includes A3A, A3C, A3DE and A3H (Jarmuz et al., 2002; Dang et al., 2006; OhAinle et al., 2006). These latter four proteins are unable to inhibit HIV-1Δ Vif replication effectively, although hA3DE does show weak inhibitory activity (Dang et al., 2006). However, A3A has been shown to be a potent inhibitor of a wide range of retrotransposons (Bogerd et al., 2006a, 2006b; Chen et al., 2006; Muckenfuss et al., 2006) while A3C has been reported to inhibit simian immunodeficiency virus infectivity (Yu et al., 2004a).
All members of the APOBEC3 protein family, both human and otherwise, contain either two copies (A3G, A3F, A3B and A3DE) or a single copy (A3A, A3C, A3H) of the cytidine deaminase consensus active site (CDA) HXEX23–28PCX2–4C (where X is any amino acid) (Jarmuz et al., 2002; Cullen, 2006). The cysteine and histidine residues are believed to coordinate a critical active site zinc ion while the glutamic acid residue participates directly in the deamination reaction (Betts et al., 1994). Importantly, all human APOBEC3 proteins analyzed thus far, with the possible exception of A3DE, are indeed capable of deaminating deoxycytidine to deoxyuridine and preferentially deaminate deoxycytidine residues present on single-stranded DNA (Suspène et al., 2004; Yu et al., 2004b). Initial research suggested that this deamination activity was critical for APOBEC3-mediated inhibition of HIV-1 replication as A3G, A3F and A3B all induced extensive mutagenesis of the few HIV-1Δ Vif proviruses that were able to successfully integrate into the genome of target cells after exposure to these proteins (Harris et al., 2003; Mangeat et al., 2003; Zhang et al., 2003; Bishop et al., 2004; Liddament et al., 2004; Wiegand et al., 2004; Yu et al., 2004a; Zheng et al., 2004; Doehle et al., 2005). It therefore appeared that A3G, A3F and A3B were being selectively incorporated into retroviral virions (Alce et al., 2004; Cen et al., 2004; Schäfer et al., 2004; Wiegand et al., 2004; Zennou et al., 2004; Doehle et al., 2005; Khan et al., 2005) and then, during a subsequent infection, were inducing massive mutagenesis of the provirus by dC to dU editing of the nascent DNA minus strand during reverse transcription. This led either to the production of a lethally mutated provirus or, more commonly, to the premature termination of reverse transcription and/or the degradation of reverse transcription intermediates.
More recent research has challenged this model based on the finding that some A3G and A3F mutants that appear incapable of catalyzing deamination of deoxycytidine nevertheless retain substantial inhibitory activity against HIV-1 (Newman et al., 2005; Bishop et al., 2006; Jónsson et al., 2006; Holmes et al., 2007). In addition, A3A and A3B mutants that lack the ability to induce cytidine deamination have been shown to effectively inhibit the mobility of the retrotransposons (Bogerd et al., 2006a, 2006b; Stenglein and Harris, 2006).
In this manuscript, we have sought to shed further light on the role of cytidine deamination in the inhibition of retrovirus and retrotransposon function by human APOBEC3 proteins. We have analyzed the effect of mutational inactivation of either one or both of the CDAs present in A3G or A3B on their ability to inhibit the replication of HIV-1. Our data argue that these CDAs play an important but partially redundant role in mediating the observed inhibition of retroviral replication and reveal that A3B, unlike A3G and A3F, contains two enzymatically active CDAs that both contribute to the mutational editing of HIV-1 proviruses produced in the presence of A3B.
Results
Effect of A3G and A3B mutants on retroviral infectivity
As noted above, A3G and A3B contain two copies of the CDA sequence HXEX23–28 PCX2–4C, although it has been reported for both A3G and A3B that only the carboxy-terminal CDA is enzymatically active (Haché et al., 2005; Navarro et al. 2005; Newman et al., 2005; Hakata and Landau, 2006; Jónsson et al., 2006; Stenglein and Harris, 2006). To examine the contribution of the CDAs to the ability of A3G and A3B to inhibit HIV-1 infectivity, we generated six mutants of A3G and A3B that are predicted to disrupt either one or both CDA sequences. In the case of A3G, we mutated the amino-terminal active site glutamic acid (residue 67) to glutamine (A3G-E1) or the carboxy-terminal site glutamic acid (residue 259) to glutamine (A3G-E2), or changed both to glutamine (A3G-E1+2). Alternatively, we mutated the first cysteine residue of the amino-terminal CDA (residue 97) to serine (A3G-C1), the first cysteine residue of the carboxy-terminal CDA (residue 288) to serine (A3G-C2), or again changed both at once (A3G-C1+2). Based on published work, these mutations, which either mutate the key active site glutamic acid or prevent zinc binding, should disrupt the activity of either one or both CDAs present in A3G (Newman et al., 2005; Jónsson et al., 2006).
Very similar mutants were also constructed in the context of a human A3B cDNA. These mutations changed glutamic acid 68 to glutamine (A3B-E1), glutamic acid 255 to glutamine (A3B-E2), or both (A3B-E1+2). Alternatively, these mutations introduced a serine in place of cysteine 97 (A3B-C1) or cysteine 284 (A3B-C2) or both (A3B-C1+2).
We next analyzed the ability of these mutant forms of A3G or A3B to inhibit the infectivity of HIV-1 (Fig. 1) using a previously described experimental protocol (Wiegand et al., 2004; Doehle et al., 2005). As shown in Fig. 1A, under the conditions used, wild-type A3G inhibited the infectivity of HIV-1 by ~30 fold. Mutation of the amino-terminal CDA of A3G, in either A3G-E1 or A3G-C1, led to an ~3 fold inhibition in HIV-1 infectivity, i.e. 8–12 fold less than seen with wild-type A3G. Similarly, A3G mutants bearing a defective carboxy-terminal CDA, i.e. A3G-E2 and A3G-C2, inhibited HIV-1 infectivity by only ~4 fold, i.e. ~8 fold less effectively than wild-type A3G. Finally, when both CDAs in A3G were mutated in A3G-E1+2, inhibition of HIV-1 infectivity was largely lost. Although the A3G-C1+2 mutant did appear to reduce HIV-1 infectivity by up to four fold in the experiments reported in Fig. 1A, this mutant displayed ≤2 fold inhibitory activity in other experiments (data not shown) and is therefore likely to share the minimal inhibitory activity noted with the equivalent A3G-E1+2 mutant.
Fig. 1.

Effect of A3G and A3B mutants on HIV-1 infectivity and virion incorporation. A) Inhibition of HIV-1 infectivity by wild-type and mutant A3G variants was measured by co-transfecting 293T cells with the HIV-1 indicator vector pNL-LucΔ Vif and an APOBEC3 expression plasmid. At 48 h post-transfection, supernatant virions were collected and used to infect naïve 293T cells. At 24 h later, these infected cells were lysed and induced luciferase levels determined. The data are presented relative to virus derived from control cultures transfected with the parental pK vector in place of an APOBEC3 expression vector (NEG), which was set at 100% infectivity. These data derive from three independent experiments with standard deviations shown. B) Similar to panel A, except that 293T cells were co-transfected with plasmids expressing wild-type or mutant A3B proteins. C) 293T cells were co-transfected with the HIV-1 proviral plasmid pNL4-3Δ EnvΔ Vif and a plasmid expressing carboxy-terminally HA-tagged A3G or an HA-tagged cytoplasmic control protein, β-arrestin (CONT). At 48 h post-transfection, the supernatant media were harvested and filtered and virions collected by pelleting through a 20% sucrose cushion. At the time the supernatant media were harvested, we also collected the virion producing 293T cells. We then performed Western analyses of the producer cell lysates and the lysed, pelleted virions using antibodies specific for the HA-tag or specific for the HIV-1 capsid protein. D) Similar to panel C, except that the intracellular expression and virion incorporation of A3B mutants was analyzed.
Analysis of the inhibition of HIV-1 by A3B mutants revealed a slightly different picture (Fig. 1B). Like A3G, wild-type A3B inhibited HIV-1 infectivity by ~40 fold. This high level of inhibition, which we have noted previously (Doehle et al., 2005; Bogerd et al., 2006a), contrasts with the relatively modest inhibition of HIV-1 infectivity by A3B noted by others (Yu et al., 2004a; Bishop et al., 2006; Hakata and Landau, 2006; Stenglein and Harris, 2006). While the reason(s) for this consistent difference is not clear, we have noted that plasmids containing the A3B gene occasionally undergo spontaneous C to T editing in bacteria (data not shown), which could result in reduced biological activity if not detected.
Mutation of the amino-terminal CDA of A3B, in A3B-E1 or A3B-C1, had a fairly modest phenotypic effect, with the observed inhibition of the infectivity of HIV-1 virions being reduced to 10–15 fold, i.e. only 2–3 fold less than seen with wild-type A3B. Mutation of the carboxy-terminal CDA of A3B, in A3B-E2 and A3B-C2, resulted in a more severe phenotype, with the observed inhibition of HIV-1 infectivity being reduced to 4–8 fold. The simultaneous mutation of both CDAs also reduced the inhibition of HIV-1 infectivity by A3B by ~5 fold, leaving a significant residual inhibition of ~8 fold when compared to the level of HIV-1 infectivity seen in the absence of any APOBEC3B protein (Fig. 1B).
Effect of CDA mutations on virion incorporation of A3G and A3B
It has previously been reported that the amino-terminal CDA, and particularly the zinc-coordinating residues of this CDA, play an important role in mediating the specific incorporation of A3G into HIV-1 virion particles (Navarro et al., 2005). To address whether the various mutations introduced into A3G or A3B affected their incorporation into HIV-1 virions, we isolated virions produced from cells expressing these proteins by ultracentrifugation through a sucrose cushion, as previously described (Wiegand et al., 2004). This analysis also allowed us to address the important question of whether any of the introduced mutations destabilized A3G or A3B and to confirm that none of the APOBEC3 proteins tested had any effect on the production and release of HIV-1 virions (Fig. 1C and 1D).
As shown in Fig. 1D, the A3B-E2 and A3B-C2 mutants were expressed at intracellular levels that were comparable to wild-type and were also incorporated into HIV-1 virions with an efficiency that was similar to the wild-type A3B protein. The E1 and C1 mutations appeared to give rise to a slightly lower level of intracellular A3B protein expression, for both the single CDA mutants A3B-E1 and A3B-C1 and both double CDA mutants (Fig. 1D). These mutations also gave rise to a slightly lower level of incorporation of A3B into HIV-1 virions (Fig. 1D). However, when compared to the HA-tagged cytoplasmic negative control protein β-arrestin, it was clear that all A3B mutants were still being selectively incorporated into HIV-1 virions at levels that varied by no more than ~3 fold. Analysis of the incorporation of the A3G mutants into HIV-1 virions showed that all A3G mutants were expressed at comparable levels in the virion producer cells (Fig. 1C). In agreement with previous reports (Navarro et al., 2005; Hakata and Landau, 2006), the introduction of mutations into the carboxy-terminal CDA of A3G had no detectable effect on the incorporation of A3G into HIV-1 virions (Fig. 1C). In contrast, introduction of either the E1 or the C1 mutation clearly reduced A3G virion incorporation by ~4 fold. Moreover, A3G variants carrying mutations in both CDAs appeared to be essentially lost from HIV-1 virion particles (Fig. 1C).
Effect of CDA mutations on the cytidine deaminase activity of APOBEC3 proteins in bacteria
As noted above, it has been reported that the carboxy-terminal CDA is the only enzymatically active CDA for both A3G and A3B (Haché et al., 2005; Navarro et al., 2005; Newman et al., 2005; Hakata and Landau, 2006; Stenglein and Harris, 2006). We wished to confirm this result and also to measure the cytidine deaminase activity of the various A3G and A3B mutants. As one approach to this question, we used a previously described assay (Beale et al., 2004; Bogerd et al., 2006a; Jónsson et al., 2006) that measures the ability of an expressed gene product to enhance the level of mutagenesis of the RNA polymerase B (rpoB) gene in E. coli. Mutations in rpoB can be readily detected by measuring the frequency of rifampicin resistant (rifR) colonies.
We first compared the ability of a range of different human APOBEC3 proteins to induce hypermutation. A3A, A3B and A3C all increased the number of rifR bacteria by 100 to 150 fold (Fig. 2A). In contrast, expression of A3G enhanced the number of rifR colonies by ~5.4 fold while A3F only had a ~1.6 fold enhancing effect over background. Because A3B, A3G and A3F are all effective at introducing C to T mutations into HIV-1 proviruses produced in their presence (Harris et al., 2003; Mangeat et a., 2003; Bishop et al., 2004; Liddament et al., 2004; Wiegand et al., 2004; Doehle et al., 2005), these data imply that the mutational activity of APOBEC3 proteins observed in the bacterial mutator assay is not an accurate predictor of their DNA editing activity in retroviral virions. Indeed, it has recently been reported that murine APOBEC3, which is highly active in editing HIV-1 reverse transcripts produced in its presence, is inactive in the bacterial mutator assay (Jónsson et al., 2006). Importantly, as shown by Western analysis (Fig. 2A), these differences do not result from differences in the level of expression of the various APOBEC3 proteins in bacteria.
Fig. 2.
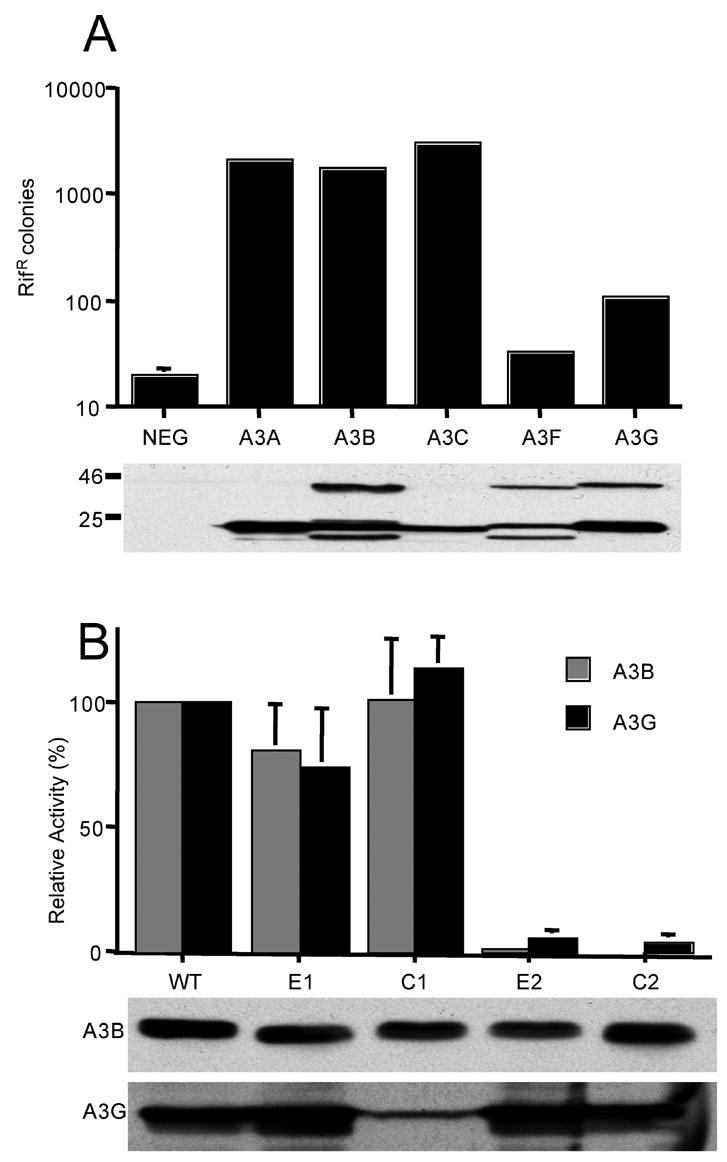
Effect of APOBEC3 protein expression on the level of mutation detected in bacteria. This analysis measures the ability of APOBEC3 proteins to induce a mutator phenotype in bacteria, as determined by acquisition of resistance to the antibiotic rifampicin (RifR). Plasmids that encode the indicated APOBEC3 proteins were introduced into E. Coli strain BW310 and their expression activated using isopropyl β-D-thiogalactoside. The level of mutation induced by each protein was then quantified by plating the bacteria on media containing rifampicin and counting the number of resistant colonies. The parental pTrc99A expression plasmid served as the negative control. A) Analysis of the mutator phenotype of wild-type human APOBEC3 proteins. Average of three experiments. The Western blot analyzing the expression of each protein in bacteria was performed using an HA-tag specific antibody. APOBEC3 proteins containing 2 CDAs, i.e. A3B, A3F and A3G, are detected in both their full-length, ~42 kDA form and as a breakdown product of ~22 kDA that likely represents the HA-tagged carboxy-terminal CDA domain. B) Analysis of the mutator phenotype of CDA mutants of A3G or A3B. Data are given as a percentage of the mutator activity seen with the wild-type A3G or A3B protein, which was set at 100%. Average of three experiments with standard deviation indicated. A Western blot showing the expression level of the full-length, ~42 kDA forms of each A3B or A3G variant in bacteria is presented at the bottom of the figure.
With the above caveat in mind, we next asked whether the very high mutator activity of A3B, or the modest but readily detectable mutator activity seen with A3G, would in fact be affected by any of the single CDA mutants. As shown in Fig. 2B, and consistent with previous reports (Navarro et al., 2005; Newman et al., 2005; Hakata and Landau, 2006; Jónsson et al., 2006; Stenglein and Harris, 2006), we found that mutation of the amino-terminal CDA of A3G or A3B, in the E1 and C1 mutants, had no significant effect on the bacterial mutator activity of these proteins. In contrast, both the E2 and the C2 mutation inhibited the ability of either A3B or A3G to induce hypermutation of bacterial DNA.
Both CDAs present in A3B are enzymatically active
Because of the clearly non-linear relationship between the editing activity of human APOBEC3 proteins in HIV-1 reverse transcription complexes and their ability to induce hypermutation in bacteria (Fig. 2A), we next directly measured the editing of HIV-1 reverse transcripts produced in the presence of wild-type or mutant variants of A3G and A3B.
In the case of A3G, the expected result (Navarro et al., 2005; Newman et al., 2005) was obtained. That is, wild-type A3G introduced a high level of C to T mutations into HIV-1 reverse transcripts (Fig. 3A) while both the A3G-E2 and the A3G-C2 mutant failed to induce a level of proviral DNA editing that was above background (Fig. 3B and C). In the case of wild-type A3B, we again detected a high level of C to T mutagenesis (Fig. 3D). Analysis of the A3B-E1 mutant produced the expected result (Hakata and Landau, 2006; Stenglein and Harris, 2006), i.e. we detected a level of C to T editing that was well over background but somewhat lower than observed with wild-type A3B (Fig. 3E), a difference that could potentially be ascribed to the slightly lower level of A3B-E1 that is packaged into HIV-1 virions (Fig. 1D). The surprising result was obtained with A3B-E2, which gave rise to a level of C to T editing of HIV-1 reverse transcripts that was both well over background and closely comparable to the level seen in A3B-E1 (Fig. 3F). To confirm that an A3B mutant lacking a functional carboxy-terminal CDA active site is indeed still capable of inducing DNA editing, we also measure editing of HIV-1 reverse transcripts produced in the presence of A3B-C2 (Fig. 3G). Again, we observed a level of C to T editing that was clearly above background. To confirm that the E1 and E2 mutations are indeed able to inactivate the CDA active sites present in A3B, we also measured editing induced by A3B-E1+E2. As shown in Fig. 3H, no significant level of C to T editing was detected. In conclusion, these data confirm earlier data (Navarro et al., 2005; Newman et al., 2005; Jónsson et al., 2006) showing that only the carboxy-terminal CDA in A3G is active and demonstrate that both CDAs in A3B have the potential to independently induce editing of HIV-1 reverse transcripts.
Fig. 3.

Analysis of C to T editing of HIV-1 reverse transcripts by A3G and A3B CDA mutants. To analyze the level of C to T editing by the various CDA mutants, we generated HIV-1 virions in the presence of each protein and used these to infect naïve 293T cells. At 16 h after infection, total cellular DNA was harvested, cleaved with DpnI and then a region of the env gene of HIV-1 amplifed using a high fidelity Taq polymerase. After cloning and sequencing, the number of introduced missense mutations were quantified and are here presented in tabular form for each wild-type or mutant APOBEC3 protein. The original HIV-1 sequence is given at left and the new sequence across the top. The total number of bases sequenced is shown at the lower right of each box.
Consensus sites for editing by A3B
An interesting aspect of the various human APOBEC3 proteins is that they all have characteristic consensus sites for editing. As shown in Fig. 4A, and previously reported, A3G prefers to edit C residues flanked by a 5′ C residue (Harris et al., 2003; Bishop et al., 2004; Wiegand et al., 2004; Khan et al., 2005). In contrast, A3B prefers to edit C residues flanked by a 5′ T residue, although a significant number of C residues flanked 5′ by C are also edited (Fig. 4B) (Bishop et al., 2004; Doehle et al., 2005). We therefore asked whether the amino- and carboxy-terminal CDA active sites of A3B might have different editing preferences. As shown in Fig. 4, the A3B-E1 mutant has a strong preference for TC while the A3B-E2 mutant prefers to edit CC.
Fig. 4.
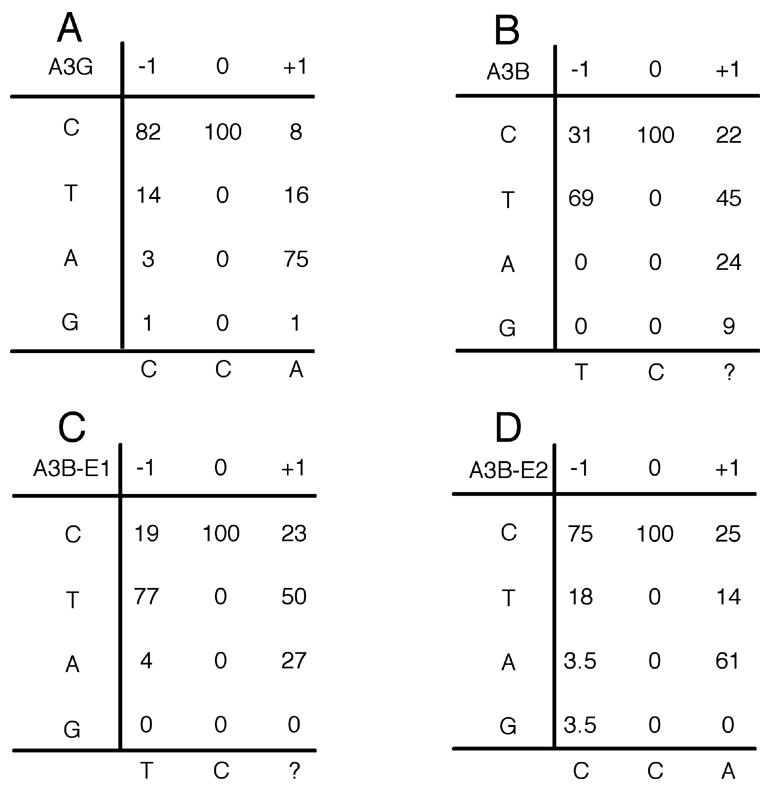
Consensus editing sequences for A3B CDA mutants. This figure presents the sequence context of the C to T mutations tabulated in Fig. 4 for each of the indicated APOBEC3 proteins. In particular, this figure shows the 5′ and 3′ flanking nucleotide around each edited C residue.
Discussion
The goal of this research was to examine the importance of the two CDAs present in the dual CDA domain APOBEC3 proteins A3G and A3B for their ability to inhibit HIV-1 infectivity. Previous research on this topic has largely focused on the ability of A3G, and to a lesser extent A3F, mutants to inhibit HIV-1 infectivity (Navarro et al., 2005; Newman et al., 2005; Jónsson et al., 2006; Holmes et al., 2007). Here, we have extended these earlier data by also examining a less well understood human APOBEC3 protein, A3B.
Mutants of A3G and A3B that are enzymatically inactive retain antiretroviral activity
An important conclusion to emerge from the research reported in this manuscript is that mutants of A3G or A3B that are inactive as cytidine deaminases nevertheless retain significant inhibitory activity against HIV-1. Specifically, the enzymatically inactive (Fig. 3) A3G-E2 and A3G-C2 mutants remained able to inhibit HIV-1 infectivity by ~4 fold (Fig. 1A) while the A3B-E1+2 and A3B-C1+2 mutants could inhibit HIV-1 infectivity by ~8 fold (Fig. 1B). Nevertheless, these mutant forms of A3G and A3B were clearly less active than wild-type. In part, at least in the case of A3B-E1+2 and A3B-C1+2, this reduced activity could reflect the somewhat reduced incorporation of these mutant proteins into HIV-1 virion particles when compared to wild-type A3G (Fig. 1D).
Our conclusion that proviral editing is not the only mechanism for inhibition of retroviral infectivity, and may not be the major mechanism, echoes conclusions reached by Malim and co-workers (Newman et al., 2005; Bishop et al., 2006; Holmes et al., 2007), although our results differ from their published data in significant ways. Specifically, Newman et al. (2005) reported that A3G mutants lacking a functional carboxy-terminal CDA, including mutants identical to A3G-E2 (E259Q) and A3G-C2 (C288S), retained almost full antiviral activity when assayed using HIV-1, yet introduced no C to T mutations into HIV-1 reverse transcripts. In contrast, our results (Fig. 1A) and work published by other groups using these same A3G mutants (Mangeat et al., 2003; Shindo et al., 2003; Hakata and Landau, 2006; Jónsson et al., 2006), argue that A3G mutants lacking a functional carboxy-terminal CDA are actually significantly attenuated for inhibition of HIV-1 infectivity. Recent data from the Malim laboratory has revealed that strong inhibition of HIV-1 by A3G-E2 (E259Q) actually is only observed when very high levels of A3G-E2 are expressed (Holmes et al., 2007). In contrast, the data presented in Fig. 1A were obtained using a fairly modest level of A3G expression that was chosen because it allowed full recovery of HIV-1 infectivity when a wild-type, i.e. Vif-expressing, HIV-1 proviral indicator construct was analyzed (Wiegand et al., 2004).
Despite these minor discrepancies among the published data, it nevertheless seems clear that A3G variants lacking an intact carboxy-terminal CDA do retain some ability to inhibit HIV-1 infectivity (Fig. 1A) (Newman et al., 2005; Jónsson et al., 2006; Holmes et al., 2007). This result has been even more apparent when mutants of A3F bearing a mutated carboxy-terminal CDA have been analyzed (Jónsson et al., 2006; Holmes et al., 2007). The data presented in this manuscript extend this earlier work by showing that enzymatically inactive forms of A3B also retain substantial inhibitory activity against HIV-1 (Fig. 1B).
A3B contains two enzymatically active CDAs
An interesting observation reported here is that A3B actually contains two enzymatically active CDAs, as revealed by the ability of not only A3B-E1 but also A3B-E2 and A3B-C2 to induce C to T mutagenesis of HIV-1 reverse transcripts produced in their presence (Fig. 3). A3G and A3F have previously been reported to contain only one enzymatically active CDA, located near the carboxy-terminus (Navarro et al., 2005; Newman et al., 2005; Haché et al., 2005; Hakata and Landau, 2006; Jónsson et al., 2006). Several non-human APOBEC3 proteins bearing two CDA domains have also been shown to contain only a single enzymatically active CDA, although in the case of murine APOBEC3 and bovine and ovine A3F, this appears to be the amino-terminal CDA (Hakata and Landau, 2006; Jónsson et al., 2006). In fact, it has previously also been reported that only the carboxy-terminal CDA of A3B is enzymatically active in mammalian cells (Kakata and Landau, 2006). We do not know the basis for this discrepancy, although we note that the single carboxy-terminal CDA mutant tested in this previous report, i.e. glutamic acid 255 to alanine, was not tested in our analysis and could be more disruptive to the structure of A3B than the more conservative A3B-E2 mutation (i.e. E255Q).
As A3B clearly contains two enzymatically active CDAs (Fig. 3), we were curious to determine whether these had different consensus editing sequence preferences. As shown in Fig. 4, and as previously reported, A3B edits cytidine residues flanked by either T or C, with the former being preferred (Bishop et al., 2004; Doehle et al., 2005). Analysis of the editing preferences of the A3B-E1 and A3B-E2 mutants indicates that the carboxy-terminal CDA of A3B strongly prefers to edit TC, while the amino-terminal CDA prefers to edit CC. These results suggest that the consensus editing sequence preference of the wild-type A3B protein may, in fact, be a composite of these two individual CDA activities.
Although the A3B-E2 and A3B-C2 mutants retained a significant ability to edit HIV-1 reverse transcripts (Fig. 3), they were unable to edit the rpoB gene in bacteria even though they were expressed at levels comparable to wild-type A3B (Fig. 2). These data recall the recent observation that the wild-type murine A3 protein, which is a potent editor of HIV-1 reverse transcripts when expressed in infected cells, is also inactive in this bacterial mutator assay (Jónsson et al., 2006). Together, these data clearly demonstrate that the inability to edit the rpoB gene in bacteria does not reliably indicate an inability to edit retroviral reverse transcripts in vivo. Why some mammalian cytidine deaminases are effective at genomic DNA editing when expressed in bacteria, while others are apparently inactive, currently remains unclear.
Materials and Methods
Molecular clones
We have previously described mammalian expression plasmids, derived from pK, that express C-terminally HA-tagged forms of A3B, A3G and β-arrestin (Bogerd et al., 2006b). Mutant forms of the pK/A3G-HA and pK/A3B-HA expression plasmids bearing single or double point mutations were generated by PCR and confirmed by DNA sequence analysis. The HIV-1 proviral derivatives pNL-LucΔ Vif and pNL4-3Δ VifΔEnv and the VSV glycoprotein expression plasmid pHIT/G have been previously described (Wiegand et al., 2004). Wild-type or mutant forms of A3B and A3G were transferred from pK into the bacterial expression plasmid pTrc99A (AB Biosciences, Piscataway, NJ) as previously described (Bogerd et al., 2006b).
Retroviral infectivity assays
Experimental determinations of the effect of APOBEC3 proteins on the infectivity of HIV-1 were performed as previously described (Wiegand et al., 2004; Doehle et al., 2005). 293T cells (35 mm plates) were transfected with 1.5 μg of pNL-LucΔ Vif, 125 ng of an APOBEC3 expression plasmid and 875 ng of pK. The pK/β-Arr-HA plasmid served as the negative control. At 48 h after transfection, supernatant media were harvested, filtered and used to infect 293T cells expressing CD4 and CXCR4. A further 24 h later, the infected cells were lysed and induced luciferase levels quantified.
For analysis of HIV-1 proviral DNA editing, 293T cells (35 mm dishes) were transfected with 1.5 μg of pNL-LucΔ Vif, 500 ng of an APOBEC3 expression plasmid, 500 ng of pK and 100 ng of pHIT/G. The resultant virions were collected at 48 h post-transfection and used to infect naïve 293T cells. After 4 h, the infected cells were washed and fresh media added. Infection was allowed to proceed for a further 12 h, at which point total DNA was harvested from the cells. Isolated DNA was digested with DpnI (New England Biolabs) to remove any contaminating plasmid DNA. A region of the proviral env gene containing DpnI sites was then amplified using Taq Plus precision polymerase (Stratagene), cloned and sequenced (Wiegand et al., 2004).
For analysis of the packaging efficiency of APOBEC3 proteins into virions, 293T cells (10 cm dishes) were transfected with 9 μg of pNL4-3Δ EnvΔ Vif together with 750 ng of an APOBEC3 expression plasmid and 5.25 μg of pK. HIV-1 virions were collected at 48 h after transfection by ultracentrifugation, as previously described (Wiegand et al., 2004; Doehle et al, 2005), and the producer cells lysed for analysis of intracellular protein expression levels. Western analyses of virion and cellular proteins were then performed using a mouse monoclonal specific for the HIV-1 capsid protein (Chesebro et al., 1992) and a mouse monoclonal specific for the HA epitope tag (Covance).
Acknowledgments
The authors thank Reuben Harris for helpful discussions and reagents used in this research. The following reagent was obtained from the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH: mouse monoclonal antibody specific for the HIV-1 capsid protein from Dr. Bruce Chesebro and Dr. Hardy Chen. This research was supported by NIH grants AI065301, AI057099 and AI064518.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alce TM, Popik W. APOBEC3G is incorporated into virus-like particles by a direct interaction with HIV-1 Gag nucleocapsid protein. J Biol Chem. 2004;279:34083–34086. doi: 10.1074/jbc.C400235200. [DOI] [PubMed] [Google Scholar]
- Beale RCL, Petersen-Mahrt SK, Watt IN, Harris RS, Rada C, Neuberger MS. Comparison of the differential context-dependence of DNA deamination by APOBEC enzymes: correlation with mutation spectra in vivo. J Mol Biol. 2004;337:585–596. doi: 10.1016/j.jmb.2004.01.046. [DOI] [PubMed] [Google Scholar]
- Betts L, Xiang S, Short SA, Wolfenden R, Carter CW., Jr Cytidine deaminase. The 2·3 Å crystal structure of an enzyme: transition-state analog complex. J Mol Biol. 1994;235:635–656. doi: 10.1006/jmbi.1994.1018. [DOI] [PubMed] [Google Scholar]
- Bishop KN, Holmes RK, Malim MH. Antiviral potency of APOBEC proteins does not correlate with cytidine deamination. J Virol. 2006;80:8450–8458. doi: 10.1128/JVI.00839-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop KN, Holmes RK, Sheehy AM, Davidson NO, Cho SJ, Malim MH. Cytidine deamination of retroviral DNA by diverse APOBEC proteins. Current Biol. 2004;14:1392–1396. doi: 10.1016/j.cub.2004.06.057. [DOI] [PubMed] [Google Scholar]
- Bogerd HP, Wiegand HL, Doehle BP, Lueders KK, Cullen BR. APOBEC3A and APOBEC3B are potent inhibitors of LTR-retrotransposon function in human cells. Nucleic Acids Res. 2006a;34:89–95. doi: 10.1093/nar/gkj416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogerd HP, Wiegand HL, Hulme AE, Garcia-Perez JL, O’Shea KS, Moran JV, Cullen BR. Cellular inhibitors of long interspersed element 1 and Alu retrotransposition. Proc Natl Acad Sci USA. 2006b;103:8780–8785. doi: 10.1073/pnas.0603313103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cen S, Guo F, Niu M, Saadatmand J, Deflassieux J, Kleiman L. The interaction between HIV-1 Gag and APOBEC3G. J Biol Chem. 2004;279:33177–33184. doi: 10.1074/jbc.M402062200. [DOI] [PubMed] [Google Scholar]
- Chen H, Lilley CE, Yu Q, Lee DV, Chou J, Narvaiza I, Landau NR, Weitzman MD. APOBEC3A is a potent inhibitor of adeno-associated virus and retrotransposons. Curr Biol. 2006;16:480–485. doi: 10.1016/j.cub.2006.01.031. [DOI] [PubMed] [Google Scholar]
- Chesebro B, Wehrly K, Nishio J, Perryman S. Macrophage-tropic human immunodeficiency virus isolates from different patients exhibit unusual V3 envelope sequence homogeneity in comparison with T-cell-tropic isolates: definition of critical amino acids involved in cell tropism. J Virol. 1992;66:6547–6554. doi: 10.1128/jvi.66.11.6547-6554.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conticello SG, Harris RS, Neuberger MS. The Vif protein of HIV triggers degradation of the human antiretroviral DNA deaminase APOBEC3G. Curr Biol. 2003;13:2009–2013. doi: 10.1016/j.cub.2003.10.034. [DOI] [PubMed] [Google Scholar]
- Cullen BR. Role and mechanism of action of the APOBEC3 family of antiretroviral resistance factors. J Virol. 2006;80:1067–1076. doi: 10.1128/JVI.80.3.1067-1076.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang Y, Wang X, Esselman WJ, Zheng YH. Identification of APOBEC3DE as another antiretroviral factor from the human APOBEC family. J Virol. 2006;80:10522–10533. doi: 10.1128/JVI.01123-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doehle BP, Schäfer A, Cullen BR. Human APOBEC3B is a potent inhibitor of HIV-1 infectivity and is resistant to HIV-1 Vif. Virology. 2005;339:281–288. doi: 10.1016/j.virol.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Haché G, Liddament MT, Harris RS. The retroviral hypermutation specificity of APOBEC3F and APOBEC3G is governed by the C-terminal DNA cytosine deaminase domain. J Biol Chem. 2005;280:10920–10924. doi: 10.1074/jbc.M500382200. [DOI] [PubMed] [Google Scholar]
- Hakata Y, Landau NR. Reversed functional organization of mouse and human APOBEC3 cytidine deaminase domains. J Biol Chem. 2006;281:36624–36631. doi: 10.1074/jbc.M604980200. [DOI] [PubMed] [Google Scholar]
- Harris RS, Bishop KN, Sheehy AM, Craig HM, Petersen-Mahrt SK, Watt IN, Neuberger MS, Malim MH. DNA deamination mediates innate immunity to retroviral infection. Cell. 2003;113:803–809. doi: 10.1016/s0092-8674(03)00423-9. [DOI] [PubMed] [Google Scholar]
- Holmes RK, Koning FA, Bishop KN, Malim MH. APOBEC3F can inhibit the accumulation of HIV-1 reverse transcription products in the absence of hypermutation: comparisons with APOBEC3G. J Biol Chem. 2007;282:2587–2595. doi: 10.1074/jbc.M607298200. [DOI] [PubMed] [Google Scholar]
- Jarmuz A, Chester A, Bayliss J, Gisbourne J, Dunham I, Scott J, Navaratnam N. An anthropoid-specific locus of orphan C to U RNA-editing enzymes on chromosome 22. Genomics. 2002;79:285–296. doi: 10.1006/geno.2002.6718. [DOI] [PubMed] [Google Scholar]
- Jónsson SR, Haché G, Stenglein MD, Fahrenkrug SC, Andrésdóttir V, Harris RS. Evolutionarily conserved and non-conserved retrovirus restruction activities of artiodactyl APOBEC3F proteins. Nucleic Acids Res. 2006;34:5683–5694. doi: 10.1093/nar/gkl721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan MA, Kao S, Miyagi E, Takeuchi H, Goila-Gaur R, Opi S, Gipson CL, Parslow TG, Ly H, Strebel K. Viral RNA is required for the association of APOBEC3G with human immunodeficiency virus type 1 nucleoprotein complexes. J Virol. 2005;79:5870–5874. doi: 10.1128/JVI.79.9.5870-5874.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddament MT, Brown WL, Schumacher AJ, Harris RS. APOBEC3F properties and hypermutation preferences indicate activity against HIV-1 in vivo. Curr Biol. 2004;14:1385–1391. doi: 10.1016/j.cub.2004.06.050. [DOI] [PubMed] [Google Scholar]
- Liu B, Sarkis PTN, Luo K, Yu Y, Yu XF. Regulation of APOBEC3F and human immunodeficiency virus type 1 Vif by Vif-Cul5-ElonB/C E3 ubiquitin ligase. J Virol. 2005;79:9579–9587. doi: 10.1128/JVI.79.15.9579-9587.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangeat B, Turelli P, Caron G, Friedli M, Perrin L, Trono D. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature. 2003;424:99–103. doi: 10.1038/nature01709. [DOI] [PubMed] [Google Scholar]
- Marin M, Rose KM, Kozak SL, Kabat D. HIV-1 Vif protein binds the editing enzyme APOBEC3G and induces its degradation. Nature Med. 2003;9:1398–1403. doi: 10.1038/nm946. [DOI] [PubMed] [Google Scholar]
- Mehle A, Strack B, Ancuta P, Zhang C, McPike MN, Gabuzda D. Vif overcomes the innate antiviral activity of APOBEC3G by promoting its degradation in the ubiquitin-proteasome pathway. J Biol Chem. 2004;279:7792–7798. doi: 10.1074/jbc.M313093200. [DOI] [PubMed] [Google Scholar]
- Muckenfuss H, Hamdorf M, Held U, Perkovic M, Löwer J, Cichutek K, Flory E, Schumann GG, Münk C. APOBEC3 proteins inhibit human LINE-1 retrotransposition. J Biol Chem. 2006;281:22161–22172. doi: 10.1074/jbc.M601716200. [DOI] [PubMed] [Google Scholar]
- Navarro F, Bollman B, Chen H, König R, Yu Q, Chiles K, Landau NR. Complementary function of the two catalytic domains of APOBEC3G. Virology. 2005;333:374–386. doi: 10.1016/j.virol.2005.01.011. [DOI] [PubMed] [Google Scholar]
- Newman ENC, Holmes RK, Craig HM, Klein KC, Lingappa JR, Malim MH, Sheehy AM. Anti-viral function of APOBEC3G can be dissociated from cytidine deaminase activity. Curr Biol. 2005;15:166–170. doi: 10.1016/j.cub.2004.12.068. [DOI] [PubMed] [Google Scholar]
- OhAinle M, Kerns JA, Malik HS, Emerman M. Adaptive evolution and antiviral activity of the conserved mammalian cytidine deaminase APOBEC3H. J Virol. 2006;80:3853–3862. doi: 10.1128/JVI.80.8.3853-3862.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schäfer A, Bogerd HP, Cullen BR. Specific packaging of APOBEC3G into HIV-1 virions is mediated by the nucleocapsid domain of the gag polyprotein precursor. Virology. 2004;328:163–168. doi: 10.1016/j.virol.2004.08.006. [DOI] [PubMed] [Google Scholar]
- Sheehy AM, Gaddis NC, Choi JD, Malim MH. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature. 2002;418:646–650. doi: 10.1038/nature00939. [DOI] [PubMed] [Google Scholar]
- Sheehy AM, Gaddis NC, Malim MH. The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nature Med. 2003;9:1404–1407. doi: 10.1038/nm945. [DOI] [PubMed] [Google Scholar]
- Shindo K, Takaori-Kondo A, Kobayashi M, Abudu A, Fukunaga K, Uchiyama T. The enzymatic activity of CEM15/Apobec-3G is essential for the regulation of the infectivity of HIV-1 virion but not a sole determinant of its antiviral activity. 2003;278:44412–44416. doi: 10.1074/jbc.C300376200. [DOI] [PubMed] [Google Scholar]
- Stenglein MD, Harris RS. APOBEC3B and APOBEC3F inhibit L1 retrotransposition by a DNA deamination-independent mechanism. J Biol Chem. 2006;281:16837–16841. doi: 10.1074/jbc.M602367200. [DOI] [PubMed] [Google Scholar]
- Stopak K, de Noronha C, Yonemoto W, Greene WC. HIV-1 Vif blocks the antiviral activity of APOBEC3G by impairing both its translation and intracellular stability. Mol Cell. 2003;12:591–601. doi: 10.1016/s1097-2765(03)00353-8. [DOI] [PubMed] [Google Scholar]
- Suspène R, Sommer P, Henry M, Ferris S, Guétard D, Pochet S, Chester A, Navaratnam N, Wain-Hobson S, Vartanian JP. APOBEC3G is a single-stranded DNA cytidine deaminase and functions independently of HIV reverse transcriptase. Nucleic Acids Res. 2004;32:2421–2429. doi: 10.1093/nar/gkh554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiegand HL, Doehle BP, Bogerd HP, Cullen BR. A second human antiretroviral factor, APOBEC3F, is suppressed by the HIV-1 and HIV-2 Vif proteins. EMBO J. 2004;23:2451–2458. doi: 10.1038/sj.emboj.7600246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Q, Chen D, König R, Mariani R, Unutmaz D, Landau NR. APOBEC3B and APOBEC3C are potent inhibitors of simian immunodeficiency virus replication. J Biol Chem. 2004a;279:53379–53386. doi: 10.1074/jbc.M408802200. [DOI] [PubMed] [Google Scholar]
- Yu Q, König R, Pillai S, Chiles K, Kearney M, Palmer S, Richman D, Coffin JM, Landau NR. Single-strand specificity of APOBEC3G accounts for minus-strand deamination of the HIV genome. Nature Structural Mol Biol. 2004b;11:435–442. doi: 10.1038/nsmb758. [DOI] [PubMed] [Google Scholar]
- Yu X, Yu Y, Liu B, Luo K, Kong W, Mao P, Yu XF. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science. 2003;302:1056–1060. doi: 10.1126/science.1089591. [DOI] [PubMed] [Google Scholar]
- Zennou V, Perez-Caballero D, Göttlinger H, Bieniasz PD. APOBEC3G incorporation into human immunodeficiency virus type 1 particles. J Virol. 2004;78:12058–12061. doi: 10.1128/JVI.78.21.12058-12061.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Yang B, Pomerantz RJ, Zhang C, Arunachalam SC, Gao L. The cytidine deaminase CEM15 induces hypermutation in newly synthesized HIV-1 DNA. Nature. 2003;424:94–98. doi: 10.1038/nature01707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng YH, Irwin D, Kurosu T, Tokunaga K, Sata T, Peterlin BM. Human APOBEC3F is another host factor that blocks human immunodeficiency virus type 1 replication. J Virol. 2004;78:6073–6076. doi: 10.1128/JVI.78.11.6073-6076.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]