

Abstract
Dendritic cells (DCs) accumulate in the CNS during inflammatory diseases, but the exact mechanism regulating their traffic into the CNS remains to be defined. We now report that MIP-1α increases the transmigration of bone marrow-derived, GFP-labeled DCs across brain microvessel endothelial cell monolayers. Furthermore, occludin, an important element of endothelial tight junctions, is reorganized when DCs migrate across brain capillary endothelial cell monolayers without causing significant changes in the barrier integrity as measured by transendothelial electrical resistance. We show that DCs produce matrix metalloproteinases (MMP) -2 and -9 and GM6001, an MMP inhibitor, decreases both baseline and MIP-1α -induced DC transmigration. These observations suggest that DC transmigration across brain endothelial cell monolayers is partly MMP dependent. The migrated DCs express higher levels of CD40, CD80, and CD86 costimulatory molecules and induce T cell proliferation, indicating that the transmigration of DCs across brain endothelial cell monolayers contributes to the maintenance of DC Ag-presenting function. The MMP dependence of DC migration across brain endothelial cell monolayers raises the possibility that MMP blockers may decrease the initiation of T cell recruitment and neuroinflammation in the CNS.
Dendritic cells (DCs)4 in the CNS are sufficient to present Ags to naive T cells during autoimmune neuroinflammatory diseases (1). Although DCs could develop from microglia in the brain (2, 3) or be found as resident cells in the CNS (4,5), they also infiltrate into the CNS from the blood (1). The mechanism of DC infiltration into the CNS is yet to be determined.
During CNS inflammation, blood-borne immune cells access the CNS via the blood-brain barrier (BBB) (6, 7). Major components of the BBB, the brain microvessel endothelial cells, are closely interconnected with special cell-cell contacts, such as tight junctions (8), and along with astrocytes, pericytes, and neurons, they form the neurovascular unit (9). The neurovascular unit is important in the maintenance of the immune-privileged nature of the CNS and regulating cellular transmigration (10, 11). In inflammation, the BBB becomes compromised and allows cellular traffic into the CNS. Although factors that regulate lymphocyte and monocyte migration into the CNS have been extensively studied (12–19), the mechanism of DC interaction with brain microvessel endothelium and DC transmigration across the BBB remains to be defined. The concordance of DC accumulation in cerebrospinal fluid throughout experimental autoimmune encephalomyelitis (16, 20–24), DC localization to the proximity of inflamed microvessels in multiple sclerosis (MS) lesions (25), and the production of astrocyte-derived chemokines that promote DC recruitment into the CNS (26) make it likely that brain microvessel endothelial cells regulate DC recruitment into the CNS. Although DC transmigration across HUVEC and epithelial cell monolayers has been studied (27–31), the mechanism of DC migration across brain microvascular endothelium has not been investigated.
To explore the mechanism of DC transmigration across the brain endothelium, primary cultures of brain microvascular endothelial cells were used (32–34). Transmigration of DCs across brain microvascular cell monolayers was augmented in the presence of the MIP-1α chemokine and partly mediated by matrix metalloproteinases (MMP). DCs developed numerous filopodia and induced the rearrangement of occludin, an endothelial tight junction protein, without causing a detectable decrease of transendothelial electrical resistance (TEER) values, indicating that endothelial monolayers stayed intact during DC migration. DC transmigration across brain microvessel endothelial cell monolayers led to the up-regulation of CD40, CD80, and CD86 costimulatory molecules on DCs. Following their transmigration across brain microvessel endothelial cells, DCs induced Ag-specific T cell activation. Our findings reveal that DC transmigration across brain microvessel endothelium is regulated by MIP-1α chemokine, MMP, and occludin perturbation. Because the transmigration of DCs across brain microvessel endothelial cells contributes to the activation of Ag-specific T cells, the BBB may be a key element in the local restimulation of T cell responses in the proximity of brain microvessels and in the amplification of T cell-mediated immunity in the CNS.
Materials and Methods
Mice
Four- to 6-wk-old female C57BL/6 mice were obtained from The Jackson Laboratory. GFP-transgenic (Tg) mice were a gift from Dr. P. Marrack (National Jewish Center for Immunology and Respiratory Medicine, Denver, CO) (35). OVA257–264 (SINFEKL) peptide-specific TCR Tg mice (OT-1) (36, 37) were provided by Dr. K. Hogquist (University of Minnesota, Minneapolis, MN). Both Tg strains were bred on the C57BL/6 background. The animals were housed according to the guidelines of the National Institutes of Health and University of Wisconsin-Madison Research Animals Resource Center.
Abs and secondary reagents
Abs anti-CD205 (clone NLDC-145) and anti-CD11c (clone N418) were purified from hybridoma cell supernatants (American Type Culture Collection) and conjugated with the Cy5 fluorochrome (Amersham Biosciences) and biotin (Sigma-Aldrich). Anti-CD11a (clone M17/4), anti-CD8 (clone 53.6-7), anti-Vα2 (B20.1), anti-I-Ab (clone AF6-120.1), anti-CD40 (HM40-3), anti-CD80 (16-10A1), anti-CD86 (GL1), and anti-CD195 (CCR5) (clone C34-3448) Abs, streptavidin-CyChrome, and streptavidin-allophycocyanin conjugates were purchased from BD Pharmingen. Streptavidin-Alexa 568 conjugate was purchased from Molecular Probes and monoclonal goat anti-mouse MMP9 (AF909) was purchased from R&D Systems.
Brain capillary endothelial cells (BCEC)
Mouse BCEC were isolated and cultured according to Deli et al. (38, 39). In brief, cerebra of 4- to 6-wk-old mice were mechanically homogenized and microvessels were isolated by two rounds of collagenase digestion and density centrifugation. Capillary fragments were directly seeded onto Falcon inserts (3-μm pore; Fisher Scientific) coated with collagen type IV/fibronectin (10 μg/ml; Sigma-Aldrich) in DMEM with 10% FBS (Fisher Scientific), 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 50 μg/ml gentamicin (Sigma-Aldrich). Isolated capillaries were cultured for 2 days in puromycin-supplemented medium (4 μg/ml; Sigma-Aldrich) to eliminate contaminating perivascular smooth muscle cells, followed by culture in medium supplemented with basic fibroblast growth factor (2 ng/ml; Roche) (40, 41). After reaching confluence, cells were cultured in serum-free DMEM/Ham’s F12 (Sigma-Aldrich) containing 2 mM l-glutamine, 100 U/ml penicillin/streptomycin, and 50 μg/ml gentamicin (Sigma-Aldrich) supplemented with hydrocortisone (550 nM; Sigma-Aldrich) (32). Cells were maintained under serum-free conditions until use on days 4 and 5. Primary cultured monolayers of BCEC were characterized by immunostaining against BBB markers including transport and tight junction proteins (42–44). The BBB properties of primary isolated BCEC such as high electrical resistance, tight junction protein expression, and low sucrose permeability were maintained by serum-free culture conditions and hydrocortisone application (32, 34, 45, 46).
Dendritic cells
DCs were generated as previously described (47, 48). Briefly, bone marrow obtained from femurs and tibias was washed and plated in 24-well plates in RPMI 1640 with 5% FBS supplemented with 100 U/ml penicillin/streptomycin and 20 ng/ml GM-CSF. GM-CSF was titrated from supernatants of the GM-CSF secreting X63 cell line (gift from Dr. A. Erdei, Eotvos University, Budapest, Hungary). On day 7, the nonadherent and loosely adherent cells were removed and replated in the absence of GM-CSF. Following overnight incubation, the nonadherent cells were collected for use. Purity of DC cultures was >80% as determined by flow cytometry demonstrating high expression of CD11c, CD205, and MHC class II proteins as previously described (48).
Flow cytometry
Cells (0.5–1 × 106) were processed for flow cytometric analysis as previously described (48). Single-cell suspensions were incubated for 30 min on ice with saturating concentrations of Abs in the presence of unlabelled FcγRII/FcγRIII-specific Ab (clone 2.4G2) to block FcR-mediated, nonspecific binding. Cell surface staining was acquired on a four-color FACSCalibur flow cytometer and analyzed with CellQuest software version 3.1 (BD Immunocytometry Systems) and FlowJo (Tree Star) software version 5.4.5.
TEER
Endohm tissue resistance measurement chamber (World Precision Instruments) was used to measure the electrical resistance of endothelial cell monolayers. TEER values (Ω × cm2) were calculated, following the manufacturer’s instructions, by subtracting the contribution of bare filters and medium and multiplying by the surface area of the membrane.
DC transendothelial migration assay
DC migration across in vitro BCEC monolayers was analyzed using the QCM 24-well invasion assay (Chemicon International). A total of 2.5 × 105 DCs in 0.1 ml of prewarmed serum-free DMEM/Ham’s F12 medium was added to the top of the endothelial monolayers and 0.6 ml of medium supplemented with 200 ng/ml MIP-1α (R&D Systems) was added in the outer chamber of the inserts. After 24 h, DCs were removed by Cell Detachment Solution from upper and lower chambers and incubated with CyQuant GR nucleic acids dye. Fluorescent intensity was proportional to the number of migrated DCs and could be measured at 480/520 nm. Migration index was calculated as a percentage of migrated DCs from the whole cell population. The broad MMP inhibitor, GM6001, or its inactive control peptide (Sigma-Aldrich) was applied according to the manufacturer’s suggestion at 100 μM to block DC transendothelial migration. Experiments were repeated five times using three separate migration chambers and data are presented as the geometric mean ± SD.
Confocal laser scanning microscopy (CLSM)
Expression of endothelial occludin and DC distribution and localization on monolayers of BCEC were studied using CLSM. Confluent monolayers of endothelial cells were cocultured with GFP-labeled DCs for 3 h, rinsed with PBS supplemented with Ca2+ and Mg2+, and fixed in 1% paraformaldehyde in PBS. Samples were permeabilized in 0.1% Triton X-100 and 1% BSA in PBS and labeled with rabbit anti-occludin (4 μg/ml; Zymed Laboratories) pAb, followed by Alexa 568-conjugated goat anti-rabbit IgG (Chemicon International). The samples were analyzed by CLSM (MRC-1024; Bio-Rad), equipped with argon (488 nm) and helium-neon (568 nm) lasers. Serial sections were collected with a step increment of 0.1 μm in the z-axis starting at the apical surface. Cross-sections of xz and yz images were rendered using Visbio (version 2.31) software (University of Wisconsin, Madison, WI).
Scanning electron microscopy
BCEC monolayers cocultured with DCs for 3 h were fixed with 1% glutaraldehyde (Sigma-Aldrich) and 1% tannic acid (Mallinckrodt) in 0.1 M phosphate buffer (pH 7.4) for 10 min at room temperature and incubated in the same buffer overnight at 4°C. The monolayers were then washed with phosphate buffer and gradually dehydrated with 30, 50, 70, 80, 90, 95, and 100% (three times) of ethanol at room temperature. The insert membranes were carefully cut out and placed into the critical point dryer to exchange the ethanol content of the cells against CO2. The membranes then were coated with Au/Pd by TM200 (VCR Group) ion-beam sputter-coating device and analyzed by an Hitachi S-570 scanning electron microscope.
Zymography
The amount of secreted MMP in the cell culture supernatants was estimated by zymography. Gelatin (1 mg/ml; Sigma-Aldrich) was incorporated into polyacrylamide gel (10%). Concentrated cell samples (YM-10; Centricon) were mixed with an equal volume of 2× sample buffer (126 mM Tris-HCl (pH 6.8), 20% (v/v) glycerol, 0.01% (w/v) bromphenol blue; 4% (w/v) SDS), and electrophoresed for 3 h through the substrate gel. The gel was renatured in 2.7% (v/v) Triton X-100 for 1 h at room temperature and incubated overnight at 37°C in developing buffer (50 mM Tris-HCl (pH 7.5), 200 mM NaCl, 5 mM CaCl2, and 0.02% (v/v) Brij-35). The gel was stained with 0.25% (w/v) Coomassie Brilliant Blue in 25% isopropanol and 10% acetic acid and destained in 50% methanol and 10% acetic acid until bands with diminished staining appeared on a uniformly stained background. The molecular mass (kDa) of the gelatinases was estimated against markers of known molecular mass (Chemicon International). Gels were scanned (Hewlett-Packard Scanjet 5400c) and analyzed with a Scion Image program (a version of NIH Image; Scion Corporation).
Ag presentation assays
Splenocytes were isolated from OT-1 mice and labeled with CFSE as described previously (49) with minor modifications (50). In brief, OT-1 spleen cells were depleted of RBC, washed, CFSE (1 μM; Molecular Probes) labeled for 5 min at 37°C, and quenched with 20% FBS. DCs were pulsed with OVA (DC-OVA, 50 μg/ml; Sigma-Aldrich) for 16 h at 37°C, washed three times with PBS, and applied (2.5 × 105) to the luminal side of BBB endothelial cells, or the membrane inserts alone. Migrated DC-OVA were collected after 24 h and combined in a ratio of 1:10 (DCs:T cells) with OVA-specific CFSE-labeled OT-1 TCR Tg splenocytes in culture. After 3 days, OT-1 cells were collected and analyzed by flow cytometry. The proliferation and activation of OT-1-specific TCR Tg T cells was measured by analyzing CFSE staining intensity and the level of CD11a/ CD18 (LFA-1) expression. Fluorescent intensity was analyzed on gated Tg CD8+Vα2+ T cells as was described previously (48, 51).
Statistical data analysis
Each data point was run in triplicate in three to five independent experiments as indicated and data were analyzed using two-tailed t tests. All values are expressed as mean ± SD. FACS measurements were also run in triplicates. Bivariate dot plots or probability contour plots were generated upon data reanalysis to display the frequencies of, and patterns by which, individual cells coexpressing certain levels of cell surface Ags. Histograms were generated based on these contour plots with CellQuest software version 3.1 (BD Immunocytometry Systems) and FlowJo (Tree Star) software version 5.4.5. Descriptive statistics were applied by calculating raw means and percentages.
Results
MIP-1α increases DC transmigration across brain microvascular endothelial cell monolayers
Because MIP-1α (also known as CCL3) has been suggested to play a critical role in the migration of DCs into peripheral tissues (52–55), and this chemokine is up-regulated under inflammatory conditions in the CNS (54, 56– 60), we first sought to define the role of MIP-1α in DC migration across BCEC monolayers. As shown in Fig. 1A, GM-CSF-matured DCs were mobile and migrated across BCEC monolayers with high efficiency, even in the absence of MIP-1α. However, there was a linear increase in DC transmigration across BCEC monolayers when increasing amounts of MIP-1α were applied (Fig. 1A). In the presence of MIP-1α, DCs strongly adhered to microvascular endothelial cells within 3 h and developed numerous filopodia (Fig. 1B, arrow), some of which reached across the endothelial cell monolayers (Fig. 1B, arrowheads). In the absence of MIP-1α, DCs adherence to brain microvessel endothelial cells was significantly compromised during the same time period (data not shown). These data indicate that GM-CSF-maturated DCs can migrate through brain microvessel endothelial cell monolayers and this migration is augmented in the presence MIP-1α.
FIGURE 1. Accelerated transmigration of DCs across brain microvessel endothelial cell monolayers in response to MIP-1α.
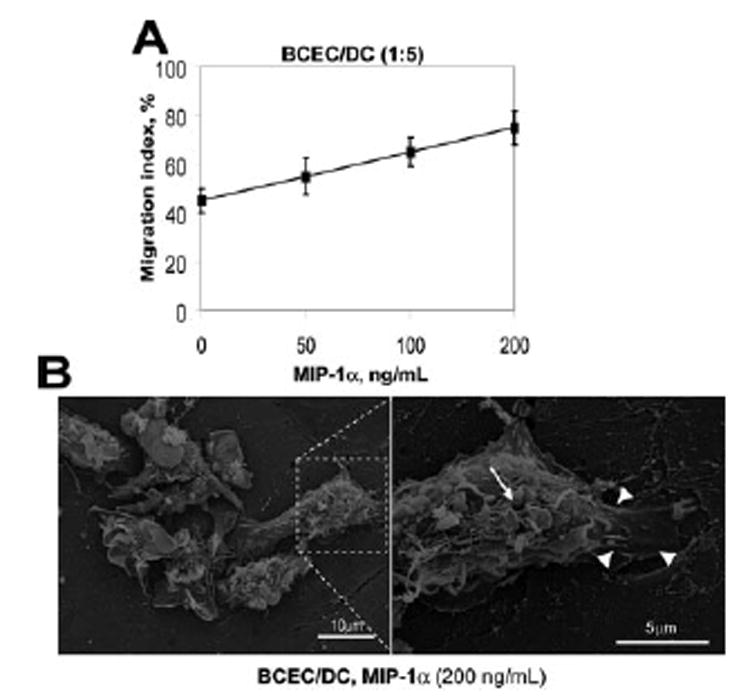
A, DC transmigration across in vitro BCEC monolayers was analyzed using QCM 24-well invasion assay. Migration index was calculated as the percentage of migrated DC number from the total cell population number (2.5 × 105) added to the upper compartment of the migrating chambers for 24 h. Data presented are the mean ± SD of five independent experiments. Each data point was run in triplicate and data were analyzed using two-tailed t tests. B, BCEC monolayers were cocultured with DCs for 3 h, fixed with 1% glutaraldehyde, and the phenotype of adhering DCs was analyzed according to Materials and Methods. Scanning electron microscopy shows that in the presence of chemokine MIP-1α there is a close contact between DCs and the BCEC monolayer, and DCs develop numerous filopodia (arrows) and protrusions (arrowheads; original magnification, ×1000). Inset, Higher magnification of DC morphology (original magnification, ×5000). The images shown are representative of three independent assays with similar observations.
Increased MMP2/MMP9 secretion and occludin tight junction molecule reorganization correlate with DC migration across BCEC monolayers
During inflammatory diseases of the CNS, MMP have been implicated in the disruption of the BBB and regulation of cellular infiltration (61–64). Therefore, we studied whether DC-secreted MMP could contribute to the migration of DCs across brain endothelial cell monolayers. To evaluate the level of MMP secretion by DCs, supernatants from GM-CSF maturated DCs were analyzed at 4, 6, 7, and 9 days of culture using gelatin zymography. As shown in Fig. 2, A and B, this method revealed a significant production of the latent or proform of MMP2 and MMP9 by DCs that accumulated in increasing amounts in the supernatants in a time-dependent manner. At day 4 of this in vitro culture period, DCs also expressed neutrophil gelatinase B-associated lipocalin (NGAL, 130-kDa band), which is known to covalently attach to MMP9 (65). Although NGAL production is predominantly associated with neutrophils, DC expression of this enzyme has also been suggested (66). In parallel with increased MMP production, NGAL expression declined in these cultures by time and was undetectable by day 6 of culture (Fig. 2A).
FIGURE 2. DCs express MMP2 and MMP9 which can be blocked by MMP inhibitor GM6001.
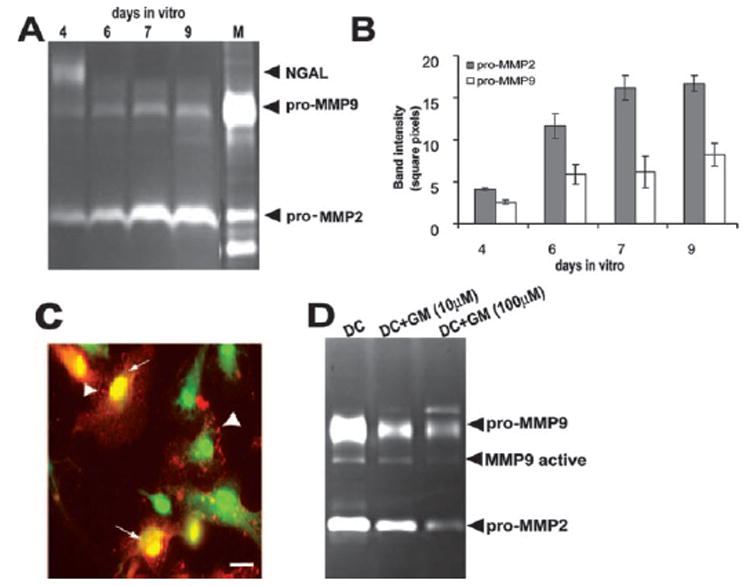
A, Zymography shows pro-MMP2 and pro-MMP9 detected in DC supernatants of cells cultured for 4, 6, 7, and 9 days in vitro. NGAL detected after 4 days of culture disappears by day 6 in vitro. M, MMP-specific marker. B, Zymography band intensity was calculated for three experiments and depicted as mean intensity ± SD (n ≥ 5). C, Immunostaining of GFP-DC (42) with mAb goat anti-mouse MMP9 (red) demonstrates that DCs store MMP9 in vesicles (arrows) and distribute them next to the plasma membrane for rapid release into the cell culture (arrowheads). Scale bar, 25 μm. D, Zymography of DC supernatants shows that DC expression of MMP is diminished by a broad MMP inhibitor, GM6001 (GM). The inhibition of both MMP2 and MMP9 was dependent on the concentration of the inhibitor applied. Zymography gel illustrates one representative result of three.
To determine MMP intracellular distribution in DCs, we performed immunocytochemistry and demonstrated punctate (vesicular) (Fig. 2C, arrowheads) as well as cytoplasmic localization of MMP9 (Fig. 2C, arrows). This is consistent with observations by Nguyen et al. (67) suggesting an accumulation of active MMP9 molecules in secretory vesicles that become effective during cell migration. We further demonstrated that MMP2 and MMP9 secretion by DCs could be blocked using an MMP-specific broad inhibitor, GM6001 (Fig. 2D). Zymography assays performed using DC culture supernatants indicated that GM6001 inhibited both MMP2 and MMP9 expression in a concentration-dependent manner (Fig. 2D).
It has been suggested that tight junction proteins can be degraded by MMP (68). To determine whether tight junction proteins were disrupted during DC transmigration across BCEC monolayers, we analyzed occludin expression and distribution on BCEC during DC migration. To measure the intercellular junctional integrity of the BCEC, the paracellular permeability of the monolayers was analyzed by measuring the TEER. After culture for 3 days in serum-supplemented medium, BCEC formed confluent monolayers that were well suited for studying the transcellular migration in vitro. The effect of DC coculture with BCEC on endothelial cell monolayer integrity was monitored from day 3 until day 5, after which experiments were performed (Fig. 3A). We also followed the effect of MIP-1α on electrical resistance of endothelial cell monolayers in the absence of DCs from day 3 until day 7 of in vitro culture (Fig. 3B). Both MIP-1α and DC interaction with BCEC caused no detectable changes in TEER values, suggesting that DCs were capable of diapedesis across the BCEC without disturbing the functional integrity of the barrier (Fig. 3).
FIGURE 3. Barrier integrity is not changed by MIP-1α alone or by MIP-1α-induced DC transmigration.
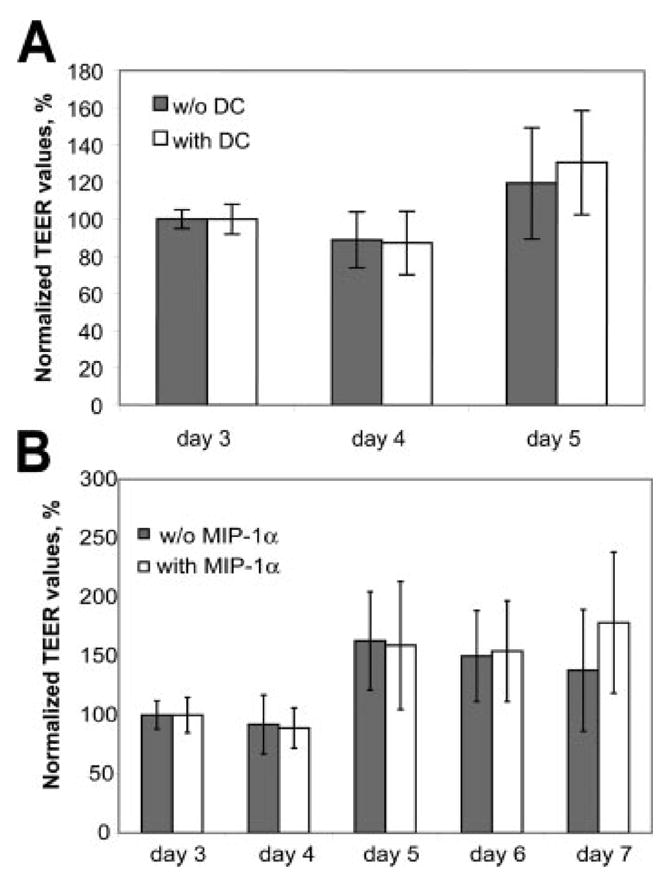
The integrity of the BCEC monolayer was monitored before and after coculture with DC (A) and in the presence or absence of MIP-1α (B) by measuring the TEER. Confluent BCEC monolayers (resistance ~50 Ω × cm2) after 3 days of culture were combined with DCs or cultured in medium, with or without MIP-1α, alone. TEER values were normalized to 100% of the initial TEER and monitored for 2–4 days following challenge. A, The TEER values of endothelial monolayers cultured with or without DCs in the presence of MIP-1α were similar and reached the highest values after DIV5 (150–200 Ω × cm2). B, Likewise, MIP-1α treatment alone had no effect on TEER values, indicating that MIP-1α does not influence the integrity of BCEC monolayers during MIP-1α-induced DC diapedesis. Data represent mean ± SD (n = 7–10).
To further analyze the mechanism of DC diapedesis across BCEC monolayers, we studied the distribution of tight junction molecules in the presence and absence of DCs. As shown in Fig. 4A, brain microvessel endothelial cells formed well-organized monolayers on basement membrane-coated filter inserts, presenting a uniform belt-like distribution of occludin tight junction molecules between endothelial cells. This appearance did not change in the presence of MIP-1α (Fig. 4B). Testing of MIP-1α treatment on zonula occludens-1 (ZO-1) molecule immunostaining also indicated that similarly to the distribution of occludin. MIP-1α had no affect on ZO-1 tight junction molecule distribution (data not shown). However, within 3 h after DCs were placed on the top of brain microvessel endothelial cell monolayers, discontinuous and poor organization of endothelial occludin was observed in the areas of DC interaction with brain microvessel endothelial cells (Fig. 4, C and D, arrows). In the absence of MIP-1α, there were fewer DCs adhering to microvascular endothelium (Fig. 4, C, arrowheads, and E). When DCs adhered to brain microvascular endothelial cell monolayers in the presence of MIP-1α, occludin distribution was different than that observed when endothelial cells were grown without DCs (Fig. 4D, arrows and xz/yz sections). Consistent with previous observations, DCs (42) also expressed occludin (yellow) in the presence of MIP-1α (69) (Fig. 4D, arrowhead). We applied the MMP inhibitor GM6001 to further understand the effect of MMP on occludin tight junction molecule distribution. We demonstrated that in the presence of GM6001, DCs (Fig. 4F, green and arrowheads) did not invade the endothelial tight junctions, leaving occludin distribution intact (Fig. 4F, right panel, arrows). These data indicate that MIP-1α-induced DC transmigration across brain microvessel endothelial cell monolayers is partly regulated by MMP and leads to occludin tight junction molecule redistribution. Because fewer DCs attached to the BCEC monolayer in the absence of MIP-1α, DC-mediated occludin perturbation could not be investigated under these conditions. However, the finding that endothelial occludin distribution did not change significantly in the presence of MIP-1α without DCs (Fig. 4B) makes it likely that DCs and not MIP-1α chemokine play a critical role in modulating occludin distribution during DC diapedesis.
FIGURE 4. MIP-1α -induced DC migration across brain microvessel endothelial cells is associated with occludin perturbation.
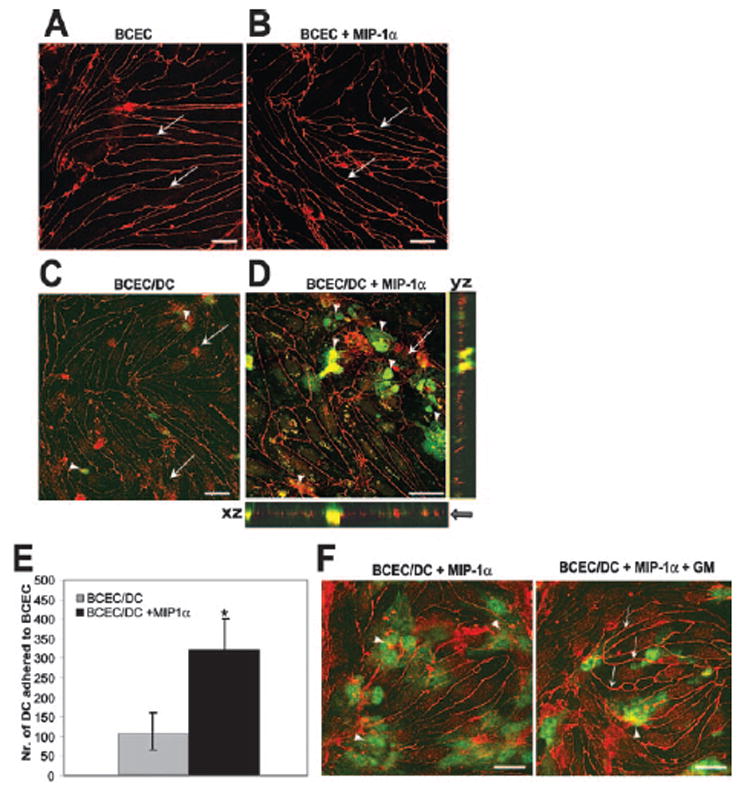
A–C, Immunostaining of the tight junction protein occludin on primary isolated BCEC grown in serum-free medium was performed as previously described (24). A, Occludin immunostaining shows that BCEC cultured without DCs form a tight monolayer with spindle-shaped morphology and without gaps in the absence of MIP-1α. B, MIP-1α does not change the structural integrity of BCEC monolayers based on occludin staining. C, Endothelial occludin expression after BCEC coculture with GFP-DC in the absence of MIP-1α. Low numbers of occludin-expressing DCs were detected on endothelial cell monolayers in the absence of MIP-1α (arrowheads). D, Distribution of occludin tight junction molecules on endothelial cells following coculture with GFP-DC in the presence of MIP-1α. Occludin immunostaining shows a punctate and discontinuous organization of endothelial occludin (arrows) in the areas of dendritic and endothelial cell interaction. The xz and yz sections exhibit strong yellow colocalization staining where GFP-DCs are partially migrated across brain microvessel endothelial cell monolayers and appear as green above and below the line of red occludin staining. This demonstrates that DCs are both above and below the BCEC layer (arrow). E, MIP-1α increases DC adhesion to BCEC monolayer by 70%. Adhered DCs were counted on three fields (original magnification, ×10) per filter on a total of four filters. Mean count ± SD is presented. *, p < 0.005. F, MMP inhibitor improves endothelial occludin distribution during DC transmigration across BCEC monolayers. Immunostaining of occludin (red) on BCEC cocultured with GFP-DC (green) was performed in the presence of inactive GM6001 control peptide (left panel) and active peptide (right panel). Images were taken in yz sections derived from the area of GFP-DC interacting with endothelium of the xy plane with a step of 1 μm. Both images represent the same time of coculture and the same plane of section. Left panel, A deep invasion of DCs into BCEC monolayer in the control sample (arrowhead). Right panel, A surface contact between DCs and BCEC at the beginning of their adhesion (arrowhead). Endothelial occludin distribution is more distinct and well organized during DC transmigration in the presence of MMP inhibitor (right panel, arrows). Images are representative of three experiments with similar results. Scale bars, 20 μm.
DC migration across BCEC monolayers can be partly blocked by MMP-specific inhibitor
Having established that GM-CSF-matured DCs produce MMP2 and MMP9 (Fig. 2), we next analyzed whether DCs would express MMP or induce MMP production by endothelial cells during their migration across BCEC monolayers. We have evaluated MMP production from BCEC supernatants cultured alone (BCEC) or with DCs (BCEC/DC) using gelatin zymography. As shown in Fig. 5, A and B, there was a significant increase of pro-MMP9 production measured in supernatants from BCEC/DC cocultures as compared with DCs cultured alone. This might indicate that MMP9 production by endothelial cells is induced by DCs. Three hours following medium exchange, endothelial cells grown alone did not produce pro-MMP9 but expressed low levels of active MMP9 and detectable amounts of pro-MMP2.
FIGURE 5. DC transmigration across brain microvessel endothelial cell monolayers can be blocked by MMP inhibitor.
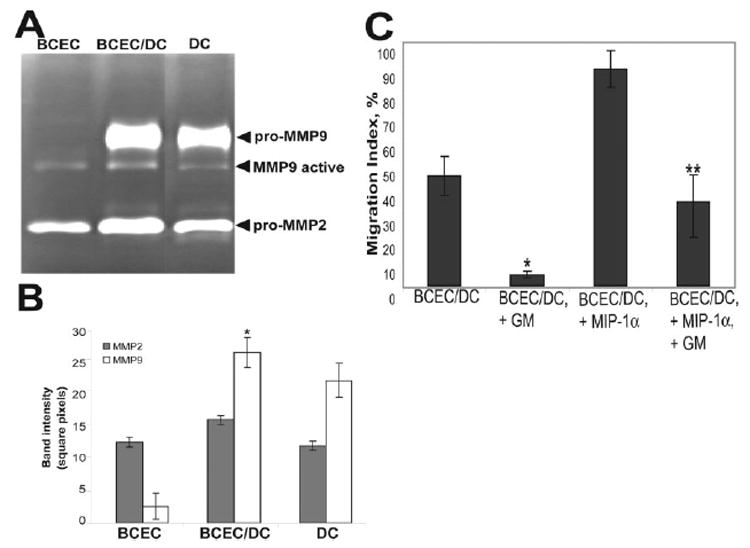
A, Zymography gel shows the amount of secreted MMP2 and MMP9 detected in the supernatants of BCEC or DCs individually, as well as in the supernatant of coculture of BCEC and DCs, for 3 h following medium change. One representative experiment of three with similar results is shown. B, Band intensities from zymography gels were calculated for three experiments and depicted as mean intensity ± SD (n ≥ 5), *, p < 0.01; **, p < 0.05. C, Migration index of DCs across BCEC monolayers in the presence and absence of MIP-1α (200 nM) was calculated and compared during treatment by control and active GM6001 peptides (GM, 100 μM). Data presented are the mean ± SD of three independent experiments. *, p < 0.001, compared with baseline DC migration across BCEC and **, p < 0.05, compared with MIP-1α-induced DC migration across BCEC under inactive GM6001 treatment.
To determine whether secreted MMP facilitate DC transendothelial migration in addition to disruption of occludin, we analyzed DC traffic across the BCEC monolayers in the presence of control and active GM6001 peptide before and after MIP-1α treatment. Fig. 5C demonstrates that baseline DC migration across BCEC monolayers is significantly impaired in the presence of GM6001. MIP-1α-induced DC migration across BCEC monolayers can also be inhibited by GM6001 peptide, resulting in a return to nearly baseline level of DC migration. This suggests that both baseline and MIP-1α-induced migration of DCs across brain microvessel endothelial cell monolayers is partly MMP dependent. However, additional mechanisms should also be considered, since both baseline and MIP-1α-induced migration of DCs across BCEC monolayers could not be entirely inhibited by MMP inhibitor (Fig. 5C).
DCs activate naive CD8+ T cells upon their migration across brain endothelial cell monolayers
Previous studies have shown that DCs might be sufficient to present Ags to naive T cells and promote epitope spreading locally in the CNS (1). Whether DCs that migrate across brain microvessel endothelial cell monolayers would be capable of stimulating naive T cell responses was uncertain. To answer this question, we analyzed the Ag-presenting capability of OVA-pulsed DCs following their transendothelial migration. OVA-pulsed DCs were allowed to migrate across brain endothelial cell monolayers in the presence of MIP-1α for 24 h. As shown in Fig. 6A, DCs upon their migration expressed elevated levels of costimulatory molecules CD40, CD80, and CD86. Increased expressions of CD40, CD80, and CD86 costimulatory molecules on migrating DCs were not directly the result of MIP-1α treatment, because the expression of these molecules on DCs did not increase in the absence of brain microvessel endothelial cells (Fig. 6B, □). The CD80 (B7-1) molecule was up-regulated by MIP-1α on DCs in the presence of BCEC (Fig. 6B,
 ); however, the expression of CD80 remained unchanged after DC transmigration across brain microvessel monolayers (Fig. 6B, ■). These data suggest that CD40, CD80, and CD86 costimulatory molecules are up-regulated upon the migration of DCs across brain microvessel cells and that this up-regulation is not dependent on the presence of MIP-1α. To further address the question of whether MIP-1α directly influences DC maturation, we measured the expression of the CD195/CCR5 chemokine receptor on DCs before and after migration in the presence and absence of MIP-1α. As we show in Fig. 6C, MIP-1α treatment did not up-regulate CCR5 expression on nonmigrating DCs (or on DCs in in vitro cultures without brain endothelial cells). Some up-regulation of CCR5 on migrating DCs after MIP-1α treatment was shown; however, this up-regulation was not statistically significant when compared to nontreated cells. In conclusion, our data demonstrate that MIP-1α treatment alone is not sufficient for inducing DC maturation.
); however, the expression of CD80 remained unchanged after DC transmigration across brain microvessel monolayers (Fig. 6B, ■). These data suggest that CD40, CD80, and CD86 costimulatory molecules are up-regulated upon the migration of DCs across brain microvessel cells and that this up-regulation is not dependent on the presence of MIP-1α. To further address the question of whether MIP-1α directly influences DC maturation, we measured the expression of the CD195/CCR5 chemokine receptor on DCs before and after migration in the presence and absence of MIP-1α. As we show in Fig. 6C, MIP-1α treatment did not up-regulate CCR5 expression on nonmigrating DCs (or on DCs in in vitro cultures without brain endothelial cells). Some up-regulation of CCR5 on migrating DCs after MIP-1α treatment was shown; however, this up-regulation was not statistically significant when compared to nontreated cells. In conclusion, our data demonstrate that MIP-1α treatment alone is not sufficient for inducing DC maturation.
FIGURE 6. DCs that transmigrated across brain microvessel endothelial cells express high levels of costimulatory molecules.
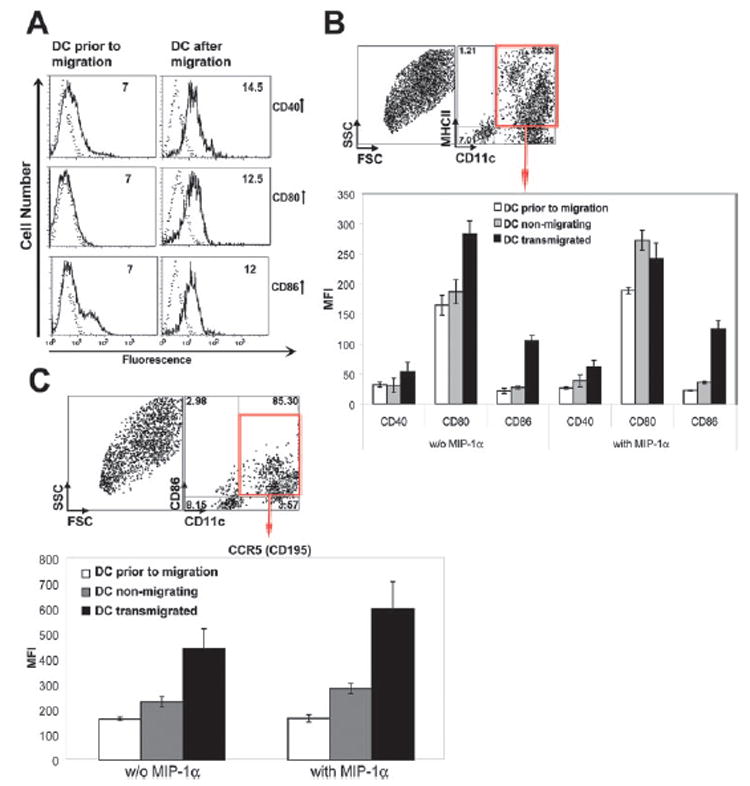
A, DCs were cocultured with BCEC in Transwell chambers as described in Materials and Methods. After 24 h, DCs were collected from the bottom chambers of Transwells and these cells were analyzed by flow cytometry for expression of CD11c, MHC class II, CD40, CD80, and CD86 molecules (right panel). DCs were also analyzed before the migration assays (left panel). Dotted lines indicate control isotype staining. Numbers indicate the geometric mean fluorescence intensity (MFI) of CD11c+CD205+MHCIIhigh DCs expressing CD40, CD80, and CD86 costimulatory molecules. B, The expression of CD40, CD80, and CD86 was analyzed on MHCII+CD11c+ DCs before and after their transmigration across brain microvessel endothelium in the presence and absence of MIP-1α. Numbers in dot plots indicate percentage of CD11c+ and class II+ cells that were analyzed from the total population. The data are represented in the geometric MFI of CD40, CD80, and CD86 molecules expressing on DCs. The level of expression of costimulatory molecules was compared for DC prior to migration, nonmigrating, and transmigrated DC (lower panel). C, The expression of CCR5 was analyzed on DCs before and after their transmigration across BCEC in the presence and absence of MIP-1α. CCR5 expression was analyzed on CD86+CD11c+ cells (upper panel) and presented in MFI (lower panel). Numbers in dot plots indicate percentage of CD11c+ and CD86+ cells that were analyzed from the total cell population. FSC, Forward scatter.
We further investigated whether DCs that migrated across brain microvessel endothelial cells were capable of presenting Ags to naive T cells. To accomplish this, we cocultured OVA-pulsed DCs collected from the lower chamber of migration wells with CFSE-labeled TCR Tg OT-1 T cells for 3 days in vitro. Cell division rapidly dilutes the CFSE signal from OT-1 progeny cells, and cell expansion could be measured by defining CSFE content using cytofluorimetry. As shown in Fig. 7 (right panel), migrated and OVA-pulsed DCs induced OT-1 T cell proliferation (black solid line) more efficiently then cultured, OVA-pulsed DCs (gray solid line), whereas CFSE-labeled OT-1 T cells cultured in the absence of DCs did not proliferate (dashed line, gray filled area). Up-regulation of LFA-1 activation marker on Ag-specific OT-1 T cells also indicated that DCs, upon their migration across brain microvessel endothelial cell monolayers, could induce T cell activation (Fig. 7, left panel). Taken together, these results indicate that DC transmigration across brain microvessel endothelium contributes to costimulatory molecule up-regulation and efficient Ag presentation by DCs.
FIGURE 7. DCs that transmigrated across BCEC monolayers present OVA Ag to Ag-specific cells.
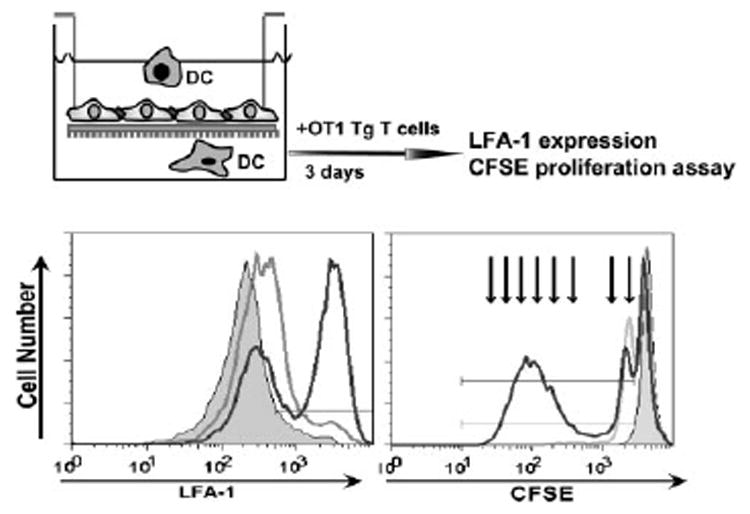
Migrated, OVA-pulsed DCs were collected and cocultured for 3 days with OVA-specific, CFSE-labeled TCR Tg T cells (OT-1 T cells). OT-1 T cells were assessed by flow cytometry. Activation of Vα2+CD8+ OT-1 T cells in the presence of migrated (black line) and cultured (gray line) OVA-pulsed DCs is demonstrated by increased numbers of LFA-1high expressing cells as compared with naive OT-1 T cells (dashed area, left panel). Proliferation of Vα2+CD8+ OT-1 T cells is shown by decreased numbers of CFSEbright cells after stimulation by migrated DCs (black line) and cultured DCs (gray line) as compared with samples in the absence of DC-OVA (dashed area, right panel). Arrows, OT-1 T cell division cycles in vitro. Data are representative of three independent experiments with similar results.
Discussion
DCs are critical during the initiation, development, and maintenance of CNS autoimmunity and inflammation (1, 22, 25, 70). Previously, it was shown that DCs could be recruited into the CNS in multiple infectious and autoimmune diseases (1, 71–73); however, the pathway of their recruitment has not yet been elucidated. In this study, we have defined a role for MMP and MIP-1α in DC transmigration across brain microvessel endothelial cell monolayers. More importantly, we show that once DCs transmigrated across brain microvessel endothelial cell monolayers, they induced the activation of naive Ag-specific T cells.
Transmigration of immune cells from the blood into the CNS is critical in CNS inflammatory responses, and it is largely regulated by the BBB. Previous studies showed that multiple factors, including chemokines and adhesion molecules, play a role in leukocyte migration into the CNS (reviewed in Ref. 6 and Ransohoff et al. (11)), but factors regulating DC migration across the BBB are poorly understood. The mechanisms involved in DC interaction with nonbrain endothelial or epithelial cells have been examined by several groups (27, 30, 69, 74, 75). DC transmigration across mouse tracheal epithelial cells has been demonstrated to be severely impaired in MMP9-deficient mice (27). In these studies, similar to our findings, MIP-1α and MIP-3β elicited DC migration across tracheal epithelial cells. We show that the migration of DCs across the BCEC monolayers is very substantial in the absence of MIP-1α and increases in the presence of this chemokine. It was already shown that MIP-1α could have an effect on both endothelial cells and DCs. For example, Solanilla et al. (76) have demonstrated that MIP-1α and TNF-α stimulate endothelial production of membrane-bound and soluble Flt3 ligand (76). A number of adhesion molecules on endothelial cells also have been shown to be up-regulated in the presence of MIP-1α and other chemokines (17). It is also known that immature DCs undergo chemotaxis in response to MIP-1α and up-regulate the CCR5 receptor that binds to this chemokine (77–80). In our work, we demonstrated that MIP-1α increases DC adhesion to BCEC and accelerates DC migration across the BBB.
MMPs were also shown to play a critical role in human DC migration across the Matrigel extracellular matrix, and the dominant MMP involved in this process was MMP9 (28). A physiologic function for p-glycoprotein (MDR-1) during the migration of DCs from skin via afferent vessels was also demonstrated (29). Because MDR-1 is a well-known transporter that mediates efflux of chemotherapeutic agents from the cytoplasm in brain endothelial cells, it is possible that this molecule also participates in DC transmigration across the BBB. Interestingly, it has also been reported that DCs express tight junction proteins, including occludin, claudin 1, and ZO-1 (69). It was proposed that DCs open the tight junctions between gut epithelial cells, send dendrites outside the epithelium, and directly sample bacteria in the lumen of the gut without compromising the integrity of the gut epithelial barrier (69). Our data confirm that DCs express tight junction occludin molecules (Fig. 4D, yellow), and DC transmigration across brain endothelial monolayers leads to a discontinuous expression of occludin molecules. Some evidence indicates that occludin contains a cleavage site for MMP in the first extracellular loop and, due to this, MMP-induced occludin degradation might be important in DC migration across tight junction-forming endothelial or epithelial cell monolayers (81). This is further supported by several groups demonstrating that occludin molecules are degraded by secreted MMP (27, 68, 82, 83). Because we showed that DCs in culture express MMP, these data suggest that DCs could use these proteases to alter occludin and traverse brain microvessel endothelial cell monolayers. Although we did not demonstrate occludin degradation by DC-secreted MMP biochemically, our data show poor and discontinuous occludin appearance at DC-endothelium contact sites and an improved immunostaining of occludin on BCEC during DC diapedesis in the presence of an MMP inhibitor, GM6001 (Fig. 4D).
Based on the observation that DCs have a distinct pattern of MMP9 expression compared with BCEC, it is likely that DC-derived MMP9 is involved in DC migration. It has been published by others that circulating DCs in the blood express MMP. Kouwenhoven et al. (62) have demonstrated that both blood monocytederived immature DC and mature DC express, produce, and secrete functionally active MMP-1, -2, -3, and -9 and their inhibitors tissue inhibitor of metalloproteinase 1 and tissue inhibitor of metalloproteinase 2 (84). The expression of the MMP and their inhibitors defines the migratory capabilities of DCs and the potency of these cells to induce immune responses (28, 84). Our data demonstrate that bone marrow GM-CSF-differentiated DCs express MMP upon their maturation and migration across the BCEC. The role of MMP in DC transmigration across in vitro brain microvessel monolayers is further supported by the blocking effect of MMP inhibitor on DC migration. Because both baseline and MIP-1α -induced DC migration could only be partly impaired using the MMP inhibitor GM6001, we suggest that other than MMP-dependent regulation of DC migration should be considered (Fig. 5C). This suggests that although MMP play a significant role in DC migration across BCEC, other mechanisms might also contribute to this process.
Recently, it has been shown that during inflammatory pain, disruption of BBB permeability is accompanied by alteration of occludin expression (85), which also suggests that loss of occludin integrity is related to inflammatory cell migration into the CNS in vivo. Although it is important to note that MIP-1α, among the other chemokines expressed by glial cells and macrophages around inflammatory plaques in MS and experimental autoimmune encephalomyelitis (86), is known to alter the integrity of the BBB, it has been previously shown that MIP-1α cannot be transported across the BBB (87) and does not affect occludin expression between epithelial cells (27). Our observations indicate that MIP-1α treatment alone does not result in perturbation of endothelial occludin (Fig. 4B) and ZO-1 (data not shown) expression in brain microvessel endothelial monolayers. Thus, discontinuous occludin expression during DC diapedesis is likely to be the result of DC transmigration and not MIP-1α chemokine treatment of BCEC in vitro.
Finally, we show that transmigration of DCs across brain microvessel endothelial cell monolayers allows Ag presentation to naive Ag-specific T cells. It has been shown previously that monocytes cultured with HUVEC differentiated into DCs within 2 days of culture, particularly after phagocytosing particles in the subendothelial collagen (29). In this study, we show that transmigration of GM-CSF-differentiated immature DCs through brain microvessel endothelial cell monolayers led to increased expression of costimulatory molecules CD40, CD80, and CD86 on the migrated DCs and facilitated their Ag presentation capability (Figs. 6 and 7). Because the bone marrow is the best known source for DC isolation, we used GM-CSF to differentiate DCs from the bone marrow to generate a sufficient number of cells to perform in vitro studies. It has been shown previously that GM-CSF-differentiated DCs resemble the phenotype of immature DCs circulating in the blood (88).
Recently, it was proposed that perivascular DCs in the CNS are critical in the initiation and maintenance of CNS inflammatory reactions (1, 89–91). Our data suggest that within the perivascular space of the CNS, Ag-specific T cells could re-encounter Ags presented by DCs. Additionally, if a small frequency of naive T cells accesses the perivascular space, activation of these cells could also happen. Although in our current model for T cell migration into the CNS the major prerequisite is that only activated T cells home to the nervous tissue, it has also been suggested that nonstimulated naive T cells are also capable of migrating into the brain (92). Our data show that after transmigration, DCs exhibit a mature phenotype with increased expression of costimulatory molecules (CD80, CD86, and CD40) and these cells retain their functional ability to activate naive CD8+ T cells in vitro.
In conclusion, we have demonstrated that DC transmigration across brain endothelial cell monolayers is increased in the presence of MIP-1α and partly dependent on MMP. Transmigrated DCs expressed high levels of costimulatory molecules and activated naive Ag-specific CD8+ T cells. Understanding the mechanism of DC transmigration across brain microvessel endothelial cells might lead to the design of targeted therapies to modify DC traffic in the CNS and thus to inhibit immune-mediated inflammatory diseases of the CNS, such as MS.
Acknowledgments
We thank Dr. C. Weidenfeller for technical help with primary endothelial cells. We are also grateful to Philip Oshel (Biological and Biomaterials Preparation, Imaging and Characterization Laboratory, University of Wisconsin-Madison) for help with scanning electron microscopy and Khen Macvilay for flow cytometry.
Abbreviations used in this paper
- DC
dendritic cell
- BBB
blood-brain barrier
- MS
multiple sclerosis
- MMP
matrix metalloproteinase
- TEER
transendothelial electrical resistance
- BCEC
brain capillary endothelial cell
- SEM
scanning electron microscopy
- ZO-1
zona occludens-1
- Tg
transgenic
- NGAL
neutrophil gelatinase B-associated lipocalin
- CLSM
confocal laser scanning microscopy
Footnotes
This work was supported by National Institutes of Health Grant RO1-NS 37570-01A2 (to Z.F.).
DisclosuresThe authors have no financial conflict of interest.
References
- 1.Greter M, Heppner FL, Lemos MP, Odermatt BM, Goebels N, Laufer T, Noelle RJ, Becher B. Dendritic cells permit immune invasion of the CNS in an animal model of multiple sclerosis. Nat Med. 2005;11:328–334. doi: 10.1038/nm1197. [DOI] [PubMed] [Google Scholar]
- 2.McQualter JL, Darwiche R, Ewing C, Onuki M, Kay TW, Hamilton JA, Reid HH, Bernard CC. Granulocyte macrophage colony-stimulating factor: a new putative therapeutic target in multiple sclerosis. J Exp Med. 2001;194:873–882. doi: 10.1084/jem.194.7.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Santambrogio L, Belyanskaya SL, Fischer FR, Cipriani B, Brosnan CF, Ricciardi-Castagnoli P, Stern LJ, Strominger JL, Riese R. Developmental plasticity of CNS microglia. Proc Natl Acad Sci USA. 2001;98:6295–6300. doi: 10.1073/pnas.111152498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pashenkov M, Huang YM, Kostulas V, Haglund M, Soderstrom M, Link H. Two subsets of dendritic cells are present in human cerebrospinal fluid. Brain. 2001;124:480–492. doi: 10.1093/brain/124.3.480. [DOI] [PubMed] [Google Scholar]
- 5.Pashenkov M, Soderstrom M, Huang YM, Link H. Cerebrospinal fluid affects phenotype and functions of myeloid dendritic cells. Clin Exp Immunol. 2002;128:379–387. doi: 10.1046/j.1365-2249.2002.01850.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hickey WF. Migration of hematogenous cells through the blood-brain barrier and the initiation of CNS inflammation. Brain Pathol. 1991;1:97–105. doi: 10.1111/j.1750-3639.1991.tb00646.x. [DOI] [PubMed] [Google Scholar]
- 7.Tsukada N, Matsuda M, Miyagi K, Yanagisawa N. Cytotoxicity of T cells for cerebral endothelium in multiple sclerosis. J Neurol Sci. 1993;117:140–147. doi: 10.1016/0022-510x(93)90166-v. [DOI] [PubMed] [Google Scholar]
- 8.Reese TS, Karnovsky MJ. Fine structural localization of a blood-brain barrier to exogenous peroxidase. J Cell Biol. 1967;34:207–217. doi: 10.1083/jcb.34.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hawkins BT, Davis TP. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev. 2005;57:173–185. doi: 10.1124/pr.57.2.4. [DOI] [PubMed] [Google Scholar]
- 10.Fabry Z, Raine CS, Hart MN. Nervous tissue as an immune compartment: the dialect of the immune response in the CNS. Immunol Today. 1994;15:218–224. doi: 10.1016/0167-5699(94)90247-X. [DOI] [PubMed] [Google Scholar]
- 11.Ransohoff RM, Kivisakk P, Kidd G. Three or more routes for leukocyte migration into the central nervous system. Nat Rev Immunol. 2003;3:569–581. doi: 10.1038/nri1130. [DOI] [PubMed] [Google Scholar]
- 12.Ancuta P, Moses A, Gabuzda D. Transendothelial migration of CD16+ monocytes in response to fractalkine under constitutive and inflammatory conditions. Immunobiology. 2004;209:11–20. doi: 10.1016/j.imbio.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 13.Biernacki K, Prat A, Blain M, Antel JP. Regulation of cellular and molecular trafficking across human brain endothelial cells by Th1- and Th2-polarized lymphocytes. J Neuropathol Exp Neurol. 2004;63:223–232. doi: 10.1093/jnen/63.3.223. [DOI] [PubMed] [Google Scholar]
- 14.Dzenko KA, Song L, Ge S, Kuziel WA, Pachter JS. CCR2 expression by brain microvascular endothelial cells is critical for macrophage transendothelial migration in response to CCL2. Microvasc Res. 2005;70:53–64. doi: 10.1016/j.mvr.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 15.Engelhardt B. Molecular mechanisms involved in T cell migration across the blood-brain barrier. J Neural Transm. 2006;113:477–485. doi: 10.1007/s00702-005-0409-y. [DOI] [PubMed] [Google Scholar]
- 16.Kivisakk P, Mahad DJ, Callahan MK, Sikora K, Trebst C, Tucky B, Wujek J, Ravid R, Staugaitis SM, Lassmann H, Ransohoff RM. Expression of CCR7 in multiple sclerosis: implications for CNS immunity. Ann Neurol. 2004;55:627–638. doi: 10.1002/ana.20049. [DOI] [PubMed] [Google Scholar]
- 17.Maslin CL, Kedzierska K, Webster NL, Muller WA, Crowe SM. Transendothelial migration of monocytes: the underlying molecular mechanisms and consequences of HIV-1 infection. Curr HIV Res. 2005;3:303–317. doi: 10.2174/157016205774370401. [DOI] [PubMed] [Google Scholar]
- 18.Prat A, Biernacki K, Antel JP. Th1 and Th2 lymphocyte migration across the human BBB is specifically regulated by interferon β and copolymer-1. J Autoimmun. 2005;24:119–124. doi: 10.1016/j.jaut.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 19.Wolburg H, Wolburg-Buchholz K, Engelhardt B. Involvement of tight junctions during transendothelial migration of mononuclear cells in experimental autoimmune encephalomyelitis. Ernst Schering Res Found Workshop. 2004;47:17–38. doi: 10.1007/978-3-662-05426-0_2. [DOI] [PubMed] [Google Scholar]
- 20.Fischer HG, Bielinsky AK. Antigen presentation function of brain derived dendriform cells depends on astrocyte help. Int Immunol. 1999;11:1265–1274. doi: 10.1093/intimm/11.8.1265. [DOI] [PubMed] [Google Scholar]
- 21.Fischer HG, Bonifas U, Reichmann G. Phenotype and functions of brain dendritic cells emerging during chronic infection of mice with Toxoplasma gondii. J Immunol. 2000;164:4826–4834. doi: 10.4049/jimmunol.164.9.4826. [DOI] [PubMed] [Google Scholar]
- 22.Fischer HG, Reichmann G. Brain dendritic cells and macrophages/microglia in central nervous system inflammation. J Immunol. 2001;166:2717–2726. doi: 10.4049/jimmunol.166.4.2717. [DOI] [PubMed] [Google Scholar]
- 23.Juedes AE, Ruddle NH. Resident and infiltrating central nervous system APCs regulate the emergence and resolution of experimental autoimmune encephalomyelitis. J Immunol. 2001;166:5168–5175. doi: 10.4049/jimmunol.166.8.5168. [DOI] [PubMed] [Google Scholar]
- 24.Serafini B, Columba-Cabezas S, Di Rosa F, Aloisi F. Intracerebral recruitment and maturation of dendritic cells in the onset and progression of experimental autoimmune encephalomyelitis. Am J Pathol. 2000;157:1991–2002. doi: 10.1016/S0002-9440(10)64838-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Serafini B, Rosicarelli B, Magliozzi R, Stigliano E, Capello E, Mancardi GL, Aloisi F. Dendritic cells in multiple sclerosis lesions: maturation stage, myelin uptake, and interaction with proliferating T cells. J Neuropathol Exp Neurol. 2006;65:124–141. doi: 10.1097/01.jnen.0000199572.96472.1c. [DOI] [PubMed] [Google Scholar]
- 26.Ambrosini E, Remoli ME, Giacomini E, Rosicarelli B, Serafini B, Lande R, Aloisi F, Coccia EM. Astrocytes produce dendritic cell-attracting chemokines in vitro and in multiple sclerosis lesions. J Neuropathol Exp Neurol. 2005;64:706–715. doi: 10.1097/01.jnen.0000173893.01929.fc. [DOI] [PubMed] [Google Scholar]
- 27.Ichiyasu H, McCormack JM, McCarthy KM, Dombkowski D, Preffer FI, Schneeberger EE. Matrix metalloproteinase-9-deficient dendritic cells have impaired migration through tracheal epithelial tight junctions. Am J Respir Cell Mol Biol. 2004;30:761–770. doi: 10.1165/rcmb.2003-0370OC. [DOI] [PubMed] [Google Scholar]
- 28.Osman M, Tortorella M, Londei M, Quaratino S. Expression of matrix metalloproteinases and tissue inhibitors of metalloproteinases define the migratory characteristics of human monocyte-derived dendritic cells. Immunology. 2002;105:73–82. doi: 10.1046/j.0019-2805.2001.01349.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Randolph GJ, Beaulieu S, Pope M, Sugawara I, Hoffman L, Steinman RM, Muller WA. A physiologic function for p-glycoprotein (MDR-1) during the migration of dendritic cells from skin via afferent lymphatic vessels. Proc Natl Acad Sci USA. 1998;95:6924–6929. doi: 10.1073/pnas.95.12.6924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Randolph GJ, Inaba K, Robbiani DF, Steinman RM, Muller WA. Differentiation of phagocytic monocytes into lymph node dendritic cells in vivo. Immunity. 1999;11:753–761. doi: 10.1016/s1074-7613(00)80149-1. [DOI] [PubMed] [Google Scholar]
- 31.Randolph GJ, Sanchez-Schmitz G, Angeli V. Factors and signals that govern the migration of dendritic cells via lymphatics: recent advances. Springer Semin Immunopathol. 2005;26:273–287. doi: 10.1007/s00281-004-0168-0. [DOI] [PubMed] [Google Scholar]
- 32.Weidenfeller C, Schrot S, Zozulya A, Galla HJ. Murine brain capillary endothelial cells exhibit improved barrier properties under the influence of hydrocortisone. Brain Res. 2005;1053:162–174. doi: 10.1016/j.brainres.2005.06.049. [DOI] [PubMed] [Google Scholar]
- 33.Zozulya A, Weidenfeller C, Galla HJ. Blood-Brain Barriers: From Ontogeny to Artificial Interfaces. Wiley; New Jersey: 2006. Induction of blood-brain barrier properties in cultured endothelial cells 2 volumes; pp. 357–371. [Google Scholar]
- 34.Hoheisel D, Nitz T, Franke H, Wegener J, Hakvoort A, Tilling T, Galla HJ. Hydrocortisone reinforces the blood-brain properties in a serum free cell culture system. Biochem Biophys Res Commun. 1998;247:312–315. [PubMed] [Google Scholar]
- 35.Schaefer BC, Schaefer ML, Kappler JW, Marrack P, Kedl RM. Observation of antigen-dependent CD8+ T-cell/dendritic cell interactions in vivo. Cell Immunol. 2001;214:110–122. doi: 10.1006/cimm.2001.1895. [DOI] [PubMed] [Google Scholar]
- 36.Hogquist KA, Jameson SC, Heath WR, Howard JL, Bevan MJ, Carbone FR. T cell receptor antagonist peptides induce positive selection. Cell. 1994;76:17–27. doi: 10.1016/0092-8674(94)90169-4. [DOI] [PubMed] [Google Scholar]
- 37.Kelly JM, Sterry SJ, Cose S, Turner SJ, Fecondo J, Rodda S, Fink PJ, Carbone FR. Identification of conserved T cell receptor CDR3 residues contacting known exposed peptide side chains from a major histocompatibility complex class I-bound determinant. Eur J Immunol. 1993;23:3318–3326. doi: 10.1002/eji.1830231239. [DOI] [PubMed] [Google Scholar]
- 38.Deli MA, Joo F. Cultured vascular endothelial cells of the brain. Keio J Med. 1996;45:183–198. doi: 10.2302/kjm.45.183. [DOI] [PubMed] [Google Scholar]
- 39.Deli MA, Abraham CS, Niwa M, Falus A. N,N-diethyl-2-[4-(phenylmethyl)phenoxy]ethanamine increases the permeability of primary mouse cerebral endothelial cell monolayers. Inflamm Res. 2003;52:S39–S40. doi: 10.1007/s000110300045. [DOI] [PubMed] [Google Scholar]
- 40.Demeuse P, Fragner P, Leroy-Noury C, Mercier C, Payen L, Fardel O, Couraud PO, Roux F. Puromycin selectively increases mdrla expression in immortalized rat brain endothelial cell lines. J Neurochem. 2004;88:23–31. doi: 10.1046/j.1471-4159.2003.02071.x. [DOI] [PubMed] [Google Scholar]
- 41.Perriere N, Demeuse P, Garcia E, Regina A, Debray M, Andreux JP, Couvreur P, Scherrmann JM, Temsamani J, Couraud PO, et al. Puromycin-based purification of rat brain capillary endothelial cell cultures: effect on the expression of blood-brain barrier-specific properties. J Neurochem. 2005;93:279–289. doi: 10.1111/j.1471-4159.2004.03020.x. [DOI] [PubMed] [Google Scholar]
- 42.Orlowski M, Sessa G, Green JP. γ-Glutamyl transpeptidase in brain capillaries: possible site of a blood-brain barrier for amino acids. Science. 1974;184:66–68. doi: 10.1126/science.184.4132.66. [DOI] [PubMed] [Google Scholar]
- 43.Skalli O, Pelte MF, Peclet MC, Gabbiani G, Gugliotta P, Bussolati G, Ravazzola M, Orci L. α-Smooth muscle actin, a differentiation marker of smooth muscle cells, is present in microfilamentous bundles of pericytes. J Histochem Cytochem. 1989;37:315–321. doi: 10.1177/37.3.2918221. [DOI] [PubMed] [Google Scholar]
- 44.Wagner DD, Marder VJ. Biosynthesis of von Willebrand protein by human endothelial cells: processing steps and their intracellular localization. J Cell Biol. 1984;99:2123–2130. doi: 10.1083/jcb.99.6.2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Franke H, Galla H, Beuckmann CT. Primary cultures of brain microvessel endothelial cells: a valid and flexible model to study drug transport through the blood-brain barrier in vitro. Brain Res Brain Res Protoc. 2000;5:248–256. doi: 10.1016/s1385-299x(00)00020-9. [DOI] [PubMed] [Google Scholar]
- 46.Franke H, Galla HJ, Beuckmann CT. An improved low-permeability in vitro-model of the blood-brain barrier: transport studies on retinoids, sucrose, haloperidol, caffeine and mannitol. Brain Res. 1999;818:65–71. doi: 10.1016/s0006-8993(98)01282-7. [DOI] [PubMed] [Google Scholar]
- 47.Inaba K, Inaba M, Romani N, Aya H, Deguchi M, Ikehara S, Muramatsu S, Steinman RM. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med. 1992;176:1693–1702. doi: 10.1084/jem.176.6.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Karman J, Ling C, Sandor M, Fabry Z. Initiation of immune responses in brain is promoted by local dendritic cells. J Immunol. 2004;173:2353–2361. doi: 10.4049/jimmunol.173.4.2353. [DOI] [PubMed] [Google Scholar]
- 49.Lyons AB, Parish CR. Determination of lymphocyte division by flow cytometry. J Immunol Methods. 1994;171:131–137. doi: 10.1016/0022-1759(94)90236-4. [DOI] [PubMed] [Google Scholar]
- 50.Swanson BJ, Baiu DC, Sandor M, Fabry Z, Hart MN. A small population of vasculitogenic T cells expands and has skewed T cell receptor usage after culture with syngeneic smooth muscle cells. J Autoimmun. 2003;20:125–133. doi: 10.1016/s0896-8411(02)00113-0. [DOI] [PubMed] [Google Scholar]
- 51.Ling C, Sandor M, Suresh M, Fabry Z. Traumatic injury and the presence of antigen differentially contribute to T-cell recruitment in the CNS. J Neurosci. 2006;26:731–741. doi: 10.1523/JNEUROSCI.3502-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kutzler MA, Weiner DB. Developing DNA vaccines that call to dendritic cells. J Clin Invest. 2004;114:1241–1244. doi: 10.1172/JCI23467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sumida SM, McKay PF, Truitt DM, Kishko MG, Arthur JC, Seaman MS, Jackson SS, Gorgone DA, Lifton MA, Letvin NL, Barouch DH. Recruitment and expansion of dendritic cells in vivo potentiate the immunogenicity of plasmid DNA vaccines. J Clin Invest. 2004;114:1334–1342. doi: 10.1172/JCI22608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Trifilo MJ, Lane TE. The CC chemokine ligand 3 regulates CD11c+CD11b+CD8α− dendritic cell maturation and activation following viral infection of the central nervous system: implications for a role in T cell activation. Virology. 2004;327:8–15. doi: 10.1016/j.virol.2004.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang N, Rogers TJ, Caterina M, Oppenheim JJ. Proinflammatory chemokines, such as C-C chemokine ligand 3, desensitize μ-opioid receptors on dorsal root ganglia neurons. J Immunol. 2004;173:594–599. doi: 10.4049/jimmunol.173.1.594. [DOI] [PubMed] [Google Scholar]
- 56.Cardona AE, Gonzalez PA, Teale JM. CC chemokines mediate leukocyte trafficking into the central nervous system during murine neurocysticercosis: role of γδ T cells in amplification of the host immune response. Infect Immun. 2003;71:2634–2642. doi: 10.1128/IAI.71.5.2634-2642.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hofmann N, Lachnit N, Streppel M, Witter B, Neiss WF, Guntinas-Lichius O, Angelov DN. Increased expression of ICAM-1, VCAM-1, MCP-1, and MIP-1α by spinal perivascular macrophages during experimental allergic encephalomyelitis in rats. BMC Immunol. 2002;3:11. doi: 10.1186/1471-2172-3-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jalonen TO, Pulkkinen K, Ukkonen M, Saarela M, Elovaara I. Differential intracellular expression of CCR5 and chemokines in multiple sclerosis subtypes. J Neurol. 2002;249:576–583. doi: 10.1007/s004150200067. [DOI] [PubMed] [Google Scholar]
- 59.Matejuk A, Dwyer J, Ito A, Bruender Z, Vandenbark AA, Offner H. Effects of cytokine deficiency on chemokine expression in CNS of mice with EAE. J Neurosci Res. 2002;67:680–688. doi: 10.1002/jnr.10156. [DOI] [PubMed] [Google Scholar]
- 60.Sano R, Tessitore A, Ingrassia A, d’Azzo A. Chemokine-induced recruitment of genetically modified bone marrow cells into the CNS of GM1-gangliosidosis mice corrects neuronal pathology. Blood. 2005;106:2259–2268. doi: 10.1182/blood-2005-03-1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Avolio C, Filippi M, Tortorella C, Rocca MA, Ruggieri M, Agosta F, Tomassini V, Pozzilli C, Stecchi S, Giaquinto P, et al. Serum MMP-9/ TIMP-1 and MMP-2/TIMP-2 ratios in multiple sclerosis: relationships with different magnetic resonance imaging measures of disease activity during IFN-β-1a treatment. Mult Scler. 2005;11:441–446. doi: 10.1191/1352458505ms1193oa. [DOI] [PubMed] [Google Scholar]
- 62.Kouwenhoven M, Ozenci V, Gomes A, Yarilin D, Giedraitis V, Press R, Link H. Multiple sclerosis: elevated expression of matrix metalloproteinases in blood monocytes. J Autoimmun. 2001;16:463–470. doi: 10.1006/jaut.2001.0505. [DOI] [PubMed] [Google Scholar]
- 63.Leppert D, Lindberg RL, Kappos L, Leib SL. Matrix metalloproteinases: multifunctional effectors of inflammation in multiple sclerosis and bacterial meningitis. Brain Res Rev. 2001;36:249–257. doi: 10.1016/s0165-0173(01)00101-1. [DOI] [PubMed] [Google Scholar]
- 64.Rosenberg GA. Matrix metalloproteinases in neuroinflammation. Glia. 2002;39:279–291. doi: 10.1002/glia.10108. [DOI] [PubMed] [Google Scholar]
- 65.Rudd PM, Mattu TS, Masure S, Bratt T, Van den Steen PE, Wormald MR, Kuster B, Harvey DJ, Borregaard N, Van Damme J, et al. Glycosylation of natural human neutrophil gelatinase B and neutrophil gelatinase B-associated lipocalin. Biochemistry. 1999;38:13937–13950. doi: 10.1021/bi991162e. [DOI] [PubMed] [Google Scholar]
- 66.Sitia G, Isogawa M, Iannacone M, Campbell IL, Chisari FV, Guidotti LG. MMPs are required for recruitment of antigen-nonspecific mononuclear cells into the liver by CTLs. J Clin Invest. 2004;113:1158–1167. doi: 10.1172/JCI21087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nguyen M, Arkell J, Jackson CJ. Active and tissue inhibitor of matrix metalloproteinase-free gelatinase B accumulates within human microvascular endothelial vesicles. J Biol Chem. 1998;273:5400–5404. doi: 10.1074/jbc.273.9.5400. [DOI] [PubMed] [Google Scholar]
- 68.Asahi M, Wang X, Mori T, Sumii T, Jung JC, Moskowitz MA, Fini ME, Lo EH. Effects of matrix metalloproteinase-9 gene knock-out on the proteolysis of blood-brain barrier and white matter components after cerebral ischemia. J Neurosci. 2001;21:7724–7732. doi: 10.1523/JNEUROSCI.21-19-07724.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rescigno M, Urbano M, Valzasina B, Francolini M, Rotta G, Bonasio R, Granucci F, Kraehenbuhl JP, Ricciardi-Castagnoli P. Dendritic cells express tight junction proteins and penetrate gut epithelial monolayers to sample bacteria. Nat Immunol. 2001;2:361–367. doi: 10.1038/86373. [DOI] [PubMed] [Google Scholar]
- 70.Pashenkov M, Link H. Dendritic cells and immune responses in the central nervous system. Trends Immunol. 2002;23:69–70. doi: 10.1016/s1471-4906(01)02114-7. [DOI] [PubMed] [Google Scholar]
- 71.Matyszak MK, Perry VH. The potential role of dendritic cells in immune-mediated inflammatory diseases in the central nervous system. Neuroscience. 1996;74:599–608. doi: 10.1016/0306-4522(96)00160-1. [DOI] [PubMed] [Google Scholar]
- 72.Prineas JW. Multiple sclerosis: presence of lymphatic capillaries and lymphoid tissue in the brain and spinal cord. Science. 1979;203:1123–1125. doi: 10.1126/science.424741. [DOI] [PubMed] [Google Scholar]
- 73.Raine CS, Mokhtarian F, McFarlin DE. Adoptively transferred chronic relapsing experimental autoimmune encephalomyelitis in the mouse. Neuropathologic analysis. Lab Invest. 1984;51:534–546. [PubMed] [Google Scholar]
- 74.Ratzinger G, Stoitzner P, Ebner S, Lutz MB, Layton GT, Rainer C, Senior RM, Shipley JM, Fritsch P, Schuler G, Romani N. Matrix metalloproteinases 9 and 2 are necessary for the migration of Langerhans cells and dermal dendritic cells from human and murine skin. J Immunol. 2002;168:4361–4371. doi: 10.4049/jimmunol.168.9.4361. [DOI] [PubMed] [Google Scholar]
- 75.Qu C, Edwards EW, Tacke F, Angeli V, Llodra J, Sanchez-Schmitz G, Garin A, Haque NS, Peters W, et al. Role of CCR8 and other chemokine pathways in the migration of monocyte-derived dendritic cells to lymph nodes. J Exp Med. 2004;200:1231–1241. doi: 10.1084/jem.20032152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Solanilla A, Grosset C, Lemercier C, Dupouy M, Mahon FX, Schweitzer K, Reiffers J, Weksler B, Ripoche J. Expression of Flt3-ligand by the endothelial cell. Leukemia. 2000;14:153–162. doi: 10.1038/sj.leu.2401635. [DOI] [PubMed] [Google Scholar]
- 77.Sozzani S, Sallusto F, Luini W, Zhou D, Piemonti L, Allavena P, Van Damme J, Valitutti S, Lanzavecchia A, Mantovani A. Migration of dendritic cells in response to formyl peptides, C5a, and a distinct set of chemokines. J Immunol. 1995;155:3292–3295. [PubMed] [Google Scholar]
- 78.Rubbert A, Combadiere C, Ostrowski M, Arthos J, Dybul M, Machado E, Cohn MA, Hoxie JA, Murphy PM, Fauci AS, Weissman D. Dendritic cells express multiple chemokine receptors used as coreceptors for HIV entry. J Immunol. 1998;160:3933–3941. [PubMed] [Google Scholar]
- 79.Greaves DR, Wang W, Dairaghi DJ, Dieu MC, Saint-Vis B, Franz-Bacon K, Rossi D, Caux C, McClanahan T, Gordon S, et al. CCR6, a CC chemokine receptor that interacts with macrophage inflammatory protein 3α and is highly expressed in human dendritic cells. J Exp Med. 1997;186:837–844. doi: 10.1084/jem.186.6.837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Granelli-Piperno A, Moser B, Pope M, Chen D, Wei Y, Isdell F, O’Doherty U, Paxton W, Koup R, Mojsov S, et al. Efficient interaction of HIV-1 with purified dendritic cells via multiple chemokine coreceptors. J Exp Med. 1996;184:2433–2438. doi: 10.1084/jem.184.6.2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Van Itallie CM, Anderson JM. Occludin confers adhesiveness when expressed in fibroblasts. J Cell Sci. 1997;110:1113–1121. doi: 10.1242/jcs.110.9.1113. [DOI] [PubMed] [Google Scholar]
- 82.Caron A, Desrosiers RR, Beliveau R. Ischemia injury alters endothelial cell properties of kidney cortex: stimulation of MMP-9. Exp Cell Res. 2005;310:105–116. doi: 10.1016/j.yexcr.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 83.Lohmann C, Krischke M, Wegener J, Galla HJ. Tyrosine phosphatase inhibition induces loss of blood-brain barrier integrity by matrix metalloproteinase-dependent and -independent pathways. Brain Res. 2004;995:184–196. doi: 10.1016/j.brainres.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 84.Kouwenhoven M, Ozenci V, Tjernlund A, Pashenkov M, Homman M, Press R, Link H. Monocyte-derived dendritic cells express and secrete matrix-degrading metalloproteinases and their inhibitors and are imbalanced in multiple sclerosis. J Neuroimmunol. 2002;126:161–171. doi: 10.1016/s0165-5728(02)00054-1. [DOI] [PubMed] [Google Scholar]
- 85.Brooks TA, Hawkins BT, Huber JD, Egleton RD, Davis TP. Chronic inflammatory pain leads to increased blood-brain barrier permeability and tight junction protein alterations. Am J Physiol. 2005;289:H738–H743. doi: 10.1152/ajpheart.01288.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Simpson JE, Newcombe J, Cuzner ML, Woodroofe MN. Expression of monocyte chemoattractant protein-1 and other β-chemokines by resident glia and inflammatory cells in multiple sclerosis lesions. J Neuroimmunol. 1998;84:238–249. doi: 10.1016/s0165-5728(97)00208-7. [DOI] [PubMed] [Google Scholar]
- 87.Banks WA, Kastin AJ. Reversible association of the cytokines MIP-1α and MIP-1β with the endothelia of the blood-brain barrier. Neurosci Lett. 1996;205:202–206. doi: 10.1016/0304-3940(96)12410-1. [DOI] [PubMed] [Google Scholar]
- 88.Duddy ME, Dickson G, Hawkins SA, Armstrong MA. Monocyte-derived dendritic cells: a potential target for therapy in multiple sclerosis (MS) Clin Exp Immunol. 2001;123:280–287. doi: 10.1046/j.1365-2249.2001.01433.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Heppner FL, Greter M, Marino D, Falsig J, Raivich G, Hovelmeyer N, Waisman A, Rulicke T, Prinz M, Priller J, Becher B, Aguzzi A. Experimental autoimmune encephalomyelitis repressed by microglial paralysis. Nat Med. 2005;11:146–152. doi: 10.1038/nm1177. [DOI] [PubMed] [Google Scholar]
- 90.McMahon EJ, Bailey SL, Castenada CV, Waldner H, Miller SD. Epitope spreading initiates in the CNS in two mouse models of multiple sclerosis. Nat Med. 2005;11:335–339. doi: 10.1038/nm1202. [DOI] [PubMed] [Google Scholar]
- 91.Platten M, Steinman L. Multiple sclerosis: trapped in deadly glue. Nat Med. 2005;11:252–253. doi: 10.1038/nm0305-252. [DOI] [PubMed] [Google Scholar]
- 92.Brabb T, von Dassow P, Ordonez N, Schnabel B, Duke B, Goverman J. In situ tolerance within the central nervous system as a mechanism for preventing autoimmunity. J Exp Med. 2000;192:871–880. doi: 10.1084/jem.192.6.871. [DOI] [PMC free article] [PubMed] [Google Scholar]