

Abstract
The RNA subunit (P RNA) of the bacterial RNase P ribonucleoprotein is a ribozyme that catalyzes the Mg-dependent hydrolysis of pre-tRNA, but it requires an essential protein cofactor (P protein) in vivo that enhances substrate binding affinities and catalytic rates in a substrate dependent manner. Previous studies of Bacillus subtilis RNase P, containing a Type B RNA subunit, showed that its cognate protein subunit increases the affinity of metal ions important for catalysis, but the functional role of these ions is unknown. Here, we demonstrate that the Mg2+ dependence of the catalytic step for Escherichia coli RNase P, which contains a more common Type A RNA subunit, is also modulated by its cognate protein subunit (C5), indicating that this property is fundamental to P protein. To monitor specifically the binding of active site metal ions, we analyzed quantitatively the rescue by Cd2+ of an inhibitory Rp phosphorothioate modification at the pre-tRNA cleavage site. The results show that binding of C5 protein increases the apparent affinity of the rescuing Cd2+, providing evidence that C5 protein enhances metal ion affinity in the active site, and thus is likely to contribute significantly to rate enhancement at physiological metal ion concentrations.
Keywords: ribozyme, catalysis, ribonucleoprotein, RNase P, active site
INTRODUCTION
The biological functions of RNA and protein are intimately related and extensive structural and functional studies reveal that RNA binding proteins can modulate RNA function by binding to and stabilizing specific conformations (e.g., Weeks and Cech 1995; Ho and Waring 1999), by facilitating necessary conformational changes (Jankowsky and Bowers 2006; Caprara et al. 2007), as well as by providing unique surfaces for RNP targeting and regulation (Egea et al. 2005; Hopper 2006). The coordinated function of RNA and protein in enzyme catalysis is fundamental to ribosome catalyzed protein synthesis as well as numerous intron splicing and RNA processing reactions. Because of this essential functional interplay, understanding in detail how RNA and proteins form specific complexes and collaborate to achieve specific physiological function is an important goal.
A ribonucleoprotein enzyme in which the protein subunit plays a key role in specificity and catalysis is ribonuclease P, which catalyzes the 5′-end maturation of tRNA (Hsieh et al. 2004; Kazantsev and Pace 2006). In bacteria, RNase P is composed of an ∼400 nucleotide (nt) RNA subunit (P RNA) and an ∼100 amino acid protein subunit (termed C5 in Escherichia coli) (Fig. 1; Evans et al. 2006). The P RNA subunit is able to recognize pre-tRNAs and catalyze the hydrolysis reaction in vitro by itself; but substrate affinity, specificity, and catalytic rate are strongly dependent on assembly with its cognate protein subunit (Loria et al. 1998; Niranjanakumari et al. 1998; Kurz and Fierke 2000; Buck et al. 2005a; Sun et al. 2006). Binding of P protein dramatically increases pre-tRNA affinity, in part by making direct contact to the 5′-leader sequence (Niranjanakumari et al. 1998). However, the E. coli C5 protein subunit can enhance tRNA binding even though it is thought to only contact the 5′-leader sequence directly. Comparative equilibrium binding analyses of different E. coli pre-tRNAs indicate that the protein has differential effects on the thermodynamic contribution of the tRNA and leader portions of the substrate and that low affinity of some tRNA bodies is compensated for by combining them with 5′ leaders that increase affinity more than others in order to reach uniformity of binding (Sun et al. 2006). Additionally, the protein subunit of E. coli RNase P was shown to significantly enhance the rate of the substrate cleavage step, which for some substrates can be >800-fold. Such effects on tRNA interactions, catalysis, and specificity likely result both from direct contacts to the 5′ leader as well as from protein-induced conformational changes that occur in the catalytic core of the ribozyme (Buck et al. 2005b). Our understanding of the mechanistic basis for these effects is currently limited by a lack of detailed information on the structural consequences of protein binding, and as well as a quantitative description of the accompanying changes in enzyme specificity and catalysis.
FIGURE 1.

(A) Secondary structure diagram of E. coli P RNA. Helices are designated as P (paired) and numbered from the 5′ end of the RNA. Segments of the RNA connecting helices are designated J (joining) and numbered according to the helices they connect. The helices P1–P18 that make up the secondary structure of E. coli P RNA are labeled. The secondary structure is organized according to the three-dimensional structure of the P RNA subunit, and arrows depict connections that define the path of the RNA chain. (B) Cartoon diagram of the three-dimensional structure of a Type A P RNA with pre-tRNA bound. The structure diagram is based on the crystal structure of Thermotoga maritime P RNA (Torres-Larios et al. 2005). Individual helices are shown as cylinders that are colored according to the secondary structure diagram in panel A. The bound pre-tRNA is depicted by a black ribbon and the P protein subunit is shown as a sphere. The 5′-leader sequence of pre-tRNA is shown as a dashed line. (C) Sequence and secondary structure of pre-tRNAMet608. The location of the RNase P cleavage site between nucleotides N(+1) and N(−1) is indicated by an arrow. (D) Detail of the proposed active site metal ion interactions and the reactive phosphate of pre-tRNA in the transition state. As described in the text, two metal ions are proposed, both of which interact with the pro-Rp phosphate oxygen of the reactive phosphate. This position is substituted by a sulfur atom in the Rp(+1) pre-tRNA substrate.
P RNA is considered to be a metalloenzyme and a key insight has been the demonstration that the protein subunit of Bacillus subtilis RNase P increases the apparent affinity of metal ions important for catalysis (Kurz and Fierke 2002). Although it is a foundational structural and kinetic system, the B. subtilis enzyme represents a minor subclass of RNase P enzyme in that it contains a variant Type B RNA subunit (Haas et al. 1994). Relatively small changes in RNA structure can result in significantly different Mg2+ requirements for folding and catalysis (Lehman and Joyce 1993; Frank and Pace 1997; Nelson et al. 2005; Roychowdhury-Saha and Burke 2006). Thus, an important question is whether the previously documented contributions of RNase P protein to catalysis by the E. coli enzyme, which contains the more common Type A RNA subunit, are due to an enhancement of Mg2+ binding. If so, it follows that this enhancement could be due to a direct effect on increasing affinities of active site metal ions.
Indeed, several lines of evidence indicate that P RNA positions active site metal ions. Detailed structural and functional studies of protein endoribonucleases that catalyze the same chemistry as P RNA show the presence of one or more active site metal ions (Cowan 1998). Additionally, the presence of active site metal ions in the Group I intron ribozyme, which catalyzes similar intermolecular nucleophilic attack of a phosphodiester, is strongly supported by detailed biochemical and high resolution structural data (Hougland et al. 2005; Stahley and Strobel 2006). Indeed, rate enhancement by P RNA is clearly dependent on the Mg2+ ion concentration under conditions in which the RNA subunit is folded and enzyme concentrations are saturating (Smith et al. 1992; Beebe et al. 1996). Furthermore, phosphorothioate modifications at the pre-tRNA cleavage site inhibit P RNA catalysis, and the effects of pro-Rp phosphate oxygen modification by sulfur can be rescued in part by addition of the thiophilic metal ion Cd2+ (Warnecke et al. 1996). A general two-metal ion mechanism for RNase P catalysis is assumed (see Fig. 5, below) in which the two catalytic metal ions are positioned to coordinate the pro-Rp oxygen of the reactive phosphate (Steitz and Steitz 1993; Warnecke et al. 1996). One of the ions helps to position a water molecule as the nucleophile and assist its deprotonation, and the second is proposed to coordinate and stabilize the 3′O leaving group. Analyses of the bonding environment of the nucleophilic water in the transition state using kinetic isotope effects is consistent with direct metal ion catalysis in nucleophilic activation (Cassano et al. 2004). Thus, if the previously observed effects of C5 protein on enhancing catalytic rates results from a general property of P protein on influencing the metal ion dependence of catalysis, then it raises the possibility that these effects could be related to enhanced active site metal ion affinity.
FIGURE 5.

Comparison of k rel as a function of Cd2+ concentration for P RNA and RNase P holoenzyme shows that C5 enhances the binding affinity of catalytic metal ion. (A) Relative reaction rates (k rel) calculated from the ratio of observed reaction rates for substrates with and without the sulfur substitution (i.e., k rel=k S obs/k O obs) are plotted versus Cd2+ concentrations. Data for the holoenzyme reaction is represented by (•) and P RNA alone is represented by (○). Fitting to the Hill equation reveals that n=2 for the holoenzyme reaction, and K Cd (holo)=9 mM and K Cd (P RNA alone)>30mM. (B) Interpretation of the concentration dependent rescue of the Rp phosphorothioate modification by Cd2+. Because the k rel analysis controls for nonspecific effects of Cd2+, the cooperative nature of the Cd2+-dependent rescue suggests that two rescuing metal ions interact with the single sulfur modification, which reflects two metal ions binding in the P RNA active site for the native, unmodified substrate.
However, the contributions of metal ions to RNA function are complex, and relating the binding of specific ions to specific functional roles continues to be one of the most vexing problems in the field. RNAs bind numerous metal ions in solution, which can interact site specifically by direct coordination and H-bonding or by electrostatic interactions (DeRose 2003; Draper et al. 2005). Both classes can make significant contributions to function that are reflected in Mg2+ titration experiments, which are difficult to interpret in terms of the thermodynamics of individual metal ion interactions. Additionally, it is well established in other ribozyme systems that Mg2+ binding can increase catalytic rate constants indirectly by stabilizing active RNA conformations or by promoting substrate docking steps (e.g., Shan and Herschlag 2002; Tinsley et al. 2004). Thus, while the contribution of the protein subunit to P RNA catalysis appears to be related to the binding of Mg2+ ions, the binding sites and functional roles of these ions are fundamentally difficult to establish.
To address these issues, we compared the pH and Mg2+ dependence of the catalytic step for the P RNA subunit of E. coli RNase P in the presence and absence of its cognate protein subunit (C5 protein) under identical reaction conditions. Additionally, we used quantitative analysis of the Cd2+ rescue of a cleavage site Rp phosphorothioate (PS) modification to isolate the binding thermodynamics of active site metal ions (Peracchi et al. 1997; Shan et al. 2001). The comparative kinetic analyses show that C5 protein reduces the apparent affinity of Mg2+ ion essential for catalysis without changing the rate-limiting step. Importantly, binding of the C5 protein to P RNA results in higher apparent affinity for the binding of rescuing Cd2+ ions to a substrate phosphorothioate. Thus, these data provide evidence that an important function of the bacterial RNase P protein subunit is to enhance the rate of catalysis by increasing metal ion affinity in the active site of the catalytic RNA subunit.
RESULTS
To quantify the contribution of the P protein subunit to rate enhancement by RNase P, we determined the effect of C5 protein binding on the single turnover rate constant for cleavage of E. coli pre-tRNAMet608. The pre-tRNAMet608 substrate was chosen because it is representative of a “typical” E. coli pre-tRNA substrate in that it contains all of the optimal recognition elements that have been defined by sequence comparisons and by structure function studies. Thus, the mechanistic features of substrate recognition and catalysis defined by using this substrate are anticipated to reflect general features of RNase P enzymology. Analyses were performed typically under conditions of 100 mM NaCl, 17.5 mM Mg2+, which was determined to be optimal for the holoenzyme multiple turnover reaction (Guo et al. 2006). Reaction pH and Mg2+ concentration were varied as described below. As shown in Figure 2A, under identical reaction conditions the holoenzyme has an ∼10-fold higher rate constant for cleavage of pre-tRNAMet608 compared to P RNA alone (0.14 min−1 versus 1.3 min−1). The lower rate constant for P RNA alone under these conditions could be due to a slower, noncatalytic rate-limiting step that is enhanced by the protein subunit. In this scenario the protein subunit could change the mechanism to one in which substrate cleavage, rather than a slower upstream step, is rate limiting. Alternatively, the protein subunit could enhance catalysis in a more direct fashion by altering the affinity of metal ions necessary for catalysis as suggested previously by results from B. subtilis RNase P.
FIGURE 2.

Kinetic analyses of the pH and Mg2+ dependence of the single turnover rate constants for P RNA and RNase P holoenzyme cleavage of pre-tRNAMet608. (A) Single turnover reactions for RNase P holoenzyme (•) and P RNA alone (○) are compared at saturating enzyme concentration in: MES 50 mM (pH 5.75), NaCl 100 mM, MgCl2 17.5 mM, and Triton 0.005%. (B) Plots of log(k obs) versus pH for RNase P (•) and P RNA (○) are fit to a linear equation (slope=1.1 and 1.2 for RNase P and P RNA alone reactions, respectively). (C) Plot of k obs values for P RNA and RNase P holoenzyme versus Mg2+ concentration. The data are fit to the equation for a cooperative binding mechanism (Equation [2], Materials and Methods). The Hill coefficients are unity for both reactions; however, the apparent dissociation constant for holoenzyme is smaller than that for P RNA alone (K Mg [RNase P]=32 mM and K Mg [P RNA alone] >250 mM). The inset shows a closeup of the data at low Mg2+ concentrations.
To first assess if the protein changes the rate-limiting step of the pre-tRNA cleavage reaction, we compared the pH dependence of the single turnover rate constants for P RNA in the presence and absence of C5 protein. Analysis of the pH dependence of the cleavage rate constant as a probe for the rate limiting step of the RNase P reaction is based on the rationale that deprotonation to yield a hydroxide ion nucleophile is an essential step in the RNase P catalyzed phosphodiester bond hydrolysis reaction. Accordingly, the rate of substrate cleavage increases with increasing hydroxide ion concentration and follows a rate law for specific base catalysis (Smith and Pace 1993; Cassano et al. 2002; Persson et al. 2003). Thus, plots of log(k obs) versus pH that are linear with a slope of unity suggest that the chemical step is rate limiting, whereas an alteration in the rate-limiting step of the reaction is likely to result in the absence of or a significant decrease in pH dependence. As shown in Figure 2B, the rates of both P RNA and holoenzyme cleavage are pH sensitive with a log-linear relationship between k obs and pH. The slopes for both reactions are approximately equal to 1 (1.1 and 1.2 for RNase P and P RNA, respectively), consistent with the deprotonation of a functional group in the transition state for both reactions. Thus, these data indicate that there is essentially no difference in the rate-limiting steps in the holoenzyme and P RNA reactions under these reaction conditions.
Next, we tested whether binding of C5 protein influences the Mg2+ concentration dependence of the single turnover rate constant for cleavage of pre-tRNAMET608. As in the pH titration experiments, reactions were performed at a saturating concentration of enzyme in order to minimize the contribution of binding steps to the observed rate constant. Additionally, titrations were performed at Mg2+ concentrations greater than the concentration required for P RNA folding (>5 mM) as indicated by sensitivity to chemical and oligonucleotide probing (Zarrinkar et al. 1996; Pan and Sosnick 1997; Kent et al. 2000). As shown in Figure 2C, the observed single turnover rate constants for both the holoenzyme and the P RNA alone reactions increase as Mg2+ concentration increases. The concentrations of monovalent ions were not adjusted to maintain constant ionic strength, and thus both site-specific and diffuse metal ion interactions will contribute to the observed metal ion dependence. For P RNA alone, the apparent Mg2+ affinity is comparatively low, and thus only an estimate of the apparent affinity (Kd,Mg) and the rate at saturating Mg2+ concentrations (k obs(max)) could be obtained. Also, binding of the protein subunit is sensitive to very high ionic strength (>0.5 M), limiting the upper range at which data can be collected (Day-Storms et al. 2004). Nonetheless, these data indicate that the k obs(max) values for the P RNA and holoenzyme reactions differ by less than twofold (∼0.07 s−1 for holoenzyme; ∼0.13 s−1 for P RNA alone). Importantly, the Mg2+ titration data for both reactions fit to a noncooperative binding model and the E. coli holoenzyme displays a significantly lower overall Kd,Mg (32 mM) compared to the P RNA-alone reaction (>250 mM). Since the highest concentration tested for P RNA was only twofold higher than the apparent Kd,Mg this value represents a lower limit. At 10 mM Mg2+, the rate enhancement due to the presence of C5 protein becomes >55-fold, and, at lower concentrations, such as those encountered in a cellular environment (∼1 mM) (e.g., Froschauer et al. 2004; Farruggia et al. 2005), the extent of enhancement will be greater.
While these data are important for establishing the contributions of C5 protein to RNase P function in modulating apparent metal ion affinity, they raise obvious questions regarding the specific role (or roles) these metal ion interactions play in enzyme catalysis and where they bind in the enzyme–substrate complex. Previous studies of both large and small ribozymes clearly demonstrate that metal ions that bind at a distance from the active site and promote folding can provide a large apparent contribution to catalysis (DeRose 2003; Draper et al. 2005). At the concentrations of divalent ions used in Figure 2 (17.5 mM), the Mg2+ requirement for P RNA global folding is satisfied (∼5 mM) (Zarrinkar et al. 1996; Fang et al. 2000); however, it is possible that C5 protein enhances the affinity of Mg2+ ions necessary for a local folding transition that affects the active site or one that is necessary for substrate positioning (Christian et al. 2006). Additionally, as described above, a simple model would be that C5 protein directly affects the affinity of metals within the ribozyme active site.
A strategy for observing the effect of the protein subunit on the specific binding of catalytic metal ions is suggested by the observation that replacing the pro-Rp oxygen of the substrate phosphate with sulfur results in a >1000-fold defect in catalysis, and that adding a “thiophilic” metal ion, like Mn2+ or Cd2+, into the reaction rescues cleavage (Warnecke et al. 1996; Chen et al. 1997). Under the appropriate conditions such a result can be interpreted as indicating that the rescuing ion is coordinated directly to the site-specific sulfur modification and by analogy that Mg2+ ions are coordinated to the analogous oxygen atom in the unmodified RNA (Pecoraro et al. 1984; Piccirilli et al. 1993). For the Group I intron, Group II intron, and hammerhead ribozymes, quantitative analysis of metal ion dependent PS rescue has provided a means for analyzing the relative affinity of specific active site metal ions (Peracchi et al. 1997; Shan et al. 1999; Gordon and Piccirilli 2001). In this analysis, nonspecific effects on reaction rate caused by the presence of Cd2+ is accounted for by evaluation of k rel, which is the ratio of the observed rates for the oxygen-containing substrate and sulfur-containing substrate (k rel=k S obs/k O obs). Plotting k rel versus the concentrations of rescuing Cd2+ provides a thermodynamic signature for binding of these ions to the substrate cleavage site (Shan et al. 1999, 2001). Thus, comparison of the concentration dependence of Cd2+ rescue for the holoenzyme and the P RNA alone reactions, in principle, should permit the contribution of C5 protein to active site metal affinity to be measured.
To interpret such data in terms of specific metal ion binding, several criteria must be met: First, the addition of Cd2+ must provide significant rescue for the modified substrate in the background of Mg2+ ions; second, the added Cd2+ should not cause miscleavage that limits the accuracy of measured rate constants; third, there must be no change in the rate limiting step in the presence and absence of rescuing metal ion, as this would invalidate the quantitative evaluation of Cd2+ binding thermodynamics (Christian 2002, and references therein). To first test whether Cd2+ can rescue the catalytic defect caused by substrate modification, single turnover cleavage reactions were performed in the presence or absence of Cd2+ in the background of 17.5 mM Mg2+ to ensure appropriate RNA folding and to minimize the effects of Cd2+ on folding. For these experiments, a substrate containing a Rp phosphorothioate modification at the N(+1) position of pre-tRNAMet608 (Rp[+1] pre-tRNA) was created by ligating an 11 nt oligo encompassing the cleavage site to a 3′ fragment containing the remainder of the tRNA portion of the substrate. As shown in Figures 3 and 4, the PS modification results in a significant defect in the rate of substrate cleavage (k O obs/k S obs>1000) for the Rp(+1) pre-tRNA substrate. This defect is not due to a deleterious effect on substrate binding, which was assessed by measuring substrate dissociation constants by gel filtration. Apparent Kd were ∼1 nM for binding of pre-tRNA and Rp(+1) pre-tRNA, respectively, to RNase P. The modification had only a minor effect on binding for P RNA alone as well (Kd of 1 μM and 3 μM for pre-tRNA and Rp[+1] pre-tRNA, respectively). Importantly, this catalytic defect can be rescued by addition of Cd2+ into both P RNA and holoenzyme reactions. These data further illustrate that the second criterion is also met in that the presence of Cd2+ does not result in significant miscleavage (Fig. 3). However, the presence of the phosphorothioate modification in the Rp(+1) pre-tRNA does engender some miscleavage in the absence of Cd2+. This result is important, since miscleavage arises from parallel reactions that would by definition obscure the intrinsic cleavage rate (Loria and Pan 1999; Zahler et al. 2005). Thus, the rate constants determined in the absence of Cd2+ contain an inherent inaccuracy. Nonetheless, because these rate constants are so small relative to the rate constants for cleavage of the unmodified substrate (>1000-fold), this inaccuracy in the 0 mM Cd2+ data point has little effect on the subsequent k rel analysis. Moreover, micromolar concentrations of Cd2+ entirely suppress miscleavage; therefore, comparison of rate constants determined under these conditions will accurately reflect the intrinsic substrate cleavage rate. In addition, to test whether the presence of Cd2+ changes the rate limiting step for cleavage of Rp(+1) pre-tRNA and unmodified pre-tRNA, the effect of pH on the rate constants for these substrates was examined. In these experiments the catalytic rate constant is still pH dependent in the presence of Cd2+ up to 15 mM for both reactions, indicating that there is no change in rate limiting step (data not shown).
FIGURE 3.

Cd2+ rescues the cleavage of a N(+1) PS substrate modification by P RNA and RNase P holoenzyme. Single turnover reactions were performed under standard conditions (50 mM MES at pH 5.75, 100 mM NaCl, 17.5 mM MgCl2, and 0.005% Triton X-100) in the absence (panels 1,3) or presence of 5 mM Cd2+ (panels 2,4) for RNase P holoenzyme (panels 3,4) and P RNA (panels 1,2) reactions. Aliquots are removed at predetermined time points and analyzed in 20% PAGE. The positions of products resulting from correct cleavage (−1/+1) and miscleavage (−2/−1) are indicated. The product marked by an asterisk results from correct cleavage of a minor population of substrate containing an additional nucleotide at its 5′ end.
FIGURE 4.

Quantitative analysis of Cd2+ dependent rescue of the (+1 Rp) PS substrate modification. Single turnover rate constants were determined at a series of increasing Cd2+ concentrations in the background of 17.5 mM Mg2+. (A) Plots of k obs (RNase P) versus Cd2+ concentration are shown for PS modified (○) and unmodified (•) substrates. (B) Plots of k obs (P RNA alone) versus Cd2+ concentration are shown for modified (○) and unmodified (•)substrates. (C) The k obs (P RNA alone) data for PS modified substrates are shown separately for a better view of Cd2+ stimulation at low Cd2+ concentrations. Data represent the average values for at least three independent trials.
With the criteria of Cd2+ dependent rescue, accurate cleavage, and no change in mechanism for the Rp(+1) pre-tRNA substrate established, it is possible to employ analysis of k rel (see above) to probe active site metal ion affinities for P RNA in the presence and absence of C5 protein. Therefore, we determined the effect of Cd2+ on the rate constant for cleavage of the native oxygen containing substrate (k O obs) by P RNA in the presence and absence of C5 protein. As shown in Figure 4, Cd2+ inhibits both reactions in a concentration-dependent, saturable fashion. The tritation data for both RNase P and P RNA fit to a cooperative binding mechanism in which two classes of Cd2+ ions bind simultaneously and inhibit RNase P catalysis. Furthermore, the apparent affinity for the inhibitory Cd2+ ions is similar for both P RNA and RNase P (Kd,Cd=1.9 mM and 3.0 mM for P RNA and RNase P, respectively). In contrast, Cd2+ results in an increase in the rate constant for cleavage of Rp(+1) pre-tRNA (k S obs) for both P RNA and holoenzyme. Because of the large degree of inhibition of the P RNA alone reaction, the stimulatory effect of Cd2+ on k S obs is most clearly observed at lower concentrations (Fig. 4C). Thus, the effect of Cd2+ ions on the rate constant for cleavage of Rp(+1) pre-tRNA is the combination of both the site-specific rescue effect and inhibition.
To isolate the thermodynamics of the binding of the rescuing metal ions, k rel is utilized as described above (k rel=k S obs/k O obs), which normalizes the inhibitory effect of Cd2+ on the cleavage reactions catalyzed by P RNA in the presence and absence of C5 protein. A plot of k rel versus the concentration of rescuing Cd2+ ions fits well to the cooperative binding model for both holoenzyme and P RNA alone reactions (Fig. 5A). The apparent dissociation constant for catalytic metal ion binding (Kd,app) for the holoenzyme reaction is 9.3 mM and the Hill coefficient (n) is 2, indicating that two metal ions interact with the Rp sulfur modification in Rp(+1) pre-tRNA, consistent with the two metal ion model as described above (Fig. 5B). For P RNA alone, a Cd2+ concentration significantly higher than 15 mM is required to reach saturation. However, precipitation of RNA in higher Cd2+ concentrations limits the precise measurement of the metal ion affinity and cooperativity, and the Kd,app is estimated to be significantly >30 mM. Thus, the apparent dissociation constant for active site metal ion binding is at least threefold higher for P RNA than the RNA–protein complex.
DISCUSSION
If the Cd2+ rescue experiments accurately reflect the interactions of Mg2+ interactions with the reactive phosphate, then the observed increase in apparent Mg2+ affinity observed in Figure 2 in part reflects active site metal ion binding. The close correspondence in the metal ion interactions in the Group I intron ribozyme active site identified by quantitative metal ion rescue experiments and high resolution X-ray crystallography indicates that this assumption is likely to be valid (Shan et al. 1999; Stahley and Strobel 2005). Thus, a simple interpretation of these data supports a model for RNase P catalysis in which two divalent metal ions bind to the reactive phosphate in the enzyme–substrate complex that are essential for catalysis. In this model the protein subunit has a significant effect on catalysis by influencing the affinity of these active site metal ions, thus increasing the apparent rate constant for pre-tRNA cleavage (Fig. 6).
FIGURE 6.

P protein increases the affinity of metal ions in the active site of P RNA. Interpretation of the Cd2+ rescue data supports a model in which two Mg2+ ions bind to the active site of the enzyme–substrate complex in the ground state. The apparent affinities of the rescuing ion interactions are indicated. However, the affinities of corresponding active site Mg2+ ions may be significantly different. However, as indicated in the text, evaluation of the Mg2+ titration data are consistent with cooperative binding of a high affinity class of metal ions that contributes to catalysis as indicated in the diagram.
An important observation regarding the differential functional effects of RNA metal ion interactions is the significant inhibition of both P RNA and RNase P holoenzyme by increasing concentrations of Cd2+. When the PS-modified substrate is used, both activation and inhibition contribute to the observed Cd2+ dependence and the k rel approach permits the affinity of activating ion binding to be deconvoluted. However, the underlying quantitative relationship between the observed inhibitory Cd2+ binding affinity and active site metal affinity for the PS-modified substrate is difficult to evaluate. Inhibition could arise from competition with active site metal ions or indirectly by affecting active site geometry at a distance. Figure 4 also shows that there is a significant different in the magnitude of the effect of increasing Cd2+ concentration on the rate constants for the P RNA and RNase P reactions. A first order interpretation is that the geometry of the RNA with Cd2+ bound is different when C5 protein is also bound to P RNA, and this difference in structure accounts for the difference in the degree of inhibition that is observed. Additionally, titration data alone do not distinguish whether there is a single inhibitory Cd2+ ion bound or multiple Cd2+ ions binding noncooperatively with similar affinity. Thus, a second possibility is that the P RNA has additional inhibitory Cd2+ binding interactions that are distinct from the RNase P holoenzyme.
A further illustration of the multicomponent nature of RNA–metal ion interactions is provided by the differences in apparent binding thermodynamics of “catalytic” metal ion binding for total Mg2+ titration versus active site metal ion binding obtained by k rel analysis. The apparent binding thermodynamics of the rescuing Cd2+ ions is cooperative, while metal ion binding in bulk Mg2+ titration experiments, which should contain a significant contribution from active site metal ion binding, are not. In this regard it is important to note that if additional Mg2+ ions are necessary for catalysis, even if they bind within the active site, they will not be detected in the PS rescue experiments, which only report on the interaction of metal ions with the introduced sulfur modification. Based on physical studies of model systems, it is likely that the bulk Mg2+ titration results reflect the binding of multiple classes of metal ions, including diffuse ions that do not interact with the RNA in a site-specific fashion. The data in Figure 2 for bulk Mg2+ titration fit to a model for binding of a single class of activating Mg2+ ions in a noncooperative manner. Inspection of the titration data at low Mg2+ ion concentrations reveals that the observed rate constants could also be accounted for by a more complex model in which two classes of ions are necessary for catalysis: a high affinity class that binds cooperatively and a lower affinity class that binds in a noncooperative fashion. However, inaccuracies in the current data set for measuring small rate constants and in controlling for depletion of Mg2+ by the high concentrations of P RNA required to achieve saturation make it difficult to distinguish between these models. Additionally, differences in the size and charge distribution of Cd2+ versus Mg2+ or changes in the electrostatic environment of the active site due to introduction of the PS modification could also result in differences in apparent Mg2+ and Cd2+ affinity. Nonetheless, these results provide the first evidence for a specific effect of the RNase P protein subunit on active site metal ion binding that at physiological divalent metal ion concentrations (0.5–1 mM) (e.g., Froschauer et al. 2004; Farruggia et al. 2005) will be very large.
Given the importance of understanding the cooperative function of RNA and protein, the mechanism by which RNase P protein acts to enhance active site metal ion affinity is of significant interest. The effect of protein binding on active site metal ion affinity could in principle result from P protein contributing protein functional groups that serve as additional ligands for metal ions in the active site. This possibility seems unlikely given that there are substrates and reaction conditions under which C5 protein can make relatively little contribution to catalysis (Beebe and Fierke 1994; Sun et al. 2006) and the low degree of sequence conservation of P protein compared to the catalytic core of P RNA (Haas and Brown 1998; Jovanovic et al. 2002). However, due to the presence of unique structural motifs in the protein cofactor, it is conceivable that the polypeptide backbone itself might contribute to metal ion binding. Alternatively, P protein could act indirectly, inducing a conformational change in P RNA that results in a more optimal alignment of RNA functional groups that contact metal ions. Previous studies have indeed shown that the protein decreases the Mg2+ requirement for folding (Buck et al. 2005a). However, these effects are manifest at lower concentrations than the titration analyses presented here and chemical and oligonucleotide probing confirm that the P RNA is folded over the range of Mg2+ concentrations tested here (Zarrinkar et al. 1996; Sun et al. 2006). Further, the single turnover analyses were performed at saturating concentrations of enzyme that should control for differences in the active fraction of enzyme at different metal ion concentrations. Alternatively, the protein could contribute to a substrate docking step, after initial substrate binding, in which the binding sites for active site metal ions are formed. If catalysis is slow relative to equilibration between the docked and undocked forms, then the pH dependence will be the same for the P RNA and holoenzyme reactions since phosphodiester bond cleavage is rate limiting in both cases (Loria and Pan 1999), consistent with the data reported here. Since in this model docking results in formation of metal ion binding sites associated with catalysis, then shifting the equilibrium to the docked state should result in a decrease in the concentration of Mg2+ required to reach saturation, again consistent with the observations reported herein. Previous studies by Fierke and colleagues showed that shortening the leader sequence of a model pre-tRNA substrate decreased the apparent affinity of metal ion interactions necessary for cleavage (Kurz and Fierke 2002). However, preliminary results show that truncation of the 5′-leader sequence has no significant effect on the extent of catalytic enhancement due to C5 protein binding to P RNA (L. Sun, F.E. Campbell, and M.E. Harris, in prep.) consistent with an indirect effect of the protein rather than a direct effect due to contribution of functional groups or by substrate positioning interactions.
The ability of RNA-binding proteins to alter the apparent metal ion affinity of their cognate RNA ligands is well documented, yet providing direct experimental linkages between metal ion binding and function remains challenging. Both Group I and Group II self-splicing introns require essential Mg2+ ion cofactors, and binding of maturase proteins essential for processing in vivo (Lambowitz and Zimmerly 2004; Stoddard 2006) results in significant decreases in the levels of Mg2+ necessary to achieve optimal catalytic rates in vitro. While such effects are generally attributable to metal ions involved in RNA folding, they are necessarily coupled to active site metal ion affinity, since formation of the active site obviously requires folding. While these proteins result in a single round of self-splicing, differential effects of P protein on the cleavage step for different substrates could provide a mechanism for substrate-dependent regulation. Clearly, the integral roles of P protein in both substrate recognition and catalysis rationalize the necessity of the protein subunit in vivo. However, the mechanism by which the protein alters catalytic metal ion affinity is not yet determined. Understanding this mechanistic basis is likely to help illuminate the important interplay between protein binding and RNA function.
MATERIALS AND METHODS
The E. coli P RNA and pre-tRNAMet608 were generated by in vitro transcription as previously described (Sun et al. 2006). The C5 protein was expressed in E. coli and isolated by affinity purification as described (Guo et al. 2006). The (+1 Rp) PS modified pre-tRNA (Rp[+1] pre-tRNA) was generated by ligation of a (−5, +6) 11 nt oligo (Dharmacon) and a (+7, +74) 3′ fragment of pre-tRNAMet generated by in vitro transcription (Zahler et al. 2005). As shown in Figure 3 this substrate gives rise primarily to cleavage at the correct phosphodiester bond 5′ to N(+1) of tRNA. An additional product migrating slower than the correct cleavage product was also observed, which is marked with an asterisk in Figure 3. We established that this product is an artifact arising from the ligation reaction. Observation of this specifies is independent of the presence of the phosphorothioate modification and is not observed with substrates generated by in vitro transcription. Formation of this product correlates with the appearance of the correct cleavage product. Because it is a minor product (<5%), rate determinations were not corrected to account of its formation.
For kinetic and thermodynamic studies, pre-tRNAs were 5′-end labeled with [γ-32P]-ATP (MP Biomedicals) and T4 polynucleotide kinase after dephosphorylation by alkaline phosphatase. Single turnover kinetic analyses were performed essentially as described (Sun et al. 2006) with the following modifications. Rates were measured under the following conditions, with pH and metal ion concentration varied as indicated in text: 50 mM MES (pH 5.75), 100 mM NaCl, 17.5 mM MgCl2, and 0.005% Triton X-100. Enzyme concentrations were determined to be saturating if the observed change in rate upon double the enzyme was less than 10%. Enzyme (1 μM RNase P or 10–30 μM P RNA) and 5′-32P-end-labeled pre-tRNA (1–4 nM) were renatured separately in the absence of divalent metal ion by incubation at 95°C for 3 min, followed by decreasing the temperature to 37°C at 0.2°C per second and incubating for 10 min. MgCl2 was then added to the desired concentration and the mixture was further incubated for 10 min. For holoenzyme reactions, C5 protein was then added (final concentration equal to P RNA), and the incubation was continued for another 10 min. Enzyme and substrate were mixed together to start the reaction, and aliquots were removed at predetermined intervals and quenched with EDTA at at least twice the concentration of divalent metal ion in the reaction. Reaction products were resolved by denaturing PAGE (20%). The conversion of substrate to product was quantified by phosphorimaging using a Molecular Dynamics system and ImageQuant software (Amersham). The data were plotted versus time using KaleidaGraph software (Synergy) and fit to a single exponential function:
where Fc is the fraction cleaved, A is the maximal extent of the reaction, B is the amplitude of the exponential, k is the observed cleavage rate constant (k obs), and t is time. Rate constants reported are the average of at least three independent determinations, each with <10% error in the curve fit to the primary data.
Cleavage reactions in presence of increasing (Mg2+) were performed essentially as described above. k obs was then plotted as a function of (Mg2+) concentrations and fitted to a form of the Hill equation:
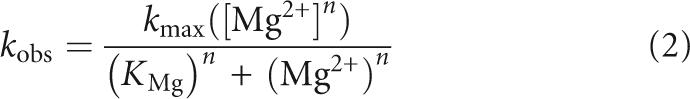 |
where k obs is the rate constant measured at each Mg2+ concentration, k max is the cleavage rate constant at saturating Mg2+ concentration, (Mg2+) is the Mg2+ concentration, K Mg is the apparent dissociation constant, and n is the Hill coefficient.
The pseudo-first-order rate constants were determined as described above in a series of increasing [Cd2+]. The relative rate k rel was calculated from the ratio of the observed reaction rates of substrates with and without the sulfur substitution (e.g., k rel = k S obs/k O obs). Plots of kobs versus (Cd2+) were fit to the Hill equation as described above for Mg2+ titration analyses.
ACKNOWLEDGMENTS
We thank Drs. James Hougland, Vernon Anderson, Eric Christian, and Frank Campbell for advice and comments on the manuscript. We are particularly grateful to Drs. Eric Christian and James Hougland for advice on interpretation of quantitative PS rescue experiments. This work was supported by NIH grant GM56742 (to M.E.H.).
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.571007.
REFERENCES
- Beebe, J.A., Fierke, C.A. A kinetic mechanism for cleavage of precursor tRNAAsp catalyzed by the RNA component of Bacillus subtilis ribonuclease P. Biochemistry. 1994;33:10294–10304. doi: 10.1021/bi00200a009. [DOI] [PubMed] [Google Scholar]
- Beebe, J.A., Kurz, J.C., Fierke, C.A. Magnesium ions are required by Bacillus subtilis ribonuclease P RNA for both binding and cleaving precursor tRNAAsp . Biochemistry. 1996;35:10493–10505. doi: 10.1021/bi960870m. [DOI] [PubMed] [Google Scholar]
- Buck, A.H., Dalby, A.B., Poole, A.W., Kazantsev, A.V., Pace, N.R. Protein activation of a ribozyme: The role of bacterial RNase P protein. EMBO J. 2005a;24:3360–3368. doi: 10.1038/sj.emboj.7600805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck, A.H., Kazantsev, A.V., Dalby, A.B., Pace, N.R. Structural perspective on the activation of RNAse P RNA by protein. Nat. Struct. Mol. Biol. 2005b;12:958–964. doi: 10.1038/nsmb1004. [DOI] [PubMed] [Google Scholar]
- Caprara, M.G., Chatterjee, P., Solem, A., Brady-Passerini, K.L., Kaspar, B.J. An allosteric-feedback mechanism for protein-assisted group I intron splicing. RNA. 2007;13:211–222. doi: 10.1261/rna.307907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassano, A.G., Anderson, V.E., Harris, M.E. Evidence for direct attack by hydroxide in phosphodiester hydrolysis. J. Am. Chem. Soc. 2002;124:10964–10965. doi: 10.1021/ja020823j. [DOI] [PubMed] [Google Scholar]
- Cassano, A.G., Anderson, V.E., Harris, M.E. Analysis of solvent nucleophile isotope effects: Evidence for concerted mechanisms and nucleophilic activation by metal coordination in nonenzymatic and ribozyme-catalyzed phosphodiester hydrolysis. Biochemistry. 2004;43:10547–10559. doi: 10.1021/bi049188f. [DOI] [PubMed] [Google Scholar]
- Chen, Y., Li, X., Gegenheimer, P. Ribonuclease P catalysis requires Mg2+ coordinated to the pro-RP oxygen of the scissile bond. Biochemistry. 1997;36:2425–2438. doi: 10.1021/bi9620464. [DOI] [PubMed] [Google Scholar]
- Christian, E.L. Identification and characterization of metal ion binding by thiophilic metal ion rescue. In: Hartmann R.K., et al., editors. Handbook of RNA biochemistry. Wiley-VCH; Berlin: 2002. pp. 319–341. [Google Scholar]
- Christian, E.L., Smith, K.M., Perera, N., Harris, M.E. The P4 metal binding site in RNase P RNA affects active site metal affinity through substrate positioning. RNA. 2006;12:1463–1467. doi: 10.1261/rna.158606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowan, J.A. Metal activation of enzymes in nucleic acid biochemistry. Chem. Rev. 1998;98:1067–1088. doi: 10.1021/cr960436q. [DOI] [PubMed] [Google Scholar]
- Day-Storms, J.J., Niranjanakumari, S., Fierke, C.A. Ionic interactions between PRNA and P protein in Bacillus subtilis RNase P characterized using a magnetocapture-based assay. RNA. 2004;10:1595–1608. doi: 10.1261/rna.7550104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRose, V.J. Metal ion binding to catalytic RNA molecules. Curr. Opin. Struct. Biol. 2003;13:317–324. doi: 10.1016/s0959-440x(03)00077-0. [DOI] [PubMed] [Google Scholar]
- Draper, D.E., Grilley, D., Soto, A.M. Ions and RNA folding. Annu. Rev. Biophys. Biomol. Struct. 2005;34:221–243. doi: 10.1146/annurev.biophys.34.040204.144511. [DOI] [PubMed] [Google Scholar]
- Egea, P.F., Stroud, R.M., Walter, P. Targeting proteins to membranes: Structure of the signal recognition particle. Curr. Opin. Struct. Biol. 2005;15:213–220. doi: 10.1016/j.sbi.2005.03.007. [DOI] [PubMed] [Google Scholar]
- Evans, D., Marquez, S.M., Pace, N.R. RNase P: Interface of the RNA and protein worlds. Trends Biochem. Sci. 2006;31:333–341. doi: 10.1016/j.tibs.2006.04.007. [DOI] [PubMed] [Google Scholar]
- Fang, X., Littrell, K., Yang, X.J., Henderson, S.J., Siefert, S., Thiyagarajan, P., Pan, T., Sosnick, T.R. Mg2+-dependent compaction and folding of yeast tRNAPhe and the catalytic domain of the B. subtilis RNase P RNA determined by small-angle X-ray scattering. Biochemistry. 2000;39:11107–11113. doi: 10.1021/bi000724n. [DOI] [PubMed] [Google Scholar]
- Farruggia, G., Iotti, S., Prodi, L., Montalti, M., Zaccheroni, N., Savage, P.B., Trapani, V., Sale, P., Wolf, F.I. 8-Hydroxyquinoline derivatives as fluorescent sensors for magnesium in living cells. J. Am. Chem. Soc. 2005;128:344–350. doi: 10.1021/ja056523u. [DOI] [PubMed] [Google Scholar]
- Frank, D.N., Pace, N.R. In vitro selection for altered divalent metal specificity in the RNase P RNA. Proc. Natl. Acad. Sci. 1997;94:14355–14360. doi: 10.1073/pnas.94.26.14355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Froschauer, E.M., Kolisek, M., Dieterich, F., Schweigel, M., Schweyen, J.J. Fluorescence measurements of free (Mg) by use of mag-fura 2 in Salmonella enterica . FEMS Microbiol. Lett. 2004;237:49–55. doi: 10.1016/j.femsle.2004.06.013. [DOI] [PubMed] [Google Scholar]
- Gordon, P.M., Piccirilli, J.A. Metal ion coordination by the AGC triad in domain 5 contributes to group II intron catalysis. Nat. Struct. Biol. 2001;8:893–898. doi: 10.1038/nsb1001-893. [DOI] [PubMed] [Google Scholar]
- Guo, X., Campbell, F.E., Sun, L., Christian, E.L., Anderson, V.E., Harris, M.E. RNA-dependent folding and stabilization of C5 protein during assembly of the E. coli RNase P holoenzyme. J. Mol. Biol. 2006;360:190–203. doi: 10.1016/j.jmb.2006.05.001. [DOI] [PubMed] [Google Scholar]
- Haas, E.S., Brown, J.W. Evolutionary variation in bacterial RNase P RNAs. Nucleic Acids Res. 1998;26:4093–4099. doi: 10.1093/nar/26.18.4093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas, E.S., Brown, J.W., Pitulle, C., Pace, N.R. Further perspective on the catalytic core and secondary structure of ribonuclease P RNA. Proc. Natl. Acad. Sci. 1994;91:2527–2531. doi: 10.1073/pnas.91.7.2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho, Y., Waring, R.B. The maturase encoded by a group I intron from Aspergillus nidulans stabilizes RNA tertiary structure and promotes rapid splicing. J. Mol. Biol. 1999;292:987–1001. doi: 10.1006/jmbi.1999.3070. [DOI] [PubMed] [Google Scholar]
- Hopper, A.K. Cellular dynamics of small RNAs. Crit. Rev. Biochem. Mol. Biol. 2006;41:3–19. doi: 10.1080/10409230500405237. [DOI] [PubMed] [Google Scholar]
- Hougland, J.L., Kravchuk, A.V., Herschlag, D., Piccirilli, J.A. Functional identification of catalytic metal ion binding sites within RNA. PLoS Biol. 2005;3:e277. doi: 10.1371/journal.pbio.0030277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh, J., Andrews, A.J., Fierke, C.A. Roles of protein subunits in RNA–protein complexes: Lessons from ribonuclease P. Biopolymers. 2004;73:79–89. doi: 10.1002/bip.10521. [DOI] [PubMed] [Google Scholar]
- Jankowsky, E., Bowers, H. Remodeling of ribonucleoprotein complexes with DExH/D RNA helicases. Nucleic Acids Res. 2006;34:4181–4188. doi: 10.1093/nar/gkl410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovanovic, M., Sanchez, R., Altman, S., Gopalan, V. Elucidation of structure–function relationships in the protein subunit of bacterial RNase P using a genetic complementation approach. Nucleic Acids Res. 2002;30:5065–5073. doi: 10.1093/nar/gkf670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazantsev, A.V., Pace, N.R. Bacterial RNase P: A new view of an ancient enzyme. Nat. Rev. Microbiol. 2006;4:729–740. doi: 10.1038/nrmicro1491. [DOI] [PubMed] [Google Scholar]
- Kent, O., Chaulk, S.G., MacMillan, A.M. Kinetic analysis of the M1 RNA folding pathway. J. Mol. Biol. 2000;304:699–705. doi: 10.1006/jmbi.2000.4263. [DOI] [PubMed] [Google Scholar]
- Kurz, J.C., Fierke, C.A. Ribonuclease P: A ribonucleoprotein enzyme. Curr. Opin. Chem. Biol. 2000;4:553–558. doi: 10.1016/s1367-5931(00)00131-9. [DOI] [PubMed] [Google Scholar]
- Kurz, J.C., Fierke, C.A. The affinity of magnesium binding sites in the Bacillus subtilis RNase P x pre-tRNA complex is enhanced by the protein subunit. Biochemistry. 2002;41:9545–9558. doi: 10.1021/bi025553w. [DOI] [PubMed] [Google Scholar]
- Lambowitz, A.M., Zimmerly, S. Mobile group II introns. Annu. Rev. Genet. 2004;38:1–35. doi: 10.1146/annurev.genet.38.072902.091600. [DOI] [PubMed] [Google Scholar]
- Lehman, N., Joyce, G.F. Evolution in vitro of an RNA enzyme with altered metal dependence. Nature. 1993;361:182–185. doi: 10.1038/361182a0. [DOI] [PubMed] [Google Scholar]
- Loria, A., Pan, T. The cleavage step of ribonuclease P catalysis is determined by ribozyme–substrate interactions both distal and proximal to the cleavage site. Biochemistry. 1999;38:8612–8620. doi: 10.1021/bi990691f. [DOI] [PubMed] [Google Scholar]
- Loria, A., Niranjanakumari, S., Fierke, C.A., Pan, T. Recognition of a pre-tRNA substrate by the Bacillus subtilis RNase P holoenzyme. Biochemistry. 1998;37:15466–15473. doi: 10.1021/bi9816507. [DOI] [PubMed] [Google Scholar]
- Nelson, J.A., Shepotinovskaya, I., Uhlenbeck, O.C. Hammerheads derived from sTRSV show enhanced cleavage and ligation rate constants. Biochemistry. 2005;44:14577–14585. doi: 10.1021/bi051130t. [DOI] [PubMed] [Google Scholar]
- Niranjanakumari, S., Stams, T., Crary, S.M., Christianson, D.W., Fierke, C.A. Protein component of the ribozyme ribonuclease P alters substrate recognition by directly contacting precursor tRNA. Proc. Natl. Acad. Sci. 1998;95:15212–15217. doi: 10.1073/pnas.95.26.15212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan, T., Sosnick, T.R. Intermediates and kinetic traps in the folding of a large ribozyme revealed by circular dichroism and UV absorbance spectroscopies and catalytic activity. Nat. Struct. Biol. 1997;4:931–938. doi: 10.1038/nsb1197-931. [DOI] [PubMed] [Google Scholar]
- Pecoraro, V.L., Hermes, J.D., Cleland, W.W. Stability constants of Mg2+ and Cd2+ complexes of adenine nucleotides and thionucleotides and rate constants for formation and dissociation of MgATP and MgADP. Biochemistry. 1984;23:5262–5271. doi: 10.1021/bi00317a026. [DOI] [PubMed] [Google Scholar]
- Peracchi, A., Beigelman, L., Scott, E.C., Uhlenbeck, O.C., Herschlag, D. Involvement of a specific metal ion in the transition of the hammerhead ribozyme to its catalytic conformation. J. Biol. Chem. 1997;272:26822–26826. doi: 10.1074/jbc.272.43.26822. [DOI] [PubMed] [Google Scholar]
- Persson, T., Cuzic, S., Hartmann, R.K. Catalysis by RNase P RNA: Unique features and unprecedented active site plasticity. J. Biol. Chem. 2003;278:43394–43401. doi: 10.1074/jbc.M305939200. [DOI] [PubMed] [Google Scholar]
- Piccirilli, J.A., Vyle, J.S., Caruthers, M.H., Cech, T.R. Metal ion catalysis in the Tetrahymena ribozyme reaction. Nature. 1993;361:85–88. doi: 10.1038/361085a0. [DOI] [PubMed] [Google Scholar]
- Roychowdhury-Saha, M., Burke, D.H. Extraordinary rates of transition metal ion-mediated ribozyme catalysis. RNA. 2006;12:1846–1852. doi: 10.1261/rna.128906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan, S.O., Herschlag, D. Dissection of a metal-ion-mediated conformational change in Tetrahymena ribozyme catalysis. RNA. 2002;8:861–872. doi: 10.1017/s1355838202020216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan, S., Yoshida, A., Sun, S., Piccirilli, J.A., Herschlag, D. Three metal ions at the active site of the Tetrahymena group I ribozyme. Proc. Natl. Acad. Sci. 1999;96:12299–12304. doi: 10.1073/pnas.96.22.12299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan, S., Kravchuk, A.V., Piccirilli, J.A., Herschlag, D. Defining the catalytic metal ion interactions in the Tetrahymena ribozyme reaction. Biochemistry. 2001;40:5161–5171. doi: 10.1021/bi002887h. [DOI] [PubMed] [Google Scholar]
- Smith, D., Pace, N.R. Multiple magnesium ions in the ribonuclease P reaction mechanism. Biochemistry. 1993;32:5273–5281. doi: 10.1021/bi00071a001. [DOI] [PubMed] [Google Scholar]
- Smith, D., Burgin, A.B., Haas, E.S., Pace, N.R. Influence of metal ions on the ribonuclease P reaction. Distinguishing substrate binding from catalysis. J. Biol. Chem. 1992;267:2429–2436. [PubMed] [Google Scholar]
- Stahley, M.R., Strobel, S.A. Structural evidence for a two-metal-ion mechanism of group I intron splicing. Science. 2005;309:1587–1590. doi: 10.1126/science.1114994. [DOI] [PubMed] [Google Scholar]
- Stahley, M.R., Strobel, S.A. RNA splicing: Group I intron crystal structures reveal the basis of splice site selection and metal ion catalysis. Curr. Opin. Struct. Biol. 2006;16:319–326. doi: 10.1016/j.sbi.2006.04.005. [DOI] [PubMed] [Google Scholar]
- Steitz, T.A., Steitz, J.A. A general two-metal-ion mechanism for catalytic RNA. Proc. Natl. Acad. Sci. 1993;90:6498–6502. doi: 10.1073/pnas.90.14.6498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoddard, B.L. Homing endonuclease structure and function. Q. Rev. Biophys. 2006;38:49–95. doi: 10.1017/S0033583505004063. [DOI] [PubMed] [Google Scholar]
- Sun, L., Campbell, F.E., Zahler, N.H., Harris, M.E. Evidence that substrate-specific effects of C5 protein lead to uniformity in binding and catalysis by RNase P. EMBO J. 2006;25:3998–4007. doi: 10.1038/sj.emboj.7601290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinsley, R.A., Harris, D.A., Walter, N.G. Magnesium dependence of the amplified conformational switch in the trans-acting hepatitis delta virus ribozyme. Biochemistry. 2004;43:8935–8945. doi: 10.1021/bi049471e. [DOI] [PubMed] [Google Scholar]
- Torres-Larios, A., Swinger, K.K., Krasilnikov, A.S., Pan, T., Mondragon, A. Crystal structure of the RNA component of bacterial ribonuclease P. Nature. 2005;437:584–587. doi: 10.1038/nature04074. [DOI] [PubMed] [Google Scholar]
- Warnecke, J.M., Furste, J.P., Hardt, W.D., Erdmann, V.A., Hartmann, R.K. Ribonuclease P (RNase P) RNA is converted to a Cd2+ ribozyme by a single Rp-phosphorothioate modification in the precursor tRNA at the RNase P cleavage site. Proc. Natl. Acad. Sci. 1996;93:8924–8928. doi: 10.1073/pnas.93.17.8924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeks, K.M., Cech, T.R. Protein facilitation of group I intron splicing by assembly of the catalytic core and the 5′-splice-site domain. Cell. 1995;82:221–230. doi: 10.1016/0092-8674(95)90309-7. [DOI] [PubMed] [Google Scholar]
- Zahler, N.H., Sun, L., Christian, E.L., Harris, M.E. The pre-tRNA nucleotide base and 2′-hydroxyl at N(−1) contribute to fidelity in tRNA processing by RNase P. J. Mol. Biol. 2005;345:969–985. doi: 10.1016/j.jmb.2004.10.080. [DOI] [PubMed] [Google Scholar]
- Zarrinkar, P.P., Wang, J., Williamson, J.R. Slow folding kinetics of RNase P RNA. RNA. 1996;2:564–573. [PMC free article] [PubMed] [Google Scholar]