

Abstract
Given two organisms, how can one distinguish whether they belong to the same species or not? This might be straightforward for two divergent organisms, but can be extremely difficult and laborious for closely related ones. A molecular marker giving a clear distinction would therefore be of immense benefit. The internal transcribed spacer 2 (ITS2) has been widely used for low-level phylogenetic analyses. Case studies revealed that a compensatory base change (CBC) in the helix II or helix III ITS2 secondary structure between two organisms correlated with sexual incompatibility. We analyzed more than 1300 closely related species to test whether this correlation is generally applicable. In 93%, where a CBC was found between organisms classified within the same genus, they belong to different species. Thus, a CBC in an ITS2 sequence-structure alignment is a sufficient condition to distinguish even closely related species.
Keywords: CBC, compensatory base change; ITS2, internal transcribed spacer 2; phylogeny; species concept
INTRODUCTION
“A species is a reproductive community of populations (reproductively isolated from others), which occupies a specific niche in nature” (Mayr 1982). Even if several objections could be raised against this “definition,” it is maybe the best-known statement in modern biology. “In fact, it is an indicator hypothesis: it does not tell us what a biospecies is but how to recognize it, namely by observing reproduction or else by failing to observe the latter. Neither [mutual] reproduction nor [sexual] isolation are defining properties of a species but, at best, properties of organisms that may be used as symptoms of the latters' membership in a particular species. In other words two organisms do not belong to the same species because they mate and reproduce, but they only are able to do so because they belong to the same species” (Mahner and Bunge 1997). In this study we are looking for a molecular classifier that might indicate that two organisms belong to different species. We are interested in an indicator hypothesis that is easy to work upon and additionally yields a certain probability that two organisms belong to distinct species. Compensatory base changes (CBCs) in the internal transcribed spacer 2 region (ITS2) of the nuclear rRNA cistron have been suggested as such a classifier. CBCs occur in a paired region of a primary RNA transcript when both nucleotides of a paired site mutate, while the pairing itself is maintained (e.g., G-C mutates to A-U). According to Coleman and Vacquier (2002), “…in all […] eukaryote groups where a broad array of species has been compared for both [rRNA] ITS2 sequence secondary structure and tested for any vestige of interspecies sexual compatibility, an interesting correlation has been found. When sufficient evolutionary distance has accumulated to produce even one CBC in the relatively conserved pairing positions of the ITS2 transcript secondary structure, taxa differing by the CBC are observed experimentally to be totally incapable of intercrossing” (see also Coleman 2003, 2007). With the advent of the ITS2 Database (Schultz et al. 2005, 2006; Wolf et al. 2005a) currently consisting of 65,000 ITS2 sequences and their individual secondary structures as well as of 4SALE, a program for synchronous sequence and secondary structure alignment and editing (Seibel et al. 2006), we are now able to test this hypothesis by a large-scale analysis. Here, we show that albeit the fact that a lack of CBCs in ITS2 secondary structures is not an indicator of two organisms belonging to the same species, at least one CBC is a classifier with a 93.11% reliability at least for plants and fungi that indicates two organisms belonging to distinct species. Because CBCs in ITS2 secondary structures are found to correlate strongly with distinct biological species, one can use this molecular indicator for determining at least the minimal number of distinct species from a set of ITS2 secondary structures of a clade, e.g., in analyses of environmental samples or for metagenomics. Moreover, the correlation between CBCs and the species concept occurs independent of reproduction and mating affinities, i.e., should work out similarly well for sexual and asexual species.
RESULTS
The taxonomic distribution of all alignments is shown in Table 1. Because of the underrepresentation of data from animals, primarily for lack of ITS2 sequences in GenBank, conclusions can currently only be made for fungi and plants. The CBC distribution for the subselection of alignments from the same species versus alignments from different species in the same genus (nonspecies) is shown in Table 2. The data imply an overall CBC rate of 0.0525 in species versus an almost 14-fold rate of 0.71 in nonspecies. To calculate the probability for the occurrence of at least one CBC in a global pairwise species sequence-structure alignment, we normalized the absolute frequencies by the overall sum. With these data we could calculate the probability that two sequences belong to different species given at least one CBC in the pairwise global sequence-structure alignment as follows:
TABLE 1.
Alignment distribution

TABLE 2.
CBC distribution

 |
Here, we have calculated a conditional probability of 93.11%, with an error rate of 6.89%, that two species are distinguished in current taxonomy, given that at least one CBC is observed. All further associated probabilities are shown below:
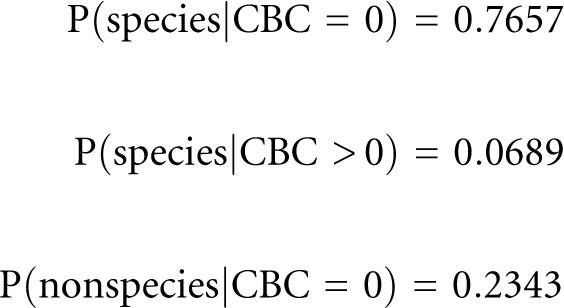 |
To estimate these probabilities for unbalanced samples, we count 66% species and 33% nonspecies alignments, and vice versa. The probability P(non-species|CBC > 0) changed to 87% and 97%, respectively.
If the emergence of CBCs is mainly caused by the amount of time two sequences evolved independently, the number of CBCs between two sequences should be dependent on their overall divergence. We analyzed the CBC rate in relation to the evolutionary distance by the Jukes Cantor Correction formula for all nonspecies sequence-structure alignments. Figure 1 shows a direct linear proportional relation between sequence divergence and the mean accumulated CBCs as indicated by the robust local regression curve (lowess) and the linear fit of the regression line. Its slope of 2.3 indicates that with one expected mutation per site, 2.3 CBCs can be expected between two sequences. At least one CBC can be expected between two sequences with a Jukes-Cantor distance of at least 0.42, although with a high variance. Complementarily, if on average every 2.5th site is mutated in the pairwise alignment, approximately one CBC can be expected. The behavior of CBCs on the structural level of the ITS2 as a marker for reconstructing phylogenies scales like the complete primary ITS2 sequence with its identities and mutations.
FIGURE 1.

Correlation of the expected number of mutations (Jukes Cantor distance) to CBCs. This figure correlates the evolutionary distances of pairwise ITS2 sequences from genus alignments to CBC frequencies. The straight line shows the robust linear regression, while the curve shows the local robust regression (lowess). While the curve and the line are fairly similar, we claim there is a real linear relation between the evolutionary distance and the CBCs mean for genus alignments. The mean differences of the lowess function and the regression line is 0 (P <0.001). The residuals reflect that the lowess function is consistently below (0, 0.5) and above (0.5, 1) the regression line.
DISCUSSION
In a large-scale analysis, we tested the hypothesis that taxa differing by at least one CBC represent different species. In 93.11% of the cases where two sequences taken from the same genus show a CBC, they belong to different species. When interpreting this percentage, it has to be taken into account that classifying species of the same genus is the most challenging scenario, as they are highly related to each other. An even better classification can be expected when comparing species of different orders or even families. This result is also influenced by the distribution of species belonging to one genus or to different genera. Depending on the sampling distribution, we therefore conclude that a CBC is indeed (on a 93.11% level) a sufficient marker for the identification of species. For the exceptional cases, the CBCs are equally distributed over all four helices. A list of all taxonomic vagaries concerning NCBIs species designations is now open for discussion (for Supplemental material, see http://its2.bioapps.biozentrum.uni-wuerzburg.de/TaxonomicVagaries.pdf), i.e., it has to be tested that in these cases the NCBI taxonomy fits mating affinities of the respective organisms.
In contrast, the absence of a CBC between two taxa predicted that in only 76.57% of the cases they belong to the same species. There was a significant correlation between the average number of CBCs and the general divergence, measured as the Jukes Cantor distance, between two ITS2 sequences. There is no causal relationship between the existence of a CBC and speciation. The CBC is rather a measure of elapsed evolutionary time, indicating that sufficient time has passed to make a speciation event very likely and is not a necessary criterion.
The expected number of CBCs depends on the degree of divergence, the sequence length (complete or partial), and on the CBC rate per site. We suggest using the whole ITS2 sequence as a basis for species classification. This raises the question as to whether the divergence within a phylogenetic marker like the ITS2 could be used directly as a marker for speciation. We found a large variance of the distance between species of the same genus in this marker. Therefore, any cut-off for classification will be inherently error prone. In contrast, the presence of a CBC is a handy and binary classifier (yes or no) with a known error rate (6.89%) that could be routinely used by tools like 4SALE (Seibel et al. 2006), the CBCAnalyzer (Wolf et al. 2005b), or by the ITS2 Database (Schultz et al. 2006). Due to concerted evolution in rRNA repeats, a CBC may thus primarily be a molecular indicator for no genetic exchange between two populations. However, how CBCs evolve inside the repeats remains unclear. It is a limitation of our study that currently sufficient data exist only for plants and fungi. It would be interesting to collect ITS2 data for animals also and to analyze whether our results can be transferred to this group. If so, we suggest using CBCs of ITS2 as a general marker for the identification and classification of eukaryotic species.
MATERIAL AND METHODS
ITS2 sequences and their predicted structures were retrieved from the ITS2 database v1.0 (Schultz et al. 2005, 2006; Wolf et al. 2005a) together with their taxonomic classification via the SOAP interface. Taxon-specific global multiple sequence-structure alignments were generated with 4SALE—a tool for synchronous sequence and secondary structure alignment and editing (Seibel et al. 2006) using an ITS2-specific scoring matrix (Wolf et al. 2005a). Alignments with at least three nonidentical sequences were subdivided into species and nonspecies alignments. The latter contained sequences from different but closely related species of the same genus. In this study, species designations are clearly not based on mating affinities, or any other species concept, but on NCBIs taxonomy as implemented in the ITS2 Database. In total, 1373 species and 400 nonspecies alignments were generated, manually checked, and curated. Four hundred species alignments were randomly chosen as a database for a balanced statistical analysis of 800 alignments. The CBCAnalyzer (Wolf et al. 2005b) as implemented in 4SALE (Seibel et al. 2006) was used to count CBCs. All statistical analyses were performed using the statistical environment R (Ihaka and Gentleman 1996). The robust linear regression was performed with the rlm function as implemented in the MASS package (Venables and Ripley 2002).
ACKNOWLEDGMENTS
We thank the DFG (German Research Foundation) for financial support (Mu-2831/1-1) and Philipp Seibel (Würzburg, Germany) for his help with 4SALE.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.617107.
REFERENCES
- Coleman, A.W. ITS2 is a double-edged tool for eukaryote evolutionary comparisons. Trends Genet. 2003;19:370–375. doi: 10.1016/S0168-9525(03)00118-5. [DOI] [PubMed] [Google Scholar]
- Coleman, A.W. Pan-eukaryote ITS2 homologies revealed by RNA secondary structure. Nucleic Acids Res. 2007;35:3322–3329. doi: 10.1093/nar/gkm233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman, A.W., Vacquier, V.D. Exploring the phylogenetic utility of ITS sequences for animals: A test case for abalone (Haliotis) J. Mol. Evol. 2002;54:246–257. doi: 10.1007/s00239-001-0006-0. [DOI] [PubMed] [Google Scholar]
- Ihaka, R., Gentleman, R. R: A language for data analysis and graphics. J. Comput. Graph. Statist. 1996;5:299–314. [Google Scholar]
- Mahner, M., Bunge, M. Foundations of biophilosophy. Springer; Berlin: 1997. [Google Scholar]
- Mayr, E. The growth of biological thought. Harvard University Press; Cambridge, MA: 1982. [Google Scholar]
- Schultz, J., Maisel, S., Gerlach, D., Muller, T., Wolf, M. A common core of secondary structure of the internal transcribed spacer 2 (ITS2) throughout the Eukaryota. RNA. 2005;11:361–364. doi: 10.1261/rna.7204505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz, J., Muller, T., Achtziger, M., Seibel, P.N., Dandekar, T., Wolf, M. The internal transcribed spacer 2 database– A web server for (not only) low level phylogenetic analyses. Nucleic Acids Res. 2006;34:W704–W707. doi: 10.1093/nar/gki129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seibel, P.N., Muller, T., Dandekar, T., Schultz, J., Wolf, M. 4SALE–a tool for synchronous RNA sequence and secondary structure alignment and editing. BMC Bioinformatics. 2006;7:498. doi: 10.1186/1471-2105-7-498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venables, W.N., Ripley, B.D. Modern applied statistics with S. Springer; Berlin: 2002. [Google Scholar]
- Wolf, M., Achtziger, M., Schultz, J., Dandekar, T., Muller, T. Homology modeling revealed more than 20,000 rRNA internal transcribed spacer 2 (ITS2) secondary structures. RNA. 2005a;11:1616–1623. doi: 10.1261/rna.2144205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf, M., Friedrich, J., Dandekar, T., Muller, T. CBCAnalyzer: Inferring phylogenies based on compensatory base changes in RNA secondary structures. In Silico Biol. 2005b;5:291–294. [PubMed] [Google Scholar]