

Abstract
Objective
It has been reported that dehydroepiandrosterone is a pulmonary vasodilator and inhibits chronic hypoxia-induced pulmonary hypertension. Additionally, dehydroepiandrosterone has been shown to improve systemic vascular endothelial function. Thus, we hypothesized that chronic treatment with dehydroepiandrosterone would attenuate hypoxic pulmonary hypertension by enhancing pulmonary artery endothelial function.
Methods and Results
Rats were randomly assigned to five groups. Three groups received food containing 0, 0.3, or 1% dehydroepiandrosterone during a 3-wk-exposure to simulated high altitude (HA). The other 2 groups were kept at Denver’s low altitude (LA) and received food containing 0 or 1% dehydroepiandrosterone. Dehydroepiandrosterone dose-dependently inhibited hypoxic pulmonary hypertension (mean pulmonary artery pressures after treatment with 0, 0.3, and 1% dehydroepiandrosterone = 45 ± 5, 33 ± 2*, and 25 ± 1*# mmHg, respectively. *P<0.05 vs 0% and # vs 0.3%). Dehydroepiandrosterone (1%, 3 wks) treatment started after rats had been exposed to 3-wk hypoxia also effectively reversed established hypoxic pulmonary hypertension. Pulmonary artery rings isolated from both LA and HA rats treated with 1% dehydroepiandrosterone showed enhanced relaxations to acetylcholine and sodium nitroprusside, but not to 8-bromo-cGMP. In the pulmonary artery tissue from dehydroepiandrosterone-treated LA and HA rats, soluble guanylate cyclase, but not endothelial nitric oxide synthase, protein levels were increased.
Conclusion
These results indicate that the protective effect of dehydroepiandrosterone against hypoxic pulmonary hypertension may involve upregulation of pulmonary artery soluble guanylate cyclase protein expression and augmented pulmonary artery vasodilator responsiveness to nitric oxide.
Keywords: endothelial function, nitric oxide, cGMP, estradiol
INTRODUCTION
Chronic generalized alveolar hypoxia, such as occurs in chronic obstructive lung disease (COPD) and chronic mountain sickness, results in sustained pulmonary hypertension that can lead to right ventricular failure and death (1). The pathogenesis of hypoxic pulmonary hypertension comprises sustained abnormal vasoconstriction and structural remodeling of pulmonary arteries (2,3). Although long-term oxygen therapy benefits some patients with hypoxemic COPD, no simple, safe, and effective treatment of hypoxic pulmonary hypertension has been established, and new therapeutic strategies need to be identified (1,4).
Dehydroepiandrosterone (DHEA) is a C-19 steroid that is synthesized mainly by the human adrenal cortex. DHEA and its sulfated ester, DHEA sulfate (DHEAS), are produced in higher levels than any other circulating steroid hormone. Most circulating DHEA is in the form of DHEAS, which functions as an inactive reservoir for DHEA. Epidemiological observations and animal experiments indicate that DHEA has a wide variety of beneficial biological and physiological effects, including prevention of cardiovascular disease (5,6). However, the mechanisms responsible for the cardiovascular protective effects of DHEA are largely unknown.
Farrukh et al. (7) and Gupte et al. (8) have shown in vitro that DHEA is a pulmonary vasodilator and inhibits acute hypoxic pulmonary vasoconstriction due at least partly to opening of potassium channels. More recently, Hampl et al. (9) and Bonnet et al. (10) report that chronic treatment with DHEAS or DHEA inhibits and reverses hypoxia-induced pulmonary hypertension in rats, and Bonnet et al. show that the DHEA treatment is associated with increased expression and function of pulmonary artery Ca2+ activated K+ channels. In addition, several studies in the systemic circulation indicate that DHEA enhances vascular endothelial function (11-14). Because endothelial dysfunction is considered to play an important role in the pathogenesis of hypoxic pulmonary hypertension (15,16), we investigated if the protective effect of chronic DHEA treatment against hypoxic pulmonary hypertension in rats involved enhancement of pulmonary artery endothelial function. Our initial results showed that DHEA blocked hypoxic pulmonary hypertension and enhanced isolated pulmonary artery vasodilator responsiveness to both acetylcholine (ACH) (endothelium-dependent) and sodium nitroprusside (SNP) (endothelium-independent), and we further investigated the mechanisms downstream of nitric oxide (NO) that were responsible for the enhanced vasodilator responsiveness.
METHODS
All experiments were approved by the institutional animal care and use committee. The investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health.
Experimental groups
-
(A)
Prevention Study: Adult male Sprague-Dawley rats were randomly divided into five groups. Three groups were exposed to simulated high altitude (5,400 m, inspired O2 tension ~76 mmHg) for 3 wks (HA rats) in a hypobaric chamber flushed continuously with room air to prevent accumulation of CO2, NH3, and H2O. Hypobaric exposure was 24h/day, except when the chamber was opened for 10-15min every 2 days to remove rats or clean cages and replenish food and water. All rats were exposed to a 12:12-h light-dark cycle and allowed free access to rat food and water. One day before hypoxic exposure, two groups of HA rats were started on either a high (1%; HAD1) or a low (0.3%; HAD0.3) concentration of DHEA (Sigma) containing food that was given throughout the study. One HA group received normal rat food (HAC). The other two groups of rats were kept at Denver’s low altitude (LA; 1,600 m, inspired O2 tension ~120 mmHg) and received either normal rat food (LAC) or food containing1%DHEA (LAD1).
-
(B)
Reversal Study: Two groups of rats were exposed to 6 wks of HA. One group received normal rat food for 6 wks (CON), and the other group received normal food for the first 3 wks and then food containing 1% DHEA for the next 3 wks (DHEA).
Conscious catheterized rats
After 3-wk exposure to normoxia or hypoxia, or 6-wk exposure to hypoxia, rats were anesthetized with ketamine (100 mg/kg, im) and xylazine (15 mg/kg, im) for placement of 3 catheters in the right jugular vein and pulmonary (via jugular vein and right ventricle) and right carotid arteries (17). Blood samples (0.1 ml) were taken for measurement of hematocrit. The rats were allowed to recover for 48 h in room air at Denver’s altitude under the same dietary treatment as before catheterization. After recovery, conscious rats were placed in a ventilated plastic box (room air), and mean pulmonary (MPAP) and systemic arterial pressures (MSAP) were measured with pressure transducers. Cardiac output (CO) was determined by a standard dye-dilution method, and cardiac index (CI) and stroke volume (SV) were calculated as dividing cardiac output by the rat’s body weight and heart rate, respectively. Total pulmonary (TPRI) and systemic (TSRI) resistance indexes were calculated by dividing MPAP and MSAP by CI. All hemodynamic measurements were performed in the morning and values were taken after pressures and heart rate were stabilized (within 15 min). Blood samples were then collected for measurement of plasma steroid levels by enzyme immunoassay kits (Diagnostic Systems Laboratories). The heart was removed for assessment of right heart hypertrophy [right ventricle (RV)/left ventricle plus septum (LV+S) wet weight ratio], and the lungs were prepared for morphometric analysis.
Isolated pulmonary artery rings
In a separate series of experiments, vessel rings of the first left branch of the extralobar and intralobar 4th - 5th branch pulmonary arteries were prepared from LAC, LAD1, HAC, and HAD1 rats as previously described (17, 25). Briefly, after anesthesia (pentobarbital sodium, 30mg ip) and heparinization (100 IU), the heart and lungs were removed en bloc and the pulmonary artery rings were carefully isolated so as to cause minimal damage to the endothelium [the rest of pulmonary arteries and lungs were snap frozen in liquid N2 and stored at −80°C for subsequent Western blot analysis and measurements of soluble guanylate cyclase (sGC) activity and endothelin-1 (ET-1) levels]. Pulmonary artery rings were then placed on steel wires attached to a force displacement transducer for measurement of changes in the isometric force, and suspended in baths containing 10 ml physiological salt solution (Earle’s balanced salt solution, Sigma) gassed with 21 % O2-5% CO2-74 % N2 at 37°C. Resting passive force was adjusted to a previously determined optimal tension (750 and 1500 mg for extralobar pulmonary arteries from LA and HA rats, and 400 and 800 mg for intralobar pulmonary arteries from LA and HA rats, respectively) (17,25). After 60-min equilibration, all rings were depolarized with 80 mM KCl for 30 min. Following washout of KCl, relaxant responses to ACH (10-9 – 10-4 M), SNP (10-10 – 10-5 M), and 8-bromo-cGMP (8-Br-cGMP; 10-6 – 10-4 M) were assessed in separate phenylephrine (0.5 μM)-contracted pulmonary artery rings. Percent relaxation was expressed as relaxation (g)/0.5 μM phenylephrine–induced contraction (g) ×100.
Morphological analysis
Histological changes of pulmonary artery were quantified by morphometry (barium-gelatin method) as previously described (18). In each tissue section at least 30 consecutive barium-filled arteries were analyzed at × 400. %Wall thickness (WT) was expressed as medial thickness/external diameter × 100 (%).
Western blot analysis
Western blots were performed as previously described (19) with 30 μg of protein from pulmonary artery with appropriate antibodies to endothelial nitric oxide synthase (eNOS) (Transduction Laboratories), or α or β1 subunit of soluble guanylate cyclase (sGC α and β1) (Cayman Chemicals).
Lung ET-1 levels
Lung tissue ET-1 peptide levels were measured in 50 mg of tissue using an ET-1 peptide ELISA kit (Cayman Chemicals) (20).
Pulmonary artery sGC enzyme activity
Pulmonary artery extracts (30 μg protein) were incubated at 37°C for 10 min in a reaction mixture containing 50 mM Tris-HCl, pH 7.5, 4 mM Mg Cl2, 0.5 mM 1-methyl-3-isobutyl-xanthine, 1 mM GTP, and 1 mM SNP. The reaction was stopped by addition of 0.9 ml HCl (0.05 N) and boiling for 3 min. Newly synthesized cGMP was measured in the reaction mixture using an ELISA kit (R&D Systems) (21,22).
Statistical analysis
Values are expressed as means ± SEM. Comparisons between groups were made with Student’s t-test, analysis of variance (ANOVA) with Scheffé’s post-hoc test for multiple comparisons, or repeated measure ANOVA. Differences were considered significant at p<0.05.
RESULTS
Body weight gain in HAC rats was less than that in LAC rats (Table 1). Treatment with DHEA caused loss of body weight in both LA and HA rats, which is consistent with previous reports (23,24). Food consumption was not different between DHEA-treated and –untreated groups (~18 to 22 g/day/rat, which can be translated that rats received approximately 60 and 200 mg/day DHEA by 0.3 and 1% DHEA treatments, respectively). DHEA treatment had no effect on the hypoxia-induced polycythemia (Table 1).
TABLE 1.
Body weight and hematocrit
| GROUP | n | BW BEFORE (g) | BW AFTER (g) | HCT (%) |
|---|---|---|---|---|
| LAC | 5 | 300 ± 18 | 380 ± 17 | 50 ± 1 |
| LAD | 5 | 308 ± 14 | 279 ± 12*† | 49 ± 1 |
| HAC | 5 | 336 ± 16 | 352 ± 14* | 69 ± 2* |
| HAD 0.3 | 4 | 309 ± 2 | 288 ± 8 *† | 64 ± 1* |
| HAD 1 | 9 | 311 ± 3 | 251 ± 8*†# | 66 ± 2* |
Values are means ± SE. BW, body weight before (BEFORE) and after (AFTER) the treatment; HCT, hematocrit after the treatment; LAC, low altitude untreated control; LAD, low altitude 1%DHEA treated; HAC, high altitude untreated control; HAD 0.3 and HAD 1, high altitude 0.3 and 1% DHEA treated groups, respectively.
P<0.05 vs. LAC;
P<0.05 vs. HAC;
P<0.05 vs. HAD 0.3.
In LA rats, 3-wk treatment with DHEA (1%) increased the plasma levels of DHEA, DHEAS, and estradiol but not that of testosterone (Table 2). In HA rats, the high dose DHEA (1%) increased plasma levels of all four steroid hormones, while the low dose DHEA (0.3%) increased only the level of DHEA.
TABLE 2.
Plasma steroid hormone levels
| GROUP | n | DHEA (ng/ml) | DHEAS (ng/ml) | ESTRADIOL (pg/ml) | TESTOSTERONE (ng/ml) |
|---|---|---|---|---|---|
| LAC | 4 | 3 ± 1 | 148 ± 16 | 16 ± 13 | 2 ± 1 |
| LAD | 5 | 99 ± 27*† | 1325 ± 417*† | 55 ± 5 † | 10 ± 5 |
| HAC | 5 | 4 ± 1 | 132 ± 28 | 8 ± 5 | 0.5 ± 0.1 |
| HAD 0.3 | 4 | 35 ± 7*† | 483 ± 122 | 11 ± 9 | 8 ± 4 |
| HAD 1 | 6 | 103 ± 29*†# | 1467 ± 531*† | 116 ± 18*†# | 30 ± 8*†# |
Values are means ± SE. LAC, low altitude untreated control; LAD, low altitude 1%DHEA treated; HAC, high altitude untreated control; HAD 0.3 and HAD 1, high altitude 0.3 and 1% DHEA treated groups, respectively.
P<0.05 vs. LAC;
P<0.05 vs. HAC;
P<0.05 vs. HAD 0.3.
Results of hemodynamic and heart weight measurements are summarized in Figure 1 and Table 3. Chronic treatment with DHEA did not have significant hemodynamic effects in LA rats, except it decreased CO [although when corrected by body weight (CI), there was no significant reduction]. Chronic exposure to hypoxia markedly increased MPAP and slightly decreased CI with no change in MSAP. Calculated TPRI was markedly, and TSRI slightly, increased in HAC rats. DHEA caused a dose-dependent and marked inhibition of the increases in MPAP and TPRI but had no effects on MSAP, TSRI, or CI (DHEA caused decreases in CO which were accompanied by losses of body weight) in HA rats. Right ventricular hypertrophy as assessed by RV/LV+S was dose-dependently inhibited by DHEA (Figure 1F). The high dose DHEA (1%) treatment normalized the hypoxic increases in MPAP and RV/LV+S.
Figure 1.
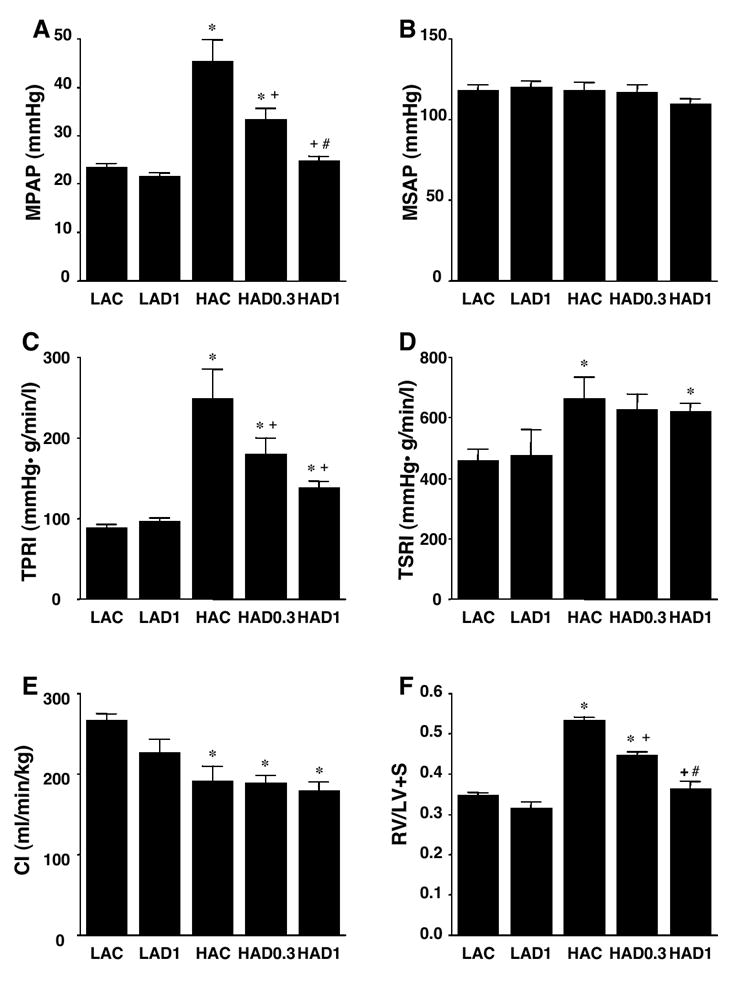
Effects of chronic DHEA treatment on hemodynamics (A: mean pulmonary artery pressure: MPAP, B: mean systemic artery pressure: MSAP, C: total pulmonary resistance index: TPRI, D: total systemic resistance index: TSRI, E: cardiac index: CI) and ratio of right ventricle to left ventricle + septum weights (RV/LV+S: F). LAC, low altitude untreated control (n=5); LAD1, low altitude 1%DHEA treated (n=5); HAC, high altitude untreated control (n=5); HAD0.3 and HAD1, high altitude 0.3 (n=4) and 1% DHEA treated (n=9) groups. Values are means ± SE; * P < 0.05 vs. LAC, + P < 0.05 vs. HAC, # P < 0.05 vs. HAD0.3.
TABLE 3.
Cardiac output, calculated stroke volume, and heart rate
| GROUP | n | CO (ml/min) | SV (ml) | SV/BW (ml/kg) | HR (beats/min) |
|---|---|---|---|---|---|
| LAC | 5 | 101 ± 4 | 0.24 ± 0.02 | 0.64 ± 0.04 | 404 ± 11 |
| LAD | 5 | 62 ± 3* | 0.15 ± 0.01* | 0.55 ± 0.03 | 430 ± 21 |
| HAC | 5 | 67 ± 6* | 0.17 ± 0.02* | 0.48 ± 0.06* | 402 ± 23 |
| HAD 0.3 | 4 | 55 ± 3*† | 0.13 ± 0.01*† | 0.44 ± 0.03* | 414 ± 10 |
| HAD 1 | 9 | 45 ± 3*†# | 0.11 ± 0.01*† | 0.44 ± 0.03* | 422 ± 25 |
Values are means ± SE. CO, cardiac output; SV, stroke volume; BW, body weight; HR, heart rate; LAC, low altitude untreated control; LAD, low altitude 1% DHEA treated; HAC, high altitude untreated control; HAD 0.3 and HAD 1, high altitude 0.3 and 1% DHEA treated groups, respectively.
P<0.05 vs. LAC;
P<0.05 vs. HAC;
P<0.05 vs. HAD 0.3.
Chronic hypoxia increased the percent medial wall thickness of small pulmonary arteries, which was dose-dependently and markedly inhibited by 3-wk treatment with DHEA (Figure 2).
Figure 2.
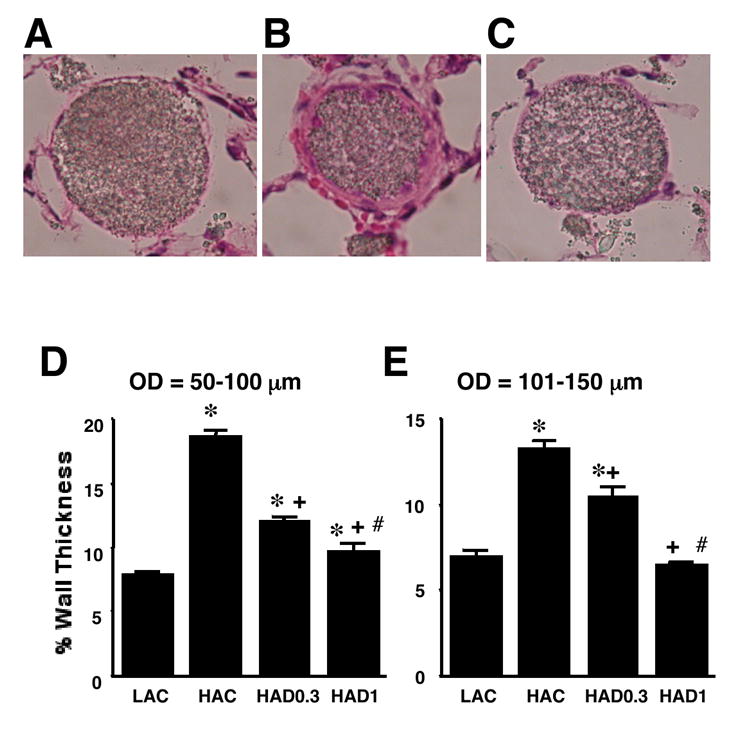
Effects of chronic DHEA treatment on medial wall thickness of pulmonary arteries accompanying terminal and respiratory bronchioles in rats exposed to low altitude (LA) or high altitude (HA) with low dose (D0.3) and high dose (D1) and without DHEA (C) treatment. Upper panels: Representative light micrographs of pulmonary arteries (50-100 μm in diameter) from LAC (A), HAC (B) and HAD1 (C). Lower panels: % wall thickness of pulmonary arteries of 50-100 μm (D) and 101-150 μm (E) in outer diameter. Medial thickness at two places of each artery was measured and medial wall thickness was expressed as medial thickness/external diameter × 100 (%). Values are means ± SE; * P < 0.05 vs. LAC, + P < 0.05 vs. HAC, # P < 0.05 vs. HAD0.3.
To determine if DHEA would reverse, as well as prevent, hypoxic pulmonary hypertension, we performed a reversal study. Three weeks of DHEA (1%) treatment begun after pulmonary hypertension had been established by a preceding 3 wks of exposure to hypoxia effectively reversed the increases in MPAP, RV/LV+S ratio, and %WT, but did not alter MSAP or CI as compared to the untreated (0% DHEA) hypoxic (6 wks) control group (Figure 3). DHEA treatment also caused body weight loss in this group similar to that observed in the prevention study group (data not shown).
Figure 3.
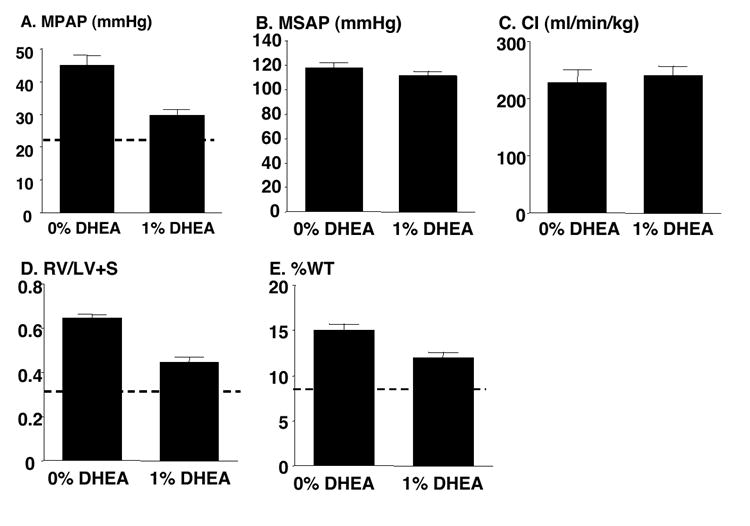
Chronic DHEA (1%) treatment reverses chronic hypoxia-induced increases in mean pulmonary artery pressure (MPAP: A), ratio of right ventricle to left ventricle + septum weights (RV/LV+S: D), % pulmonary artery wall thickness (%WT: E, 50-100 μm in diameter) without effects on mean systemic pressure (MSAP: B) and cardiac index (CI: C). Dashed lines indicate normal values of low altitude untreated rats from the prevention study. Values are means ± SE of n=7 for 0% DHEA-treated (0% DHEA) and 6 for 1% DHEA-treated (1% DHEA) groups, except for CI which was measured in 5 (0% DHEA) and 3 rats (1% DHEA); * P < 0.05 vs. 0% DHEA.
To test if the DHEA-induced inhibition of hypoxic pulmonary hypertension was associated with enhancement of pulmonary artery endothelial function, we evaluated effects of chronic DHEA (1%) treatment on the NO-cGMP pathway activity pharmacologically using isolated pulmonary artery rings (Figure 4). In both extralobar and intralobar pulmonary artery rings from LA rats, LAD rings showed enhanced relaxant responses to both ACH (endothelium-dependent) and SNP (endothelium-independent), but not to 8-Br-cGMP (sGC-independent). Consistent with previous reports, chronic hypoxia impaired pulmonary artery responses to ACH and SNP in extralobar pulmonary artery rings (17,25), but did not impair those responses in intralobar arteries (25). Chronic DHEA treatment improved the impaired responses of extralobar pulmonary arteries to near normal levels, and enhanced the responses of intralobar pulmonary arteries. DHEA treatment did not augment the response of HA pulmonary artery rings to 8-Br-cGMP.
Figure 4.
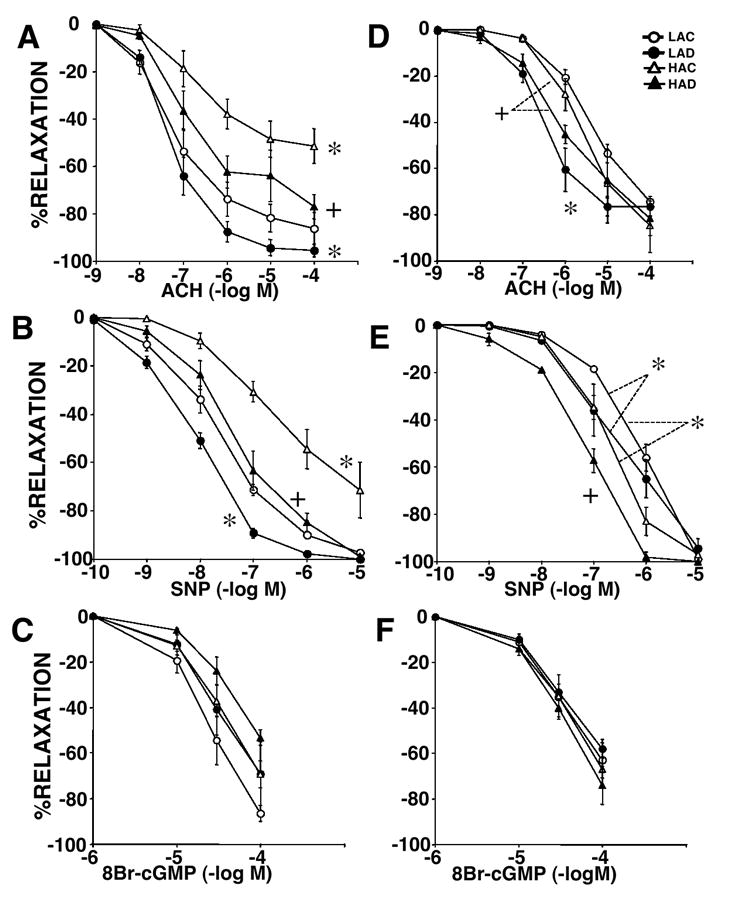
Concentration relaxation curves to acetylcholine (ACH: A and D), sodium nitroprusside (SNP: B and E), and 8-Bromo-cGMP (8-Br-cGMP: C and F) in pulmonary artery rings from rats exposed to low altitude with (LAD: closed circle) or without 1% DHEA treatment (LAD: open circle) and high altitude with (HAD: closed triangle) or without DHEA treatment (HAC: open triangle). Panels A – C show data of extralober large pulmonary artery and D – F show those of intrapulmonary small arteries. Values are means ±SE of n=4~5/group; * P < 0.05 vs. LAC, + P < 0.05 vs. HAC.
Since the above results unexpectedly indicated that DHEA treatment augmented pulmonary artery responsiveness to the NO-mediated vasodilators ACH and SNP (which depend on sGC), but not to 8-Br-cGMP (which acts downstream of sGC), we investigated effects of DHEA on sGC expression and activity. In pulmonary artery tissues from LA rats, DHEA (1%) increased sGC β1 and tended to increase sGC α protein expression, but had no effect on eNOS level (Figure 5). Chronic hypoxia increased eNOS, did not alter sGCα , and decreased sGC β1 expression. DHEA treatment of hypoxic rats markedly increased both sGCα and β1 protein expression, and eliminated the hypoxia-induced upregulation of eNOS protein in two out of three rats. DHEA treatment markedly and slightly increased sGC activity (SNP-stimulated cGMP production) in pulmonary arteries from LA and HA rats, respectively (Figure 6).
Figure 5.
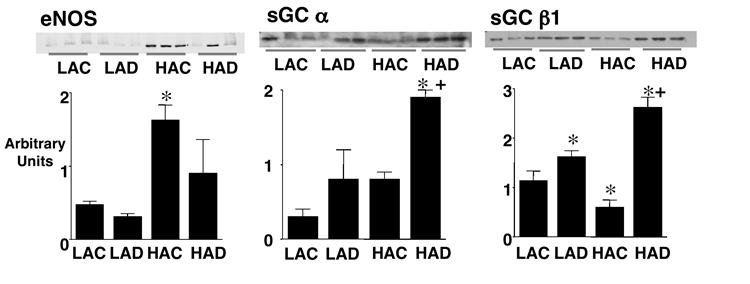
Protein levels of endothelial nitric oxide synthase (eNOS), soluble guanylate cyclase (sGC) α and β1 in main pulmonary arteries from low altitude rats without (LAC) and with 1% DHEA treatment (LAD), and high altitude rats without (HAC) and with 1% DHEA treatment (HAD). Upper pulmonary arterynels show immunoblots and lower pulmonary arterynels show densitometric assessment. Values are means ± SE of n=3; * P < 0.05 vs. LAC, + P < 0.05 vs. HAC.
Figure 6.
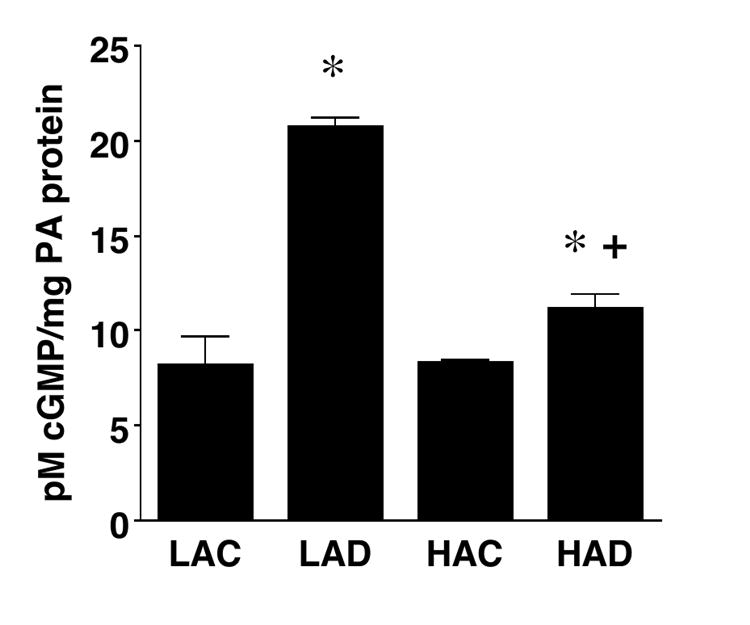
Sodium nitroprusside-stimulated soluble guanylate cyclase activity in main pulmonary arteries from low altitude rats without (LAC) and with 1% DHEA treatment (LAD), and high altitude rats without (HAC) and with 1% DHEA treatment (HAD). Values are means ± SE of n=3; * P < 0.05 vs. LAC, + P < 0.05 vs. HAC.
Lung tissue ET-1 peptide levels were increased after exposure to chronic hypoxia, and DHEA treatment had no effect on the levels in either LA or HA rats (Table 4).
TABLE 4.
Lung tissue ET-1 levels
| GROUP | n | ET-1 (μg /50mg tissue) |
|---|---|---|
| LAC | 5 | 4.8 ±1.0 |
| LAD | 4 | 4.7 ± 2.9 |
| HAC | 6 | 14.4 ± 1.9 * |
| HAD 0.3 | 3 | 11.6 ± 5.3 * |
| HAD 1 | 5 | 19.5 ± 4.9 * |
Values are means ± SE. LAC, low altitude untreated control; LAD, low altitude 1%DHEA treated; HAC, high altitude untreated control; HAD 0.3 and HAD 1, high altitude 0.3 and 1% DHEA treated groups, respectively.
P<0.05 vs. LAC.
DISCUSSION
This study showed that dietary treatment of rats exposed to chronic hypobaric hypoxia with 0.3 and 1% DHEA caused a dose-dependent inhibition of the development of pulmonary hypertension without causing systemic hypotension. Long-term treatment with 1% DHEA essentially normalized the chronic hypoxia-induced pulmonary hypertension RV hypertrophy, and pulmonary artery medial thickening with no effects on SAP. These impressive effects of DHEA treatment were associated with improved vasodilator responsiveness to ACH and SNP and with upregulation of sGC protein expression and activity in isolated pulmonary arteries. Chronic DHEA treatment also effectively reversed the severity of established hypoxic pulmonary hypertension. These results, which confirm and extend earlier reports (9,10), suggest that dietary DHEA is highly effective in preventing and reversing hypoxic pulmonary hypertension, which is due at least partly to enhancing the pulmonary artery cGMP signaling activity through increasing sGC expression and activity.
Numerous studies support an important role for impaired endothelium-derived NO activity in the pathogenesis of hypoxic pulmonary hypertension (15,16). The beneficial effects of NO are mediated largely by production of cGMP via activation of sGC and subsequent stimulation of cGMP-dependent kinase (cGK). Although loss of NO itself may not cause severe pulmonary hypertension, dysfunction of this pathway facilitates sustained pulmonary vasoconstriction and pulmonary artery remodeling associated with development and progression of severe pulmonary hypertension (16,26). Conversely, enhancement of this pathway is protective against pulmonary hypertension. DHEA has been shown to increase the production of NO and cGMP and to improve systemic vascular endothelial function either directly (11,14) or through conversion to estrogen (12). We found in this study that chronic treatment of LA (normal) rats with DHEA augmented relaxation responses to ACH and SNP but not to 8-Br-cGMP in pulmonary artery rings. Exposure to chronic hypoxia impaired the responses to ACH and SNP in extralobar but not intralobar pulmonary artery rings. Concomitant DHEA treatment nearly normalized and enhanced these relaxation responses in extralobar and intralobar pulmonary artery rings, respectively. These findings suggested the possibility that DHEA augmented the pulmonary artery smooth muscle cell responsiveness to NO by altering expression and/or activity of a certain factor or factors downstream of NO production and upstream of cGK, rather than that DHEA enhanced pulmonary artery endothelial function. To further investigate this possibility, we measured eNOS and sGC protein expression, and found that sGC α and β1, but not eNOS, were upregulated by DHEA in the pulmonary artery tissue from LA (where there was only a trend for increased sGC α) and HA rats. We also found that the stimulated enzyme activity was enhanced by DHEA treatment in pulmonary artery tissues from both LA and HA rats. Although our results showed that both sGC expression and activity were increased in the pulmonary artery tissue from both LAD and HAD rats, there was a discrepancy between the levels of expression and activity in LAD vs. HAD rats; i.e., whereas sGC protein expression was higher in pulmonary artery from HAD rats (Fig. 5), SNP-stimulated activity was greater in pulmonary artery from LAD rats (Fig. 6). We have no clear explanation for this discrepancy. Regardless, taken together, these results indicated that chronic DHEA treatment enhanced pulmonary artery responsiveness to NO by upregulating sGC protein expression and activity.
Our findings that DHEA treatment had no effect on eNOS expression in LA pulmonary artery and reduced the increased expression in HA pulmonary arteries differ from those of previous studies in systemic vessels in which DHEA has increased eNOS activity/expression (11,12). It is not clear whether this difference simply reflects the difference between the systemic and pulmonary circulations. However, the downregulation of eNOS expression in DHEA-treated HA pulmonary artery might have been due to the normalized pulmonary arterial pressure, because there is evidence that hemodynamic factors such as increased shear force, rather than hypoxia per se, may cause the upregulation of pulmonary artery eNOS in this model (27,29), although controversy exists (19).
There are essentially two ways to augment the pulmonary vascular NO-cGMP-cGK pathway activity. One is to increase NO itself, such as by inhaled NO (29), NO synthase gene transfer (30), or L-arginine supplementation (31). The other is to increase cGMP levels by controlling downstream modulator/effector activity, such as by inhibiting phosphodiesterase type 5 (PDE5) or activating sGC. Because increased NO may have adverse effects (as a free radical), and its effects are tightly regulated by downstream effector/modulator activity, inhibition of PDE5 and activation of sGC might be safer and more efficient than increasing NO itself. In fact, PDE5 inhibition by sildenafil appears promising for the clinical treatment of pulmonary hypertension (32), and recent studies indicate that pharmacological activation of sGC may also be an effective therapeutic intervention in pulmonary hypertension (33,34).
Several human and animal studies have shown that DHEA has cardiovascular protective effects (35,36). Although mechanisms responsible for the cardiovascular protective effects of DHEA are not fully understood, it seems that multiple actions are involved; these include enhancement of vascular endothelial function (11-14), inhibition of vascular smooth muscle cell proliferation (37), prevention of platelet aggregation (38), suppression of oxidative stress (39), and conversion of DHEA to estrogen (12). In addition, we and others have recently shown in vitro that DHEA induces acute pulmonary vasodilation and inhibits acute hypoxic pulmonary vasoconstriction due at least partly to opening of K+ channels (both Kv and KCa channels) through decreasing NADPH production and altering tissue redox status (7,40). All of these actions could reduce the severity of hypoxic pulmonary hypertension. In fact, in a recent report, Bonnet et al. demonstrated that DHEA prevented and reversed hypoxic pulmonary hypertension and restored the chronic hypoxia-induced impaired KCa channel function and expression via a redox-dependent pathway (10). It is thus speculated that DHEA’s marked protective effect against hypoxic pulmonary hypertension is multifunctional, including upregulation of sGC and modulation of K+ channel function/expression, both of which can potentially inhibit vasoconstriction and vascular remodeling in hypoxic pulmonary hypertension. We (41-44) and others (45,46) have recently found that RhoA/Rho kinase-mediated vasoconstriction is an important pathogenic component of of pulmonary hypertension in various animal models, and future studies should investigate if DHEA inhibits RhoA/Rho kinase signaling through increased activity of the pulmonary artery NO/sGC pathway (47,48) and/or other mechanisms, such as inhibition of 3-hydroxy-3-methylglutaryl-CoA reductase (49).
Some of the beneficial effects of DHEA could be exerted via its conversion to estradiol, which has a protective effect against the development of hypoxic pulmonary hypertension by suppressing erythropoietin expression and ET-1 production (50-52). Whereas plasma estradiol levels were elevated in the high dose DHEA-treated rats, no effects related to estradiol (i.e., decreased ET-1 production or suppressed polycythemia) were observed in our study. In addition, while there was no increase in plasma estradiol concentration in the low dose DHEA-treated rats, this dose still reduced the development of pulmonary hypertension. Thus, although this study did not completely rule out the possible involvement of increased conversion of DHEA to estradiol, it is unlikely that estradiol played a major role in preventing the development and causing the reversal of hypoxic pulmonary hypertension by DHEA in this study. Plasma testosterone levels were also elevated in HA rats treated with the high dose of DHEA. However, it is also unlikely that testosterone played a significant role, because male sex hormones have no protective effect against the development of hypoxic pulmonary hypertension (50,51).
DHEA treatment caused weight loss of rats in this study, which is consistent with previous reports (23,24). While some studies suggest that food restriction is responsible for DHEA-induced body weight loss (53), others indicate food consumption is not changed by DHEA, and an alteration in fat metabolism causes the loss of body weight (23). Although severe dietary restriction has been reported to protect against the development of monocrotaline-induced pulmonary hypertension (54), this mechanism was unlikely in our study since we did not find any difference in food consumption between the DHEA-treated and –untreated rat groups.
A major advantage for DHEA treatment is that this drug has already been approved by the US Food and Drug Administration as a food supplement and is used extensively in human studies without any major side effects (55). Several clinical trials have provided evidence that even pharmacological doses (up to 2,250 mg/day for 12 weeks) of DHEA have no serious adverse effects (55). However, because DHEA can be metabolized to both androgens and estrogens, there is a possibility that hormone-dependent cancer (such as breast cancer in women and prostate cancer in men) may be promoted in long-term DHEA treatments. Thus, future studies should evaluate the effectiveness of DHEA’s synthetic analogues, such as 16 alpha-fluoro-5-androsten-17-one, which do not have any sex steroid activity but retain the anti-proliferative and cancer preventive activity of DHEA (56).
There are limitations of this study. First, our data do not directly determine how much the observed upregulation of sGC expression and activity contributed to the protective effect of DHEA against hypoxic pulmonary hypertension. Second, since pulmonary blood flow and resistance were not directly measured, this study can not exclude the possibility that the decreased CO and SV in DHEA-treated HA rats (Table 3) may have contributed to the reduction in pulmonary artery pressure even though our data show that CI and SV/body weight were unchanged. Whether the decrease in CO was related directly to the decrease in body weight or to some other effect of the DHEA treatment on cardiac function is unknown and will require further study to address.
In summary, this study demonstrated that DHEA, a naturally occurring steroid hormone, dose-dependently and completely blocked the development of hypoxic pulmonary hypertension, which was accompanied by upregulation of sGC protein expression and activity and improved pulmonary artery vasodilator responsiveness to NO. It also showed that DHEA treatment effectively reversed established hypoxic pulmonary hypertension. Based on results of this and previous studies (9,10), we propose that DHEA may be effective in preventing and reversing hypoxic and possibly other forms of pulmonary hypertension via multiple important actions, including upregulation of sGC and improving K+ channel activity/expression.
Acknowledgments
The authors thank Kenneth G Morris and Sandi Walchak for their technical assistance.
This study was supported by grants from the National Institutes of Health (HL 14985 and HL 07171) and the American Heart Association (SDG 0335208N and Desert Mountain Affiliate).
Part of this study was presented at the 2003 International Conference of the American Thoracic Society and published in abstract form (Am J Respir Crit Care Med 2003;167:A824).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Barberà JA, Peinado VI, Santos S. Pulmonary hypertension in chronic obstructive pulmonary disease. Eur Respir J. 2003;21:892–905. doi: 10.1183/09031936.03.00115402. [DOI] [PubMed] [Google Scholar]
- 2.Jeffery TK, Wanstall JC. Pulmonary vascular remodeling: a target for therapeutic intervention in pulmonary hypertension. Pulmonary arteryrmacology & Therapeutics. 2001;92:1–20. doi: 10.1016/s0163-7258(01)00157-7. [DOI] [PubMed] [Google Scholar]
- 3.Shimoda LA, Sham JS, Sylvester JT. Altered pulmonary vasoreactivity in the chronically hypoxic lung. Physiol Res. 2000;49:549–60. [PubMed] [Google Scholar]
- 4.Voelkel NF, Cool CD. Pulmonary vascular involvement in chronic obstructive pulmonary disease. Eur Respir J. 2003;22(Suppl 46):28s–32s. doi: 10.1183/09031936.03.00000503. [DOI] [PubMed] [Google Scholar]
- 5.Williams JR. The effects of dehydroepiandrosterone on carcinogenesis, obesity, the immune system, and aging. Lipids. 2000;35:325–31. doi: 10.1007/s11745-000-0529-7. [DOI] [PubMed] [Google Scholar]
- 6.Barrett-Oconnor E, Goodman-Gruen The epidemiology of DHEAS and cardiovascular diseases. Ann NY Acad Sci. 1995;774:259–70. doi: 10.1111/j.1749-6632.1995.tb17386.x-i1. [DOI] [PubMed] [Google Scholar]
- 7.Farrukh IS, Peng W, orlinska U, Hoidal JR. Effect of dehydroepiandrosterone on hypoxic pulmonary vasoconstriction: a Ca2+ -activated K+ -channel opener. Am J Physiol Lung Cell Mol Physiolol. 1998;274:L186–95. doi: 10.1152/ajplung.1998.274.2.L186. [DOI] [PubMed] [Google Scholar]
- 8.Gupte SA, Li K-X, Okada T, Sato K, Oka M. Inhibitors of pentose phosphate pulmonary arterythway cause vasodilation: Involvement of voltage-gated potassium channels. J Pharmacol Exp Ther. 2002;301:299–305. doi: 10.1124/jpet.301.1.299. [DOI] [PubMed] [Google Scholar]
- 9.Hampl V, Bíbová J, Povysilová, Herget J. Dehydroepiandrosterone sulfate reduces chronic hypoxic pulmonary hypertension in rats. Eur Respir J. 2003;21:862–5. doi: 10.1183/09031936.03.00084503. [DOI] [PubMed] [Google Scholar]
- 10.Bonnet S, Dumas-de-La-Roque E, Bégueret H, Marthan R, Fayon M, Santos PD, et al. Dehydroepiandrosterone (DHEA) prevents and reverses chronic hypoxic pulmonary hypertension. Proc Natl Acad Sci. 2003;100:9488–93. doi: 10.1073/pnas.1633724100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Simoncini T, Mannella P, Fornari L, Varone G, Caruso A, Genazzani AR. Dehydroepiandrosterone modulates endothelial nitric oxide synthesis via direct genomic and nongenomic mechanisms. Endocrinology. 2003;144:3449–55. doi: 10.1210/en.2003-0044. [DOI] [PubMed] [Google Scholar]
- 12.Hayashi T, Esaki T, Muto E, Kano H, Asai Y, Thakur NK, et al. Dehydroepiandrosterone retards atherosclerosis formation through its conversion to estrogen; The possible role of nitric oxide. Arterioscler Thromb Vasc Biol. 2000;20:782–92. doi: 10.1161/01.atv.20.3.782. [DOI] [PubMed] [Google Scholar]
- 13.Kawano H, Yasue H, Kitagawa A, Hirai N, Yoshida T, Soejima H, et al. Dehydroepiandrosterone supplementation improves endothelial function and insulin sensitivity in men. J Clin Endocrinol Metab. 2003;88:3190–5. doi: 10.1210/jc.2002-021603. [DOI] [PubMed] [Google Scholar]
- 14.Liu D, Dillon JS. Dehydroepiandrosterone activates endothelial cell nitric-oxide synthase by specific plasma membrane receptor coupled to Gαi2,3. J Biol Chem. 2002;277:21379–88. doi: 10.1074/jbc.M200491200. [DOI] [PubMed] [Google Scholar]
- 15.Le Cras TD, McMurtry IF. Nitric oxide production in the hypoxic lung. Am J Physiol Lung Cell Mol Physiol. 2001;280:L575–82. doi: 10.1152/ajplung.2001.280.4.L575. [DOI] [PubMed] [Google Scholar]
- 16.Fagan KA, McMurtry IF, Rodman DM. Nitric oxide synthase in pulmonary hypertension: lessons from knockout mice. Physiol Res. 2000;49:539–48. [PubMed] [Google Scholar]
- 17.Oka M, Hasunuma K, Webb SA, Stelzner TJ, Rodman DM, McMurtry IF. EDRF suppresses an identified vasoconstrictor mechanism in hypertensive rat lungs. Am J Physiol Lung Cell Mol Physiolol. 1993;264:L587–97. doi: 10.1152/ajplung.1993.264.6.L587. [DOI] [PubMed] [Google Scholar]
- 18.Hanasato N, Oka M, Muramatsu M, Nishino M, Adachi H, Fukuchi Y. E-4010, a selective phosphodiesterase 5 inhibitor, attenuates hypoxic pulmonary hypertension in rats. Am J Physiol Lung Cell Mol Physiol. 1999;277:L225–32. doi: 10.1152/ajplung.1999.277.2.L225. [DOI] [PubMed] [Google Scholar]
- 19.Le Cras TD, Tyler RC, Horan MP, Morris KG, Tuder RM, McMurtry IF, et al. Effects of chronic hypoxia and altered hemodynamics on endothelial nitric oxide synthase expression in the adult rat lung. J Clin Invest. 1998;101:795–801. doi: 10.1172/JCI786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hocher B, Schwarz A, Fagan KA, Thöne-Reineke C, El-Hag K, Kusserow H, et al. Pulmonary Fibrosis and Chronic Lung Inflammation in ET-1 Transgenic Mice. Am J Respir Cell Mol Biol. 2000;23:19–26. doi: 10.1165/ajrcmb.23.1.4030. [DOI] [PubMed] [Google Scholar]
- 21.Mittal KC. Determination of adenylate cyclase and guanylate cyclase activities in cells of the immune system. Methods Enzymol. 1986;132:422–8. doi: 10.1016/s0076-6879(86)32027-5. [DOI] [PubMed] [Google Scholar]
- 22.Hassoum PM, Filippov G, Fogel M, Donaldson C, Kayyali US, Shimoda LA, et al. Hypoxia decreases expression of soluble guanylate cyclase in cultured rat pulmonary artery smooth muscle cells. Am J Respir Cell Mol Biol. 2004;30:908–13. doi: 10.1165/rcmb.2003-0287OC. [DOI] [PubMed] [Google Scholar]
- 23.Hansen PULMONARYARTERY, han DH, Nolte LA, Chen M, Holloszy JO. DHEA protects against visceral obesity and muscle insulin resistance in rats fed a high-fat diet. Am J Physiol Regul Integr Comp Physiol. 1997;273:R1704–8. doi: 10.1152/ajpregu.1997.273.5.R1704. [DOI] [PubMed] [Google Scholar]
- 24.Yen TT, Allan JA, Peason DV, Acton JM. Control of obesity in Avy/a mice by 5a-androstan-17-one. Lipids. 1977;12:409–13. doi: 10.1007/BF02533624. [DOI] [PubMed] [Google Scholar]
- 25.Oka M. Phosphodiesterase 5 inhibition restores impaired ACh relaxation in hypertensive conduit pulmonary arteries. Am J Physiol Lung Cell Mol Physiol. 2001;280:L432–5. doi: 10.1152/ajplung.2001.280.3.L432. [DOI] [PubMed] [Google Scholar]
- 26.Michelakins ED. The role of the NO axis and its therapeutic implications in pulmonary arterial hypertension. Heart Failure Rev. 2003;8:5–21. doi: 10.1023/a:1022150819223. [DOI] [PubMed] [Google Scholar]
- 27.Resta TC, Chicoine LG, Omdahl JL, Walker BR. Maintained upregulation of pulmonary eNOS gene and protein expression during recovery from chronic hypoxia. Am J Physiol Heart Circ Physiol. 1999;276:H699–708. doi: 10.1152/ajpheart.1999.276.2.H699. [DOI] [PubMed] [Google Scholar]
- 28.Blumberg FC, Wolf K, Arzt M, Lorenz C, Riegger GA, Pfeifer M. Effects of ET-A receptor blockade on eNOS gene expression in chronic hypoxic rat lungs. J Appl Physiol. 2003;94:446–52. doi: 10.1152/japplphysiol.00239.2002. [DOI] [PubMed] [Google Scholar]
- 29.Hasuda T, Satoh T, Shimouchi A, Sakamaki F, Kyotani S, Matsumoto T, et al. Improvement in exercise capacity with nitric oxide inhalation in patients with precapillary pulmonary hypertension. Circulation. 2000;101:2066–70. doi: 10.1161/01.cir.101.17.2066. [DOI] [PubMed] [Google Scholar]
- 30.Janssens SP, Bloch KD, Nong Z, Gerard RD, Zoldhelyi P, Collen D. Adenoviral-mediated transfer of the human endothelial nitric oxide synthase gene reduces acute hypoxic pulmonary vasoconstriction in rats. J Clin Invest. 1996;98:317–24. doi: 10.1172/JCI118795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mehta S, Stewart DJ, Langleben D, Levy RD. Short-term pulmonary vasodilation with L-arginine in pulmonary hypertension. Circulation. 1995;92:1539–45. doi: 10.1161/01.cir.92.6.1539. [DOI] [PubMed] [Google Scholar]
- 32.Michelakis ED, Tymchak W, Noga M, Webster L, Wu XC, Lien D, et al. Long-term treatment with oral sildenafil is safe and improves functional capulmonary arterycity and hemodynamics in pulmonary arterytients with pulmonary arterial hypertension. Circulation. 2003;108:2066–9. doi: 10.1161/01.CIR.0000099502.17776.C2. [DOI] [PubMed] [Google Scholar]
- 33.Evgenov OV, Ichinose F, Evgenov NV, Gnoth MJ, Falkowski GE, Chang Y, et al. Soluble guanylate cyclase activator reverses acute pulmonary hypertension and augments the pulmonary vasodilator response to inhaled nitric oxide in awake lambs. Circulation. 2004;110:2253–9. doi: 10.1161/01.CIR.0000144469.01521.8A. [DOI] [PubMed] [Google Scholar]
- 34.Deruelle P, Grover TR, Storme L, Abman SH. Effects Of Bay 41-2272, A Soluble Guanylate Cyclase Activator, On Pulmonary Vascular Reactivity In The Ovine Fetus. Am J Physiol Lung Cell Mol Physiol. 2005;288:L727–33. doi: 10.1152/ajplung.00409.2004. [DOI] [PubMed] [Google Scholar]
- 35.Barrett-Connor E, Goodman-Gruen D. The epidemiology of DHEAS and cardiovascular diseases. Ann NY Acad Sci. 1995;774:259–70. doi: 10.1111/j.1749-6632.1995.tb17386.x-i1. [DOI] [PubMed] [Google Scholar]
- 36.Porsova-Dutoit I, Sulcova J, Starka L. Do DHEA/DHEAS play a protective role in coronary heart disease? Physiol Res. 2000;49:S43–S56. [PubMed] [Google Scholar]
- 37.Williams MRI, Ling S, Dawood T, Hashimura K, Dai A, Li H, et al. Dehydroepiandrosterone inhibits human vascular smooth muscle cell proliferation independent of Ars and Ers. J Clin Endocrinol Metab. 2002;87:176–81. doi: 10.1210/jcem.87.1.8161. [DOI] [PubMed] [Google Scholar]
- 38.Jesse RL, Loesser K, Eich DM, Qian YZ, Hess ML, Nestler JE. Dehydroepiandrosterone inhibits human platelet aggregation in vitro and in vivo. Ann NY Acad Sci. 1995;774:281–90. doi: 10.1111/j.1749-6632.1995.tb17388.x-i1. [DOI] [PubMed] [Google Scholar]
- 39.Aragno M, Pulmonary arteryrola S, Brignardello E, Mauro A, Tamagno E, Manti R, et al. Dehydroepiandrosterone prevents oxidative injury induced by transient ischemia/reperfusion in the brain of diabetic rats. Diabetes. 2000;49:1924–31. doi: 10.2337/diabetes.49.11.1924. [DOI] [PubMed] [Google Scholar]
- 40.Peng W, Hoidal JR, Farrukh IS. Role of a novel Kca opener in regulating K+ channels of hypoxic human pulmonary vascular cells. Am J Respir Cell Mol Biol. 1999;20:737–45. doi: 10.1165/ajrcmb.20.4.3390. [DOI] [PubMed] [Google Scholar]
- 41.Nagaoka T, Morio Y, Casanova N, Bauer N, Gebb S, McMurtry I, et al. Rho/Rho kinase signaling mediates basal pulmonary vascular tone in chronically hypoxic rats. Am J Physiol Lung Cell Mol Physiol. 2004;287:L665–72. doi: 10.1152/ajplung.00050.2003. [DOI] [PubMed] [Google Scholar]
- 42.Nagaoka T, Fagan KA, Gebb SA, Morris KG, Suzuki T, Shimokawa H, et al. Inhaled Rho kinase inhibitors are potent and selective vasodilators in rat pulmonary hypertension. Am J Respir Crit Care Med. 2005;171:494–9. doi: 10.1164/rccm.200405-637OC. [DOI] [PubMed] [Google Scholar]
- 43.Fagan KA, Oka M, Bauer NR, Gebb SA, Ivy DD, Morris KG, et al. Attenuation of acute hypoxic pulmonary vasoconstriction and hypoxic pulmonary hypertension in mice by inhibition of Rho-kinase. Am J Physiol Lung Cell Mol Physiol. 2004;287:L656–64. doi: 10.1152/ajplung.00090.2003. [DOI] [PubMed] [Google Scholar]
- 44.Nagaoka T, Gebb SA, Karoor V, Homma N, Morris KG, McMurtry IF, et al. Involvement of RhoA/Rho kinase signaling in pulmonary hypertension of the fawn-hooded rat. J Appl Physiol. 2006;100:996–1002. doi: 10.1152/japplphysiol.01028.2005. [DOI] [PubMed] [Google Scholar]
- 45.Abe K, Shimokawa H, Morikawa K, Uwatoku T, Oi K, Matsumoto Y, et al. Long-term treatment with a rho-kinase inhibitor improves monocrotaline-induced fatal pulmonary hypertension in rats. Circ Res. 2004;94:385–93. doi: 10.1161/01.RES.0000111804.34509.94. [DOI] [PubMed] [Google Scholar]
- 46.Hyvelin JM, Howell K, Nichol A, Costello CM, Preston RJ, McLoughlin P. Inhibition of Rho-kinase attenuates hypoxia-induced angiogenesis in the pulmonary circulation. Circ Res. 2005;97:185–91. doi: 10.1161/01.RES.0000174287.17953.83. [DOI] [PubMed] [Google Scholar]
- 47.Jernigan NL, Walker BR, Resta TC. Chronic hypoxia augments protein kinase G-mediated Ca2+ desensitization in pulmonary vascular smooth muscle through inhibition of RhoA/Rho kinase signaling. Am J Physiol Lung Cell Mol Physiol. 2004;287:L1220–9. doi: 10.1152/ajplung.00196.2004. [DOI] [PubMed] [Google Scholar]
- 48.Guilluy C, Sauzeau V, Rolli-Derkinderen M, Guerin P, Sagan C, Pacaud P, et al. Inhibition of RhoA/Rho kinase pulmonary arterythway is involved in the beneficial effect of sildenafil on pulmonary hypertension. Br J Pharmacol. 2005;146:1010–8. doi: 10.1038/sj.bjp.0706408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pulmonary arteryscale RM, Simile MM, De Miglio MR, Nufris A, Seddaiu MA, Muroni MR, Danni O, Rao KN, Feo F. Inhibition of 3-hydroxy-3-methylglutaryl-CoA reductase activity and gene expression by dehydroepiandrosterone in preneoplastic liver nodules. Carcinogenesis. 1995;16:1537–42. doi: 10.1093/carcin/16.7.1537. [DOI] [PubMed] [Google Scholar]
- 50.Ou LC, Sardella GL, Leiter JC, Brinck-Johnson T, Smith RP. Role of sex hormones in development of chronic mountain sickness in rats. J Appl Physiol. 1994;77:427–33. doi: 10.1152/jappl.1994.77.1.427. [DOI] [PubMed] [Google Scholar]
- 51.Moore LG, McMurtry IF, Reeves JT. Effects of sex hormones on cardiovascular and hematologic responses to chronic hypoxia in rats. Proc Soc Exp Biol Med. 1978;158:658–62. doi: 10.3181/00379727-158-40268. [DOI] [PubMed] [Google Scholar]
- 52.Earley S, Resta TC. Estradiol attenuates hypoxia-induced pulmonary endothelin-1 gene expression. Am J Physiol Lung Cell Mol Physiolol. 2002;283:L86–93. doi: 10.1152/ajplung.00476.2001. [DOI] [PubMed] [Google Scholar]
- 53.Catalina F, Milewich L, Kumar V, Bennet M. Dietary dehydroepiandrosterone inhibits bone marrow and leukemia cell transplants: role of food restriction. Exp Biol Med (Maywood) 2003;228:1303–20. doi: 10.1177/153537020322801109. [DOI] [PubMed] [Google Scholar]
- 54.Hacker AD. Dietary restriction, polyamines and monocrotaline-induced pulmonary hypertension. Biochem Pharmacol. 1993;45:2475–81. doi: 10.1016/0006-2952(93)90229-p. [DOI] [PubMed] [Google Scholar]
- 55.Tummala S, Svec F. Correlation between the administration dose of DHEA and serum levels of DHEA and DHEA-S in human volunteers: analysis of published data. Clin Biochem. 1999;32:355–61. doi: 10.1016/s0009-9120(99)00021-1. [DOI] [PubMed] [Google Scholar]
- 56.Schwartz AG, Pashko LL. Cancer prevention with dehydroepiandrosterone and non-androgenic structural analogs. J Cell Biochem Suppl. 1995;22:210–7. doi: 10.1002/jcb.240590826. [DOI] [PubMed] [Google Scholar]