

Abstract
Modified vaccinia virus Ankura (MVA), a highly attenuated strain of vaccinia virus (VV) that is unable to replicate in most mammalian cells, was evaluated as an expression vector for a model tumor associated antigen (TAA) and as a potential anti-cancer vaccine. We employed an experimental murine model in which an adenocarcinoma tumor line, CT26.CL25, was stably transfected with a model TAA, β-galactosidase (β-gal). Mice injected intrumuscularly with a recombinant MVA (rMVA) expressing β-gal (MVA-LZ), were protected from a lethal intravenous (i. v.) challenge with CT26.CL25. In addition, splenocytes from mice primed with MVA-LZ were therapeutically effective upon adoptive transfer to mice bearing pulmonary metastases of the CT26.CL25 tumor established 3 days earlier. Most importantly, i.v. inoculation with MVA-LZ resulted in significantly prolonged survival of mice bearing three day old pulmonary metastases. This prolonged survival compared favorably to mice treated with a replication competent recombinunt VV expressing β-gal. These findings indicate that rMVA is an efficacious alternative to the more commonly used replication competent VVfor the development of new recombinant anti-cancer vaccines.
Keywords: modified vaccinia virus Ankara (MVA), vaccinia virus, cancer immunotherapy
The identification and cloning of several cellular and viral tumor associated antigens (TAA) that are specifically recognized by T lymphocytes has made it possible to design recombinant and synthetic anti-cancer vaccines1-3. A large number of viral and nonviral vectors could be genetically engineered to express TAA. A suitable vector would be nononcogenic, have a low probability of causing disseminated infection in a recipient individual, have no capacity to cause contagious infection in a population of individuals and stably express the TAA, in that expression of heterologous protein is not lost or mutated upon serial passage of the vector. Other qualities that would make the vector attractive for use in clinical trials include ease of expansion and purification of the recombinant to sufficient quantities for testing and use, and a pre-established record of safety and efficacy in the clinic. Most importantly, an effective anti-cancer vaccine must be able to elicit the kind of powerful immune responses against heterologously expressed TAA that could protect against and contribute to the elimination of an established cancer.
One vector which has already become a central component of a number of experimental anti-cancer vaccines is vaccinia virus (VV). A member of the poxvirus family, VV was used successfully in the worldwide eradication of smallpox4. It has more recently been used as a versatile eukaryotic expression vecto5,6. In mouse models, recombinant VV (rVV) have also been used to induce anti-tumor responses to human tumor associated differentiation antigens, CEA7 and p978, mutated forms of p539 and viral TAAs; HPV16 E6/E710 and polyoma virus LT or MT antigens11. It is presently being used in human clinical trials as an anti-cancer vaccine by our group and others.
A highly attenuated strain of VV, designated modified vaccinia virus Ankara (MVA), was derived as a safe smallpox vaccine12. There were no serious complications in over 120000 recipients, many of them young children or aged individuals. MVA became extensively host range restricted during over 570 passages in primary chick embryo fibroblast cells13. More recent studies using MVA have shown that infected mammalian cells efficiently produce products of both early and late viral genes, as the block in viral replication occurs during virion assembly14. The ability to produce large quantities of recombinant material under the control of late promoters, in nonpermissive mammalian cells, distinguishes MVA from other host range-restricted poxvirus vectors14. MVA has significant advantages when compared with replication competent VV in that it could be used in immunocompromised patients, such as those with hematologic malignancies, human immunodeficiency virus (HIV) or those undergoing treatment with chemotherapy. Furthermore, the lack of assembly of infectious MVA in mammalian cells makes the chance of spread to nonvaccinated individuals or to the general environment minimal. Nevertheless, like the replication-competent VV strain western reserve (WR), the large double-stranded DNA genome is likely to accommodate over 25 kb of foreign DNA15.
A recombinant MVA (rMVA) expressing influenza virus structural proteins was recently shown to induce protective immunity to lethal influenza virus challenge in mice comparable to that obtained with a replication competent strain of VV16,17. Although both antibody and T-cell responses were demonstrated, protection is primarily due to neutralizing antibody in this system. In addition, immunization of macaques with rMVA expressing SIV env-gag-pol, provided better protection than a similar replication competent rVV against an SIV challenge18. Following challenge, the animals mounted a rapid neutralizing antibody response which probably contributed to protection.
To explore the potential uses of MVA as a vaccine in the treatment of established cancer, in which a cytotoxic T cell (CTL) response is most important, we employed a tumor system using β-galactosidase (β-gal) as a model TAA19,20. β-gal is a better model TAA for “nonself” antigens consisting of mutated21 or frameshift22 antigens than it is for the nonmutated “self” proteins that comprise many of the recently discovered melanoma associated antigens. The β-gal model will be particularly instructive for the design of recombinant vaccines for the treatment of virally induced cancer, for example cervical carcinomas caused by human papilloma virus23, hepatocellular carcinomas associated with the hepatitis B virus24, Burkitt’s lymphoma and nasopharyngea1 carcinomas correlated with Epstein–Barr Virus25, and adult T cell leukemias thought to be caused by human T cell lymphotropic virus (HTLV)26. The β-gal model also has the advantage of being an enzyme, which aids in its detection. In addition, a variety of vectors containing 1acZ (lacZ, Escherickia coli 1acZ gene) have previously been constructed and are readily available for evaluation in this tumor therapy system.
In this model an undifferentiated murine colon adenocarcinoma cell line, CT26.CL25, that stably expresses β-gal is used. The growth rate in vitro and in vivo and the lethality of the tumor are identical to that of the parental CT26.WT clone from which the β-gal transfectant was made. Here we show that MVA expressing β-gal can protect against and actively treat established pulmonary metastases expressing the same antigen.
MATERIALS AND METHODS
Tumor cell line
CT26 is an N-nitroso-N-methylurethrane induced undifferentiated colon carcinoma of BALB/c (H-2d) origin27. The generation of the β-gal expressing CT26 cell line has been described elsewhere19. Briefly, a clone of CT26 (CT26.WT) was stably transfected using a retrovirus vector containing the lacZ gene under control of the retrovirus LTR. Subclones were evaluated for β-gal expression and their ability to be lysed in a 51Cr release assay by CTL specific for β-gal. The subclone CT26.CL25 was selected for use in all studies due to its stable expression of both β-gal and the MHC class I molecule H-2 Ld. CT26.WT and CT26.CL25 were maintained in RPMI 1640, 10% heat inactivated fetal calf serum (FCS) (Biofluids, Rockville, MD), 0.03% L-glutamine, 100 μg ml−1 streptomycin, 100 μg ml−1 penicillin, and 50 μg ml−1 gentamicin sulfate (NIH Media Center, MD). CT26.CL25 was maintained in medium containing 400 μg ml−1 G418 (GIBCO BRL laboratories, Gaithersburg, MD).
Recombinant poxviruses
Construction of the three recombinant poxviruses, used in this study, employed the principles of homologous DNA recombination as previously described5,6. Recombinant MVA expressing β-gal14 was constructed such that the lacZ gene, under transcriptional control of the VV strong late 11 k promoter, is inserted within the deletion region III of MVA28. The replication competent rVV, VJS6 (J. Sisler and B. Moss. NIH, MD, unpublished data) was constructed by insertion of the lacZ gene, under control of the VV early late 7.5 k promoter29 into the thymidine kinase gene of wild type WR. In addition this virus expresses the HPV E6 gene. Recombinant FPV.bg40k (FPV, fowlpox virus) was constructed by Therion Biologics Inc. (MA) such that the lacZ gene is inserted into the BamHIJ region of the FPV (strain POXVAC-TC, Schering Corp.) genome and under transcriptional control of the vaccinia early late H530 promoter. Both MVA and FPV based viruses were propagated on primary chick embryo fibroblast (CEF) cells whereas WR based viruses were grown on Hela cells. High titered viral stocks were purified by ultra centrifugation through a 36% sucrose cushion31. CEF and Hela cells were grown in MEM (NIH Media Center) supplemented with 10% FCS (Gibco BRL, Gaithersburg), 0.03% glutamine, 100 μg ml−1 streptomycin, and 100 μg ml−1 of penicillin (NIH Media Center).
In vivo protection
Female BALB/c (H-2d) mice, 8–12 weeks old, obtained from the Frederick Cancer Research Center (Frederick, MD) were used for all animal experiments. Mice were inoculated intravenously (i.v.) or intramuscularly (i.m.) with recombinant virus. Approximately 21 days postinoculation, mice were challenged i.v. with a lethal dose (5 × 105) of tumor cells. On day 12 post-tumor challenge, mouse lungs were removed and stained by inflating them with a solution of India ink. Before lung removal, mice were randomized such that metastatic lung nodules were enumerated in a blinded manner.
In vivo active treatment
Mice were inoculated i.v. on day 0 with either 1 × 105 or 5 × 105 tumor cells. On day 3 or days 3 and 10, mice were inoculated i.v. with either 2 × 107 or 5 × 107 plaque forming units (p.f.u.) of virus. Mice were checked daily for mortality.
Adoptive immunotherapy
Splenectomies were performed on mice that were inoculated 21 days previously, i.v. or i.m., with recombinant virus. Single splenocyte cell suspensions were prepared and cells were incubated at a concentration of 4 × 106 ml−1 with 10 μg ml−1 of an MHC class I-restricted peptide with the sequence TPHPARIGL, corresponding to residues 876–884 of the full-length β-gal protein. Six days post-peptide stimulation, spleen cells were washed and inoculated i.v. (2 × 106 or 2 × 107) into mice that had been injected i.v. with 5 × 105 tumor cells 3 days previously. Nine days after adoptive transfer of splenocytes, lung metastases were enumerated as described previously.
Splenocyte cytokine profiles
Splenocyte suspensions from mice inoculated 21 days previously with recombinant virus were prepared as previously described. Cells were incubated at ca 4 × 106 ml−1 with 10 μg ml−1 of either an MHC I restricted peptide TPHPARIGL, derived from β-gal or a control peptide, corresponding to residues 35–43 from the PIA TAA and having the sequence LPYLGWLVF32. For 6 days, culture supernatant samples were taken daily and stored at −20°C. Supernatants were analyzed for cytokine activity using quantitative ELISA kits specific for interferon-α (IFN-γ), tumor necrosis factor-α (TNF-α), granulocyte-macrophage-colony stimulating factor (GM-CSF), interleukin-2 (IL-2), IL-4, (Endogen, MA), and IL-10 (Genzyme, MA).
X-gal staining of pulmonary tumors
Lungs from mice inoculated i.v. with tumor, as described above, were removed and fixed in a solution containing 2% formaldehyde (v/v), 0.2% gluteraldehyde (v/v) in phosphate-buffered saline (PBS) for 30 min. The lungs were then washed with PBS three times. After washing, lungs were incubated for ca 4 h at 37°C in a solution of X-gal (l mg ml−1 X-gal (Gold Biotechnology, MO) 5 mM potassium ferricyanide, 5 mM potassium ferracyanide, 2 mM MgCl in PBS). After staining in X-gal solution, the lungs were rinsed and stored in PBS containing 0.01% sodium azide.
Statistical analysis
Survival was analyzed with standard Kaplan–Mier survival curve33. p2 values are presented.
RESULTS
Intramuscular inoculation of MVA-LZ induces protective immunity to tumor challenge
In the previously reported MVA influenza virus protection study the i.v. route of immunization was shown to be effective in inducing protective immunity16. To evaluate the efficiency of MVA as a vaccine vector for the induction of a protective immune response to β-gal, mice were inoculated with MVA-LZ and subsequently challenged with a lethal dose of CT26.CL25 (Figure 1). BALB/c mice inoculated i.v. with 5 × 105 cells of the adenocarcinoma line CT26.WT or the subclone CT26.CL25, stably transfected with β-gal, form over 500 macroscopic pulmonary metastases 12 days post-tumor inoculation, and result in animal death within 16–25 days18,19. MVA-LZ inoculated either i.v or i.m. induced effective protective immunity to CT26.CL25 with 14 out of 15 mice, displaying lungs with no metastases. The protection response was β-gal specific: the lungs of mice inoculated with nonrecombinant MVA, or mice bearing CT26.WT (which did not express β-gal) had 500 metastases.
Figure 1.
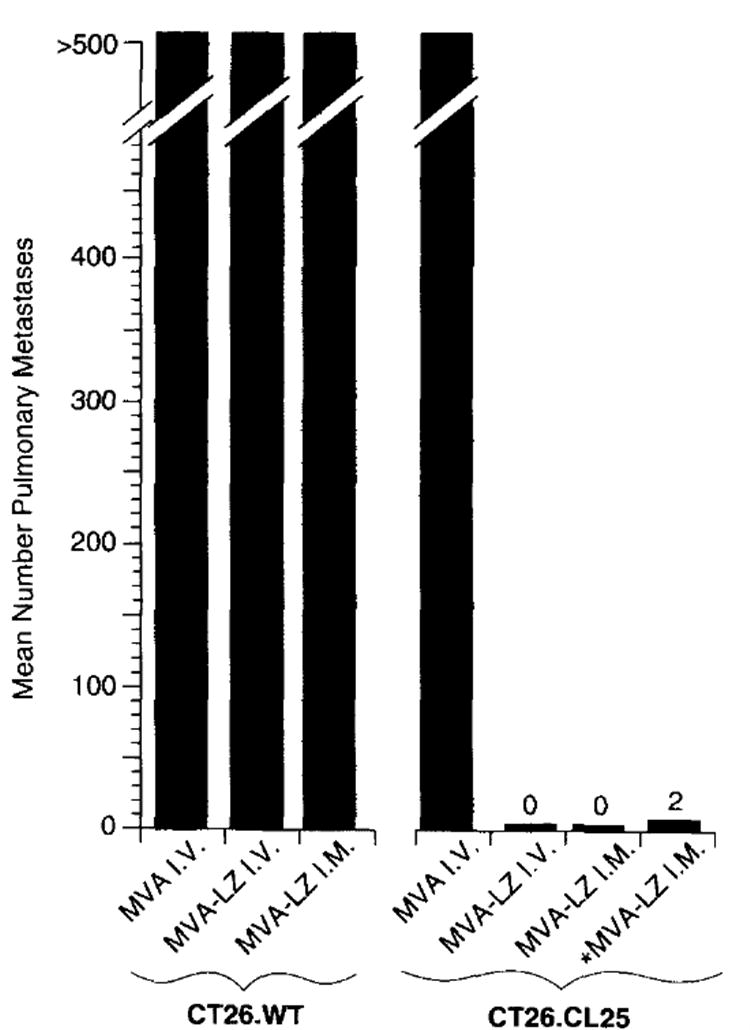
Intramuscular inoculation of MVA-LZ induces protective immunity to tumor challenge with CT26.CL25. BALB/c mice were inoculated with 108 or 106 (designated *) p.f.u. per mouse of MVA-WT or MVA-LZ. Twenty-one days later, mice received an i.v. challenge with 5×105 tumors cells. Twelve days after tumor inoculation, pulmonary metastases were enumerated in a blinded fashion. A maximum of 500 metastases per lung was recorded. Shown are average numbers of metastases obtained using five mice per group. In a repeat experiment i.m. inoculation with MVA-LZ and VJS6 showed similar results
Adoptive immunotherapy of pulmonary metastases established for 3 days using MVA-LZ primed splenocytes
One potential immunotherapeutic strategy involves the use of a recombinant immunogen to prime T lymphocytes in vivo. This priming is thought to both activate and increase the absolute numbers of anti-tumor T lymphocytes. In this scenario, in vivo priming is followed by ex vivo expansion and subsequent adoptive transfer back into the tumor bearing host.
To ascertain the ability of MVA-LZ to prime lymphocytes, mice were inoculated i.v. with MVA-LZ (Figure 2). The positive control was an i.v. inoculation with a replication competent (WR) rVV expressing β-gal (VJS6), previously shown to be capable of priming splenocytes for adoptive transfer34. Twenty-one days after priming, splenocytes were cultured for 6 days with a synthetic peptide from β-gal, previously shown to be presented by the Ld MHC class I molecule. These cultured splenocytes were then adoptively transferred to mice bearing pulmonary metastases established for 3 days. Splenocytes primed with a single i.m. inoculation of MVA-LZ were able to reduce the number of lung metastases to an average of 2.7 (Figure 2). Specificity controls consisted of antigen negative tumor as well as β-gal negative virus. Splenocytes from mice primed with MVA-LZ displayed no therapeutic activity against mice bearing the β-gal negative tumor line, CT26.WT. In addition, splenocytes from mice primed with the β-gal negative MVA failed to treat mice bearing CT26.CL25. All mice in these control groups had 500 pulmonary metastases. Interestingly, a one log reduction in the number of adoptively transferred splenocytes abrogated the observed therapeutic activity (data not shown).
Figure 2.

Successful adoptive immunotherapy of pulmonary metastases established for 3 days using MVA-LZ primed splenocytes. BALB/c mice were inoculated i.v. with 5×105 CT26.WT or CT26.CL25 (β-gal transfected) tumor cells. Three days later, tumorbearing mice were treated with the adoptive transfer of 2×107 splenocytes obtained from mice primed i.m. with 107 p.f.u. of either MVA or MVA-LZ. 107 p.f.u. of VJS6, delivered i.v., was used as a positive control. Twenty-one days after priming, adoptively transferred splenocytes were cultured in the presence of 10 μg ml−1 of the MHC class I-restricted peptide with the sequence TPHPARIGL, corresponding to residues 876–884 of the full-length β-gal protein, for 6 days before adoptive transfer into tumor bearing mice. Mice were killed 12 days after adoptive transfer of splenocytes, and lung metastases were enumerated in a coded blinded fashion. The experiment was repeated and similar results were obtained
Cytokine profile of therapeutic splenocytes
One measure of the potential in vivo anti-tumor reactivity of T lymphocytes is their ability to specifically secrete cytokines in vitro when exposed to β-gal. In order to understand the effects of in vitro MHC class I restricted β-gal peptide stimulation of the therapeutic T-cells, we analyzed the spectrum of cytokines produced by these cells. Mice were primed with MVA-LZ or the positive control VJS6. The negative control was non-recombinant MVA. Splenocytes were cultured in the presence of the MHC class I-restricted (H-2 Ld) β-gal peptide or a control peptide encoded by another TAA, designated PlA, and also restricted by H-2 Ld. Supernatants taken on the second day of culture were analyzed by ELISA for IL-2, IFN-γ, IL-10, GM-CSF, IL-4, and TNF-α.
The general cytokine profile of splenocytes from mice primed with either MVA-LZ or VJS6 revealed elevated levels of IL-2 and IFN-γ and low levels of IL-10, GM-CSF, IL-4, and TNF-α (Figure 3). Though levels of GM-CSF were low they were specific for splenocytes from mice previously inoculated with MVA-LZ and VJS6 and stimulated with the β-gal peptide. Splenocytes primed with MVA-LZ and activated with a negative control peptide (MHC class I restricted PlA) and splenocytes primed with MVA and cultured with β-gal peptide, revealed considerable specificity of the measured reactivity.
Figure 3.
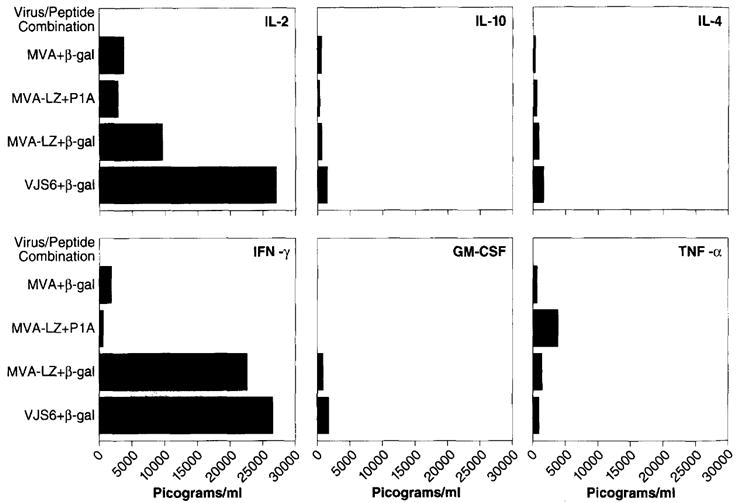
Cytokine profile of splenocytes cultured with MHC class I restricted peptides. Splenocytes were prepared from mice that had previously been inoculated i.m. with 107 p.f.u. of the MVA or MVA-LZ viruses. The same dose of VJS6 given i.v. was used as a positive control. Three weeks later, splenocytes were removed and cultured together with 10 μg ml−1 of the class I-restricted β-gal peptide, TPHPARIGL. Two days later, supernatants of the samples were collected and analyzed by ELISA for IL-2, IFN-γ, IL-10, GM-CSF, IL-4, and TNF-α. Results are shown in picograms per ml of supernatant. Note that each flask contained 30 ml of supernatant
Active immunotherapy of mice with established pulmonary metastases, using nonreplicating rMVA
Anti-cancer vaccines could be greatly effective in the prevention of infection with tumor inducing viruses, for example, HPV and EBV. However, an important target in the development of recombinant anti-cancer vaccines is the treatment of growing cancer cells.
To determine the therapeutic effectiveness of a rMVA in an active immunotherapy setting, mice were inoculated with 5 × 105 tumor cells, then treated either 3 days, or 3 and 10 days later with 5 × 107 p.f.u. of recombinant virus expressing the TAA, β-gal (Figure 4). In addition to MVA-LZ we also used the replication competent VJS6 and a FPV expressing β-gal, FPV.bg.40k. The latter two viruses were used as positive controls but not for direct comparison purposes since they have different promoters regulating lacZ. Mice treated with Hank’s balanced salt solution (HBSS), or with viruses that did not express the model TAA died within 28 days (Figure 4A). Mice receiving treatment with either of the three β-gal expressing poxvirus recombinants displayed a significant increase in survival when compared to control groups inoculated with β-gal negative virus (P<0.05) (Figure 4C). In this experiment the active treatment efficacy of VJS6 when compared to HBSS was not statistically significant, however, a repeat experiment did show a significant difference. There appeared to be no statistically significant advantage when mice received a second treatment inoculation 10 days after tumor inoculation (Figure 4B, P>0.05), though a trend was noted.
Figure 4.
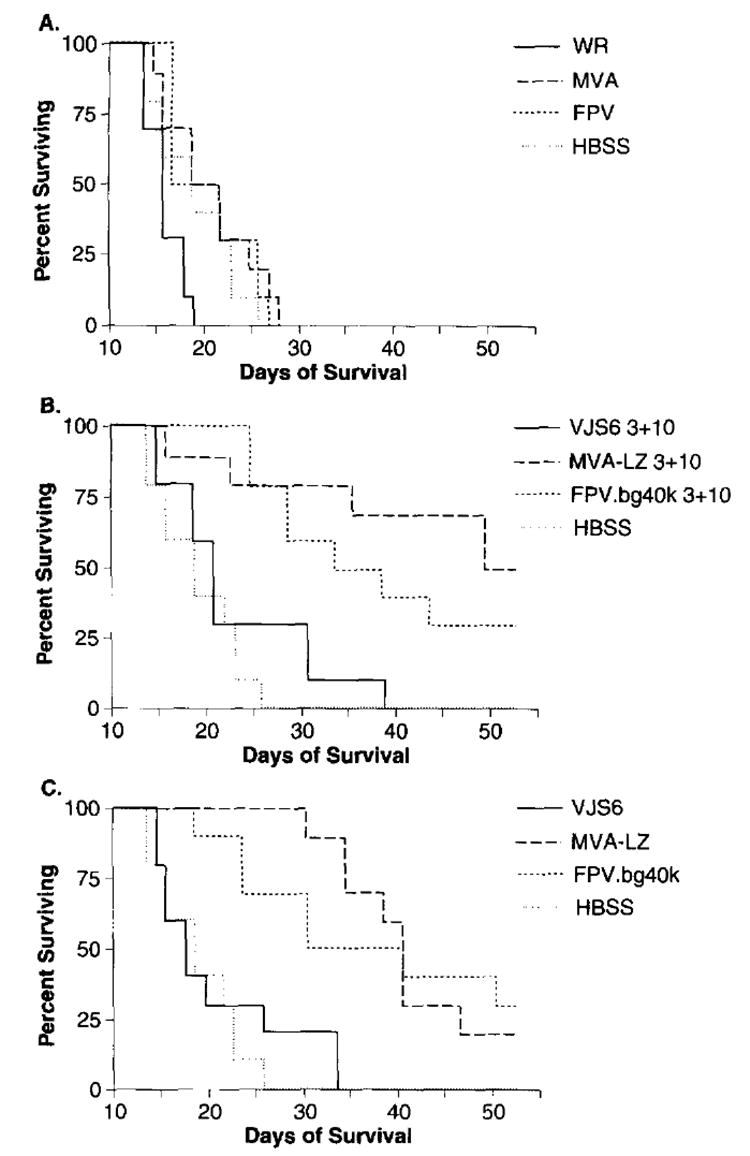
Active immunotherapy of pulmonary metastases established for 3 days. BALB/c mice were inoculated with 5×105 CT26.CL25 tumor cells. Three, or 3 and 10 days after tumor inoculation, mice were inoculated with 5×105 p.f.u. i.v. of the designated virus and checked daily for survival. Results shown are from a single experiment, although data is shown in three panels for clarity. Shown are mice receiving two immunizations, 3 and 10 days after tumor inoculation in (A) and (B). Mice receiving a single immunization 3 days after tumor inoculation are shown in C). An independent repeat experiment showed similar results
Homologous vs heterologous boosting
The ability of VV recombinants to elicit efficient immunogenic responses to heterologous protein has been shown to be greatly impaired in those recipients that have pre-existing immunity to VV35,36,37. With this in mind we analyzed the effects of boosting 10 days after tumor inoculation with a different poxvirus vector to that employed 1 week earlier, when the tumors of the mice had been established for 3 days. Mice primed with MVA-LZ 3 days after tumor inoculation and boosted with FPV.bg40k 10 days after tumor inoculation showed a noticeable improvement in the therapeutic effect compared to two inoculations with MVA-LZ or FPV.bg40k (data not shown). However, the same improvement in the efficiency of treatment was not observed when mice were initially inoculated with FPV.bg40k and boosted with MVA-LZ. Due to the aggressive nature of the tumor it is perhaps not surprising that treatment at a relatively late stage of tumor growth would not have a dramatic effect on survival even if heterologous boosting was advantageous.
Mechanisms of tumor escape
In an attempt to determine if mice that survived for extended times died due to down regulation of model TAA and thus escape from specific β-gal CTLs, the lungs of dead mice were stained with a solution of X-gal. The effects of active treatment with MVA-LZ on the down regulation of β-gal by the tumor are shown in Figure 5. The lungs from mice inoculated with the parental tumor line CT26.WT typically showed no blue colouration after staining with X-gal. Figure 5A depicts a lung from a mouse inoculated with CT26.WT, and treated with MVA-LZ, and displays the usual pattern of intense tumor growth which usually leads to death by day 16–25. This lung was used as a control for the nonspecific staining sometimes seen with extended periods of incubation with X-gal. As can be seen in Figure 5B, the lungs from a mouse inoculated with CT26.CL25 tumor stained strongly blue with X-gal, indicating high levels of β-gal expression. This mouse received treatment with a β-gal negative FPV. In contrast, the lungs from a mouse inoculated with CT26.CL25 and treated with MVA-LZ showed significantly decreased numbers of tumors which poorly stained for β-gal suggesting down regulation of antigen expression as a mode of tumor escape from a TAA specific immune response. All three mice died on day 24. This pattern of tumor suppression in active treatment groups was observed in a number of mice.
Figure 5.

Down-regulation of β-gal expression of CT26.CL25 pulmonary metastases. In a second survival experiment (data not shown) mice were inoculated with 2×107 tumor cells and treated 3 and 10 days later with 2×107 p.f.u. i.v. with the relevant virus. When mice died their lungs were removed and stained for β-gal expression as described in Materials and Methods. (A) Mouse inoculated with CT26.WT and treated with MVA-LZ, (B) mouse inoculated with CT26.CL25 and treated with FPV, (C) mouse inoculated with CT26.CL25 and treated with MVA-LZ
DISCUSSION
In this report we have shown a potential use of recombinant MVA as an immunogen in cancer immunotherapy. The superior safety of MVA, compared to replication competent poxviruses, makes this vector highly attractive for use as a recombinant immunogen. The dose used for the different viruses in the survival study (Figure 4), 5 × 107 p.f.u., had been previously optimized for the rVV. While these doses of replication-competent rVV are nearly lethal in immunocompetent mice and result in cachexia and profound splenomegaly, a dose of 108 MVA has no morbidity, even in suckling mice13.
Although the β-gal expressing rVV and rFPV were used as positive controls, we noted that the survival of mice bearing 3 day old pulmonary metastases was significantly longer when the mice received active immunization with the two nonreplicating recombinant poxviruses, MVA-LZ and FPV.bg40k, compared with treatment with the replication competent VJS6 (P<0.05). In a repeat experiment (data not shown) using 105 tumor cells and 2 × 107 p.f.u. of recombinant virus (eight mice per group), a similar pattern was observed with MVA-LZ displaying significantly longer survival times compared to VJS6. One could hypothesize that the differences in promoter strength and kinetics affected the efficacy of the different poxviruses. However, in similar active treatment survival experiments (V. Bronte et al., PNAS, in press) the 7.5 k promoter driving expression of the model TAA in the VJS6 construct has been shown to be more efficient than the VV late promoter utilized in MVA-LZ. The promoter used in the FPV construct was also an early late promoter similar to that used in the WR based VJS6. In fact studies suggest that some antigens expressed at late times of VV infection do not induce effective CTL response38. However, this phenomena can usually be overcome by expression of the antigen at early times using a VV early promoter38. One explanation is that VV infection shuts off host protein synthesis, thereby creating a scarcity of MHC-I molecules for peptides processed at late times of infection. Thus, a bias of the promoter used should work against MVA-LZ.
One explanation why MVA and FPV appeared to perform more efficiently than the replication competent WR strain, as vectors in our active treatment model is the reported expression of immune evasion or suppression molecules by poxviruses. Vaccinia virus WR and other members of the poxvirus genus are known to express a spectrum of host immune evasion factors, many of which are soluble secreted homologues of cytokine receptors including IFN-α, IFN-γ, and IL-lβ,39,40,41,42,43. It is possible that the absence of immune evasion molecules may enhance the resulting immune response induced by a target antigen expressed by a VV recombinant. MVA has endured over 500 passages in CEF cells during which time it has lost over 30 kb of its genomic DNA28. Deletions or point mutations within many of the immune evasion genes may have occurred, especially as these gene products are not thought to be beneficial for replication in vitro. Furthermore, if the avian FPV possesses such immune evasion molecules it is unlikely that they will be functional in a mammalian host. These differences may be one explanation why MVA and FPV appear to be more efficient vectors in active treatment in our pulmonary metastases model.
When a monolayer of mammalian cells (eg mouse NIH 3T3) were infected for 72 h with VV strain WR. large plaques were visible consisting of 150 infected cells, however, in the case of MVA only single cells were infected16. This difference in viral replication and spread is due to the block in MVA assembly14. These differing growth characteristics will probably be evident in vivo. It has been suggested that the kinetics of CTL induction and longevity of activated CTL is dependent on the persistence properties of the infecting virus44. In mice inoculated with vesicular stomatitis virus (VSV), CTL are rapidly activated but their numbers, like the virus itself, decrease quickly usually within 2 days. However, in the case of infection with lymphocytic choriomeningitis virus (LCMV), in which the virus persists. the kinetics of CTL activation are generally reversed (R. Zinkernagel, Zurich University, Switzerland, personal communication) i.e. slower rate of induction but levels stay high for longer periods45. Perhaps the cytopathic properties of WR induce rapid clearance of infected cells and virus, whereas, MVA and FPV cause less cellular destruction and express recombinant antigen longer and thus extend the length of activated CTL activity.
In the tumor model we also noticed signs of immune evasion by down regulation of β-gal, human tumors have been reported to employ similar mechanisms46. This problem may be partially overcome by constructing recombinant poxviruses expressing several defined TAA In the case of HPV associated cervical cancer this phenomena should not arise as expression of the viral antigen is associated with cellular transformation47, however, the reported incidence of down regulation of MHC I by some tumors may still facilitate immune evasion.
A potential advantage of MVA over FPV is that the block in abortive replication is at the stage after late gene expression14 whereas FPV replication is usually blocked prior to this stage48. Early poxvirus promoter expression levels can be only a few percent of those of late promoters. The co-expression of cytokines (IL-2 or IL-12) and TAA have been shown to enhance the active treatment properties of VV recombinants in this tumor model (M.W. Carroll et al., manuscript in preparation)20. In both cases the cytokines are under transcriptional control of strong VV late promoters as it is thought that these cytokines work in a dose dependent manner. In addition, high level expression of immune costimulatory molecules, for example, B7 may be advantageous in the design of therapeutic poxviruses.
Pre-existing VV immunity has been shown to be detrimental to the induction of specific cellular responses to recombinant proteins expressed by rVV35,36,37. Recombinant FPV does not appear to be affected by pre-existing immunity to VV19. Many putative recipients of cancer immunotherapy treatment will have received VV inoculation as smallpox vaccination was administered until the early 1970s. It will be important to determine if immunity to VV, induced 20 years previously, will hinder the effectiveness of a recombinant virus based on MVA, to induce a therapeutic TAA CTL response. Perhaps a combination of several noncross reacting poxvirus vectors will be used in future therapy regimes.
It is important to note that this tumor model has a number of artificial properties compared to many naturally occurring cancers, in that it has a retroviral promoter driving expression of the TAA and the only tissue expressing the β-gal antigen is the target tissue which has been introduced into a mature mouse. In the case of melanoma, many of the TAA (e.g. gp100) are expressed by all melanocytes and even cells in the retina. This brings into question whether a vaccinia recombinant could break tolerance to a natural nonmutated “self antigen” and if so would this lead to autoimmunity? To address this question we are currently developing new tumor models using B16 melanoma cells, C57-BL mice and vaccinia recombinants expressing the mouse homologues of the unmutated TAA gp100, MART-l, tyrosinase, and TRP-1. However, this β-gal model is relevant to tumor cells that constitutively express “nonself antigens” as seen in many virally induced tumors (e.g. HPV E6/E7 induced cervical cancer), or cancers that express mutated self antigens (e.g. mutated P53 induced breast cancer).
In summary, MVA has now been compared, to replication competent vaccinia strains, in three animal models, including influenza virus16 and SIV18. In each case it has performed as efficiently, if not more so, than the commonly used replicating VV strains WR and Wyeth. MVA possesses all the advantages of a versatile and efficient expression vector with an extensive history as a safe vaccine vector. These properties should allow its use in human cancer therapy trials in the near future.
References
- 1.Boon T, Gajewski TF, Coulie PG. From defined human tumour antigens to effective immunization? Immunol Today. 1995;16:334–336. doi: 10.1016/0167-5699(95)80149-9. [DOI] [PubMed] [Google Scholar]
- 2.Boon T, Cerottini J, Van den Eynde B, van der Bruggen P, Van Pel A. Tumour antigens recognized by T lymphocytes. A Rev Immunol. 1994;12:337–365. doi: 10.1146/annurev.iy.12.040194.002005. [DOI] [PubMed] [Google Scholar]
- 3.Rosenberg SA. The development of new cancer therapies based on the molecular identification of cancer regression antigens. Cancer J. 1995;1:90–l00. [PubMed] [Google Scholar]
- 4.Arita I. Virological evidence for the success of the smallpox eradication programme. Nature. 1979;279:293–298. doi: 10.1038/279293a0. [DOI] [PubMed] [Google Scholar]
- 5.Mackett M, Smith GL, Moss B. Vaccinia virus: a selectable eukaryotic cloning and expression vector. Proc Natl Acad Sci USA. 1982;79:7415–7419. doi: 10.1073/pnas.79.23.7415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Panicali D, Paoletti E. Construction of poxviruses as cloning vectors: insertion of the TK gene from herpes simplex virus into the DNA of infectious vaccinia virus. Proc Natl Acad Sci USA. 1982;79:4927–4931. doi: 10.1073/pnas.79.16.4927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kantor J, Irvine K, Abrams S, Kaufman H, DiPietro J, Schlom J. Antitumour activity and immune responses induced by a recombinant carcinoembryonic antigen-vaccinia virus vaccine. J Natl Cancer Inst. 1992;84:1084–1091. doi: 10.1093/jnci/84.14.1084. [DOI] [PubMed] [Google Scholar]
- 8.Estin CD, Stevenson US, Plowman GD, et al. Recombinant vaccinia virus vaccine against the human melanoma antigen p97 for use in immunotherapy. Proc Natl Acad Sci USA. 1988;85:1052–1056. doi: 10.1073/pnas.85.4.1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roth J, Dittmer D, Rea D, Tartaglia L, Paoletti E, Levine AJ. p53 as a target for cancer vaccines: recombinant canarypox virus vectors expressing p53 protect mice against lethal tumour cell challenge. Proc Natl Acad Sci USA. 1996;93:4781–4786. doi: 10.1073/pnas.93.10.4781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meneguzzi G, Cemi C, Kieny MP, Lathe R. Immunization against human papillomavirus type 16 tumour cells with recombinant vaccinia viruses expressing E6 and E7. Virology. 1991;181:62–69. doi: 10.1016/0042-6822(91)90470-v. [DOI] [PubMed] [Google Scholar]
- 11.Lathe R, Kieny MP, Gerlinger P, et al. Tumour prevention and rejection with recombinant vaccinia. Nature. 1987;326:878–880. doi: 10.1038/326878a0. [DOI] [PubMed] [Google Scholar]
- 12.Manhel H, Mayr A. Erfahrungen bei der schutzimpfung gegen orthopocken von mensch und tier mit dem impfstamm MVA. Berlin München tierärztliche wochenschrift. 1994;107:253–256. [PubMed] [Google Scholar]
- 13.Mayr A, Hochstein-Mintzel V, Stickl H. Abstammung, eigenschaften und verwendung des attenuierten vacciniastammes MVA. Infection. 1975;3:6–14. [Google Scholar]
- 14.Sutter G, Moss B. Nonreplicating vaccinia virus vector efficiently expresses recombinant genes. Proc Natl Acad Sci USA. 1992;89:10847–10851. doi: 10.1073/pnas.89.22.10847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smith GL, Moss B. Infectious poxvirus vectors have capacity for at least 25 000 base pairs of foreign DNA. Gene. 1983;25:21–28. doi: 10.1016/0378-1119(83)90163-4. [DOI] [PubMed] [Google Scholar]
- 16.Sutter G, Wyatt L, Foley P, Bennink JR, Moss BA. recombinant vector derived from the host range-restricted and highly attenuated MVA strain of vaccinia virus stimulates protective immunity in mice to influenza virus. Vaccine. 1994;12:1032–1040. doi: 10.1016/0264-410x(94)90341-7. [DOI] [PubMed] [Google Scholar]
- 17.Moss B, Carroll MW, Wyatt LS, et al. Host range restricted, non-replicating vaccinia virus vectors as vaccine candidates. In: Cohen S, Shafferman A, editors. Novel Strategies in the Design and Production of Vaccines; Proceedings of the 39th Oholo conference; New York: Plenum Publishing Corporation; 1996. pp. 7–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hirsch VN, Fuerst TR, Sutter G, et al. Patterns of viral replication correllate with outcome in simian immunodeficiency virus-infected macaques: effects prior to immunization with a trivalent SIV vaccine in modified vaccinia virus Ankara (MVA) J Virol. 1996;70:3741–3752. doi: 10.1128/jvi.70.6.3741-3752.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang M, Bronte V, Chen PW, et al. Active immunotherapy of cancer with a nonreplicating recombinant fowlpox virus encoding a model tumour-associated antigen. J Immunol. 1995;154:4685–4692. [PMC free article] [PubMed] [Google Scholar]
- 20.Bronte V, Tsung K, Rao JB, et al. IL-2 enhances the function of recombinant poxvirus-based vaccines in the treatment of established pulmonary metastases. J Immunol. 1995;154:5282–5292. [PMC free article] [PubMed] [Google Scholar]
- 21.Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science. 1991;253:49. doi: 10.1126/science.1905840. [DOI] [PubMed] [Google Scholar]
- 22.Townsend A, Ohlen C, Rogers M, Edwards J, Mukhedee S, Bastin J. Source of unique tumour antigens. Nature. 1994;371:662. doi: 10.1038/371662a0. [DOI] [PubMed] [Google Scholar]
- 23.Beverley PC, Sadovnikova E, Zhu X, et al. Vaccines Against virally Induced Cancers. Wiley; Chichester, UK: 1994. Strategies for studying mouse and human immune responses to human papillomavirus type 16; pp. 78–96. [DOI] [PubMed] [Google Scholar]
- 24.Milich DR, Jones J, Hughes J, Maruyama T. Vaccines Against Virally Induced Cancers. Wiley; Chichester, UK: 1994. Hepatitis B virus infection, the immune response and hepatocellular carcinoma; pp. 113–131. [DOI] [PubMed] [Google Scholar]
- 25.Moss DJ, Burrows SR, Suhrbier A, Khanna R. Vaccines Against Virally Induced Cancers. Wiley; Chichester, UK: 1994. Potential antigenic targets on Epstein–Barr virus-associated tumours and the host response; pp. l–20. [DOI] [PubMed] [Google Scholar]
- 26.de The G, Bomford R, Kazanji M, Ibrahim F. Vaccines Against Virally Induced Cancers. Wiley; Chichester, UK: 1994. Human T cell lymphotropic virus: necessity for and feasibility of a vaccine; pp. 47–60. [DOI] [PubMed] [Google Scholar]
- 27.Brattain MG, Strobel-Stevens J, Fine D, Webb M, Sarrif AM. Establishment of mouse colonic carcinoma cell lines with different metastatic properties. Cancer Res. 1980;40:2142–2146. [PubMed] [Google Scholar]
- 28.Meyer H, Sutter G, Mayr A. Mapping of deletions in the genome of the highly attenuated vaccinia virus MVA and their influence on virulence. J Gen Virol. 1991;72:1031–1038. doi: 10.1099/0022-1317-72-5-1031. [DOI] [PubMed] [Google Scholar]
- 29.Weir JP, Moss B. Determination of the promoter region of an early vaccinia virus gene encoding thymidine kinase. Virology. 1987;158:206–210. doi: 10.1016/0042-6822(87)90254-6. [DOI] [PubMed] [Google Scholar]
- 30.Rosel JL, Earl PL, Weir JP, Moss B. Conserved TAAATG sequence at the transcriptional and translational initiation sites of vaccinia virus late genes deduced by structural and functional analysis of the Hind III H genome fragment. J Virol. 1986;60:436–449. doi: 10.1128/jvi.60.2.436-449.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Earl PL, Cooper N, Moss B. Preparation of cell cultures and vaccinia virus stocks. In: Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith LA, editors. Current Protocols in Molecular Biology. Greene Publishing Associates and Wiley Interscience; New York: 1991. pp. 16.16.1–16.16.17. [Google Scholar]
- 32.Irvine KR, McCabe BJ, Rosenberg SA, Restifo NP. Synthetic oligonucleotide expressed by a recombinant vaccinia virus elicits therapeutic CTL. J Immunol. 1995;154:4651–4657. [PMC free article] [PubMed] [Google Scholar]
- 33.Kaplan, Meier Non-parametric estimation from incomplete observations. J Am Stat Assoc. 1958;53:457–481. [Google Scholar]
- 34.Wang M, Chen PW, Bronte V, Rosenberg SA, Restifo NP. Anti-tumour activity of cytotoxic T lymphocytes elicited with recombinant and synthetic forms of a model tumour associated antigen. J lmmunother. 1995;18:139–146. doi: 10.1097/00002371-199510000-00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rooney JF, Wohlenberg CR, Cremer KJ, Moss B, Notkins AL. Immunization with a vaccinia virus recombinant expressing herpes simplex virus type I glycoprotein D: long-term protection and effect of revaccination. J Virol. 1988;62:1530–1534. doi: 10.1128/jvi.62.5.1530-1534.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kundig TM, Kalberer CP, Hengartner H, Zinkernagel RM. Vaccination with two different vaccinia recombinant viruses: long-term inhibition of secondary vaccination. Vaccine. 1993;11:1154–1158. doi: 10.1016/0264-410x(93)90079-d. [DOI] [PubMed] [Google Scholar]
- 37.Cooney EL, Collier AC, Greenberg PD, et al. Safety of and immunological response to a recombinant vaccinia virus vaccine expressing HIV envelope glycoprotein. Lancet. 1991;337:567–572. doi: 10.1016/0140-6736(91)91636-9. [DOI] [PubMed] [Google Scholar]
- 38.Coupar BEH, Andrew ME, Both GW, Boyle DB. Temporal regulation of influenza hemagglutinin expression in vaccinia virus recombinants and effects on the immune response. Eur J Immunol. 1986;16:1479–1487. doi: 10.1002/eji.1830161203. [DOI] [PubMed] [Google Scholar]
- 39.Symons JA, Alcami A, Smith GL. Vaccinia virus encodes a soluble type I interferon receptor of novel structure and broad species specificity. Cell. 1995;81:551–560. doi: 10.1016/0092-8674(95)90076-4. [DOI] [PubMed] [Google Scholar]
- 40.Alcami A, Smith GL. Vaccinia, cowpox, and camelpox viruses encode soluble gamma interferon receptors with novel broad species specificity. J Virol. 1995;69:4633–4639. doi: 10.1128/jvi.69.8.4633-4639.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alcami A, Smith GL. A soluble receptor for interleukin-1 beta encoded by vaccinia virus: a novel mechanism of virus modulation of the host response to infection. Cell. 1992;71:153–l67. doi: 10.1016/0092-8674(92)90274-g. [DOI] [PubMed] [Google Scholar]
- 42.Smith GL. Vaccinia virus glycoproteins and immune evasion. The sixteenth Fleming Lecture. J Gen Virol. 1993;74:1725–1740. doi: 10.1099/0022-1317-74-9-1725. [DOI] [PubMed] [Google Scholar]
- 43.McFadden G. Viroceptors, Virokines and Related Immune Modulators Encoded by DNA Viruses. R.G Landes Company; 1994. [Google Scholar]
- 44.Zinkernagel RM. The role of antigen in maintaining T cell memory. Dev Biol Stand. 1994;82:189–191. [PubMed] [Google Scholar]
- 45.Lau LL, Jamieson BD, Somasundaram T, Ahmed R. Cytotoxic T-cell memory without antigen. Nature. 1994;369:648–652. [PubMed] [Google Scholar]
- 46.Lehmann F, Marchand M, Hainaut P, et al. Differences in the antigens recognized by cytoloytic T Cells on two successive metastases of a melanoma patient are consistent with immune selection. Eur J Immunol. 1995;25:340–347. doi: 10.1002/eji.1830250206. [DOI] [PubMed] [Google Scholar]
- 47.Chen L, Ashe S, Singhal MC, Galloway DA, Hellstrom I, Hellstrom KE. Metastatic conversion of cells by expression of human papillomavirus type 16 E6 and E7 genes. Proc Natl Acad Sci USA. 1993;90:6523–6527. doi: 10.1073/pnas.90.14.6523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Somogyi P, Frazier J, Skinner MA. Fowlpox virus host range restriction: gene expression, DNA replication, and morphogenesis in nonpermissive mammalian cells. Virology. 1993;197:439–444. doi: 10.1006/viro.1993.1608. [DOI] [PubMed] [Google Scholar]