

Abstract
Capacitative Ca2+ entry is a component of the inositol-lipid signaling in which depletion of inositol 1,4,5-trisphosphate (InsP3)-sensitive Ca2+ stores activates Ca2+ influx by a mechanism that is still unknown. This pathway plays a central role in cellular signaling, which is mediated by many hormones, neurotransmitters, and growth factors. Studies of Drosophila photoreceptors provided the first putative capacitative Ca2+ entry mutant designated transient receptor potential (trp) and a Drosophila gene encoding TRP-like protein (trpl). It is not clear how the Ca2+ store depletion signal is relayed to the plasma membrane and whether both TRP and TRPL participate in this process. We report here that coexpressing Drosophila TRP and TRPL in Xenopus oocytes synergistically enhances the endogenous Ca2+-activated Cl− current and produces a divalent inward current. Both of these currents are activated by Ca2+ store depletion. In the absence of Ca2+, Mg2+ is the main charge carrier of the divalent current. This current is characterized by lanthanum sensitivity and a voltage-dependent blocking effect of Mg2+, which is relieved at both hyperpolarizing (inward rectification) and depolarizing (outward rectification) potentials. The store-operated divalent current is neither observed in native oocytes nor in oocytes expressing either TRP or TRPL alone. The production of this current implicates a cooperative action of TRP and TRPL in the depletion-activated current.
Keywords: signal transduction, phosphoinositide signaling, trp mutant, calcium stores
Stimulation of many cell types by hormones, neurotransmitters, and growth-factors activates the inositol-lipid pathway leading to release of Ca2+ from intracellular stores. This Ca2+ release is followed by an influx of extracellular Ca2+ via a capacitative Ca2+ entry (CCE) mechanism (1). It has been suggested that activation of the surface membrane Ca2+ channels is caused by the depletion of Ca2+ in the internal stores and not by the release per se (1, 2, 3, 4, 5, 6, 7, 8). Although CCE has been described in a large variety of cells and tissues (summarized in refs. 5 and 9), its mechanism of activation and molecular components is largely unknown. Visual transduction in Drosophila, which is triggered by the inositol lipid signaling (reviewed in refs. 10, 11, 12, 13) has been suggested by Minke and Selinger (14) as a powerful model system to study CCE. This suggestion was based on the properties of a Drosophila mutant designated transient receptor potential trp (15, 16). In the trp mutant the photoreceptor potential declines to baseline during prolonged intense illumination and renders the photoreceptor cell inactive. The receptor potential recovers within 1 min in the dark (17). Because lanthanum (La3+), a known non-specific blocker of Ca2+ channels, mimics many aspects of the trp phenotype in wild-type flies (18, 19), it has been suggested that trp has defects in Ca2+ entry into the photoreceptor cells (14). This prediction has been strongly supported by subsequent experiments demonstrating that the high Ca2+ permeability of the light-activated channels (20, 21) is reduced by about 10-fold in the trp mutant (22). Furthermore, fluorimetric measurements of cellular Ca2+ (23, 24) and measurements of a reduction in extracellular Ca2+ (25) have shown that Ca2+ influx is reduced by about 3-fold in the trp mutant relative to wild-type Drosophila, suggesting that TRP takes part in the main route of Ca2+ entry into the photoreceptor cells (10, 13, 14).
Molecular cloning of Drosophila trp (26, 27) and a related Drosophila gene designated transient receptor potential-like, trpl (28), revealed that their putative transmembrane domain exhibits weak but significant similarity to the α-subunit of the voltage-gated Ca2+ channel (28). Recently, a trpl mutant lacking TRPL has been isolated by Zuker and colleagues (29). The trpl mutant has a receptor potential similar to wild type and it reveals a strong phenotype only in a trp background. Thus, the double mutant trpl;trp is almost totally unresponsive to light (29). This study has suggested that TRP and TRPL are capable of responding to light activation independently of each other, but it does not exclude the possibility that TRP and TRPL form a multimeric channel (29). Additional subunit might exist but its function should depend on the presence of TRP and TRPL. Molecular and physiological data on TRP led a number of investigators to express the Drosophila TRP or TRPL in insect Sf9 cells (30, 31) or human homologue genes of trp (32, 33) in COS, Chinese hamster ovary, and Sf9 cells (9, 34). The heterologously expressed TRPL forms a constitutively active non-selective cation conductance, which could be enhanced by activation of the inositol lipid cascade (31, 35, 36). Heterologous expression of Drosophila TRP forms a conductance, which is activated by Ca2+ store depletion following treatment with the microsomal Ca2+ ATPase inhibitor, thapsigargin (30, 37, 38). Expression of Drosophila TRP in Xenopus oocytes enhances the endogenous Ca2+-activated Cl− conductance following depletion of the inositol 1,4,5-trisphosphate (InsP3)-sensitive Ca stores (37). Recently, six trp-related genes of the mouse genome were identified in addition to new human homologues of trp. The expressed human gene products enhanced Ca2+ influx following Ca2+ store depletion (9, 34). Furthermore, expression in L cells of small portions of the mouse trp genes, in antisense orientation, suppressed the endogenous capacitative Ca2+ entry (9).
Here we report that heterologous coexpression of Drosophila TRP and TRPL in Xenopus oocytes synergistically enhanced the endogenous Ca2+-activated Cl− current upon Ca2+ stores depletion. In addition, the coexpressed TRP and TRPL produced a novel divalent cation current, which was activated by store depletion but could not be similarly activated when either TRP or TRPL were individually expressed. These findings strongly suggest a cooperative action of TRP and TRPL in the depletion-activated current.
MATERIALS AND METHODS
Analysis of Heterologous Expression of TRP and TRPL.
The trp (26, 27) and trpl (28) cDNAs were subcloned into PGEMHE expression vector, which was constructed for expression in Xenopus oocytes (39, 40). Capped cRNA was synthesized in vitro and tested by translation in a rabbit reticulocyte lysate system before injection into oocytes. An amount of 0.2 mg/ml trp, 0.02 mg/ml trpl, and 0.2 mg/ml trp in combination with 0.02 mg/ml trpl cRNA, all in final volume of 50 nl, were injected into oocytes. The efficiency of translation of the trp and trpl cRNAs in oocytes was tested by Western blots on days 3–5 after injection. Aliquots equivalent to three oocytes were used for Western blot analyses. Aliquots equivalent to three Drosophila heads were used as markers for TRP and TRPL proteins (Fig. 1). Monoclonal anti-TRP antibody (mAb83F6) (41) and affinity-purified rabbit polyclonal anti-TRPL antibodies raised against the C-terminal hexadeca peptide of TRPL and enhanced chemiluminescense (Amersham) were used to visualized the TRP and TRPL proteins (Fig. 1).
Figure 1.
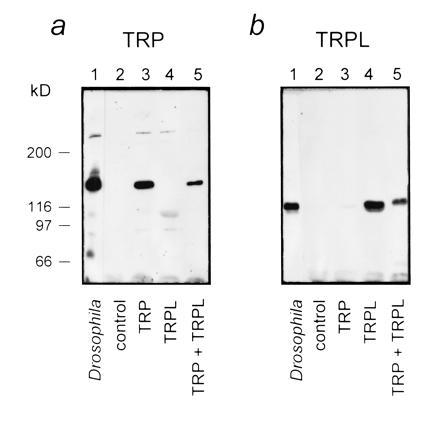
Heterologous expression of TRP and TRPL gives rise to large amounts of TRP and TRPL proteins. Western blots show the expression of TRP (a) and TRPL (b) in oocytes injected with cRNA of trp, trpl, and trp+trpl, as indicated. Control (uninjected oocytes) and extract of three wild-type Drosophila heads (Drosophila) show that the equivalent molecular sizes of the Drosophila TRP and TRPL were produced in the oocytes, which cannot be confused with endogenous oocyte protein of similar structure and size. The data of this figure were highly reproducible in oocytes from different frogs (n = 8).
Fluorescent Measurements of Ca2+ Changes.
Fluorescent confocal Ca2+ measurements and electrophysiological studies were carried out 3–5 days after injection of cRNAs into oocytes. Oocytes were maintained and examined as described (42). Non-injected (control) oocytes or oocytes injected with cRNA encoding TRP, TRPL, and their combination (TRP+TRPL) were loaded with 50 μM of fluo-3 and 50 μM Fura Red (Molecular Probes). Control oocytes injected with 50 nl of cRNA buffer gave results indistinguishable from those of non-injected oocytes. Fluorescent dyes were injected approximately 20 min before the measurements together with 10 μM inositol 1,4,5,-trisphosphate 3-deoxy-3-fluoro (InsP3-F) (final concentration, Sigma). The oocytes were bathed in Ca2+-free ND96 medium containing: 96 mM NaCl, 2 mM KCl, 5 mM Hepes, 10 mM MgCl2, 0.2 mM EGTA. Intracellular Ca2+ changes were measured before and during Ca2+ application and after washout of the Ca2+-containing solution. In the Ca2+-containing solution, EGTA was replaced with 2 mM CaCl2 and MgCl2 was reduced to 1 mM. Data acquisition was performed using the Sarastro confocal laser scanning microscope where the excitation light was 488 nm, the dichroic mirror was LP595, and the emission filters were 640DF35 and 530DF30 (Omega Optical, Brattleboro, VT) for the Fura Red and fluo-3 fluorescence, respectively. Scans included 512 × 512 pixels, with a pixel size of 2 μm using the Pl Fl 10/0.30 (Leitz) objective lens and a pinhole of 100 mm. Ratios were calculated as F640/F530 fluorescence. All oocytes were also tested electrophysiologically after the fluorescence measurements and gave results similar to those presented in Fig. 3.
Figure 3.
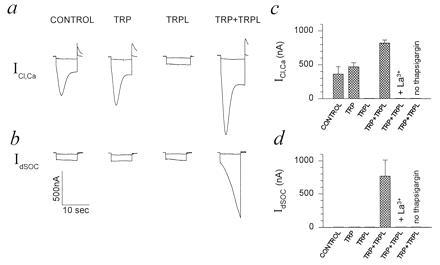
Functional coexpression of TRP+TRPL in Xenopus oocytes produced a capacitative Ca2+ entry system revealed by ICl,Ca and IdSOC. Shown is a single experiment, employing several oocytes from a single frog that were maintained and treated together. (a) Measurements of currents (ICl,Ca) in thapsigargin-treated oocytes (1 μM in Ca2+-free solution for 1.5–2 hr). ICl,Ca was activated by stepping the holding voltage from −10 mV to −30 mV (upper traces) and to −120 mV (bottom traces) to show the relatively small instantaneous leak current in solution containing 1 mM Ca2+ (see Fig. 2). Oocytes were injected with cRNA 5 days before the measurements. (b) Measurements of IdSOC were carried out as described in a. The same oocytes were perfused with Ca2+-free ND96 solution (10 mM Mg2+). In some of the measurements, 2 mM EGTA was injected into the oocytes 1–2 hr before the recordings, but no significant effect on IdSOC was found. IdSOC was totally and reversibly blocked by addition of 1 mM La3+ to the perfusate (c and d). IdSOC was also blocked reversibly by 500 μM, but not by 50 μM, La3+ (n = 12). (c and d) Histograms summarizing the results from all the oocytes of the same experimental run of a and b. Five to 10 oocytes were used for each of the experimental groups of a and b. The histograms present the mean and SEM of the peak ICl,Ca (c) and maximal IdSOC (d) measured at −120-mV holding potentials after the instantaneous leak currents were subtracted from all current traces. The TRP+TRPL group was significantly different from the other oocyte groups of c (P < 0.01). The control and TRP groups were not significantly different (P > 0.05).
Electrophysiological Measurements Using Voltage-Clamped Oocytes.
For electrophysiology, oocytes were impaled with two glass microelectrodes, which were filled with 3 M KCl with a resistance of 0.5–2.0 MΩ. The cells were voltage clamped using the standard two electrode-voltage-clamp technique. Drugs were added externally to the perfusate, while InsP3-F (10 mM, final concentration) was injected into the oocyte with a Drummond 10 ml microdispenser. The small differences between the histograms of Fig. 3 c and d and the summary histogram of Fig. 5 arise from differences in the quality of the oocytes used in the various experiments. The most significant results were obtained in oocyte groups showing no deterioration with age and minimal leak currents.
Figure 5.
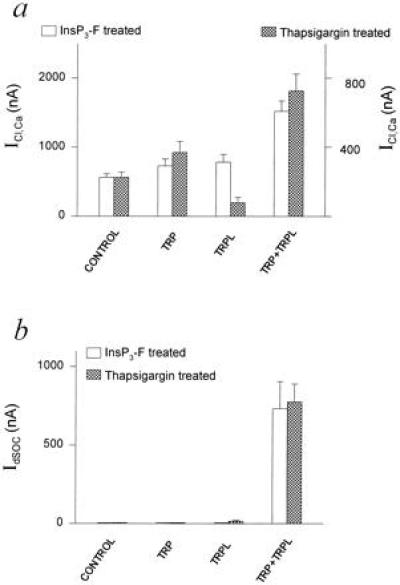
Histograms summarizing ICl,Ca and IdSOC in TRP, TRPL, and TRP+TRPL-expressing oocytes in experiments similar to those in Fig. 3. (a) ICl,Ca measured in InsP3-F-treated oocytes (left ordinate) or thapsigargin-treated oocytes (right ordinate). Eleven independent experiments used 16–64 oocytes in each group. The TRP+TRPL groups were significantly different from the other oocyte groups (P < 0.01), whereas the other oocyte groups were not significantly different in the InsP3-F-treated oocytes (P > 0.05). In the thapsigargin-treated oocytes all groups were significantly different from each other (P < 0.01 for a comparison between the control TRPL and TRP+TRPL groups; P < 0.05 for a comparison between the control and TRP group). (b) IdSOC measured in InsP3-F or thapsigargin-treated oocytes. Seven independent experiments used 8–22 oocytes in each group.
Whole-Cell Recordings of Light-Induced Current (LIC) in Drosophila.
Current-voltage relationship of the leak-subtracted peak LIC were plotted from responses to identical 100 ms light flashes of orange light (OG 590, Schott edge filter). The LICs were measured from Drosophila isolated ommatidia during whole-cell voltage clamp recordings as described (22, 23). Bath solution contained: 120 mM NaCl, 5 mM KCl, 10 mM TES buffer (pH 7.15), 30 mM sucrose, 8 MgSO4, with no Ca2+ added. The whole-cell recording pipette contained: 100 mM CsCl, 15 mM TEA, 2 mM MgSO4, 10 mM TES buffer (pH 7.15), 4 mM MgATP, 0.4 mM Na2GTP.
RESULTS
Western Blot Analysis Showing Expression of TRP and TRPL.
Expression of TRP and TRPL proteins in Xenopus oocytes microinjected with cRNA to their respective genes was demonstrated by Western blot analysis using monoclonal anti-TRP antibody (41) (Fig. 1a) or affinity-purified anti-TRPL antibody (Fig. 1b). We have found that high levels of TRPL expression largely reduced the survival time of the oocytes and, therefore, the level of TRPL expression should be carefully controlled. In contrast, the expression level of TRP had no effect on the survival time of the oocytes. Accordingly, we injected a 10-fold larger amount of cRNA encoding TRP than TRPL. The significant reduction in expression of TRP or TRPL in oocytes coexpressing TRP+TRPL is probably due to competition on the protein synthesis system of the oocyte.
Fluorescent Measurements of Ca2+ Changes Reveal Enhanced Ca2+ Permeability in TRP+TRPL-Expressing Oocytes.
Fluorescent Ca2+ indicators were used to detect intracellular Ca2+ changes in oocytes expressing TRP, TRPL, or both (TRP+TRPL, Fig. 2). CCE was induced by depletion of the InsP3-sensitive Ca2+ stores and measured as a change in intracellular Ca2+ following application of Ca2+ to the external medium. Confocal images of Fura Red/fluo-3 ratio difference revealed an enhanced rise in cytoplasmic Ca2+ in TRP+TRPL-expressing oocytes (Fig. 2a). Spatial averages of fluorescence ratios at different times are shown in Fig. 2b. The oocytes expressing TRP+TRPL showed a 2- to 3-fold larger increase in cytoplasmic Ca2+ than did control oocytes or oocytes individually expressing either TRP or TRPL, suggesting a larger Ca2+ permeability of the TRP+TRPL-expressing oocytes (Fig. 2b). The Western blots (Fig. 1) indicate that the large response of the TRP+TRPL system (Fig. 2) could not be due to excessive expression of either TRP or TRPL.
Figure 2.
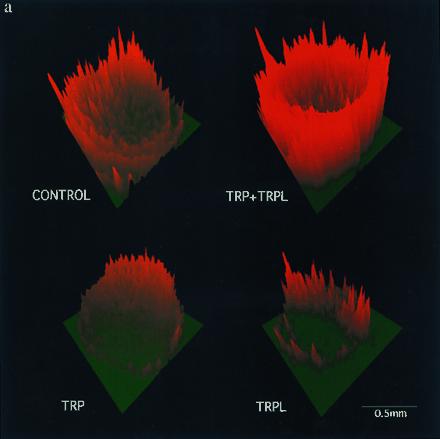
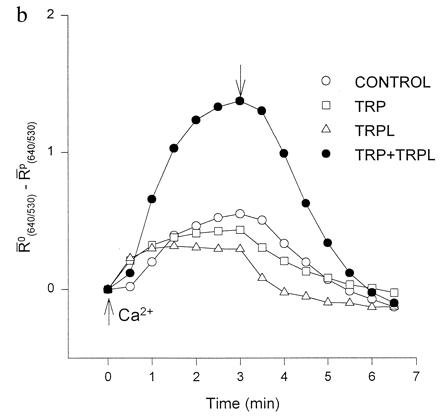
Coexpression of TRP and TRPL largely enhances Ca2+ influx into Ca2+ stores-depleted oocytes. (a) Confocal images of ratio changes between resting and peak Ca2+ levels during application of Ca2+ containing solution (2 mM). Changes of ratios were coded as the green-to-red gradient together with the z-axis magnitude. One pair of optical sections across the oocyte of about 70 μm deep was analyzed to form each image. The “ring” shape of the image is due to the melanin pigmentation, which interferes with the fluorescence detection from the center of the oocyte. (b) A plot of changes in fluorescence ratio as a function of time in the oocytes shown in a. The ordinate plots the average ratio difference (R̄o(640/530) − R̄P(640/530)) where R̄ is the averaged fluorescence ratio of the scan before (R̄o) or during and after (R̄P) Ca2+ application. The ratio of the pixels was averaged for a whole scan after threshold noise reduction. The normalized average ratio difference of TRP+TRPL was 2.08 ± 0.23 (n = 5) times the averaged control (n = 9), as compared with TRPL alone at 0.82 ± 0.10 (n = 5) times the control. The TRP+TRPL group was significantly different from the other oocyte groups (P < 0.01), whereas the other oocyte groups were not significantly different (P > 0.05). The initial Ca2+ level was variable among oocytes; therefore, ratio differences were used to demonstrate consistent results similar to those shown in Fig. 3 b and c. The magnitude of the ratio differences varied in different experiments; therefore, the summary results were normalized. The time of Ca2+ application and removal is indicated by up- and down-pointing arrows, respectively.
Functional Coexpression of TRP+TRPL Produced Capacitative Ca2+ Entry Currents.
To analyze CCE oocytes were bathed in Ca2+-free medium and the InsP3-sensitive Ca2+ stores were depleted of Ca2+ either by treatment with thapsigargin (1 μM) for ≈2 hr before the measurements, or by intracellular injection of InsP3-F (10 μM). This procedure totally eliminated intracellular calcium as verified by subsequent injection of InsP3-F (10 μM), which failed to induce any response (n = 8) (43, 44). An increase in cellular Ca2+ produces a large native Ca2+-activated Cl− current (ICl,Ca), which interferes with direct Ca2+ current measurements (37, 44, 45, 46). Therefore, two independent procedures were used to assess divalent cation permeability: (i) endogenous ICl,Ca was used as a sensitive reporter for an increase in cellular Ca2+ (Fig. 3a), (ii) a novel inward current carried mainly by Mg2+, (referred to as Drosophila store-operated current, IdSOC) was measured in the absence of external and internal Ca2+. A prior injection of 2 mM EGTA (final concentration, n = 7, see Fig. 3b) ensured that the internal Ca2+ stores were totally depleted.
ICl,Ca was activated by a Ca2+ pulse, followed by a voltage step from holding voltage of −10 mV to −120 mV in Ca2+ store-depleted oocytes. The voltage step increased the driving force for Ca2+ influx. An instantaneous leak current followed by a transient inward ICl,Ca current with well-described characteristics was measured (44, 45). A relatively small ICl,Ca was observed in non-injected (control) oocytes or in oocytes expressing TRP (Fig. 3a, but see Fig. 5a). Oocytes expressing TRPL revealed a dependence of ICl,Ca on the chemical agent used for Ca2+ store depletion (see below, Figs. 3 a and c and 5a). Oocytes expressing TRP+TRPL showed the largest ICl,Ca (Figs. 3 a and c and 5a).
Properties of the Depletion-Activated Divalent Current.
To measure IdSOC in Ca2+ store-depleted oocytes the holding voltage (−10 mV) was stepped to −120 mV in the presence of 10 mM external Mg2+ and Ca2+-free medium. The store-depletion-activated IdSOC was observed only in oocytes expressing TRP+TRPL (Figs. 3 b and d and 5b). Lanthanum, an efficient blocker of the trp-dependent conductance in Drosophila photoreceptors (19, 22) completely and reversibly blocked ICl,Ca and IdSOC (Fig. 3 c and d). IdSOC had a slow rise time of ≈3 min (n = 6); also, it did not decline during the negative voltage step. These properties of IdSOC may arise from a partial voltage-dependent blocking effect of Mg2+ (see below) that was slowly removed by the large hyperpolarizing voltage step and from the absence of Ca2+-mediated negative feedback, respectively. Ca2+-mediated negative feedback is typical for InsP3 systems (5, 43).
The current-voltage relationship (I-V curve) of IdSOC (Fig. 4) showed an increased inward current below −80 mV (inward rectification) and outward current above 30 mV (outward rectification). Similarly, the Drosophila LIC shows an I-V curve with inward and outward rectification (22, 41) (Fig. 4, Inset). When Na+ was replaced with the impermeable cation N-methyl-d-glucamine at 10 mM external Mg2+ no significant effect on the I-V curve was observed. This suggests that Mg2+ is the main cationic charge carrier of IdSOC (Fig. 4). However, Mg2+ had an inhibitory effect on the amplitude of IdSOC showing a decrease in inward current when external Mg2+ was increased. Thus, the dependence of IdSOC on external Mg2+ concentration is complex and requires further study. There are quantitative differences between the Drosophila LIC and IdSOC. The LIC of Drosophila is more sensitive to La3+ (ref. 22, and see legend of Fig. 3b) and has a larger Na+ permeability than IdSOC of the oocytes expressing TRP+TRPL (22) (Fig. 4).
Figure 4.
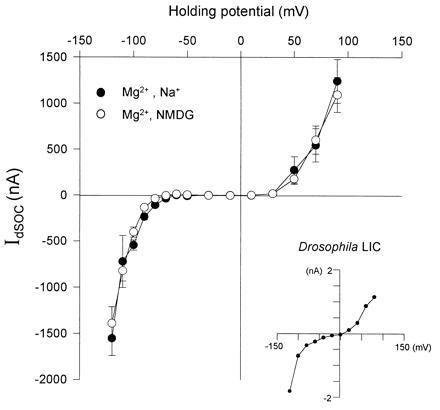
Current-voltage relationship of IdSOC in oocytes expressing TRP+TRPL. Inward and outward rectification typical of Drosophila light-activated current are shown. Current-voltage relationship (I-V curve) plotting the leak-subtracted maximal IdSOC as a function of holding voltage in InsP3-F (10 μM) or thapsigargin-treated oocytes incubated in Ca2+-free medium (see Fig. 3 a and b). Solutions are as in Fig. 3b. The I-V curves were measured before (•) and after (○) Na+ was replaced by N-methyl-d-glucamine (NMDG, 96 mM). Graphs show the average of currents obtained from nine oocytes of a single experiment. Very similar results were obtained in five other experiments. (Inset) Current-voltage relationship of the leak-subtracted peak LIC responses to identical 100-ms light flashes of orange light (OG 590, Schott edge filter) recorded from Drosophila isolated ommatidia during whole-cell voltage clamp recordings as described (22).
Without Ca2+ store depletion at non-toxic TRPL expression level (see below), neither ICl,Ca (44) nor IdSOC could be observed (Fig. 3 c and d) even with elevated external concentration of Ca2+ (10 mM, n = 9) or Mg2+ (40 mM, n = 8). Oocytes injected with a 10-fold larger dose of trpl cRNA showed a significant ICl,Ca or inward Mg2+ current similar to IdSOC; however, this current was activated by a negative voltage step without depletion of the Ca2+ stores (n = 21). The latter effect is reminiscent of a property of the currents that have been described in TRPL-expressing Sf9 cells, which show constitutively active cationic channels (30, 31, 36), but poor ability to conduct Mg2+. However, these conditions were toxic and most of the oocytes died within 4 days depending on the expression level of TRPL.
The summary histogram in Fig. 5a shows that oocytes expressing TRP or TRPL exhibited a dependence of ICl,Ca on the method of Ca2+ store depletion. In the TRPL-expressing oocytes, depletion using thapsigargin significantly reduced ICl,Ca below the control level, whereas depletion using InsP3-F enhanced the ICl,Ca current above the level of the controls (Fig. 5a). In oocytes expressing TRP alone, thapsigargin treatment exhibited a significant increase in ICl,Ca over control oocytes (37), which was not observed in InsP3-F treated oocytes (Fig. 5a). The above differential effects are reminiscent of the results reported for Sf9 cells expressing individually TRP or TRPL (30, 31, 36). At present we do not have sufficient data to explain the effects of either TRP or TRPL on ICl,Ca or the currents recorded in Sf9 cells. A possible explanation is that the effects shown in Fig. 5a are apparently due to individual interactions of TRP or TRPL with the endogenous CCE (8, 9). Such individual interactions may also account for the enhancement of the depletion-activated current described for Sf9 cells or ICl,Ca recorded in oocytes expressing TRP alone (30, 37, 38). Oocytes expressing TRP+TRPL revealed approximately 3-fold larger ICl,Ca than the controls regardless of the method used to deplete the Ca2+ stores (Fig. 5a). The summary histogram in Fig. 5b shows that under our experimental conditions the store-depletion activated IdSOC was observed only in oocytes expressing TRP+TRPL.
DISCUSSION
The induction of the IdSOC response to Ca2+ store depletion, in oocytes coexpressing Drosophila TRP+TRPL, clearly showed that these proteins are efficiently coupled to the endogenous store-depletion signal. Depletion of internal stores in Drosophila photoreceptors by thapsigargin fails to induce inward current in the dark (47). However, application of ionomycin together with Ca2+ chelators, a procedure that might be expected to deplete intracellular stores, leads to production of inward current in Drosophila photoreceptors suggesting that CCE does exist in Drosophila photoreceptors (R. C. Hardie, personal communication).
The most significant result of coexpressing TRP+TRPL was revealed by measuring IdSOC and its voltage-dependent Mg2+ current showing inward and outward rectification (Fig. 4) in thapsigargin-treated oocytes. Similarly, the LIC of Drosophila has a permeability to Mg2+ (22), as well as a voltage-dependent blocking effect of Mg2+ (48). The creation of La3+-sensitive Mg2+ current with inward rectification in the oocyte is thus similar to a unique characteristic of the Drosophila photoreceptor cell.
Unlike IdSOC, capacitative Ca2+ entry current in various vertebrate cells is carried almost exclusively by Ca2+ (3, 4, 49–51; but also see ref. 52). It therefore appears that the TRP+TRPL-dependent current has some properties different from the capacitative Ca2+ entry currents of several vertebrate cells (5, 7). IdSOC is also different from the currents found in the Sf9 cells expressing either TRP or TRPL, which are characterized by a linear I-V curve, absence of a Mg2+ block of the TRP-dependent conductance; and a constitutive activity of the TRPL-dependent conductance. The coexpressed TRP+TRPL-dependent current in Xenopus oocyte suggests that we have reconstituted a capacitative Ca2+ entry mechanism with some similarity to the native Drosophila system. Our data suggest that a cooperative action of TRP and TRPL is efficiently coupled to the endogenous Ca2+ store-depletion signal. The synergistic activity of TRP and TRPL in the production of the depletion-activated current suggest that these proteins interact with each other (for a review see ref. 13). The simplest interpretation of our results is that TRP and TRPL contribute channel subunits to form a multimeric channel. Nevertheless, our results, as well as the individual expression of Drosophila TRP, TRPL, and the human trp homologue genes (9, 30, 31, 34, 35, 36) have not demonstrated that the various TRP proteins form ionic channels. It is still possible that these expressed proteins exert their function through the endogenous CCE channels (8, 9). Other approaches, such as coimmunoprecipitation of TRP and TRPL should be used in future experiments to demonstrate that the physiological synergism found in this study is based on physical interaction between the two proteins. Also, single-channel recordings should be made to study the identity and properties of the depletion-activated channels. This study has established a useful basis for such future studies.
Acknowledgments
We thank Dr. Roger C. Hardie for the permission to quote his unpublished results, Drs. Craig Montell and Len Kelly for the cDNA of trp and trpl in bluescript vector, and Arnd Baumann for the cDNA of the above genes in PGEMHE vector. We thank Drs. U. Benjamin Kaupp, Charles S. Zuker, and Roger C. Hardie for critical comments on an earlier version of the manuscript. We also thank O. Devary, L. Alfonte, M. Eshel, and G. Barkai for technical help. This research was supported by grants from the National Institutes of Health, the Kuhne Minerva Center, the U.S.–Israel Binational Science Foundation (B.M. and Z.S.), the German-Israel Foundation (B.M.), and grants from the National Institutes of Health and National Science Foundation–Center for Light Microscope Imaging and Biotechnology (J.A.P.)
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: CCE, capacitative Ca2+ entry; InsP3, inositol 1,4,5-trisphosphate; InsP3-F, inositol 1,4,5-trisphosphate, 3-deoxy-3-fluoro; TRP, transient receptor potential protein; TRPL, TRP-like protein; LIC, light-induced current.
References
- 1.Putney J W J. Cell Calcium. 1990;11:611–624. doi: 10.1016/0143-4160(90)90016-n. [DOI] [PubMed] [Google Scholar]
- 2.Hoth M, Penner R. Nature (London) 1992;355:353–356. doi: 10.1038/355353a0. [DOI] [PubMed] [Google Scholar]
- 3.Zweifach A, Lewis R S. J Biol Chem. 1995;270:14445–14451. doi: 10.1074/jbc.270.24.14445. [DOI] [PubMed] [Google Scholar]
- 4.Lewis R S, Cahalan M D. Annu Rev Immunol. 1995;13:623–653. doi: 10.1146/annurev.iy.13.040195.003203. [DOI] [PubMed] [Google Scholar]
- 5.Berridge M J. Biochem J. 1995;312:1–11. doi: 10.1042/bj3120001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clapham D E. Cell. 1995;80:259–268. doi: 10.1016/0092-8674(95)90408-5. [DOI] [PubMed] [Google Scholar]
- 7.Clapham D E. Neuron. 1996;16:1069–1072. doi: 10.1016/s0896-6273(00)80132-4. [DOI] [PubMed] [Google Scholar]
- 8.Friel D D. Cell. 1996;85:617–619. doi: 10.1016/s0092-8674(00)81021-1. [DOI] [PubMed] [Google Scholar]
- 9.Zhu X, Jiang M S, Peyton M, Boulay G, Hurst R, Stefani E, Birnbaumer L. Cell. 1996;85:661–671. doi: 10.1016/s0092-8674(00)81233-7. [DOI] [PubMed] [Google Scholar]
- 10.Hardie R C, Minke B. Cell Calcium. 1995;18:256–274. doi: 10.1016/0143-4160(95)90023-3. [DOI] [PubMed] [Google Scholar]
- 11.Ranganathan R, Malicki D M, Zuker C S. Annu Rev Neurosci. 1995;18:283–317. doi: 10.1146/annurev.ne.18.030195.001435. [DOI] [PubMed] [Google Scholar]
- 12.Pak W L. Invest Ophthalmol Visual Sci. 1995;36:2340–2357. [PubMed] [Google Scholar]
- 13.Minke B, Selinger Z. Curr Opin Neurobiol. 1996;6:459–466. doi: 10.1016/s0959-4388(96)80050-x. [DOI] [PubMed] [Google Scholar]
- 14.Minke B, Selinger Z. Prog Retinal Res. 1991;11:99–124. [Google Scholar]
- 15.Minke B, Wu C, Pak W L. Nature (London) 1975;258:84–87. doi: 10.1038/258084a0. [DOI] [PubMed] [Google Scholar]
- 16.Cosens D J, Manning A. Nature (London) 1969;224:285–287. doi: 10.1038/224285a0. [DOI] [PubMed] [Google Scholar]
- 17.Minke B. J Gen Physiol. 1982;79:361–385. doi: 10.1085/jgp.79.3.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hochstrate P. J Comp Physiol A. 1989;166:179–187. doi: 10.1007/BF00193462. [DOI] [PubMed] [Google Scholar]
- 19.Suss-Toby E, Selinger Z, Minke B. J Gen Physiol. 1991;98:849–868. doi: 10.1085/jgp.98.4.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hardie R C. Proc R Soc London B. 1991;245:203–210. [Google Scholar]
- 21.Ranganathan R, Harris G L, Stevens C F, Zuker C S. Nature (London) 1991;354:230–232. doi: 10.1038/354230a0. [DOI] [PubMed] [Google Scholar]
- 22.Hardie R C, Minke B. Neuron. 1992;8:643–651. doi: 10.1016/0896-6273(92)90086-s. [DOI] [PubMed] [Google Scholar]
- 23.Peretz A, Suss-Toby E, Rom-Glas A, Arnon A, Payne R, Minke B. Neuron. 1994;12:1257–1267. doi: 10.1016/0896-6273(94)90442-1. [DOI] [PubMed] [Google Scholar]
- 24.Hardie R C. J Neurosci. 1996;16:2924–2933. doi: 10.1523/JNEUROSCI.16-09-02924.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peretz A, Sandler C, Kirschfeld K, Hardie R C, Minke B. J Gen Physiol. 1994;104:1057–1077. doi: 10.1085/jgp.104.6.1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Montell C, Rubin G M. Neuron. 1989;2:1313–1323. doi: 10.1016/0896-6273(89)90069-x. [DOI] [PubMed] [Google Scholar]
- 27.Wong F, Schaefer E L, Roop B C, LaMendola J N, Johnson Seaton D, Shao D. Neuron. 1989;3:81–94. doi: 10.1016/0896-6273(89)90117-7. [DOI] [PubMed] [Google Scholar]
- 28.Phillips A M, Bull A, Kelly L E. Neuron. 1992;8:631–642. doi: 10.1016/0896-6273(92)90085-r. [DOI] [PubMed] [Google Scholar]
- 29.Niemeyer B A, Suzuki E, Scott K, Jalink K, Zuker C S. Cell. 1996;85:651–659. doi: 10.1016/s0092-8674(00)81232-5. [DOI] [PubMed] [Google Scholar]
- 30.Vaca L, Sinkins W G, Hu Y, Kunze D L, Schilling W P. Am J Physiol. 1994;267:C1501–C1505. doi: 10.1152/ajpcell.1994.267.5.C1501. [DOI] [PubMed] [Google Scholar]
- 31.Harteneck C, Obukhov A G, Zobel A, Kalkbrenner F, Schultz G. FEBS Lett. 1995;358:297–300. doi: 10.1016/0014-5793(94)01455-a. [DOI] [PubMed] [Google Scholar]
- 32.Wes P D, Chevesich J, Jeromin A, Rosenberg C, Stetten G, Montell C. Proc Natl Acad Sci USA. 1995;92:9652–9656. doi: 10.1073/pnas.92.21.9652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhu X, Chu P B, Peyton M, Birnbaumer L. FEBS Lett. 1995;373:193–198. doi: 10.1016/0014-5793(95)01038-g. [DOI] [PubMed] [Google Scholar]
- 34.Zitt C, Zobel A, Obukhov A G, Harteneck C, Kalkbrenner F, Lückhoff A, Schultz G. Neuron. 1996;16:1189–1196. doi: 10.1016/s0896-6273(00)80145-2. [DOI] [PubMed] [Google Scholar]
- 35.Hu Y, Vaca L, Zhu X, Birnbaumer L, Kunze D L, Schilling W P. Biochem Biophys Res Commun. 1994;201:1050–1056. doi: 10.1006/bbrc.1994.1808. [DOI] [PubMed] [Google Scholar]
- 36.Dong Y, Kunze D L, Vaca L, Schilling W P. Am J Physiol. 1995;269:C1332–C1339. doi: 10.1152/ajpcell.1995.269.5.C1332. [DOI] [PubMed] [Google Scholar]
- 37.Petersen C C H, Berridge M J, Borgese M F, Bennett D L. Biochem J. 1995;311:41–44. doi: 10.1042/bj3110041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sinkins W G, Vaca L, Hu Y, Kunze D L, Schilling W P. J Biol Chem. 1996;271:2955–2960. doi: 10.1074/jbc.271.6.2955. [DOI] [PubMed] [Google Scholar]
- 39.Liman E R, Tytgat J, Hess P. Neuron. 1992;9:861–871. doi: 10.1016/0896-6273(92)90239-a. [DOI] [PubMed] [Google Scholar]
- 40.Baumann A, Frings S, Godde M, Seifert R, Kaupp U B. EMBO J. 1994;13:5040–5050. doi: 10.1002/j.1460-2075.1994.tb06833.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pollock J A, Assaf A, Peretz A, Nichols C D, Mojet M H, Hardie R C, Minke B. J Neurosci. 1995;15:3747–3760. doi: 10.1523/JNEUROSCI.15-05-03747.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gillo B, Lass Y, Nadler E, Oron Y. J Physiol (London) 1987;392:349–361. doi: 10.1113/jphysiol.1987.sp016784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Singer D, Boton R, Moran O, Dascal N. Pflügers Arch. 1990;416:7–16. doi: 10.1007/BF00370215. [DOI] [PubMed] [Google Scholar]
- 44.Petersen C C, Berridge M J. J Biol Chem. 1994;269:32246–32253. [PubMed] [Google Scholar]
- 45.Yao Y, Parker I. J Physiol (London) 1993;468:275–295. doi: 10.1113/jphysiol.1993.sp019771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Parekh A B, Terlau H, Stuhmer W. Nature (London) 1993;364:814–818. doi: 10.1038/364814a0. [DOI] [PubMed] [Google Scholar]
- 47.Ranganathan R, Bacskai B J, Tsien R Y, Zuker C S. Neuron. 1994;13:837–848. doi: 10.1016/0896-6273(94)90250-x. [DOI] [PubMed] [Google Scholar]
- 48.Hardie R C, Mojet M H. J Neurophysiol. 1995;74:2590–2599. doi: 10.1152/jn.1995.74.6.2590. [DOI] [PubMed] [Google Scholar]
- 49.Lewis R S, Cahalan M D. Cell Regul. 1989;1:99–112. doi: 10.1091/mbc.1.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zweifach A, Lewis R S. Proc Natl Acad Sci USA. 1993;90:6295–6299. doi: 10.1073/pnas.90.13.6295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Penner R, Fasolato C, Hoth M. Curr Opin Neurobiol. 1993;3:368–374. doi: 10.1016/0959-4388(93)90130-q. [DOI] [PubMed] [Google Scholar]
- 52.Lückhoff A, Clapham D E. Biophys J. 1994;67:177–182. doi: 10.1016/S0006-3495(94)80467-9. [DOI] [PMC free article] [PubMed] [Google Scholar]