

Abstract
The increasing demand for the identification of genetic variation responsible for common diseases has translated into a need for sophisticated methods for effectively prioritizing mutations occurring in disease-associated genetic regions. In this article, we prioritize candidate nonsynonymous single-nucleotide polymorphisms (nsSNPs) through a bioinformatics approach that takes advantages of a set of improved numeric features derived from protein-sequence information and a new statistical learning model called “multiple selection rule voting” (MSRV). The sequence-based features can maximize the scope of applications of our approach, and the MSRV model can capture subtle characteristics of individual mutations. Systematic validation of the approach demonstrates that this approach is capable of prioritizing causal mutations for both simple monogenic diseases and complex polygenic diseases. Further studies of familial Alzheimer diseases and diabetes show that the approach can enrich mutations underlying these polygenic diseases among the top of candidate mutations. Application of this approach to unclassified mutations suggests that there are 10 suspicious mutations likely to cause diseases, and there is strong support for this in the literature.
With the accelerating advancement of biomedical research, it has been widely accepted that inherited genetic variation plays an important role in the pathogenesis of common diseases, including heart disease, hypertension, diabetes, cancer, and many others, making the identification of causal genes and genetic variants the primary step toward the prevention, diagnosis, and treatment of these diseases.1–3 Family-based linkage analysis and population-based association studies have been the two major categories in which there have been remarkable successes in the identification of causal genetic variants. With use of a transmission model, linkage analysis explains the pattern of inheritance of phenotypes and genotypes exhibited in a pedigree. It works well for rare Mendelian diseases in which an individual genetic variant in a single gene is both necessary and sufficient to cause a disorder, but it has limited explanatory power when a single locus fails to explain a significant fraction of a disease.1 In contrast, association studies that test whether the frequencies of alleles in patients are significantly different from those in control individuals are most meaningful when applied to genetic variants that have clear biological relation to the disease phenotype, but they are minimally effective for whole-genome searches in large and/or mixed populations.1
The emergence of powerful yet low-cost sequencing techniques4,5 has been opening a new era in biotechnology in which the examination of all genetic variants in a large number of affected individuals and controls has become more and more feasible. For instance, the International HapMap Project has reported >1 million SNPs, including 10 completely sequenced 500-kb regions in which essentially all information about common DNA variation has been extracted.3 Another major data source, the Swiss-Prot database, has collected >20,000 nonsynonymous SNPs (nsSNPs).6 With the advent of such abundant information, traditional statistical methods confront ever-greater challenges. Linkage analysis has low power for complex diseases that are thought to be caused by the combined effect of many susceptible genetic variants and their interactions with environmental factors, whereas association studies suffer from serious multiple-hypothesis-testing problems when applied to a number of markers in a large population.1 Indeed, the demand for identification of causal genetic variants among a vast number of irrelevant ones in which they are immersed has translated into a need for sophisticated tools integrating biophysical and biochemical knowledge with statistical learning methods, to effectively prioritize genetic variants underlying complex diseases.
Typically, in the traditional approach to the study of disease mutations (fig. 1, left panel), 10–30-Mb genetic regions are initially identified after establishing statistically significant genomewide evidence of linkage or association. Then, the suspicious regions are reduced to <1 Mb by a fine-mapping procedure, and candidate genetic variants are identified by sequence analysis. Finally, causal genetic variants are determined by functional tests.2 With the emergence of current sequencing techniques,4,5 however, skipping the fine-mapping step and directly sequencing the 10–30-Mb genetic regions becomes increasingly possible. With a vast number of candidate genetic variants being sequenced, it becomes impractical to apply functional tests to determine causal genetic variants. Instead, a bioinformatics approach integrating biophysical and biochemical knowledge with statistical learning methods should be adopted (fig. 1, right panel).
Figure 1. .

The traditional and the proposed bioinformatics approaches for disease mapping. In the traditional approach, 10–30-Mb genetic regions are obtained after establishing statistically significant genomewide evidence of linkage or association. Then, a fine-mapping procedure is applied to reduce the regions to <1 Mb. Finally, sequence analysis and functional tests are applied, to determine causal genetic variants. In the proposed bioinformatics approach, the fine-mapping step is replaced by the direct sequencing of the 10–30-Mb regions. To determine causal genetic variants, a bioinformatics approach integrating biophysical and biochemical, as well as statistical learning methods, is adopted.
Genetic variants in single bases of DNA sequences yield SNPs, among which nsSNPs occurring in protein-coding regions lead to amino acid substitutions in protein products, potentially affecting protein functions and causing common diseases. Research about distinguishing deleterious amino acid substitutions from neutral nsSNPs in laboratory mutagenesis experiments of the Escherichia coli lac repressor and the bacteriophage T4 lysozyme aim mainly at assigning binary labels (deleterious/neutral) to the mutations.7–15 Recent studies about distinguishing disease-causing amino acid substitutions from neutral nsSNPs in human proteins generally focus on predicting numeric scores (indicating the likelihood of causing diseases) of the mutations.14–23 Despite notable successes, these methods have not yet reasoned from the real situation in identifying disease mutations—that is, with a number of mutations distributed in genetic regions, the task would be to prioritize (rank) them, to identify those that are most likely to cause diseases. Regardless of the diverse objectives of these studies, their methods share two common characteristics. First, all of them are based on a set of features calculated using sequence, structure, annotations, and evolutionary information of proteins, despite the variation in the definitions of features. Second, all of these methods adopt standard rule-based models and/or statistical learning models, including logistic regression models,18 Bayesian models,13 neural networks,24 decision trees,25,26 support vector machines,27,28 random forests,29 and many others. Use of protein structure information to calculate features restricts the scope of applications for many of these methods, because the availability of protein-structure information is quite limited for human proteins. Therefore, a set of sequence-based features is preferable for maximizing the scope of applications for prioritizing disease mutations. On the other hand, most of the standard statistical learning models are not designed to capture subtle characteristics of mutations; thus, a carefully designed learning model is needed to use the calculated features more effectively.
In this article, we report the principle, development, and effectiveness of a disease mutation–prioritization method, and we offer a free Web-based, interactive software tool. Compared with existing methods for classifying or predicting disease mutations, our approach provides a more realistic solution for identifying such mutations. Specifically, we prioritize mutations occurring in genetic regions, to find those that are most likely to cause diseases. We put forward a novel scheme to calculate a set of 26 features that uses only protein-sequence information and multiple-sequence alignments, and we propose a newly designed multiple selection rule voting (MSRV) learning model to capture subtle characteristics of mutations from individual types of amino acids. Then we systematically analyze the effectiveness of the feature set and the learning model, using rigorous hypothesis testing, after which we integrate the easy-to-calculate feature set and the state-of-the-art learning model and validate that our prioritization method is both effective and robust in prioritizing causal mutations for not only simple monogenic diseases but also complex polygenic diseases. As application examples, we demonstrate the successful identification of 10 suspicious mutations from currently unannotated ones. With an ever-increasing amount of candidate genetic variants examined in a large number of affected and control individuals with use of today’s powerful sequencing techniques, our approach could provide reliable in silico screening for disease-causing variants, thereby assisting in the prevention, accelerating the diagnosis, and guiding the treatment of common diseases.
Material and Methods
Data Sources
The Swiss-Prot database6 is the major data source used to train our learning model and to validate our approach. Version 50.2 (released June 27, 2006) of the Swiss-Prot database contains 26,265 amino acid–substitution entries for 4,225 human proteins, with each substitution annotated as “Disease,” “Polymorphism,” or “Unclassified.” For a clear and concise presentation, we refer to amino acid substitutions with the annotation “Disease” as disease mutations and those with the annotation “Polymorphism” as neutral nsSNPs. The International HapMap Project database3 is another data source used to validate our approach. By the end of 2005, the International HapMap Project database had collected >1 million SNPs for which accurate and complete genotypes have been obtained in 269 DNA samples from four populations.3 In addition, the Ensembl database provides a comprehensive source for large genome sequences of 19 species and integrated genome-variation data for human and mouse.30 The Pfam database31 is the data source used to obtain multiple-sequence alignments for query proteins. Version 20.0 (released May 2006) contains curated alignments and models for 8,296 protein families, and ∼74% of the known protein sequences have at least one match to Pfam. In this article, we study human proteins that have at least 20 homologous proteins in the Pfam database, and we focus on amino acid substitutions occurring in known protein domains. In total, we collected 9,640 disease mutations, 4,581 neutral nsSNPs, and 1,517 unclassified mutations in 2,165 human proteins from the Swiss-Prot database.
Principles of Prioritization in MSRV
Mutations that cause common diseases generally raise significant changes in the structures and functions of proteins. In contrast, neutral nsSNPs typically result in minor or even negligible changes in protein structures, and they hardly affect the normal functioning of proteins. This difference has been the fundamental principle for distinguishing disease mutations from neutral nsSNPs.11,13,14,16,22 Nevertheless, the availability of protein-structure information limits the applications of this principle. Recent studies of proteins related to human diseases suggest that, for disease mutations, the mutated amino acids rarely appear in the corresponding positions in homologous proteins. It has been shown that a mutation has an ∼90% possibility of causing a disease if the mutated amino acid never appears at the same position in homologous proteins, and this rule can explain ∼30% of disease mutations.23 This observation, based on the evolutionary conservation of amino acids in homologous proteins, has been adopted as a major principle for identifying disease mutations.10,17,18,21,23 In addition, the physicochemical properties of the original and the substituted amino acids, as well as the characteristics of amino acids around the substitutions, could also provide valuable information for the identification of disease mutations.23
From these principles and observations, we reason that a bioinformatics framework capable of automatically extracting the physicochemical properties of amino acids from protein sequences and calculating the evolutionary conservation information from homologous proteins might be a powerful tool for identifying mutations responsible for common diseases. On the basis of this notion, we designed a three-step algorithm—MSRV. As illustrated in figure 2, in the first step, a feature-extracting procedure is applied to candidate mutations, and a set of 26 numeric features is automatically extracted. These features are derived from protein sequences (according to the Swiss-Prot database6) and from multiple-sequence alignments (according to the Pfam database31), with no structure information involved. In the second step, mutations with known effects in the Swiss-Prot database are used to train a statistical learning model, which is composed of 20 modules, one for each type of amino acid. This step can be done offline in advance and can be automatically updated with the updates of the Swiss-Prot and Pfam databases. Finally, in the third step, candidate mutations with numeric features are evaluated by the trained learning model to receive scores, and a sorting procedure is applied to rank the candidate mutations in nonincreasing order on the basis of their scores.
Figure 2. .

The concept of the MSRV framework. In the first step, a feature-extracting procedure is applied to candidate mutations to automatically extract a set of 26 numeric features. In the second step, mutations with known effects in the Swiss-Prot database are used to train a statistical learning model, which is composed of 20 modules, one for each type of amino acid. In the third step, candidate mutations with numeric features are evaluated by the trained model to receive scores, and a sorting procedure is applied to rank the candidate mutations in nonincreasing order on the basis of their scores.
Calculation of the Feature Set
We derived a set of 26 features that are based on three physicochemical properties (molecular weight, pI value, and hydrophobicity scale) of amino acids, three relative frequencies for the occurrences of amino acids in the secondary structures (helices, strands, and turns) of proteins with known secondary structural information, and two evolutionary conservation scores. The unit of molecular weight is the Dalton. The isoelectric point (pI) is the pH at which a molecule carries no net electrical charge. The hydrophobicity scale of Kyte and Doolitle is derived from the physicochemical properties of amino acid side chains.32 The three relative frequencies are calculated by counting the occurrences of amino acids in the corresponding secondary structure of proteins with known secondary structural information. All of these six properties can be either obtained from the literature32,33 or calculated using only the sequence information of proteins.34 The conservation scores are defined as the frequencies of occurrences of the amino acids (the original or the substituted) in the corresponding position of the Pfam multiple-sequence alignment.
For a given amino acid substitution pair (Org→Sub) in a certain query protein, the above physicochemical properties and relative frequencies are calculated for the original (Org) and the substituted (Sub) amino acids, as well as in a window-sized situation that includes the neighbors of the original amino acids in the query protein sequence and in a column-weighted circumstance in which the query protein sequence is aligned with its homologous proteins. The calculation of the properties for the original and the substituted amino acids is straightforward. The window-sized properties (where W is the window size) are calculated as the average of the corresponding properties for the original amino acid and its W-1 neighbors in the query protein sequence. In this article, we set the window size at W=9 (because α helices are defined by repeated hydrogen bonds with a period of 4 aa and have 3.6 aa per turn35). The column-weighted properties are calculated as follows. For the query protein, its homologous proteins are extracted from the Pfam database.31 With the supposition that the substitution occurs at a position corresponding to the cth column of the alignment, the column-weighted properties are then calculated as the weighted average of the corresponding properties for all 20 kinds of amino acids, where the weight of a certain kind of amino acid is the frequency of its occurrence in the cth column of the alignment.
With the above properties calculated, we define the feature set that includes 24 physicochemical or relative frequency properties (with each of the six amino acid properties calculated in four different situations) and two conservation scores (for the original amino acids and the substituted amino acids), as shown in table 1.
Table 1. .
Summary of the Proposed Features[Note]
| Physicochemical |
Relative Frequency in |
||||||
| Category | Molecular Weight | pI Value | Hydrophobicity | Helices | Strands | Turns | Conservation Frequency in MSAa |
| Original | X1 | X2 | X3 | X4 | X5 | X6 | X25 |
| Substitution | X7 | X8 | X9 | X10 | X11 | X12 | X26 |
| Window sized | X13 | X14 | X15 | X16 | X17 | X18 | |
| Column weighted | X19 | X20 | X21 | X22 | X23 | X24 | |
Note.— Each of the six amino acid properties is calculated in four different situations, resulting in X1–X24. The two conservation scores for the original and the substituted amino acids become X25 and X26, respectively.
MSA = multiple-sequence alignment.
MSRV Model
The underlying reasons that different types of amino acids cause diseases vary significantly. For instance, mutations from cysteines to other amino acids are very likely to destroy disulfide bridges within polypeptides, changing structures of proteins and consequently inducing the loss-of-protein functions.23 As another example, mutations from glycines to other amino acids are likely to cause diseases because glycine is smaller than any other amino acids and prefers the turn structure second only to proline.23 With this understanding, it is reasonable to assume that each of the 20 aa is biased toward different features and that a disease mutation is likely to cause dramatic change to some of these features. This assumption immediately suggests the following multiple-selection strategy. We first partition the entire training data set into 20 subsets according to the original amino acids of the samples, and then we train a statistical learning model for each of these 20 subsets separately and combine them into one scoring system. Thus, the scoring system consists of 20 modules. In the training process, we apply a greedy feature-selection method to select the optimal subset of features in each module.
The feature-selection technique is independent of the learning model. Our objective is to select an optimal feature set for each module, to maximize certain criterion (e.g., area under the receiver operating characteristic curve [AUC]) for training samples. Because the number of possible feature subsets is exponential to the number of features (i.e., 2n for n features), it is computationally impractical to enumerate all subsets to select the best one. Instead, we adopt a sequential forward feature selection (SFS) technique, a greedy algorithm that can produce a reasonable approximation of the true optimum with much less computation time. The SFS technique is as follows.
The SFS Algorithm
-
1.
Xopt←Φ; Jopt←0; D←26;
-
2.
X←Φ; Y←(1⩽i⩽26); d←1;
-
3.
while d≤D, do
-
4.
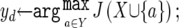
-
5.
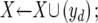
-
6.
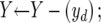
-
7.
d←d+1;
-
8.
if J(X)>Jopt, then
-
9.
Xopt←X;
-
10.
Jopt←J(X);
-
11.
end if
-
12.
end while
-
13.
return Xopt.
Let Xopt be the optimal feature set and Jopt be the optimal criterion (e.g., the maximum AUC). The basic idea of the algorithm is to start from an empty feature set (X) and repeatedly add one feature (yd) at a time (conditional on the current optimal set), to produce the next optimal subsets, until all features have been added. Among all of the subsets produced in this process, we select the one with the best criterion, Jopt. The function J(·) is calculated by taking a subset of features as input and performing 10-fold cross-validation by use of the training samples with the underlying learning model.
The learning model is independent of the feature-selection algorithm. Although, in general, any statistical learning method can be used as the underlying learning model, the ideal model is one with the best prediction power and the lightest computational burden. Recent studies of statistical learning suggest that random forests29 have preferred performance in many classification applications.23 This method is easy to implement, fast, and capable of dealing with large-scale data. On the basis of the idea of ensemble learning used in random forests, we propose a rule-voting method and use it as the underlying learning model in our scoring system. The learning model in each of the 20 modules is an ensemble of decision trees, each of which is trained on a data set bootstrapped from training samples. Decision trees are grown to the maximum size without pruning, according to the C4.5 methodology.24,26 At each node of the trees, a small number of features (typically three) is randomly selected from a corresponding module’s optimal feature set, and the one that maximizes the information gain is used to split the node. After a tree is generated, leaf nodes in the tree can be thought of as rules defined by the paths from the root of the tree to the leaf nodes. These rules claim that the mutation samples that fall into leaf nodes are disease causing, and the frequencies of disease-causing samples against all data samples fallen into the rules serve as empirical measures of the confidences of the corresponding rules. When a future test sample comes, all rules, one for each tree in the ensemble, are retrieved, and the confidences (frequencies of disease-causing samples) for the rules are averaged to provide a score indicating how likely it is that the test sample is disease causing.
Results
Greedy Feature Selection
The SFS algorithm is capable of selecting an (approximately) optimal subset of features for each of the 20 modules. To demonstrate, figure 3 shows the features selected by the algorithm with use of mutations collected in the Swiss-Prot database. This figure shows that each module has preference for its own optimal subset of features and that the numbers of features in the subsets vary significantly. For example, tryptophan has more molecular weight than any other amino acid. Consequently, only the column-weighted molecular weight (X19) and the relative frequency of the original amino acid in the multiple-sequence alignment (X25) are selected to characterize the optimal learning model for tryptophan. As another example, leucine is one of the amino acids that prefers the helix structure the most; thus, the relative frequency of the substituted amino acid in helices (X10) and the column-weighted relative frequency in helices (X22) are included in the optimal learning model for leucine.
Figure 3. .

Features selected by the SFS algorithm. Horizontal axis = indices of the features; vertical axis = modules. The selected features are marked in blue.
In detail, the original features are not included in the optimal feature sets, because modules are partitioned according to the original amino acids. The two conservation scores (X25 and X26) are the most frequently selected features (30 times in total and 0.75 times per feature for each module). The column-weighted features (X19–X24) are also frequently selected (81 times in total and 0.68 times per feature for each module). The window-sized features (X13–X18) and the substitution features (X7–X12) are less frequently selected (57 and 56 times in total, and 0.48 and 0.47 times per feature for each module, respectively). To further explore the performance of individual features, we adopted a mechanism that was similar to the one used in the random forests23,29 and obtained the relative importance of individual features in each module (fig. 4). The results confirm that the two conservation scores are of the most importance. The column-weighted features are also significantly more important than the window-sized features and the substitution features. The window-sized features are slightly more important than the substitution features.
Figure 4. .

Relative importance of individual features in each module. Horizontal axis = indices of the features; vertical axis = modules. The relative importance values range from 0.00 to 1.00, with 0.00 (white) indicating the least importance and 1.00 (red) indicating the greatest importance.
Performance of MSRV
The MSRV framework works as a scoring system, and the performance of this scoring system depends on the set of proposed features. Within available methods for extracting features, we assessed whether our feature set can outperform others in the classification of deleterious mutations occurring in the E. coli lac repressor8,9 and the bacteriophage T4 lysozyme.7 We applied two classification methods (the decision tree24 and the support vector machine27,28) to these mutagenesis experimental data, and we compared the classification accuracy with two published studies.11,12 The 10-fold cross-validation (homogeneous, heterogeneous, and mixed)12 results showed that our feature set can lead them by up to 3% when working with the decision tree and by up to 10% when working with the support vector machine, suggesting that our feature set was more effective in capturing underlying characteristics of mutations. Moreover, since our feature set used only sequence information, whereas the published one11 requires information on protein three-dimensional structure (derived from the Protein Data Bank with protein homology modeling), in practice, our feature set can be applied to almost every protein, whereas theirs will be restricted to proteins with known structures.
The performance of this scoring system also depends on the statistical learning methods used to determine scores from features. Within available methods for determining scores, we evaluated whether our approach can outperform others in the prediction of potential effects of mutations occurring in human proteins. We applied the support vector machine,27,28 the random forest,29 and the MSRV model with our feature set to mutations occurring in human proteins and collected in the Swiss-Prot database, and we compared the AUCs. The 10-fold cross-validation results showed that the AUC (±SD) for the MSRV model was 0.8484±0.0065, which was significantly higher than that for the random forest (0.8220±0.0073) and the support-vector machine (0.8058±0.0042). In other words, the MSRV model was more capable of producing reasonable scores from features.
An ideal scoring system should give high scores to disease mutations and low scores to neutral nsSNPs. Consequently, the scores for these two types of mutations are expected to be significantly different from each other. To assess whether our scoring system can distinguish disease mutations from neutral nsSNPs, we performed large-scale leave-one-out cross-validation experiments on annotated mutations in the Swiss-Prot database. In each validation run, a disease was selected, and its causal genes were collected. Mutations occurring in the causal genes were collected to form the test set, and those occurring in all other genes were used as the training set. Then, an MSRV model was trained on the training set, and mutations in the test set were scored using the trained model. Finally, a rank-sum test was applied to the scores of all mutations in the test set, and a P value was calculated to indicate the performance of the scoring system. We performed a total of three tests, as described below.
In the first test, we collected 30 diseases from 1,641 diseases in the Swiss-Prot database. All 30 diseases have comparable numbers of disease mutations and neutral nsSNPs in associated proteins. Thus, a total of 30 rank-sum tests were performed. The P values for all of the 30 diseases were <.05. In other words, the MSRV scoring system can effectively distinguish disease mutations from neutral nsSNPs. In particular, table 2 shows 11 diseases that have comparable numbers of disease mutations and neutral nsSNPs among the 30 diseases. The P values, ranging from 3.63×10-2 for age-related macular degeneration (ARMD2 [MIM 153800]) to 3.67×10-9 for osteogenesis imperfecta (OI) type II (OI-II [MIM 166210]), demonstrate that the difference between the scores of disease mutations and neutral nsSNPs is statistically significant.
Table 2. .
P Values of the Rank-Sum Tests
| No. of Mutations |
|||
| Disease/Region Name | Disease | Neutral | P |
| ARMD2 | 26 | 9 | 3.62×10-2 |
| Congenital bilateral absence of the vas deferens (MIM 277180) | 10 | 16 | 3.28×10-2 |
| Colorectal cancer (MIM 114500) | 11 | 27 | 2.43×10-2 |
| Polycystic kidney disease type I (MIM 173900) | 22 | 14 | 1.56×10-2 |
| Alport syndrome (MIM 203780) | 13 | 13 | 4.79×10-3 |
| Retinitis pigmentosa (RP [MIM 268000]) | 15 | 15 | 3.69×10-3 |
| Wilson disease (MIM 277900) | 69 | 10 | 9.33×10-4 |
| Cystic fibrosis (MIM 219700) | 111 | 16 | 1.35×10-4 |
| OI type IV (OI-IV [MIM 166220]) | 17 | 46 | 2.54×10-6 |
| OI type III (OI-III [MIM 259420]) | 22 | 28 | 6.26×10-7 |
| OI type II | 73 | 46 | 3.67×10-9 |
| STEA2_HUMAN region on chromosome 7 (10 Mb, 22 proteins) | 48 | 50 | 2.22×10-16 |
| CALU_HUMAN region on chromosome 7 (10 Mb, 7 proteins) | 10 | 33 | 1.33×10-5 |
| LHON, MELAS, and LS | 33 | 91 | 4.74×10-5 |
In the second test, we extended the leave-one-out cross-validation experiment to examine two 10-Mb genetic regions on chromosome 7, in which mutations have been densely identified and annotated. For each region, we used all mutations outside the region to train an MSRV model, scored all mutations occurring in the region using the trained model, and applied the rank-sum test to calculate the statistical significance of the scores of disease mutations and neutral nsSNPs. For the region centered at the gene of STEA2_HUMAN (MIM 605094) (position 89,678,936–99,678,936 on chromosome 7), 48 disease mutations and 50 neutral nsSNPs in 22 proteins were collected, and the difference between the scores of disease mutations and neutral nsSNPs was statistically significant (P=2.22×10-16). For the region centered at the gene of CALU_HUMAN (MIM 603420) (position 128,166,653–138,166,653 on chromosome 7), 10 disease mutations and 33 neutral nsSNPs in seven proteins were collected, and the difference between the scores of disease mutations and neutral nsSNPs was statistically significant (P=1.33×10-5).
In the third test, we applied the above experiment to the entire mtDNA. We first used all mutations occurring in nuclear DNAs to train an MSRV model. Then we scored all neutral nsSNPs occurring in the mtDNA and all mutations causing one of the following three mitochondrial diseases: Leber hereditary optic neuropathy (LHON [MIM 535000]), mitochondrial encephalopathy lactic acidosis stroke (MELAS [MIM 540000]), and Leigh syndrome (LS [MIM 256000]). Finally, we ran the rank-sum test to calculate the P value. The result (P=4.74×10-5) showed that the difference between the scores of disease mutations and neutral nsSNPs occurring in the mtDNA was statistically significant.
All three tests confirm that the difference between the scores of disease mutations and neutral nsSNPs was statistically significant, indicating that our approach can effectively distinguish disease mutations from neutral nsSNPs.
Prioritization of Mutations Causing Monogenic Diseases
Although we can directly apply our scoring system to predict disease mutations, in practical terms, investigators are more interested in screening out disease mutations from a number of suspicious mutations occurring in genetic regions (10–30 Mb in size) that are associated with the diseases of interest. To simulate this real-life situation, we selected a number of 10-Mb chromosomal regions centered at genes annotated as causing Mendelian diseases, and we performed a large-scale leave-one-out cross-validation experiment to assess whether our approach was capable of prioritizing mutations occurring in these regions.
As illustrated in figure 5, in each validation run, one disease mutation, together with all neutral nsSNPs occurring in the same genetic region, is used for the test, and all mutations occurring outside the genetic region are collected to form the training set. Then, an MSRV model is trained on the training set, and mutations in the test set are scored using the trained model. Finally, all mutations in the test set are ranked in nonincreasing order on the basis of their scores. This prioritization procedure is repeated for every disease mutation, and the number of prioritization procedures performed is then equal to the number of disease mutations in all genetic regions. From the prioritization results, we calculated the sensitivity and specificity values corresponding to different rank threshold values. The sensitivity is defined as the percentage of disease mutations ranked above a particular threshold, and the specificity is defined as the percentage of mutations ranked below this threshold.36 Considering that different regions have different numbers of mutations, we adopted the relative rank positions (the percentiles of mutations, independent of the number of mutations) instead of the raw rank positions (the ranks of mutations, depending on the number of mutations) as the rank threshold values. To obtain a comprehensive picture of the performance of our prioritization approach, we plotted its relative rank ROC curve and calculated the AUC. We also calculated the average relative rank position of disease mutations by averaging the percentiles of the disease mutations, because this number gives us a straightforward measure for the performance of the prioritization method.
Figure 5. .

The large-scale leave-one-out cross-validation experiment for assessing prioritization performance. In each validation run, the test set was formed by combining one disease mutation with all neutral nsSNPs occurring in the same genetic region. The training set was formed by all mutations occurring outside the genetic region. Then, the MSRV model was trained on the training set, and mutations in the test set were scored using the trained model. Finally, all mutations in the test set were ranked in decreasing order on the basis of their scores. This prioritization procedure was repeated for every disease mutation, so the number of prioritization procedures performed was equal to the number of disease mutations in all genetic regions.
To validate the effectiveness of the MSRV approach for prioritizing mutations that cause monogenic diseases, we selected from the Swiss-Prot database 30 chromosomal regions that contain mutations that cause Mendelian diseases. The criteria for selecting a region were that the numbers of disease mutations and neutral nsSNPs occurring in the region had to be both comparable and >10. Then we applied the leave-one-out cross-validation experiment to these regions and summarized the results in figure 6A and table 3. For a total of 1,095 disease mutations and 1,062 neutral nsSNPs occurring in the 30 genetic regions, the AUC is 86.6%, and the average relative-rank position for disease mutations is 15.9%. Both criteria suggest that MSRV is effective in putting disease mutations among the top of the ranking. Comparisons with two often-cited programs—SIFT10 and PolyPhen17—show that MSRV is more effective than both SIFT (AUC=75.2%; rank=24.8%) and PolyPhen (AUC=74.8%; rank=25.3%) in prioritizing disease mutations responsible for monogenic diseases.
Figure 6. .

The rank ROC curves for prioritizing disease mutations collected in the Swiss-Prot database. A, The rank ROC curve for the 30 monogenic diseases. B, The rank ROC curve for the 20 polygenic diseases. The zoom-in plots show that the rank ROC curve of MSRV climbs much faster toward the upper left corner of the plots than do those of SIFT and PolyPhen, suggesting that MSRV has better prioritization performance than both of them.
Table 3. .
Prioritization of Mutations Occurring in the 30 Monogenic Disease Regions
| No. of Mutations |
Rank(percentile)a |
||||
| Disease Name | Disease | Neutral | Unannotated | Swiss-Prot Only |
Swiss-Prot, HapMap, and Ensembl |
| A colon tumor (MIM 191170) | 21 | 40 | 92 | 15.0 | 23.3 |
| Antithrombin III deficiency (MIM 107300) | 44 | 75 | 35 | 22.0 | 21.2 |
| Autosomal dominant neurohypophyseal diabetes insipidus (MIM 125700) | 32 | 53 | 65 | 3.6 | 1.2 |
| Autosomal dominant RP (MIM 268000) | 40 | 21 | 32 | 22.2 | 29.3 |
| Best macular dystrophy (MIM 153700) | 86 | 44 | 108 | 17.5 | 6.0 |
| Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (MIM 125310) | 24 | 51 | 109 | 13.5 | 16.2 |
| Chronic nonspherocytic hemolytic anemia (MIM 266200) | 90 | 38 | 158 | 14.9 | 17.6 |
| Citrullinemia type 1 (MIM 215700) | 37 | 44 | 83 | 24.7 | 13.6 |
| Crouzon syndrome (MIM 123500) | 26 | 23 | 27 | 18.1 | 21.3 |
| Dystrophic epidermolysis bullosa (MIM 131750 and 226600) | 59 | 32 | 76 | 9.7 | 24.8 |
| Epidermolysis bullosa simplex Weber-Cockayne type (MIM 131800) | 23 | 87 | 222 | 9.7 | 23.5 |
| Epidermolytic hyperkeratosis (MIM 113800) | 23 | 88 | 225 | 5.9 | 14.3 |
| Familial multiple endocrine neoplasia type I (MIM 131100) | 33 | 45 | 90 | 8.6 | 25.8 |
| Familial porphyria cutanea tarda (MIM 176100) | 27 | 20 | 53 | 21.9 | 25.0 |
| Glycogen storage disease II (MIM 232300) | 32 | 13 | 80 | 16.5 | 7.4 |
| Glycogen storage disease Ib (MIM 232220) | 26 | 18 | 51 | 16.6 | 2.9 |
| Hereditary nonspherocytic hemolytic anemia (MIM 172400) | 21 | 10 | 35 | 22.5 | 2.9 |
| Long QT syndrome type 1 (MIM 192500) | 54 | 29 | 121 | 22.4 | 20.9 |
| Maturity onset diabetes of the young type III (MIM 600496) | 34 | 20 | 36 | 11.2 | 10.9 |
| Mucopolysaccharidosis type IIIB (MIM 252920) | 24 | 46 | 85 | 10.7 | 17.2 |
| Nail-patella syndrome (MIM 161200) | 30 | 15 | 76 | 10.8 | 13.8 |
| OI-III | 22 | 28 | 54 | 3.4 | 19.8 |
| Sjoegren-Larsson syndrome (MIM 270200) | 25 | 19 | 56 | 24.0 | 24.6 |
| Smith-Lemli-Opitz syndrome (MIM 270400) | 44 | 22 | 50 | 13.5 | 23.8 |
| Tay-Sachs disease (MIM 272800) | 38 | 48 | 29 | 21.2 | 20.7 |
| Usher syndrome type 1B (MIM 276903) | 24 | 13 | 23 | 15.5 | 28.0 |
| Very long chain acyl-CoA dehydrogenase deficiency (MIM 201475) | 25 | 40 | 92 | 20.6 | 28.9 |
| X-linked chronic granulomatous disease (MIM 306400) | 36 | 22 | 9 | 15.7 | 7.3 |
| X-linked nephrogenic diabetes insipidus type I (MIM 304800) | 72 | 29 | 42 | 21.2 | 25.6 |
| X-linked recessive myotubular myopathy (MIM 310400) | 23 | 29 | 42 | 12.3 | 17.0 |
| Average relative rank position | 15.9 | 17.8 | |||
The average percentiles of all disease mutations in the genetic regions.
To validate the robustness of the MSRV approach in dealing with “unknown” data, we extracted 2,256 unannotated nsSNPs in those 30 genetic regions from the International HapMap Project,3 and the Ensembl,30 databases. We mixed these unannotated mutations with the known mutations extracted from the Swiss-Prot database, to examine whether our approach was still capable of putting known disease mutations among the top of the ranking. We then repeated the leave-one-out cross-validation experiment. For a total of 1,095 disease mutations and 3,318 “neutral” nsSNPs (1,062 annotated mutations from the Swiss-Prot database and 2,256 unannotated ones from the International HapMap Project and the Ensembl databases) occurring in the 30 genetic regions, the AUC is 82.3%, and the average relative-rank position for disease mutations is 17.8% (table 3). There is no surprise that both criteria are slightly worse than prioritization results for mutations purely from the Swiss-Prot database, because some of the unannotated mutations may actually be responsible for some diseases—a situation that we did not take into consideration. Nevertheless, the performance of our approach shows its robustness.
Prioritization of Mutations Underlying Polygenic Diseases
In many cases, common diseases are not caused by a single mutation in a single gene; instead, multiple mutations occurring in more than one gene across chromosomes may have combined effects in inducing polygenic diseases.37,38 Indeed, polygenic diseases are more general in nature, but the identification of genetic variants responsible for these diseases is more challenging because of their intrinsic complexity, especially when the results of linkage analysis and association studies lack adequate repeatability and robustness.37
To validate the effectiveness of the MSRV approach to polygenic diseases, we extracted from the Swiss-Prot database 20 polygenic diseases with 40 disease-causing proteins (table 4). Using these 40 proteins and their corresponding 10-Mb genetic regions, we extracted a total of 379 disease mutations and 1,261 neutral nsSNPs, and we applied the leave-one-out cross-validation method to prioritize the disease mutations. As shown in figure 6B and table 5, the AUC is 82.3%, and the average relative-rank position for disease mutations is 17.7%. Both criteria suggest that the MSRV approach is capable of enriching mutations underlying polygenic diseases among the top of the ranking. Comparisons with SIFT (AUC=75.4%; rank=27.0%) and PolyPhen (AUC=70.3%; rank=29.7%) also indicate that MSRV is effective in prioritizing mutations underlying polygenic diseases. Application of the leave-one-out cross-validation method to prioritize the mixed data that contain additional 3,967 unannotated mutations from the International HapMap Project and the Ensembl databases shows that the AUC is 81.8%, and the average relative-rank position for disease mutations is 18.2% (table 5), suggesting the robustness of MSRV in prioritizing mutations underlying polygenic diseases.
Table 4. .
The 40 Proteins Associated with the 20 Polygenic Diseases
| Disease Name | Proteins |
| Autosomal recessive osteopetrosis (OPTB1 [MIM 259700]) | CLCN7_HUMAN and VPP3_HUMAN |
| Crohn disease (CD [MIM 266600]) | CAR15_HUMAN and IL10_HUMAN |
| Dejerine-Sottas syndrome (DSS [MIM 145900]) | MYP0_HUMAN and PMP22_HUMAN |
| Epidermolysis bullosa simplex Dowling-Meara type (DM-EBS [MIM 131760]) | K1C14_HUMAN and K2C5_HUMAN |
| Epidermolysis bullosa simplex Koebner type (K-EBS [MIM 131900]) | K1C14_HUMAN and K2C5_HUMAN |
| Fast-channel congenital myasthenic syndrome (FCCMS [MIM 608930]) | ACHA_HUMAN, ACHD_HUMAN, and ACHE_HUMAN |
| Glanzmann thrombasthenia (GT [MIM 273800]) | ITA2B_HUMAN and ITB3_HUMAN |
| Isolated growth hormone deficiency type IB (IGHD IB [MIM 262400]) | GHRHR_HUMAN and SOMA_HUMAN |
| Juvenile polyposis syndrome (JPS [MIM 174900]) | BMR1A_HUMAN and SMAD4_HUMAN |
| LS | ATP6_HUMAN, DHSA_HUMAN, and SURF1_HUMAN |
| Leukoencephalopathy with vanishing white matter (VWM [MIM 603896]) | EI2BA_HUMAN, EI2BB_HUMAN, EI2BD_HUMAN, and EI2BE_HUMAN |
| Li-Fraumeni syndrome (LFS [MIM 151623]) | CD2A1_HUMAN and P53_HUMAN |
| Loeys-Dietz aortic aneurysm syndrome (LDAS [MIM 609192]) | TGFR1_HUMAN and TGFR2_HUMAN |
| Lung cancer (MIM 211980) | BRAF1_HUMAN and EGFR_HUMAN |
| OI-II | CO1A1_HUMAN and CO1A2_HUMAN |
| OI-IV | CO1A1_HUMAN and CO1A2_HUMAN |
| Pachyonychia congenita type 1 (PC1 [MIM 167200]) | K1C16_HUMAN and K2C6A_HUMAN |
| Pachyonychia congenita type 2 (PC2 [MIM 167210]) | K1C17_HUMAN and K2C6B_HUMAN |
| Sitosterolemia (MIM 210250) | ABCG5_HUMAN and ABCG8_HUMAN |
| Tuberous sclerosis complex (TSC [MIM 191100]) | TSC1_HUMAN and TSC2_HUMAN |
Table 5. .
Prioritization of Mutations Occurring in the 40 Genetic Regions Underlying the 20 Polygenic Diseases
| No. of Mutations |
Rank(percentile)a |
||||
| Disease Name | Disease | Neutral | Unannotated | Swiss-Prot Only |
Swiss-Prot, HapMap, and Ensembl |
| OPTB1 | 12 | 91 | 317 | 16.8 | 16.6 |
| CD | 13 | 48 | 94 | 35.8 | 45.9 |
| DSS | 24 | 66 | 221 | 14.1 | 14.7 |
| DM-EBS | 21 | 91 | 373 | 9.0 | 9.8 |
| K-EBS | 15 | 91 | 373 | 22.9 | 28.0 |
| FCCMS | 8 | 86 | 373 | 29.3 | 40.5 |
| GT | 36 | 69 | 380 | 24.6 | 26.9 |
| IGHD IB | 17 | 45 | 191 | 47.3 | 37.9 |
| JPS | 9 | 23 | 84 | 13.4 | 18.6 |
| LS | 9 | 52 | 149 | 19.3 | 17.7 |
| VWM | 11 | 70 | 202 | 28.6 | 36.3 |
| LFS | 16 | 80 | 40 | 6.7 | 7.1 |
| LDAS | 11 | 19 | 48 | 8.2 | 8.6 |
| Lung cancer | 18 | 46 | 86 | 19.7 | 23.2 |
| OI-II | 73 | 46 | 86 | 4.0 | 2.0 |
| OI-IV | 17 | 46 | 86 | 3.6 | 1.9 |
| PC1 | 10 | 91 | 373 | 14.9 | 17.5 |
| PC2 | 11 | 91 | 373 | 12.0 | 13.5 |
| Sitosterolemia | 16 | 24 | 18 | 26.0 | 23.5 |
| TSC | 32 | 111 | 315 | 31.9 | 29.5 |
| Average relative rank position | 17.7 | 18.2 | |||
The average percentiles of all disease mutations in the genetic regions.
The extensive studies mentioned above suggest that, for diseases caused by combined effects of multiple mutations occurring in multiple genes, our approach is capable of enriching disease mutations among the top of the ranking. To demonstrate the prioritization of mutations underlying polygenic diseases, we collected two sets of mutation data related to Alzheimer disease and diabetes and applied the MSRV approach to prioritize suspicious mutations responsible for these diseases.
For Alzheimer disease, we collected the mutation data for familial Alzheimer disease 1 (AD1 [MIM 104300]), familial Alzheimer disease 3 (AD3 [MIM 607822]), and familial Alzheimer disease 4 (AD4 [MIM 606889]). For AD1, the prioritization results showed that two of three disease mutations were ranked at the top among a total of 20 mutations. For AD3, 70 disease mutations and 42 neutral nsSNPs were collected, and all of the top 11 mutations in the prioritization results were disease causing. For AD4, 5 disease mutations and 48 neutral nsSNPs were collected, and 2 of the disease mutations were ranked among the top 5 in the prioritization results.
For diabetes, it has been shown that several mutations occurring in PPARγ (MIM 601487) might impair tissue insulin sensitivity and result in severe insulin resistance.39–43 In our study, the common mutation P12A (mutation P→A at position 12) received a medium prediction score of 0.450 (ranking 9 of 16), suggesting that it was on the boundary of causing diseases. This result was consistent with a previous study of >3,000 individuals,39 which showed that the more common proline allele conferred a modest (1.25-fold) but statistically significant (P=.002) increase in diabetes risk. The mutations P467L and V290M both received high prediction scores (0.969 and 0.800, respectively) and top rank positions (1 of 16 and 3 of 16, respectively), indicating their high disease-causing probabilities. The literature has shown that they existed in two and one subjects, respectively, with severe insulin resistance but were absent from 314 normal alleles.40 In addition to insulin resistance, recent studies have also shown that two other mutations, F388L and R425C, both were associated with familial partial lipodystrophy (FPLD [MIM 151660]).42,43 In our studies, they received high prediction scores (0.877 and 0.881, respectively) and top rank positions (2 of 16 for both). Previous studies found that F338L existed in a family of four members with FPLD but was absent from 520 normal white alleles42 and that R425C existed in one patient with FPLD but was absent from 96 normal alleles.43
These examples demonstrate the effectiveness of the MSRV approach in prioritizing mutations underlying polygenic diseases, illustrate the consistency between our prioritization results and current knowledge of the diseases, and suggest that the potential applications of our approach have a broad scope.
Prioritization of Unclassified Mutations
To demonstrate real applications, we further applied MSRV to 1,517 unclassified mutations collected in the Swiss-Prot database (version 50.2). We first used all annotated mutations in the Swiss-Prot database to train an MSRV model. Then we scored the unclassified mutations and ranked them on the basis of their scores. Finally, we selected the 10 mutations with the highest scores, to see whether they were responsible for certain diseases as indicated in the literature, as described below.
In HBB_HUMAN (MIM 141900), four unclassified mutations, A129V, A70D, D21G, and H117P, received high prediction scores (0.9882, 0.9818, 0.9798, and 0.9773, respectively). The unstable mutation A129V was found in two unrelated black families in association with hemoglobin (Hb) S, Hb C, or β0 thalassemia. It has also been shown that Hbs with this mutation exhibited a decreased oxygen affinity.44 The mutation A70D, known as “Hb Seattle,” has been shown to be associated with a considerable decrease in oxygen affinity45 and to cause mild-to-moderate chronic hemolytic anemia in a white family in the United States,46 as well as a Ukrainian family.47 The mutation D21G, named “Hb Connecticut,” has been found in members of a family of Polish origin living in the United States. Several individuals with this mutation exhibited mild anemia. Hbs with this mutation also exhibited decreased oxygen affinity and slightly decreased effect of allosteric effectors on the oxygen equilibrium properties.48 The mutation H117P, called “Hb Saitama,” was identified in a 23-year-old Japanese female with hemolytic anemia and jaundice but was absent from other members of the family.49 From these evidences, we conclude that these mutations are likely to cause anemia.
In CD36_HUMAN (MIM 173510), three unclassified mutations, I270T, T173A, and E122K, were predicted with high scores (0.9782, 0.9717, and 0.8421, respectively). Studies have identified these mutations from 12 individuals from a malaria-endemic area in West Africa (8 with small spleens and 4 with large ones).50 Therefore, these mutations are likely to cause malaria.
In CASR_HUMAN (MIM 601199), two mutations, E767K and E127A, received high scores of 0.9785 and 0.9761, respectively. The mutation E767K was reported in a family with autosomal dominant hypocalcemia and is suggested to be associated with familial hypocalciuric hypercalcaemia (MIM 145980) and neonatal severe hyperparathyroidism (MIM 239200).51 It has also been found that the missense mutation E127A caused familial hypocalcemia in affected members of one family that is heterozygous for such a mutation.52
In CD22_HUMAN (MIM 107266), one mutation, Q152E, received a high score of 0.9789. Studies of 207 healthy Japanese individuals and 68 patients with systemic lupus erythematosus (SLE [MIM 152700])53 have shown that this mutation existed with a marginally higher frequency in patients with SLE (3 of 68 [4.4%]) than in healthy individuals (1 of 207 [0.5%]) (P=.048). This evidence suggests that this mutation is likely to cause SLE.
Discussion
In this article, we proposed a bioinformatics approach that uses a novel statistical learning model (MSRV) with a set of improved sequence-based numeric features to effectively prioritize candidate nsSNPs occurring in genetic regions. Our approach has several advantages. First, the prioritization of nsSNPs within a genetic region gives users flexibility in dealing with both monogenic and polygenic disease mutations—that is, users can select the most likely disease-causing candidates for further studies. Second, our approach does not rely on protein-structure information; instead, it is based on simple numeric features calculated from protein sequences and multiple-sequence alignments, and this maximizes the scope of the method’s applications. Third, our approach adopts a simple but powerful statistical learning model (MSRV) that captures subtle characteristics of mutations and outperforms other methods. Finally, our approach can be combined with case-control data in large-scale disease-mapping projects. Traditional statistical methods suffer from serious multiple-hypothesis-testing problems in dealing with large-scale data and polygenic diseases. In contrast, our method is based purely on biophysical and biochemical information of disease mutations and evolutionary information of the corresponding sequences; thus, it complements the traditional methods.
Certainly, our approach can be improved in the following directions. First, we currently use the Pfam multiple-sequence alignment31 to extract conserved protein domains for the query protein sequence. As a result, we are limited to the mutations occurring in known protein domains. This limitation can be overcome by using some other multiple-sequence alignment methods, such as BLAST,54 PSI-BLAST,55 and PANTHER.56 Second, partitioning the mutations into 20 categories according to the original amino acids is natural and feasible but not necessarily optimal. Third, most of the current studies (including ours) use classification models that are aimed at predicting the categorical mutation effects, because effects of mutations are given as categorical values in most mutation databases. As a long-term goal, prediction of the subtle effects of mutations causing common diseases would be a significant step in this field. The major difficulty is the lack of adequate samples for use in building statistical learning models. When the database resources are available, many of the current classification models (including ours) could be extended to regression models, to establish the relationships between the regression results and the subtle modest effects. Finally, our approach is limited to nsSNPs that have been found to be the major reason for causing diseases. However, mutations in other genome regions such as the transcriptional-factor binding sites and promoter regions are also known to cause diseases. Further studies are needed for these mutations.
Acknowledgments
We appreciate the two anonymous reviewers for suggested revisions that significantly improved the presentation of this article. We thank Dr. Pauline Ng for providing a stand-alone version of SIFT, and we thank Dr. Shamil Sunyaev for providing a stand-alone version of PolyPhen. This work was partly supported by National Institutes of Health (NIH)/National Science Foundation Joint Mathematical Biology Initiative grant DMS-0241102, NIH grants P50 HG 002790 and R01 LM008991-01, and the Alfred P. Sloan Research Fellowship.
Web Resources
The URLs for data presented herein are as follows:
- Ensembl, http://www.ensembl.org/
- International HapMap Project, http://www.hapmap.org/
- MSRV, http://msms.usc.edu/msrv/
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for ARMD2, OI-II, STEA2_HUMAN, CALU_HUMAN, LHON, MELAS, LS, AD1, AD3, AD4, PPARγ, FPLD, HBB_HUMAN, CD36_HUMAN, CASR_HUMAN, familial hypocalciuric hypercalcaemia, neonatal severe hyperparathyroidism, CD22_HUMAN, SLE, congenital bilateral absence of the vas deferens, colorectal cancer, polycystic kidney disease type I, Alport syndrome, RP, Wilson disease, cystic fibrosis, OI-IV, OI-III, A colon tumor, antithrombin III deficiency, autosomal dominant neurohypophyseal diabetes insipidus, autosomal dominant RP, Best macular dystrophy, cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy, chronic nonspherocytic hemolytic anemia, citrullinemia type 1, Crouzon syndrome, dystrophic epidermolysis bullosa, epidermolysis bullosa simplex Weber-Cockayne type, epidermolytic hyperkeratosis, familial multiple endocrine neoplasia type I, familial porphyria cutanea tarda, glycogen storage disease II, glycogen storage disease Ib, hereditary nonspherocytic hemolytic anemia, long QT syndrome type 1, maturity onset diabetes of the young type III, mucopolysaccharidosis type IIIB, nail-patella syndrome, Sjoegren-Larsson syndrome, Smith-Lemli-Opitz syndrome, Tay-Sachs disease, Usher syndrome type 1B, very long chain acyl-CoA dehydrogenase deficiency, X-linked chronic granulomatous disease, X-linked nephrogenic diabetes insipidus type I, X-linked recessive myotubular myopathy, OPTB1, CD, DSS, DM-EBS, K-EBS, FCCMS, GT, IGHD IB, JPS, VWM, LFS, LDAS, lung cancer, PC1, PC2, sitosterolemia, and TSC)
- Pfam, http://www.sanger.ac.uk/Software/Pfam/
- Swiss-Prot, http://expasy.org/sprot/
References
- 1.Lander ES, Schork NJ (1994) Genetic dissection of complex traits. Science 265:2037–2047 10.1126/science.8091226 [DOI] [PubMed] [Google Scholar]
- 2.Glazier AM, Nadeau JH, Aitman TJ (2002) Finding genes that underlie complex traits. Science 298:2345–2349 10.1126/science.1076641 [DOI] [PubMed] [Google Scholar]
- 3.The International HapMap Consortium (2005) A haplotype map of the human genome. Nature 437:1299–1320 10.1038/nature04226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen ZT, et al (2005) Genome sequencing in microfabricated high-density picolitre reactors. Nature 437:376–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blazej RG, Kumaresan P, Mathies RA (2006) Microfabricated bioprocessor for integrated nanoliter-scale Sanger DNA sequencing. Proc Natl Acad Sci USA 103:7240–7245 10.1073/pnas.0602476103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.The UniProt Consortium (2007) The universal protein resource (UniProt). Nucl Acids Res 35:D193–D197 10.1093/nar/gkl929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Renell D, Bouvier SE, Hardy LW, Poteete AR (1991) Systematic mutation of bacteriophage T4 lysozyme. J Mol Biol 222:67–87 10.1016/0022-2836(91)90738-R [DOI] [PubMed] [Google Scholar]
- 8.Markiewicz P, Kleina LG, Cruz C, Ehret S, Miller JH (1994) Genetic studies of the lac repressor XIV: analysis of 4000 altered Escherichia coli lac repressors reveals essential and non-essential residues, as well as “spacers” which do not require a specific sequence. J Mol Biol 240:421–433 10.1006/jmbi.1994.1458 [DOI] [PubMed] [Google Scholar]
- 9.Suckow YJ, Markiewicz P, Kleina LG, Miller J, Kisters-Woike B, Muller-Hill B (1996) Genetic studies of the lac repressor XV: 4000 single amino acid substitutions and analysis of the resulting phenotypes on the basis of the protein structure. J Mol Biol 261:509–523 10.1006/jmbi.1996.0479 [DOI] [PubMed] [Google Scholar]
- 10.Ng PC, Henikoff S (2001) Predicting deleterious amino acid substitutions. Genome Res 11:863–874 10.1101/gr.176601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chasman D, Adams RM (2001) Predicting the functional consequences of non-synonymous single nucleotide polymorphisms: structure-based assessment of amino acid variation. J Mol Biol 307:683–706 10.1006/jmbi.2001.4510 [DOI] [PubMed] [Google Scholar]
- 12.Krishnan VG, Westhead DR (2003) A comparative study of machine-learning methods to predict the effects of single nucleotide polymorphisms on protein function. Bioinformatics 19:2199–2209 10.1093/bioinformatics/btg297 [DOI] [PubMed] [Google Scholar]
- 13.Verzilli CJ, Whittaker JC, Stallard N, Chasman D (2005) A hierarchical Bayesian model for predicting the functional consequences of amino-acid polymorphisms. Appl Statist 54:191–206 [Google Scholar]
- 14.Saunders CT, Barker D (2002) Evaluation of structural and evolutionary contributions to deleterious mutation prediction. J Mol Biol 322:891–901 10.1016/S0022-2836(02)00813-6 [DOI] [PubMed] [Google Scholar]
- 15.Lau AY, Chasman DI (2004) Functional classification of proteins and protein variants. Proc Natl Acad Sci USA 101:6576–6581 10.1073/pnas.0305043101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sunyaev S, Ramensky V, Koch I, Lathe III W, Kondrashov AS, Bork P (2001) Prediction of deleterious human alleles. Hum Mol Genet 10:591–597 10.1093/hmg/10.6.591 [DOI] [PubMed] [Google Scholar]
- 17.Ramensky V, Bork P, Sunyaev S (2002) Human non-synonymous SNPs: server and survey. Nucl Acids Res 30:3894–3900 10.1093/nar/gkf493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Balasubramanian S, Xia Y, Freinkman E, Gerstein M (2005) Sequence variation in G-protein-coupled receptors: analysis of single nucleotide polymorphisms. Nucl Acids Res 33:1710–1721 10.1093/nar/gki311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yue P, Melamud E, Moult J (2006) SNPs3D: candidate gene and SNP selection for association studies. BMC Bioinformatics 7:166 10.1186/1471-2105-7-166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ferrer-Costa C, Orozco M, de la Cruz X (2002) Characterization of disease-associated single amino acid polymorphisms in terms of sequence and structure properties. J Mol Biol 315:771–786 10.1006/jmbi.2001.5255 [DOI] [PubMed] [Google Scholar]
- 21.Ferrer-Costa C, Orozco M, de la Cruz X (2004) Sequence-based prediction of pathological mutations. Proteins 57:811–819 10.1002/prot.20252 [DOI] [PubMed] [Google Scholar]
- 22.Bao L, Cui Y (2005) Prediction of the phenotypic effects of non-synonymous single nucleotide polymorphisms using structural and evolutionary information. Bioinformatics 21:2185–2190 10.1093/bioinformatics/bti365 [DOI] [PubMed] [Google Scholar]
- 23.Jiang R, Yang H, Sun F, Chen T (2006) Searching for interpretable rules for disease mutations: a simulated annealing bump hunting strategy. BMC Bioinformatics 7:417 10.1186/1471-2105-7-417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mitchell MT (1997) Machine learning. McGraw-Hill, New York [Google Scholar]
- 25.Breiman L, Friedman J, Olshen R, Stone C (1984) Classification and regression trees. Wadsworth, Belmont, CA [Google Scholar]
- 26.Quinlan JR (1993) C4.5: programs for machine learning. Morgan Kaufmann, San Mateo, CA [Google Scholar]
- 27.Vapnik V (1998) Statistical learning theory. John Wiley, New York [Google Scholar]
- 28.Fan RE, Chen PH, Lin CJ (2005) Working set selection using the second order information for training support vector machines. J Machine Learning Res 69:1889–1918 [Google Scholar]
- 29.Breiman L (2001) Random forests. Machine Learning 45:5–32 10.1023/A:1010933404324 [DOI] [Google Scholar]
- 30.Birney E, Andrews D, Caccamo M, Chen Y, Clarke L, Coates G, Cox T, Cunningham F, Curwen V, Cutts T, et al (2006) Ensembl 2006. Nucl Acids Res 34:D556–D561 10.1093/nar/gkj133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Robert DF, Jaina M, Berjamin SB, Sam GJ, Volker H, Timo L, Simon M, Mhairi M, Ajay K, Richard D, et al (2006) Pfam: clans, web tools and services. Nucl Acids Res 34:D247–D251 10.1093/nar/gkj149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kyte J, Doolittle RF (1982) Biological sequence analysis: probabilistic models of proteins and nucleic acids. J Mol Biol 157:105–132 10.1016/0022-2836(82)90515-0 [DOI] [PubMed] [Google Scholar]
- 33.Durbin R, Eddy S, Krogh A, Mitchison G (1998) Biological sequence analysis: probabilistic models of proteins and nucleic acids. Cambridge University Press, Cambridge, United Kingdom [Google Scholar]
- 34.Levitte M (1978) Conformational preferences of amino acids in globular proteins. Biochemistry 17:4277–4285 10.1021/bi00613a026 [DOI] [PubMed] [Google Scholar]
- 35.Berg JM, Tymoczko JL, Stryer L (2002) Biochemistry. WH Freeman, New York [Google Scholar]
- 36.Aerts S, Lambrechts D, Maity S, Van Loo P, Coessens B, De Smet F, Tranchevent LC, De Moor B, Marynen P, Hassan B, et al (2006) Gene prioritization through genomic data fusion. Nat Biotechnol 24:537–544 (erratum 24:719) 10.1038/nbt1203 [DOI] [PubMed] [Google Scholar]
- 37.Hirschhorn JN, Lohmueller K, Byrne K, Hirschhorn K (2002) A comprehensive review of genetic association studies. Genet Med 4:45–61 [DOI] [PubMed] [Google Scholar]
- 38.Thomas PD, Kejariwal A (2004) Coding single-nucleotide polymorphisms associated with complex vs Mendelian disease: evolutionary evidence for differences in molecular effects. Proc Natl Acad Sci USA 101:15398–15403 10.1073/pnas.0404380101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Altshuler D, Hirschhorn JN, Klannemark M, Lindgren CM, Vohl MC, Nemesh J, Lane CR, Schaffner SF, Bolk S, Brewer C, et al (2000) The common PPARγ Pro12Ala polymorphism is associated with decreased risk of type 2 diabetes. Nat Genet 26:76–80 10.1038/79839 [DOI] [PubMed] [Google Scholar]
- 40.Barroso I, Gurnell M, Crowley VE, Agostini M, Schwabe JW, Soos MA, Maslen GL, Williams TD, Lewis H, Schafer AJ, et al (1999) Dominant negative mutations in human PPARγ associated with severe insulin resistance, diabetes mellitus and hypertension. Nature 402:880–883 [DOI] [PubMed] [Google Scholar]
- 41.Agostini M, Schoenmakers E, Mitchell C, Szatmari I, Savage D, Smith A, Rajanayagam O, Semple R, Luan J, Bath L, et al (2006) Non-DNA binding, dominant-negative, human PPARγ mutations cause lipodystrophic insulin resistance. Cell Metab 4:303–311 10.1016/j.cmet.2006.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hegele RA, Cao H, Frankowski C, Mathews ST, Leff T (2002) PPARG F388L, a transactivation-deficient mutant, in familial partial lipodystrophy. Diabetes 51:3586–3590 10.2337/diabetes.51.12.3586 [DOI] [PubMed] [Google Scholar]
- 43.Agarwal AK, Garg A (2002) A novel heterozygous mutation in peroxisome proliferator-activated receptor-γ gene in a patient with familial partial lipodystrophy. J Clin Endocrinol Metab 87:408–411 10.1210/jc.87.1.408 [DOI] [PubMed] [Google Scholar]
- 44.Merault G, Keclard L, Garin J, Poyart C, Blouquit Y, Arous N, Galacteros F, Feingold J, Rosa J (1986) Hemoglobin la desirade alpha A2 beta 2 129(H7) Ala—Val: a new unstable hemoglobin. Hemoglobin 10:593–605 [DOI] [PubMed] [Google Scholar]
- 45.Stamatoyannopoulos G, Parer JT, Finch CA (1969) Physiologic implications of a hemoglobin with decreased oxygen affinity (hemoglobin Seattle). N Eng J Med 281:916–919 [DOI] [PubMed] [Google Scholar]
- 46.Huehns ER, Hecht F, Yoshida A, Stamatoyannopoulos G, Hartman J, Motulsky AG (1970) Hemoglobin-Seattle (α2A β276 Glu): an unstable hemoglobin causing chronic hemolytic anemia. Blood 36:209–218 [PubMed] [Google Scholar]
- 47.Chow EY, Haley LP, Krikler SH, Wadsworth LD (1994) Hb Seattle [beta 70(E14) Ala→Asp]: a report of a second kindred in a Ukrainian family. Hemoglobin 18:231–234 [DOI] [PubMed] [Google Scholar]
- 48.Moo-Penn WF, McPhedran P, Bobrow S, Johnson MH, Jue DL, Olsen KW (1981) Hemoglobin Connecticut (beta 21 (B3) Asp leads to Gly): a hemoglobin variant with low oxygen affinity. Am J Hematol 11:137–145 10.1002/ajh.2830110204 [DOI] [PubMed] [Google Scholar]
- 49.Ohba Y, Hasegawa Y, Amino H, Miwa S, Nakatsuji T, Hattori Y, Miyaji T (1983) Hemoglobin saitama or beta 117 (G19) His leads to Pro, a new variant causing hemolytic disease. Hemoglobin 7:47–56 [DOI] [PubMed] [Google Scholar]
- 50.Gelhaus A, Scheding A, Browne E, Burchard GD, Horstmann RD (2001) Variability of the CD36 gene in West Africa. Hum Mut 18:444–450 10.1002/humu.1215 [DOI] [PubMed] [Google Scholar]
- 51.Uckun-Kitapci A, Underwood LE, Zhang J, Moats-Staats B (2005) A novel mutation (E767K) in the second extracellular loop of the calcium sensing receptor in a family with autosomal dominant hypocalcemia. Am J Med Genet A 132:125–129 [DOI] [PubMed] [Google Scholar]
- 52.Pollak MR, Brown EM, Estep HL, McLaine PN, Kifor O, Park J, Hebert SC, Seidman CE, Seidman JG (1994) Autosomal dominant hypocalcaemia caused by a Ca2+-sensing receptor gene mutation. Nat Genet 8:303–307 10.1038/ng1194-303 [DOI] [PubMed] [Google Scholar]
- 53.Hatta Y, Tsuchiya N, Matsushita M, Shiota M, Hagiwara K, Tokunaga K (1999) Identification of the gene variations in human CD22. Immunogenetics 49:280–286 10.1007/s002510050494 [DOI] [PubMed] [Google Scholar]
- 54.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410 [DOI] [PubMed] [Google Scholar]
- 55.Altschul S, Madden T, Schaffer A, Zhang J, Zhang Z, Miller W, Lipman D (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucl Acids Res 25:3389–3402 10.1093/nar/25.17.3389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Thomas PD, Kejariwal A, Campbell MJ, Mi HY, Diemer K, Guo N, Ladunga I, Ulitsky-Lazareva B, Muruganujan A, Rabkin S, et al (2003) PANTHER: a browsable database of gene products organized by biological function, using curated protein family and subfamily classification. Nucl Acids Res 31:334–341 10.1093/nar/gkg115 [DOI] [PMC free article] [PubMed] [Google Scholar]