

Abstract
Many mRNAs in Caenorhabditis elegans are generated through a trans-splicing reaction that adds one of two classes of spliced leader RNA to an independently transcribed pre-mRNA. SL1 leaders are spliced mostly to pre-mRNAs from genes with outrons, intron-like sequences at the 5′-ends of the pre-mRNAs. In contrast, SL2 leaders are nearly exclusively trans-spliced to genes that occur downstream in polycistronic pre-mRNAs produced from operons. Operon pre-mRNA processing requires separation into individual transcripts, which is accomplished by 3′-processing of upstream genes and spliced leader trans-splicing to the downstream genes. We used a novel computational analysis, based on nonnegative matrix factorization, to identify and characterize significant differences in the cis-acting sequence elements that differentiate various types of functional site, including internal versus terminal 3′-processing sites, and SL1 versus SL2 trans-splicing sites. We describe several key elements, including the U-rich (Ur) element that couples 3′-processing with SL2 trans-splicing, and a novel outron (Ou) element that occurs upstream of SL1 trans-splicing sites. Finally, we present models of the distinct classes of trans-splicing reaction, including SL1 trans-splicing at the outron, SL2 trans-splicing in standard operons, competitive SL1-SL2 trans-splicing in operons with large intergenic separation, and SL1 trans-splicing in SL1-type operons, which have no intergenic separation.
Keywords: trans-splicing, polyadenylation, bioinformatics, mRNA processing, C. elegans
INTRODUCTION
More than 70% of mature mRNAs in the nematode Caenorhabditis elegans are produced through trans-splicing, which attaches one of a small number of leader sequences to the 5′-end of otherwise unique precursor transcripts (for review, see Blumenthal 2005). Caenorhabditis elegans has the additional feature of polycistronic transcripts, in which two or more adjacent, commonly oriented, but distinct, genes are initially transcribed as a polygenic precursor molecule that is further processed into individual mature transcripts (Spieth et al. 1993; Zorio et al. 1994; Blumenthal and Spieth 1996). Previous studies have shown that C. elegans spliced leader sequences can be separated into two classes, SL1, which can be trans-spliced to individual or operon-contained genes, and SL2, which in contrast, is nearly exclusively found attached to the downstream genes in operons (Blumenthal et al. 2002; Blumenthal and Gleason 2003).
Eukaryotic mRNA transcripts are typically processed at their 3′-ends in a two-step reaction (Minvielle-Sebastia and Keller 1999) (referred to as 3′-processing or polyadenylation) including cleavage of the precursor transcript, followed by the addition of a non-templated polyadenylate (poly[A]) tail of up to a few hundred nucleotides in length, depending on organism and cellular environment (Colgan and Manley 1997; Minvielle-Sebastia and Keller 1999; Zhao et al. 1999; Edmonds 2002). Creation and maintenance of the poly(A) tail has significant implications for subsequent stability, processing, and translation of the transcript (Zhao et al. 1999; Edmonds 2002; Kuersten and Goodwin 2003).
Organisms with their genes organized into operons, such as nematodes, primitive chordates, and hydrae (for review, see Blumenthal 2004), include a significant complication to the 3′-processing procedure, in that it must include mechanisms for polyadenylation of the upstream fragment and trans-splicing of a 5′-capped leader RNA to the downstream fragment (Blumenthal 1998). While typical eukaryotic 3′-end formation leads to transcription termination, within operons the presence of additional genes downstream requires both extended transcription and protection and subsequent processing of the downstream portion of the transcript. For clarity, we use the terms “internal site” to refer to a 3′-processing site on an upstream gene of a polycistronic transcript and “terminal site” to refer to 3′-processing sites on either monocistronic or 3′-terminal genes in operons.
Previous studies of C. elegans have demonstrated a mechanistic link between the upstream polyadenylation and downstream trans-splicing events (Liu et al. 2001), using the specialized SL2 small nuclear ribonuclear protein (snRNP). The SL2 snRNP has been shown to interact with the cleavage stimulation factor (CstF) (Kuersten et al. 1997; Evans et al. 2001; Huang et al. 2001; Liu et al. 2001), a three-polypeptide complex that, together with cleavage polyadenylation specificity factor (CPSF) and the poly(A) polymerase, is minimally required for the complete 3′-processing reaction (Colgan and Manley 1997; Minvielle-Sebastia and Keller 1999; Zhao et al. 1999). Studies of mammalian systems demonstrated that CstF binds to U-rich sequence elements on the precursor mRNA downstream from the 3′-processing site, specifically through an interaction of subunit CSTF2 (previously referred to as CstF64) (MacDonald et al. 1994). While the homology of the C. elegans CSTF2 is unmistakable, its sequence is diverged significantly with respect to CSTF2 in other metazoan species (Salisbury et al. 2006). Our recent studies correlated the protein sequence differences with changes in the apparent pre-mRNA binding sequences, both in content and in positioning (Salisbury et al. 2006).
Complete 3′-processing signals for metazoans other than nematodes minimally include the canonical hexamer polyadenylation signal (PAS) element, most commonly manifested as AAUAAA and positioned 10–30 nucleotides (nt) upstream (5′) of the processing site, and two downstream elements (DSEs), a proximal UG-rich element, typically positioned 5–15 nt downstream from the processing site, and a more distal U-rich element, typically positioned 15–25 nt downstream (Zhao et al. 1999; Hu et al. 2005; Salisbury et al. 2006). In contrast, C. elegans sequences show no evidence of a UG-rich element, and the U-rich element shows a broadened positioning range, typically falling between 5 and 25 nt downstream (Hajarnavis et al. 2004; Salisbury et al. 2006). Furthermore, the PAS appears to be much more tolerant of sequence divergence than in other animals: more than half of the genes have at least one mismatch in this hexamer, and ∼6% of genes have no appropriately positioned discernable match to AAUAAA at all (Blumenthal and Steward 1997; Hajarnavis et al. 2004).
Operon intergenic regions have a strong tendency to be quite short (∼100–120 nt long) (Blumenthal et al. 2002), which suggests there is an important mechanistic connection between the upstream 3′-end formation site and the SL2-specific trans-splice site. Indeed, when 3′-processing is disabled (e.g., through elimination of the AAUAAA element), SL2 trans-splicing is reduced, and when a weak site is strengthened (e.g., conversion of AGUAAA to AAUAAA), SL2 trans-splicing 100 nt downstream is increased (Evans et al. 2001; Liu et al. 2001). Furthermore, studies of the gpd-2/gpd-3 operon revealed an evolutionarily conserved U-rich (Ur) sequence element in the intergenic region that is required for SL2 trans-splicing and for any accumulation of downstream gene RNA (Huang et al. 2001). When the Ur element was eliminated in the context of an otherwise wild-type operon, no downstream gene expression was detected, suggesting that either the downstream RNA is degraded following 3′-end formation or transcription terminates. When the AAUAAA was deleted to inhibit 3′-end formation in the context of the Ur mutation, downstream mRNA was restored, but without the Ur element none of it was trans-spliced to SL2.
C. elegans also has a second type of operon, trans-spliced exclusively by SL1. These operons, termed SL1-type operons, are characterized by intergenic lengths of zero nt, with the AAUAAA signal for 3′-end formation of the upstream gene only a few nucleotides upstream of the trans-splice site of the downstream gene (Williams et al. 1999). In addition, these operons have long polypyrimidine (poly[Y]) tracts in the 3′-UTRs of the upstream genes that are required for correct operon pre-mRNA processing. The conserved poly(Y) tract is necessary for 3′-processing of the upstream gene, but not for trans-splicing to the downstream gene. The investigators concluded that the processing of this operon was possibly mutually exclusive in that standard 3′-processing of the upstream gene precluded SL1 processing of the downstream gene, and conversely, SL1 trans-splicing at least inhibits the formation of the 3′-end of the upstream gene.
The present study was initiated to better characterize the sequences responsible for directing and regulating the trans-splicing reactions, including the differentiation between internal polycistronic and terminal 3′-processing sites, and the selection between SL1 and SL2-type spliced leaders. We find that the signal that specifies internal sites can be primarily defined by a U-rich signal (core consensus UAUUUU) positioned 35–65 nt downstream from the 3′-processing site. This element presumably is the Ur element previously implicated in SL2-specific trans-splicing. In addition, we report on a novel element that occurs upstream of SL1 trans-splice sites, possibly defining the function and extent of the outrons. Integrating our findings with previous results, we present updated models for the molecular control of C. elegans trans-splicing and operon pre-mRNA processing.
RESULTS
Distinguishing classes of 3′-processing sites: Internal versus terminal
As described in Materials and Methods, we obtained the following training sets of sequences:
Putative “terminal” 3′-processing sites (931 sites), representing the 3′-ends of either monocistronic genes or 3′-most genes in operons.
Putative “internal” 3′-processing sites (182 sites), representing the 3′-ends of genes that occur in operons, excluding (1) the terminal (most 3′) genes in operons, and (2) 3′-processing sites that apparently correspond to “SL1-type” operons (Williams et al. 1999).
Putative 3′-processing sites representing internal sites in “SL1-type” operons (29 sites), which are characterized by coincidence of the upstream 3′-processing site and the downstream trans-splicing site.
All sequences were initially extracted including 200 nt up and downstream of the processing site. Sequence Logo (Schneider and Stephens 1990) representations (Fig. 1) of the single-nucleotide frequency profiles near the functional sites reveal several expected phenomena. All classes of 3′-processing sites (Fig. 1A–C) display strong A-rich elements ∼15–20 nt upstream of the processing site, characteristic of the PAS, and a general U-rich characteristic immediately up- and downstream of the PAS. A comparison of the internal 3′-processing sites with terminal sites reveals several regions of interest, including regions with apparent elevated information content (indicating nonrandom, potentially functional sequence) around positions −45 to −50, −10 to −5, +10 to +20, and +45 to +55 relative to the site of cleavage. The SL1-type operons present a more stark contrast, with strong evidence of the highly conserved 3′-splice site heptamer sequence (Kent and Zahler 2000; Hollins et al. 2005) at the joint 3′-processing/trans-splice site, and a seeming absence of any signal in the region downstream from the processing site.
FIGURE 1.

Sequence logo (Schneider and Stephens 1990) representations of the sequence environment surrounding different types of 3′-processing sites. (A) Terminal sites that imply the end of a mono- or polycistronic transcript. (B) Internal sites in an operon that include trans-splicing to downstream genes. (C) Internal sites that specifically fall into the “SL1-type” operon, in which the 3′-processing site of the upstream gene and the trans-splice site of the downstream gene are coincident.
Differential positional word counting
To better characterize the sequences that define the various functional sites, we performed a differential positional word counting (DPWC) analysis. The hypothesis in this analysis is that the sequences that define and differentiate the classes of functional site (e.g., internal vs. terminal 3′-processing sites) occur at different frequencies in the training sets. Furthermore, since RNA regulatory sequences are frequently defined both by sequence content and positioning, our analysis tests differences in frequency in specific positioning windows. We report on the analysis of pentamers (or tetramers for comparisons including the small SL1-type sequence set) in 10-nt-wide windows. The choice of parameters was a compromise between the improved characterization of functional elements afforded by longer words and smaller windows and the greater statistical robustness of shorter words and longer windows. Statistical significance was obtained through a permutation analysis, in which the sequence sets were combined and randomly resegregated into equivalent sets. P-values were calculated for Z-scores within each window as the frequency at which an equal or greater magnitude Z-score was obtained for any word in the permuted set.
Comparative analysis of the internal and terminal 3′-processing sites (Table 1, Part 1) was dominated by a group of words over-represented in the internal sequences in the sequence windows between 40 and 60 nt downstream from the 3′-processing site, including UACUU, UAUCU, UAUUU, CUUUU, and UUUCU. The position and content of these sequence elements are consistent both with the variation in single nucleotide frequencies (Fig. 1) and also with a previously described “Ur” element, demonstrated to be necessary for successful processing of the gpd-2/gpd-3 operon (Huang et al. 2001). In addition, the pentamers CAGAU (window 90–100), AGAUG (window 100–110), and GCAGA (window 190–200) are all consistent with a downstream 3′-splice site sequence necessary for trans-splicing of the SL2 leader sequence.
TABLE 1.
Differential positional word counting (DPWC) results for various classes of C. elegans 3′-processing sites

Comparative analysis with the SL1-type sequence set was hampered by the small number (29) of examples, thus we reduced the word size to tetramer for this analysis (Table 1, Parts 2 and 3). The dominant difference in comparison with both internal and terminal 3′-processing sites was a set of words (e.g., AGAU, CAGA, UCAG, UUAG) near the processing site (window −10 to 0) that reflects the presence of the SL1 trans-splicing site. Many of the additional significant word–window pairs appear to be purine-rich elements that occur downstream, and likely reflect the difference between the coding sequence present in the immediately adjacent downstream gene of SL1-type operons and the intergenic sequences in either internal or terminal sites. Finally, both comparisons reveal a single result containing a CUC trinucleotide ∼90 nt upstream of the processing site. The significance of this element is discussed further below.
Nonnegative matrix factorization (NMF)
While DPWC analysis can identify specific sequences that are differentially used between two sequence sets, it cannot give us a detailed picture of the sequence content and positioning constraints of the degenerate elements involved. For that we turn to a novel analysis based on NMF analysis of the PWC matrixes (Materials and Methods). NMF is a dimensional reduction algorithm that assumes that the matrix under analysis can be adequately approximated as a linear superposition of a small number of patterns, represented by basis vectors. The nonnegative constraint on the elements in the basis vectors results in elements that represent discrete components of the underlying data, which are sequence motifs and patterns in our work. In Figures 2 and 4, below, each basis vector is described with a line plot that shows its positioning distribution and a corresponding sequence logo (Schneider and Stephens 1990) that shows its sequence content.
FIGURE 2.

NMF models of the sequence elements surrounding the classes of 3′-processing sites: (A) terminal sites; (B) internal sites; and (C) SL1-type sites. Line plots represent position probability, while sequence logos (Schneider and Stephens 1990) represent probable sequence content. All sequence logos are plotted on a vertical scale of 0–2 bits of information.
FIGURE 4.

NMF models of the sequence elements surrounding the classes of 3′-splice sites: (A) SL1 sites; (B) SL2 sites; and (C) intron–exon junctions. Line plots represent position probability, while sequence logos (Schneider and Stephens 1990) represent probable sequence content. All sequence logos are plotted on a vertical scale of 0–2 bits of information.
We generated NMF decompositions for the terminal (Fig. 2A), internal (Fig. 2B), and SL1-type (Fig. 2C) 3′-processing sites. In each case, we modeled the aligned sequences from 200 nt upstream to 200 nt downstream from the putative 3′-processing site. To more accurately define the positioning distributions, we reduced the window size from the 10 nt used in the DPWC analysis to 5 nt, and counted tetramers rather than pentamers due to the reduced number of sequences in the reduced positioning windows (especially in the SL1-type operon set). Our studies indicate that our ability to identify strong motifs is robust with variation in both word and window size (L.N. Hutchins, S.P. Murphy, P. Singh, and J.H. Graber, in prep.).
Examination of the NMF patterns for the 3′-processing sites reveals several patterns that are consistent with the single-nucleotide and DPWC analysis described above. The classic PAS hexamer is clearly visible as Element 2 in Figure 2, A and B, and to a lesser extent in Figure 2C, where the positioning is as expected, but the sequence content is only a weak match.
The terminal 3′-processing site was described with seven elements, of which three imply strong motifs: Element 1, representing a U-rich element at or near the processing site and upstream of the PAS; Element 2, the canonical poly(A) signal, with optimal sequence AAUAAA and occurring around position −20; and Element 4, an A-rich signal with a peak in positioning probability near position +25. Elements 5–7 are representative of varying background content, given the measured combination of low information content sequence patterns and slowly varying, but skewed positioning probabilities. Element 3 is specifically positioned to occur in the putative 3′-UTR, with a low contribution to total sequence counts (based on the relative magnitude of the line plot), but a U-rich sequence pattern somewhat similar to Element 3 in the SL1 sites (Fig. 4A). Possible implications of this similarity are investigated in the Discussion.
The NMF analysis of the internal 3′-processing sites (Fig. 2B) revealed a more complex pattern compared to the terminal sites. The regions immediately surrounding and upstream of the putative 3′-processing site are similar to that of the terminal sites, with a probable PAS (Element 2) and U-rich signal (Element 1) clear from both sequence content and positioning. Element 4 is the most intriguing additional pattern, since it overlaps in both sequence content (core consensus AGAUAUUU) and positioning (peaking roughly between positions +45 and +65) with the pentamers identified as over-represented in internal sites with the DPWC analysis (Table 1, Part 1). This element is referred to as the “Ur element” for the remainder of this manuscript. In addition, Elements 5 and 6 contain evidence, both in positioning and sequence content, of the trans-splice site and initiation codon, respectively, as would be expected for downstream genes in operons processed by the SL2 snRNP. As with terminal sites, there is a U-rich element (Element 3) with a significant positioning probability in the putative 3′-UTR region.
The motifs generated by NMF analysis of the SL1-type sites are somewhat less precise than those of either the internal or terminal sites, as is to be expected with the smaller sequence set. Interestingly, while the sequence motifs are difficult to interpret, the positioning distributions show patterns similar to both 3′-processing sites (Fig. 2A,B), and also the larger set of SL1 trans-splicing sites (see Fig. 4A below). Besides the probable PAS (Element 2), a number of other putative elements can be seen, including the trans-splice site (Element 6), upstream U-rich sequences (Element 4), and a motif (Element 3) similar to the “Ou element” described in detail below.
Distinguishing classes of trans-splicing sites: SL1 versus SL2
As described in Materials and Methods, we obtained the following training sets of sequences for trans-splice sites:
Putative SL1 trans-splice sites (250 sites).
Putative SL2 trans-splice sites (1217 sites).
Unique 3′-splice sites from standard cis-splicing introns (107,755 sites). To reduce computational time, this set was further reduced by randomly selecting a subset of 3913 sites.
The three classes of 3′-splice sites (Fig. 3A–C) all display the previously described highly conserved heptamer sequence with consensus UUUUCAG (Kent and Zahler 2000; Hollins et al. 2005), along with evidence of a potential branch point, represented by the increase in A frequency around 15–20 nt upstream of the 3′-splice site. SL1 trans-splice sites appear to have two specific regions of interest, including an elevated information content (with an apparent increased pyrimidine content) from positions −50 to −45, and also a significant switch from A to G at position 0, when compared with either SL2 trans-splice or intronic 3′-splice sites. This variation holds even when controlling for AUG start codons immediately following the trans-splice site (data not shown). In addition, the 3′-splice heptamer shows elevated information content, especially reflecting increased occurrence of the preferred uracils at positions −6 and −5. The SL2 trans-splice site regions appear to be characterized by a general increased U-content over a broad range covering at least positions −60 to −20.
FIGURE 3.

Sequence logo (Schneider and Stephens 1990) representations of the sequence environment surrounding different types of 3′-splice sites: (A) SL1 trans-splicing sites, determined by EST evidence. (B) SL2 trans-splicing sites, determined by EST evidence. (C) Intron–exon junctions, implied by genomic annotation.
DPWC
Pairwise DPWC analysis of the various classes of 3′-splice sites (Table 2) revealed several differences indicative of putative control sequences. Comparison of the sequences around the SL1 and SL2 trans-splice sites (Table 2, Part 1) indicated two regions of variation:
TABLE 2.
Differential positional word counting (DPWC) results for various classes of C. elegans trans-splicing or 3′-splice sites


The base immediately downstream from the trans-splice site (position 0), which, consistent with single nucleotide analysis (Fig. 3) tends toward G in SL1 sites and toward A in SL2 sites. This tendency is reflected in multiple pentamers in the windows between −10 and +10, for example, CAGGG and CAGGU, both significantly over-represented in SL1 sites (indicated by the positive Z-scores), or AGAUG and CAGAU, both overrepresented in SL2 sites (negative Z-scores).
In the windows between positions −60 and −40, several pyrimidine-rich pentamers, specifically those containing cytosine residues (e.g., CUCUU, UCUCU, and UUCUC) are significantly more abundant near SL1 sites than SL2 sites.
To further characterize these sequences, we also performed the DPWC analysis between each of the trans-splicing site collections and our randomly selected set of 3913 intron 3′-splice sites from standard cis-spliced introns. The difference in nucleotide content, as already seen in the single nucleotide plots (Fig. 3), is strong enough such that several hundred word–window pairs pass the FDR <0.20 threshold. We report here only the top 25 scoring pairs for the SL1-intron and SL2-intron comparisons (Table 2, Parts 2 and 3, respectively). A complete list of all pentamer–window pairs that pass the FDR <0.2 criterion is available as Supplemental Material at http://harlequin.jax.org/nematode/.
As with the SL1–SL2 comparison, the SL1–intron comparison (Table 2, Part 2) reveals both the elevation of cytosine and pyrimidine-rich sequences between positions −60 and −40 near SL1 sites, as well as the preference for a guanosine base immediately following the trans-splice site.
The SL2–intron comparison (Table 2, Part 3) in the −60 to −40 range shows evidence of both the C-rich sequences favored by the SL1 site, as well as several of the pentamers identified as over-represented downstream from internal 3′-processing sites as compared with terminal sites. In addition, there is an excess of pentamers with an AUG initiation codon surrounding the SL2 trans-splice site.
NMF
We generated NMF decompositions of the SL1 (Fig. 4A), SL2 (Fig. 4B) trans-splice sites, and intron (Fig. 4C) 3′-splice sites. The NMF analysis of the three classes of 3′-splice site are not surprisingly dominated by the highly constrained 3′-splice site, shown as Element 2 in Figure 4, A–C. Similar to the single nucleotide analysis (Fig. 3), the NMF sequence motifs show the increased preference for guanosine immediately after the trans-splice site for SL1 when compared with either SL2 trans-splice or intronic 3′-splice sites. In addition, all three classes of 3′-splice sites display putative branch point patterns (Element 1 in Fig. 4A–C). Interestingly, while the AU-rich content of the putative branch point is consistent with previous characterization (Lim and Burge 2001), the specific arrangement within the putative motif is not. Previous description of the branch point indicated a consensus of UUUAAA, whereas our analysis generates a motif with the adenosines upstream of the uracils.
A striking new finding of our analysis is the characterization of an outron-specific sequence (Element 3 in Fig. 4A), referred to in the rest of this manuscript as the “Ou element.” Previous reports have not identified defining characteristics of the outron. Element 4 in the analysis of SL1 trans-splice sites shows clear evidence of the expected AUG initiation codon, and from positioning (Fig. 4A) shows a strong preference for positioning close to the trans-splice site. Elements 5–7 primarily reflect changes in background nucleotide probabilities, although Element 7 has a characteristic CAA pattern that also appears in a similar position near SL2 trans-splice sites (Elements 4 and 8) (Fig. 4B) and intron 3′-splice sites (Element 4) (Fig. 4C).
The NMF analysis of SL2 trans-splice sites (Fig. 4B) includes a strong U-rich motif (Element 3) that appears to be a composite of the Ur element and the 3′-processing DSE, which represents the putative binding site for CSTF2. The positioning distribution of this element is bimodal, with peaks ∼50 and 90 nt upstream of the trans-splice site, positions that are consistent with the standard 120-nt separation between 3′-processing and trans-splice site. The idea of a mixture is also supported by the sequence content, which is primarily U-rich, with only a small indication of the typical leading A-residue (Fig. 2B). Element 5 represents a U-rich element with a broad positioning distribution consistent with the 3′-UTR of the upstream gene, assuming the standard 100–120-nt intergenic separation. Element 4 shows clear evidence of the AUG initiation codon, while Elements 6–8 reflect changes in the sequence background.
The NMF analysis of the intronic 3′-splice sites (Fig. 4C) reveals a somewhat simpler pattern, with clear evidence of the hexamer (Fig. 4C, Element 2), branch point (Element 1), and upstream 5′-splice site (Fig. 4C, Element 3). The positioning distribution for Element 6 reflects sequences that are much more prevalent in exons than in introns.
The relationship between SL2 trans-splicing and intergenic separation
Although most operon genes are spaced only ∼100 base pairs (bp) apart, some have much greater intergenic distances. If the spacing between genes is an important instructive element of SL2 trans-splicing, we would expect that operons with greater intergenic spacing would have lower levels of SL2 trans-splicing. Previous studies revealed a class of operon in which the downstream gene can be (and shows molecular evidence of) processing to include either SL1 or SL2 spliced leaders (Blumenthal et al. 2002). These operons have shown at least anecdotal evidence of longer than expected intergenic separation. This phenomenon can be verified quantitatively with our data. We identified eight genes with trans-splice sites with evidence of both SL1 and SL2 splicing, measured the fraction of the observed EST sequences with SL1 leader sequence, and plotted the resulting values as a function of the intergenic separation (Fig. 5). A strong direct correlation (Pearson r=0.74) is obvious, verifying the previous observations: SL1 processing of downstream genes in operons is highly correlated with intergenic separation. Of particular interest are two pairs of trans-splice sites that arise in genes that lie downstream in two distinct operons (Fig. 5, F59G1.1b, represented by open circles; C30H7.2b, represented by squares). In both cases, the further distal trans-splice site shows increased SL1 processing, demonstrating directly the correlation with intergenic separation within a single operon.
FIGURE 5.

Longer intergenic regions favor SL1 trans-splicing. The fraction of ESTs trans-spliced by SL1 is plotted as a function of the distance between the upstream 3′-processing site and the trans-splice site. The two highlighted pairs of sites are generated from single genes with multiple trans-splice sites (open circles: F59G1.1b; squares: C30H7.2b). Error bars represent the 95% confidence interval of the SL1 fraction.
DISCUSSION
In this study, we identify and analyze putative 3′-processing and trans-splice sites from the nematode C. elegans. Our analysis has provided characterizations of cis-acting sequences that distinguish internal polycistronic 3′-processing sites from those that terminate the transcript. In addition, we have identified systematic differences between the trans-splice sites used by SL1 and SL2 type operons, including a novel “Ou” element located in the outron of transcripts that are spliced to the SL1 leader.
An updated model of the SL2 precursor mRNA interaction
Our studies of internal 3′-processing sites significantly extend the model presented in previous work (Huang et al. 2001), which proposed that internal sites were specified by an intergenic “Ur element.” The Ur element was characterized as an evolutionarily conserved sequence (in comparison with Caenorhabditis briggsae) in the operon that contains genes gpd-2 and gpd-3 (Huang et al. 2001). The position of the functional sequences identified through a deletion procedure is consistent with the +40 to +70 window we observe in the current analysis. The previous study included analysis of a number of additional operons. However, the positioning was measured with respect to the PAS element, rather than the 3′-processing site, and the short distances reported for a number of genes (as small as ∼25 nt) led us to conclude that some of these sequences may be 3′-processing elements, rather than Ur elements.
The positioning distribution of the Ur element has its primary mode at ∼50 nt downstream from the 3′-processing site, and a second potential mode around 90 nt downstream. The second mode is questionable, however, as the positioning and sequence content is also consistent with the most common positioning of the trans-splice site. In general, the evidence presented here, as well as previous work (Huang et al. 2001), indicates that the Ur element is constrained by the location of the 3′-processing site rather than the trans-splice site. A pattern indicative of the Ur element occurs in both the analysis of the internal 3′-processing sites (Fig. 2B) and the SL2 trans-splice sites (Fig. 4B), but the positioning has a sharper distribution near the 3′-processing sites. This apparent connection with the 3′-processing site has implications for the processing of operons with long intergenic separation, as discussed below.
Our analysis also highlights several other phenomena of interest in the comparison of internal and terminal 3′-processing sites. The PAS element, optimally manifested as AAUAAA, is conserved throughout metazoans, and to a reduced level in other eukaryotes (Graber et al. 1999a,b; Zhao et al. 1999; Brockman et al. 2005; Lee et al. 2007). Comparison of exact matches of AAUAAA in internal sites (76/182=0.418) and with terminal sites (487/931=0.523) indicates a statistically significant (p ∼0.009, large sample test of equal proportions) reduction in internal sites. Previous studies have indicated that near matches to AAUAAA result in less efficient processing (Sheets et al. 1990), thus the strength of 3′-processing may be modulated, with correspondingly modulated expression of the downstream operon genes. Alternatively, the constraints on the PAS could be reduced due to the stronger constraints on the downstream elements, including the CstF-binding site and the Ur element. This model is consistent with the observation that the overall “strength” of a 3′-processing site is determined as the net effect of all components, such that strong manifestations of one element compensate for weaker manifestations of others (Beyer et al. 1997; Graber et al. 1999b). It is reasonable that the sequence requirements for successful polycistronic processing could include strong downstream 3′-processing elements, with relaxed constraints on the PAS element. Indeed, a tetramer-based DPWC analysis (data not shown) demonstrated a significant overabundance of the UUUU tetramer in the 20 nt downstream from internal sites, a sequence and positioning consistent with CstF–mRNA interaction.
We can now delineate specific positioning and sequence content for the Ur element and update the processing model (Fig. 6A). We restrict this model to the standard cases, operons that have ∼100 nt between the upstream 3′-processing site and downstream SL2 trans-splicing site. Special models for operons with longer and shorter intergenic separation are discussed below.
FIGURE 6.

Graphical representations of the processing models for several different C. elegans pre-mRNA processing interactions. (A) Standard internal operon processing, including 3′-processing of the upstream gene, and trans-splicing of an SL2 leader to the downstream gene. (B) SL1 splicing to a monocistronic or first-in-an-operon gene. (C) Large intergenic separation genes in an operon, processed as competition between 3′-processing-coupled SL2 trans-splicing and independent SL1 trans-splicing. (D) SL1-type operons (Williams et al. 1999), modeled as a competition between 3′-processing of the upstream gene and SL1 trans-splicing to the downstream gene. An upstream pyrimidine-rich sequence that acts as an auxiliary 3′-processing element is shown. As noted previously, 3′-processing of the upstream gene removes the trans-splice site and prevents trans-splicing. Trans-splicing to SL1 leaves a cleaved upstream product that could be degraded or polyadenylated.
The principal change from the existing model (Evans et al. 2001; Huang et al. 2001) is the separation of U-rich elements into putative CstF-binding regions (typically within 20 nt of the 3′-processing site) (Salisbury et al. 2006) and the updated Ur element, typically 40–60 nt downstream from the 3′-processing site. We postulate that the CstF element is present in both internal and terminal sites, but that the U-rich consensus is more closely adhered to in internal sites, likely due to the requirements for tethering the SL2 machinery to the downstream fragment following 3′-processing.
The sequence of the Ur element is intriguingly similar to the Sm protein binding site (Hartmuth et al. 1999; Achsel et al. 2001). Sm proteins are integral components of snRNPs, including the SL snRNPs (Blumenthal 2005). Previous studies produced a consensus Sm binding sequence of RAUUUUUGA (Hartmuth et al. 1999). We find that (Fig. 2) the Ur motif is characterized as a uracil run following an adenosine. This was further confirmed with standard pattern recognition programs such as the Gibbs Sampler (data not shown; Lawrence et al. 1993). Clearly the Sm proteins would be interesting candidates for further investigation for a direct role in tethering the pre-mRNA to the SL2 snRNP for polycistronic processing.
A potential novel A-rich downstream signal in terminal 3′-processing sites
Examination of the patterns identified near the terminal 3′-processing sites indicated the potential of an A-rich element with a positioning mode ∼30 nt downstream from the 3′-processing site (Fig. 2B, Element 4). Such an element was hinted at in a previous study of downstream elements in metazoan 3′-processing sites (Salisbury et al. 2006).
The Ou element: Defining the outron and SL1 trans-splice sites
Our analysis of SL1 trans-splice sites revealed a novel element that we term the “Ou element” (Fig. 4A, Element 3) in the upstream region that can potentially help to define the boundaries of the outron (Fig. 6B). Positioning of the Ou element shows a mode around 50 nt upstream of the trans-splice site, and is characterized by a UC-rich sequence.
All classes of 3′-splice site are similarly positioned with respect to an upstream element. C. elegans introns have a tight length distribution compared with other metazoans, with the majority of the introns falling near 50 nt in length, which places the 5′-splice site at −50. As stated above, the Ou element is similarly positioned. Finally, while we believe that the Ur element is functionally tied to the 3′-processing site through its interaction with CstF, the majority of known operons have an intergenic spacing around 100–120 nt, a distance that typically places the Ur element around 50 nt upstream of the SL2 trans-splice site. The similarity of the spacing is suggestive of a common origin for these processes, as has been postulated in some models of trans-splicing (Blumenthal and Steward 1997; Guiliano and Blaxter 2006).
Large separation operon processing
The models we propose for control of SL1- and SL2-mediated trans-splicing provide insight into the variation in SL1/SL2 ratio with intergenic spacing (Fig. 5). In our model, the interaction of the SL2 snRNP with the pre-mRNA, mediated through CstF, is dominant in operons with standard short spacing (100–120 nt), possibly through steric exclusion. As the distance between the 3′-processing and trans-splicing sites grows, the dominance of SL2 processing wanes. There are several potential causes for the change (Fig. 6C): First, the increased separation reduces the efficiency of the interaction between the SL2 snRNP and the trans-splice site because of increased physical separation. Second, the increased intergenic separation makes possible the insertion of an Ou element into the spacing that can recruit the SL1 processing complex. A third possibility is that long intergenic regions could contain an internal promoter, the product of which would be expected to be entirely SL1-trans-spliced.
SL1-type operon processing
As described in the Introduction, SL1-type operons are characterized by a 0-bp-length intergenic separation, with a clear trans-splice site immediately adjacent to the apparent 3′-processing site (Williams et al. 1999). Our findings, and the models proposed above for SL1 and SL2 processing, easily accommodate this class (Fig. 6D). The positioning and sequence content that we characterize for the Ou element near SL1 trans-splice sites (Fig. 4A, Element 3) is consistent with the required pyrimidine-rich element located in the 3′-UTR of the upstream gene. NMF analysis of SL1-type processing sites resolves two distinct, but overlapping, signals upstream of the processing site (Fig. 2C). The positioning of Element 3 is consistent with the Ou element, whereas Element 4 is a U-rich element with positioning that extends up to ∼150 nt upstream of the processing site. It seems reasonable to assume that the extended poly(Y) tract represents an auxiliary 3′-processing element, necessary in SL1-type processing sites to compensate for the absence of the downstream U-rich CstF-binding site (Figs. 1C, 2C).
The processing of these operons is not necessarily mutually exclusive. CstF, which is known to bind downstream from the 3′-processing site, is required only for the cleavage step (Gilmartin and Nevins 1989; Takagaki et al. 1989; Zhao et al. 1999). CPSF, which binds upstream of the 3′-processing site, is required for both cleavage and polyadenylation (Gilmartin and Nevins 1989; Takagaki et al. 1989; Keller and Minvielle-Sebastia 1997; Zhao et al. 1999). In vitro experiments in yeast demonstrated that these processes could be uncoupled, and that CPSF can bind to a cleaved pre-mRNA and recruit the additional factors necessary for addition of the poly(A) tail. In SL1-type operons, the cleavage step is performed by the SL1 trans-splicing reaction, leaving the 5′-portion of the polycistronic pre-mRNA in a state for binding by CPSF and subsequent polyadenylation. Presumably this RNA would need to be debranched prior to polyadenylation. It is an interesting possibility that the AAUAAA may serve as the branch site in these operons.
Alternative processing of downstream genes based on selection of upstream 3′-processing sites
The EST evidence at hand provides several intriguing examples for further experimental investigation. Specifically, we have evidence of operons in which the upstream gene has alternative 3′-processing sites that imply different operon processing capacity. One interesting case is operon CEOP4304 (http://www.wormbase.org/), which includes the genes SRP54 (F21D5.7) and F21D5.6. Transcript evidence for F21D5.6 supports trans-splicing of both SL1 and SL2. ESTs support two 3′-processing sites for SRP54, a proximal site at +370 (downstream of the stop codon), and a distal site +500. The downstream site is nearly coincident with the trans-splice site for F21D5.6, implying an SL1-type operon. In contrast, the proximal 3′-processing site is situated at the typical separation ∼120 nt from the trans-splice site, and has reasonable matches to the Ur element. The intervening sequence, which can be included or excluded in the final SRP54 transcript based on 3′-processing site selection, contains four different microRNA-binding sites as predicted by miRanda (Enright et al. 2003). Such alternative operon processing provides the possibility of subtle variations in expression and subsequent post-transcriptional regulation of genes through control of the processing of the precursor transcript.
MATERIALS AND METHODS
Collection of sequences
C. elegans operons have been previously identified through analysis of ESTs or specific microarrays (Blumenthal and Steward 1997; Blumenthal 1998; Blumenthal et al. 2002). 3′-Processing sites have been identified through integrated analysis of ESTs and genomic sequence (Brockman et al. 2005; Salisbury et al. 2006). We merged these data sets to generate three sets of 3′-processing sites, representing 931 putative terminal 3′-processing sites, 182 putative internal 3′-processing sites, and 29 putative SL1-type 3′-processing sites (Williams et al. 1999). While the SL1-type sites are also internal to operons, they represent a different functional class, and have been separated accordingly.
We obtained SL1 and SL2 trans-splice sites in a two-step process. First, BLAT (Kent 2002) was used to align the SL1 and SL2 leader sequences to all C. elegans ESTs. EST sequences with full-length leader-EST alignments were subsequently aligned to the genome, and finally sequence was extracted up and downstream of the putative trans-splicing sites. We obtained 250 and 1217 SL1 and SL2 trans-splice sites, respectively. No attempt was made in this initial analysis to remove sites with evidence of both SL1 and SL2 trans-splicing activity, based on the reasoning that such sites presumably contain regulatory sequence elements for both SL1 and SL2 trans-splicing.
Standard cis-splicing 3′-splice sites were obtained from the ENSEMBL C. elegans annotations (version 43.160a) (Hubbard et al. 2007). Following reduction to unique sites, we extracted sequence up and downstream of the 3′-splice site. We extracted 107,755 unique 3′-splice sites, and subsequently randomly selected a set of 3913 for comparison with SL1 and SL2 sites.
Differential positional word counting (DPWC)
Functional sequences in pre-mRNA, for example, protein-binding sites, are frequently constrained both by sequence content and positioning with respect to a functional site. We have previously used positional word counting (PWC) techniques to characterize the functional elements across a broad range of metazoan species (Salisbury et al. 2006). In the present study, we have adapted this technique to identify sequence words that occur at different frequencies with specific positioning in two related sequence sets. In brief, the method includes the following steps:
Collect sequence sets aligned on a specific functional site, which in our case is either the putative 3′-processing site or the putative trans-splicing site.
Select an appropriate word length k and window size w, based on the number of sequences available in the smaller of the two sets (Fairbrother et al. 2004). We counted tetramers or pentamers in windows of 5 nt, where an occurrence of a specific word is counted in the window that contains its first base.
Perform the count. We choose to count the number of sequences with at least one instance word–window pair, rather than the total number of occurrences. The reasoning behind this choice is that we are searching for general regulatory elements that we expect to occur in all or nearly all of the sequences in a set. In contrast, counting all occurrences of a word–window pair could enable the overemphasis of sequences that occur multiple times in a subset of the sequences (dependent on choice of word length and window size). The count produces two two-dimensional matrixes V1 and V2, such that V1i,j (V2i,j) is the number of sequences in set 1(2) with at least one instance of word i in window j.
Normalize the V matrixes by dividing each entry by the number of sequences in their respective sets, converting count matrixes V1 and V2 into frequency matrixes F1 and F2.
Calculate a Z matrix, where element Zij is the score for word i in position window j. The Z-score is calculated according to the large sample test for equal proportions (Eq. [1]). Statistical significance for the Z-scores is obtained through a permutation analysis, in which Z is created many times (typically 200 or more) with randomization of the assignment of each individual sequence to one set or the other, preserving the sizes of the original sets. The permutation was designed explicitly to address the question, “What is the likelihood of observing an equal or greater Z-score for any sequence word in this particular positioning window under a random partitioning of the sequences into two sets?”
Zi,j scores that exceed a given threshold are reported as candidate differential word-position pairs. Multiple hypothesis testing was addressed via control of the false discovery rate (FDR) as described previously (Benjamini and Hochberg 1995), and reported results are for FDR≤0.2.
 |
In practice, we frequently find that groups of related words pass our statistical tests, representing either acceptable substitution (e.g., AAUAA and AAUGA) or positioning offsets (e.g., AAUAA and AUAAA).
Nonnegative matrix factorization (NMF) characterization of RNA regulatory signals
To obtain probabilistic models of the positioning and sequence content of the functional elements that specify RNA processing sites, we used a combination of PWC and nonnegative matrix factorization (NMF), a data reduction approach first developed for image processing (Lee and Seung 1999), which has subsequently gained popularity in processing high-dimensional biological data such as microarrays (Kim and Tidor 2003; Carmona-Saez et al. 2006; Pascual-Montano et al. 2006). NMF, similar to principal component analysis, generates “basis vectors” to reduce the dimensionality of large data sets. The NMF approach to motif characterization makes three assumptions:
The positioning profile of each sequence word can be approximated as a linear superposition of a few characteristic patterns that reflect the positioning of the underlying motifs.
The coefficients in this sum reflect the likelihood of the word occurring as part of the respective motifs.
The words generated by each motif have the same probability distribution at all positions.
Finally, our method is specifically designed to find motifs with constrained positioning. Motifs with random position distributions will not be detected. We summarize the method here: a more complete description is available elsewhere (L.N. Hutchins, S.P. Murphy, P. Singh, and J.H. Graber, in prep.).
In NMF, the two-dimensional PWC array V (size m words × n position windows) is decomposed into two new matrixes (V = WH), where W (size m words × r bases) contains the basis vectors, H (size r bases × n position windows) contains the multiplicative coefficients, and r is the number of basis vectors. We implemented an identical multiplicative update and objective function for maximization as reported previously (Lee and Seung 1999). Note that in contrast with the DPWC analysis, Vij is the total count of word i occurring in window j, rather than the number of sequences with at least one occurrence. For small sequence sets (∼100 or less), we find it useful to smooth the positioning distribution for each individual word. Following NMF, element Wij represents the contribution of the ith k-mer to the jth basis vector. Element Hjk represents the relative contribution of the jth basis vector to position k in the count vector, and as such, the r rows of H can be interpreted as the positioning probability for the regulatory motifs represented by their respective basis vectors.
The original authors reported that the nonnegativity constraint resulted in basis vectors that represented discrete components of the images. We interpret the r basis vectors as representing distinct patterns of defined sequence content and positioning. The resulting basis vectors can reflect both specific elements (such as the canonical polyadenylation hexamer) as well as any positional variation in composition (such as the change from UTR to intergenic sequence at the 3′-processing site). Selection of r, the number of basis vectors, is a complex problem (Brunet et al. 2004; Pascual-Montano et al. 2006). Using artificially generated data sets, we found that the plot of mean square difference between the original data and the NMF approximation (MSD=Σ[Vij–WHij]2/MN) shows an inflection when r matches the number of clusters used to generate the data (L.N. Hutchins, S.P. Murphy, P. Singh, and J.H. Graber, in prep.). Real data display a similar behavior; therefore we use the MSD versus r plot to select the operational value for r.
The NMF algorithm as implemented is a hill-climbing approach, guaranteed to give the best local solution, given the initial estimates of W and H. We restart the analysis at least several hundred times, keeping the solution with the maximum objective score. While this does not guarantee identification of the globally optimal solution (or even necessarily a unique solution) (Donoho and Stodden 2004), in practice we find that the dominant patterns (e.g., the AAUAAA-like PAS signal) are stably present in the solutions represented by the local maxima.
In order to convert the weighted list of k-mers for each elements to a probabilistic motif, we use the insight that the weights W can be reasonably interpreted as the expected distribution of counts for each k-mer for each basis vector. Given this assumption, the optimal motif is defined as the motif with the maximum multinomial probability (Eq. [2]) of observing the expected count distribution n as specified by the weights W. Since we are trying to maximize the probability of the motif to generate the observed weights (represented by the counts n in Eq. [2]), the constant C(n) can be neglected, allowing us to search for a motif that maximizes the logarithmic probability.
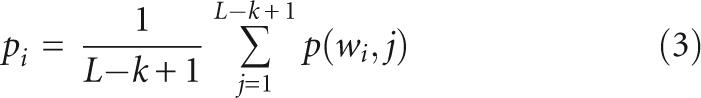 |
The calculation of pi, the probability of observing the ith k-mer, is shown in Equation (3), where wi is the ith k-mer, L is the length of the motif plus k-1 background positions padding on each side of the motif, and p(wi,j) is probability of observing wi at position j. As shown, pi is calculated as an average of the probability across the motif, including positions that span the boundary between motif and background, a choice that addresses two specific issues. First, clustered k-mers frequently include sequences that partially overlap, such as AAUAAA and AUAAAA. Second, the underlying motifs can be of variable size, larger or smaller than k. Simulations indicate that we can characterize motifs of nearly arbitrary size, regardless of choice of k (L.N. Hutchins, S.P. Murphy, P. Singh, and J.H. Graber, in prep.). We build a single-nucleotide (0-th order) model, and the final reported motif does not include the flanking background padding positions.
To optimize the search of potential motifs, we use a Markov chain Monte Carlo (MCMC) approach (Gelman et al. 1995). We start by sampling the length of the motif from a user specified range, and then randomly filling the nucleotide probabilities at each position. The initial motif is scored against the expected multinomial distribution. Following a standard MCMC approach, the motif is updated (L.N. Hutchins, S.P. Murphy, P. Singh, and J.H. Graber, in prep.), and updated motifs are automatically accepted if the multinomial probability increases, and randomly accepted according to a Boltzmann distribution if the multinomial probability decreases (Gelman et al. 1995). This process iterates until no improvement is observed for a user-specified number of iterations. In practice, we restart the MCMC process between 50 and 500 times, iterating until no improvement is seen for at least 250 iterations, keeping the best overall solution obtained.
The analysis tools described here are implemented as four distinct C++ programs. Linux binaries and source code are available from the authors by request.
ACKNOWLEDGMENTS
This work was partially supported by research grants NIH GM072706, NIH/NCRR INBRE Maine Contract P20 RR16463, NIH HD037102, NIH GM42432, and NSF contract DGE-0221625. We thank Carol Bult, Greg Cox, Erika Lasda, Peg MacMorris, and three anonymous reviewers for critical reading of the manuscript.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.596707.
REFERENCES
- Achsel, T., Stark, H., Lührmann, R. The Sm domain is an ancient RNA-binding motif with oligo(U) specificity. Proc. Natl. Acad. Sci. 2001;98:3685–3689. doi: 10.1073/pnas.071033998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini, Y., Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Methodol. 1995;85:289–300. [Google Scholar]
- Beyer, K., Dandekar, T., Keller, W. RNA ligands selected by cleavage stimulation factor contain distinct sequence motifs that function as downstream elements in 3′-end processing of pre-mRNA. J. Biol. Chem. 1997;272:26769–26779. doi: 10.1074/jbc.272.42.26769. [DOI] [PubMed] [Google Scholar]
- Blumenthal, T. Gene clusters and polycistronic transcription in eukaryotes. Bioessays. 1998;20:480–487. doi: 10.1002/(SICI)1521-1878(199806)20:6<480::AID-BIES6>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Blumenthal, T. Operons in eukaryotes. Brief. Funct. Genomic. Proteomic. 2004;3:199–211. doi: 10.1093/bfgp/3.3.199. [DOI] [PubMed] [Google Scholar]
- Blumenthal, T. Community TCeR. 2005. Trans-splicing and operons. (ed. The C. elegans Research Community). WormBook. [DOI] [PubMed] [Google Scholar]
- Blumenthal, T., Gleason, K.S. Caenorhabditis elegans operons: Form and function. Nat. Rev. Genet. 2003;4:112–120. doi: 10.1038/nrg995. [DOI] [PubMed] [Google Scholar]
- Blumenthal, T., Spieth, J. Gene structure and organization in Caenorhabditis elegans . Curr. Opin. Genet. Dev. 1996;6:692–698. doi: 10.1016/s0959-437x(96)80022-0. [DOI] [PubMed] [Google Scholar]
- Blumenthal, T., Steward, K. RNA processing and gene structure. In: Riddle D.L., editor. C. elegans II. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1997. pp. 117–145. [PubMed] [Google Scholar]
- Blumenthal, T., Evans, D., Link, C.D., Guffanti, A., Lawson, D., Thierry-Mieg, J., Thierry-Mieg, D., Chiu, W.L., Duke, K., Kiraly, M., et al. A global analysis of Caenorhabditis elegans operons. Nature. 2002;417:851–854. doi: 10.1038/nature00831. [DOI] [PubMed] [Google Scholar]
- Brockman, J.M., Singh, P., Liu, D., Quinlan, S., Salisbury, J., Graber, J.H. PACdb: PolyA cleavage site and 3′-UTR database. Bioinformatics. 2005;21:3691–3693. doi: 10.1093/bioinformatics/bti589. [DOI] [PubMed] [Google Scholar]
- Brunet, J.P., Tamayo, P., Golub, T.R., Mesirov, J.P. Metagenes and molecular pattern discovery using matrix factorization. Proc. Natl. Acad. Sci. 2004;101:4164–4169. doi: 10.1073/pnas.0308531101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmona-Saez, P., Pascual-Marqui, R.D., Tirado, F., Carazo, J.M., Pascual-Montano, A. Biclustering of gene expression data by nonsmooth nonnegative matrix factorization. BMC Bioinformatics. 2006;7:78. doi: 10.1186/1471-2105-7-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colgan, D.F., Manley, J.L. Mechanism and regulation of mRNA polyadenylation. Genes & Dev. 1997;11:2755–2766. doi: 10.1101/gad.11.21.2755. [DOI] [PubMed] [Google Scholar]
- Donoho, M., Stodden, V. When does nonnegative matrix factorization give a correct decomposition into parts? In: Thrun S., et al., editors. Advances in neural information processing systems 16. MIT Press; Cambridge, MA: 2004. pp. 1141–1148. [Google Scholar]
- Edmonds, M. A history of poly(A) sequences: From formation to factors to function. Prog. Nucleic Acid Res. Mol. Biol. 2002;71:285–389. doi: 10.1016/s0079-6603(02)71046-5. [DOI] [PubMed] [Google Scholar]
- Enright, A.J., John, B., Gaul, U., Tuschl, T., Sander, C., Marks, D.S. MicroRNA targets in Drosophila . Genome Biol. 2003;5:R1. doi: 10.1186/gb-2003-5-1-r1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans, D., Perez, I., MacMorris, M., Leake, D., Wilusz, C.J., Blumenthal, T. A complex containing CstF-64 and the SL2 snRNP connects mRNA 3′ end formation and trans-splicing in C. elegans operons. Genes & Dev. 2001;15:2562–2571. doi: 10.1101/gad.920501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairbrother, W.G., Yeo, G.W., Yeh, R., Goldstein, P., Mawson, M., Sharp, P.A., Burge, C.B. RESCUE-ESE identifies candidate exonic splicing enhancers in vertebrate exons. Nucleic Acids Res. 2004;32:W187–W190. doi: 10.1093/nar/gkh393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelman, A., Carlin, J.B., Rubin, D.B., Stern, H.S. Bayesian data analysis. Chapman & Hall/CRC; Boca Raton, FL: 1995. [Google Scholar]
- Gilmartin, G.M., Nevins, J.R. An ordered pathway of assembly of components required for polyadenylation site recognition and processing. Genes & Dev. 1989;3:2180–2190. doi: 10.1101/gad.3.12b.2180. [DOI] [PubMed] [Google Scholar]
- Graber, J.H., Cantor, C.R., Mohr, S.C., Smith, T.F. Genomic detection of new yeast pre-mRNA 3′-end-processing signals. Nucleic Acids Res. 1999a;27:888–894. doi: 10.1093/nar/27.3.888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graber, J.H., Cantor, C.R., Mohr, S.C., Smith, T.F. In silico detection of control signals: mRNA 3′-end-processing sequences in diverse species. Proc. Natl. Acad. Sci. 1999b;96:14055–14060. doi: 10.1073/pnas.96.24.14055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guiliano, D.B., Blaxter, M.L. Operon conservation and the evolution of trans-splicing in the phylum Nematoda. PLoS Genet. 2006;2:e198. doi: 10.1371/journal.pgen.0020198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajarnavis, A., Korf, I., Durbin, R. A probabilistic model of 3′ end formation in Caenorhabditis elegans . Nucl. Acids Res. 2004;32:3392–3399. doi: 10.1093/nar/gkh656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmuth, K., Raker, V.A., Huber, J., Branlant, C., Luhrmann, R. An unusual chemical reactivity of Sm site adenosines strongly correlates with proper assembly of core U snRNP particles. J. Mol. Biol. 1999;285:133–147. doi: 10.1006/jmbi.1998.2300. [DOI] [PubMed] [Google Scholar]
- Hollins, C., Zorio, D.A., MacMorris, M., Blumenthal, T. U2AF binding selects for the high conservation of the C. elegans 3′ splice site. RNA. 2005;11:248–253. doi: 10.1261/rna.7221605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, J., Lutz, C.S., Wilusz, J., Tian, B. Bioinformatic identification of candidate cis-regulatory elements involved in human mRNA polyadenylation. RNA. 2005;11:1485–1493. doi: 10.1261/rna.2107305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, T., Kuersten, S., Deshpande, A.M., Spieth, J., MacMorris, M., Blumenthal, T. Intercistronic region required for polycistronic pre-mRNA processing in Caenorhabditis elegans . Mol. Cell. Biol. 2001;21:1111–1120. doi: 10.1128/MCB.21.4.1111-1120.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard, T.J., Aken, B.L., Beal, K., Ballester, B., Caccamo, M., Chen, Y., Clarke, L., Coates, G., Cunningham, F., Cutts, T., et al. Ensembl 2007. Nucleic Acids Res. 2007;35:D610–D617. doi: 10.1093/nar/gkl996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller, W., Minvielle-Sebastia, L. A comparison of mammalian and yeast pre-mRNA 3′-end processing. Curr. Opin. Cell Biol. 1997;9:329–336. doi: 10.1016/s0955-0674(97)80004-x. [DOI] [PubMed] [Google Scholar]
- Kent, W.J. BLAT–The BLAST-like alignment tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent, W.J., Zahler, A.M. Conservation, regulation, synteny, and introns in a large-scale C. briggsae–C. elegans genomic alignment. Genome Res. 2000;10:1115–1125. doi: 10.1101/gr.10.8.1115. [DOI] [PubMed] [Google Scholar]
- Kim, P.M., Tidor, B. Subsystem identification through dimensionality reduction of large-scale gene expression data. Genome Res. 2003;13:1706–1718. doi: 10.1101/gr.903503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuersten, S., Goodwin, E.B. The power of the 3′-UTR: Translational control and development. Nat. Rev. Genet. 2003;4:626–637. doi: 10.1038/nrg1125. [DOI] [PubMed] [Google Scholar]
- Kuersten, S., Lea, K., MacMorris, M., Spieth, J., Blumenthal, T. Relationship between 3′ end formation and SL2-specific trans-splicing in polycistronic Caenorhabditis elegans pre-mRNA processing. RNA. 1997;3:269–278. [PMC free article] [PubMed] [Google Scholar]
- Lawrence, C.E., Altschul, S.F., Boguski, M.S., Liu, J.S., Neuwald, A.F., Wootton, J.C. Detecting subtle sequence signals: A Gibbs sampling strategy for multiple alignment. Science. 1993;262:208–214. doi: 10.1126/science.8211139. [DOI] [PubMed] [Google Scholar]
- Lee, D.D., Seung, H.S. Learning the parts of objects by non-negative matrix factorization. Nature. 1999;401:788–791. doi: 10.1038/44565. [DOI] [PubMed] [Google Scholar]
- Lee, J.Y., Yeh, I., Park, J.Y., Tian, B. PolyA_DB 2: mRNA polyadenylation sites in vertebrate genes. Nucleic Acids Res. 2007;35:D165–D168. doi: 10.1093/nar/gkl870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim, L.P., Burge, C.B. A computational analysis of sequence features involved in recognition of short introns. Proc. Natl. Acad. Sci. 2001;98:11193–11198. doi: 10.1073/pnas.201407298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Y., Huang, T., MacMorris, M., Blumenthal, T. Interplay between AAUAAA and the trans-splice site in processing of a Caenorhabditis elegans operon pre-mRNA. RNA. 2001;7:176–181. doi: 10.1017/s1355838201002333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald, C.C., Wilusz, J., Shenk, T. The 64-kilodalton subunit of the CstF polyadenylation factor binds to pre-mRNAs downstream of the cleavage site and influences cleavage site location. Mol. Cell. Biol. 1994;14:6647–6654. doi: 10.1128/mcb.14.10.6647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minvielle-Sebastia, L., Keller, W. mRNA polyadenylation and its coupling to other RNA processing reactions and to transcription. Curr. Opin. Cell Biol. 1999;11:352–357. doi: 10.1016/S0955-0674(99)80049-0. [DOI] [PubMed] [Google Scholar]
- Pascual-Montano, A., Carmona-Saez, P., Chagoyen, M., Tirado, F., Carazo, J.M., Pascual-Marqui, R.D. bioNMF: A versatile tool for nonnegative matrix factorization in biology. BMC Bioinformatics. 2006;7:366. doi: 10.1186/1471-2105-7-366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salisbury, J., Hutchison, K.W., Graber, J.H. A multispecies comparison of the metazoan 3′-processing downstream elements and the CstF-64 RNA recognition motif. BMC Genomics. 2006;7:55. doi: 10.1186/1471-2164-7-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider, T.D., Stephens, R.M. Sequence logos: A new way to display consensus sequences. Nucleic Acids Res. 1990;18:6097–6100. doi: 10.1093/nar/18.20.6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheets, M.D., Ogg, S.C., Wickens, M.P. Point mutations in AAUAAA and the poly(A) addition site: Effects on the accuracy and efficiency of cleavage and polyadenylation in vitro. Nucleic Acids Res. 1990;18:5799–5805. doi: 10.1093/nar/18.19.5799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spieth, J., Brooke, G., Kuersten, S., Lea, K., Blumenthal, T. Operons in C. elegans: Polycistronic mRNA precursors are processed by trans-splicing of SL2 to downstream coding regions. Cell. 1993;73:521–532. doi: 10.1016/0092-8674(93)90139-h. [DOI] [PubMed] [Google Scholar]
- Takagaki, Y., Ryner, L.C., Manley, J.L. Four factors are required for 3′-end cleavage of pre-mRNAs. Genes & Dev. 1989;3:1711–1724. doi: 10.1101/gad.3.11.1711. [DOI] [PubMed] [Google Scholar]
- Williams, C., Xu, L., Blumenthal, T. SL1 trans splicing and 3′-end formation in a novel class of Caenorhabditis elegans operon. Mol. Cell. Biol. 1999;19:376–383. doi: 10.1128/mcb.19.1.376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, J., Hyman, L., Moore, C. Formation of mRNA 3′ ends in eukaryotes: Mechanism, regulation, and interrelationships with other steps in mRNA synthesis. Microbiol. Mol. Biol. Rev. 1999;63:405–445. doi: 10.1128/mmbr.63.2.405-445.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorio, D.A., Cheng, N.N., Blumenthal, T., Spieth, J. Operons as a common form of chromosomal organization in C. elegans . Nature. 1994;372:270–272. doi: 10.1038/372270a0. [DOI] [PubMed] [Google Scholar]