

Abstract
Recent studies have used dense markers to examine the human genome in ancestrally homogeneous populations for hallmarks of selection. No genomewide studies have focused on recently admixed groups—populations that have experienced admixing among continentally divided ancestral populations within the past 200–500 years. New World admixed populations are unique in that they represent the sudden confluence of geographically diverged genomes with novel environmental challenges. Here, we present a novel approach for studying selection by examining the genomewide distribution of ancestry in the genetically admixed Puerto Ricans. We find strong statistical evidence of recent selection in three chromosomal regions, including the human leukocyte antigen region on chromosome 6p, chromosome 8q, and chromosome 11q. Two of these regions harbor genes for olfactory receptors. Interestingly, all three regions exhibit deficiencies in the European-ancestry proportion.
Whereas the vast majority of DNA sequence–level variation evolves neutrally and is maintained by a balance between the mutation process and random drift,1 some parts of the human genome have been subjected to natural selection. With use of dense genotype data on human populations, recent studies have identified regions that appear to have undergone positive selection.2–5 These results provide information about population history, as well as clues for the identification of genes with important biological functions.
Admixed populations offer special opportunities for studying recent selection. Before admixing, the ancestral populations had typically been isolated by distance, and their genomes had evolved in distinct environments. In contrast, in admixed populations of the Americas, the environmental changes have been sudden. For example, Hispanic populations resulted from recent admixing between genomes from the Old World (Africans and Europeans) and those from the New World (Native Americans). Intercontinental migration brought genomes of some ancestral populations to an unfamiliar environment in the New World and, at the same time, disturbed the local environment, introducing life-style changes as well as pathogens, such as smallpox, from the Old World. Thus, genomes from each ancestral population were presented with new challenges. This kind of selection pressure may be quite different from that faced by stationary populations, in which the local environmental changes may occur gradually, allowing for rare advantageous alleles to increase in frequency. At the same time, it is possible that a region (or regions) of the genome has been disproportionately included in the newly admixed population because of selection. Therefore, in theory, it is possible, with use of dense genotype data, to detect a signature of selection by looking in an admixed population for genomic regions that exhibit unusually large deviations in ancestry proportions compared with what is typically observed elsewhere in the genome.
The idea of detecting signatures of selection in an admixed population has a considerable history and has been explored in various studies, which compare admixture estimates at a small number of loci.6–8 Workman et al.6 examined 12 genetic markers in a sample of 1,287 African Americans from Claxton, Georgia. The average European admixture proportion at most markers was estimated to be ∼.10. A few markers, however, exhibited higher European ancestry; those included the loci for G6PD, hemoglobin, haptoglobin, and phenylthiocarbamide taste blindness. Those authors concluded that the markers had been subject to natural selection. However, evolutionary fluctuations due to both small population size (genetic drift) and sampling variability can also generate differences in ancestry proportions among loci.9 Subsequent reanalysis of these and related data argued that the observed variation in European admixture among markers can be attributed largely to evolutionary and statistical noise.9,10 In a rigorous analysis, Long9 showed that genetic drift alone could generate an interlocus ancestry distribution with SD of .044 in the Claxton population. Furthermore, because these early studies estimated ancestry proportions on the basis of a single marker at each locus, the ancestry estimates were sensitive to bias because of erroneous estimates of allele frequencies in the ancestral populations.
The recent development of high-density SNP markers now allows for accurate ancestry estimation on a global, genomewide scale.11 This allows us to revisit the question of ancestral selection in admixed populations. To do so, we employ a recently proposed statistical approach for locus-by-locus ancestry estimation on a genomewide scale.11
The Puerto Rican population arose as a result of admixing among Europeans, West Africans, and Native Americans. The indigenous people on the island were the Taíno Indians, who came from South America. In November 1493, Christopher Columbus discovered the island on his second voyage to the New World. In 1513, Africans were introduced to the island as slaves.12 Today, most Puerto Rican individuals have genomes derived from all three ancestral populations. The average genomewide individual ancestry proportions have been estimated as .66, .18, and .16, for European, West African, and Native American, respectively.13 Under neutral evolution, we expect each individual’s genome to represent an ensemble of ancestry blocks, randomly sampled, with a probability similar to that of the genomewide average. Therefore, at each genomic location, the regional ancestry proportions are also expected to follow the same probability distribution. Here, we examine the distributions of these three ancestry proportions across the genome in a sample of 192 Puerto Rican individuals, in a search for outlier regions.
The 192 Puerto Ricans were recruited from six primary-care clinics in Puerto Rico as part of the Genetics of Asthma in Latino Americans study.14 All study participants were aged 8–40 years. Individuals of European (from the United States), West African (from Nigeria), and Native American (Pima and Mayan) descent were included, to approximate the ancestral populations. With use of the Affymetrix Human Mapping 100K GeneChip set,15 192 Puerto Ricans, 42 Europeans, 37 Africans, and 30 Native Americans were genotyped. Details of data-quality assessment and statistical analysis are provided in appendix A.
Under the assumption of a trihybrid population model and of 112,584 SNPs, marker-specific ancestries were estimated, with the program SABER,11 for all autosomes for each individual. Averaged over 192 Puerto Ricans, the genomewide mean estimated European ancestry was .67, African ancestry was .18, and Native American ancestry was .15.18 For each individual, we then computed Δ ancestry by subtracting the genomewide ancestry from locus-specific ancestry for each of the three ancestry components. We then calculated average locus-specific Δ ancestry values for the 192 subjects.
Figure 1 shows the average excess ancestry at each SNP location for the 192 Puerto Rican individuals, with the genomewide ancestry as the baseline reference. At most locations, deviations are within the range of evolutionary drift and statistical fluctuation. However, a few regions exhibit extreme fluctuations that are unlikely to have occurred by chance. Most prominently, on chromosome 6p21-6p22, an excess of African ancestry reaches .14 around SNP rs169679; this level of excess was not observed in 20,000 permutations (see appendix A) and represents 6.4 SD from the mean of the distribution. Correspondingly, the European ancestry at this location is depressed by .14, whereas Native American ancestry remains unchanged. Additionally, chromosome 8 (peak at SNP rs896760) and chromosome 11 (peak at SNP rs637249) show an excess of .13 in Native American ancestry and a deficit of European ancestry (P<.001 for each by permutation test). This level of excess in Native American ancestry represents 4.6 SD from the mean of the distribution.
Figure 1. .

Genomewide variation of ancestry in Puerto Ricans. The X-axis denotes the physical location of SNPs; the Y-axis indicates the excess/deficiency in ancestry at the corresponding SNP, averaged for 192 Puerto Ricans. The red, blue, and green curves represent African, European, and Native American ancestries, respectively.
Figure 2 compares the distribution of estimated locus-specific ancestry with that simulated under genetic drift (see appendix A for population parameters). The histograms indicate that, under the assumption of a reasonable population-history model, the distribution of the locus-specific ancestry matches well with the observed distribution except for the few outliers noted (chromosomes 6p, 8q, and 11q). There is modest skewness in these distributions, but it is insufficient to account for the outliers observed in any of the three ancestry distributions. Noticeably, in 107 simulations, the African ancestry deviation exceeded .14 (in either direction) once. The Native American ancestry deviation exceeded .13 35 times in 107 simulations. Because ancestry blocks extend, on average, several centimorgans,19 these results suggest that it is unlikely that the outliers we observed are due to genetic drift, even allowing for testing of multiple chromosomal regions.
Figure 2. .

Comparison of observed (histogram) and simulated (line) variation in ancestry across the genome. A population model was assumed in which, for the first 5 generations, Europeans and Native Americans admix at a ratio of 0.82:0.18, with a total population size of 1,000. At generation 5, Africans enter the gene pool, bringing N to 1,250. This trihybrid population is then allowed to mate randomly for 10 generations at constant population size. The density curve for each population is based on 107 independent simulations.
In previous studies, locus-specific ancestry was estimated using a single marker at each locus; however, the high-density genotyping platform we used allows for inference based on multiple neighboring SNPs. Since most SNPs in the genome are not completely informative, combining information at neighboring SNPs produces more-accurate estimates than does using single markers. Furthermore, an ancestry estimate based on a single marker is sensitive to mutation or misspecification of ancestral-allele frequencies. In contrast, under evolutionary neutrality, it is unlikely that these factors affect several neighboring markers in a way that leads to biased ancestry estimates. Thus, the estimation approach we use provides some safeguards against inaccurate ancestral-allele frequencies.11,18 Similarly, our method of estimating locus-specific ancestry is more robust to systematic genotyping error or deviation from Hardy-Weinberg equilibrium (HWE) at a single marker.
We investigated more closely the three chromosome regions that achieved statistical significance. To exclude the possibility that the excess in ancestry is related to the inclusion of individuals with asthma, we examined the asthmatic cases (n=96) and controls (n=96) separately, and we found that the excess of African (or Native American) ancestry and the deficiency of European ancestry occurred identically in both groups. To eliminate the possibility that the peaks are an artifact due to a few outlier markers, we repeated the analysis separately with the even-numbered and odd-numbered markers. The resulting excess ancestry along chromosomes 6, 8, and 11 is shown in figure 3; both marker sets show comparable excess ancestry in all three regions. Figure 4 illustrates that the estimated African ancestry (shown in red) is indeed higher around chromosome 6p21-6p22 by comparing the estimated ancestries in two regions on chromosome 6: on the left is the 10-Mb window around the peak of excess African ancestry on chromosome 6p21-6p22, whereas on the right is a randomly selected region of the same physical width. The areas that are colored blue, red, and green correspond to the estimated European, African and Native American ancestries, respectively.
Figure 3. .

Extreme variations of ancestry on chromosomes 6 (a), 8 (b), and 11 (c). SNPs were divided into two sets (even-numbered vs. odd-numbered markers), which were analyzed separately with use of SABER. The red and green points are the excess African and Native American ancestries, respectively, on the two marker subsets, whereas the black points are the results with use of all available markers on that chromosome.
Figure 4. .

Display of estimated ancestry on chromosome 6. Each pair of horizontal strips represents an (unphased) individual, with the vertical height proportional to the marker-specific ancestry estimates of African (red), European (blue), and Native American (green) ancestries. Left, Ancestry estimates around the peak on 6p21-22. Right, Randomly selected region of chromosome 6.
The region on chromosome 6p harbors numerous genes; some of the most studied are those of the major histocompatibility complex (MHC). Since the human leukocyte antigen (HLA) loci have been well studied in a variety of populations, we sought independent confirmatory evidence using existing published HLA data. We compiled allele frequencies for Puerto Ricans, West Europeans, sub-Saharan Africans, and Native Amerindians for four HLA loci (HLA-A, -B, -DRB, and -DQB) from literature sources16 and the HLA Frequency database from the American Society for Histocompatibility and Immunogenetics (ASHI). The data were used to determine the maximum-likelihood estimates of Puerto Rican ancestries at the HLA loci (table 1). For all four HLA loci examined separately and combined, ancestry proportions are similar to our previous estimates for the corresponding region, with elevated African ancestral proportion and decreased European ancestral proportion, in particular, compared with genomewide ancestry estimates obtained in another study of Puerto Ricans that used 15 microsatellite markers randomly distributed across the genome.17
Table 1. .
Maximum-Likelihood Estimates of Ancestry Proportions in Puerto Ricans[Note]
| Marker Region | European | African | Native American |
| HLA-A | .51 | .32 | .17 |
| HLA-B | .59 | .28 | .13 |
| HLA-DRB | .55 | .24 | .21 |
| HLA-DQB | .43 | .37 | .20 |
| Combined (HLA) | .51 | .30 | .20 |
| Genomewide | .76 | .17 | .07 |
Note.— The top five rows are estimates for the MHC region on chromosome 6; the last row is the genomewide average ancestry estimated from 15 microsatellites.13
The region underneath our observed peak on chromosome 6p22 includes, in addition to HLA, an olfactory gene cluster. Similarly, the peak on chromosome 11q11 also harbors an olfactory gene cluster, which has been identified elsewhere as a target of ongoing positive selection.4,20 The peak on 8q23.3 harbors less obvious candidates. There is only one gene in the region, CSMD3 (CUB and sushi multiple domains 3), of which the highest expression is found in fetal brain and intermediate expression is found in adult pancreas, spleen, testis, spinal cord, and all specific adult brain regions examined. Little to no expression was detected in the other tissues examined.21
Admixed populations offer unique opportunities for the detection of signatures of selection by examination of the genomewide distribution of ancestry. It is reasonable to assume that selection occurred in recent times, subsequent to the intercontinental migration. Because the peaks are quite broad, it is difficult to identify the precise gene(s) that has been the target of selection, nor can we state with confidence the nature of the selective agent. However, we can postulate two scenarios that led to apparent regional admixture bias. First, the Africans and Europeans were exposed to an unfamiliar environment, with various new pathogens that are native to the New World. Because they are genetically more diverse, some African-admixed alleles might have conferred an advantageous phenotype. The European-derived alleles are less diverse and thus may have been less advantageous. This would have led to an excess of either African admixture (in the case of chromosome 6) or Native American admixture (in the case of chromosomes 8 and 11) and a corresponding deficiency in European admixture. Alternatively, it is possible that the massive intercontinental migration altered environmental conditions in the New World sufficiently that all three ancestral populations were challenged. Again, specific African- or Native American–derived alleles rose in frequency.
A second possibility is that various genes contributed to specific phenotypes, which have been influenced either by natural selection or by mating preferences. In the latter case, the admixture bias may reflect some type of social selection. In terms of the target of selection, it is intriguing that two regions exhibiting the strongest selection signals both harbor olfactory genes. This is consistent with the findings of Voight et al.4—namely, that genes related to olfaction are overrepresented in the regions under recent positive selection. But olfactory genes may not be the sole target. The region on chromosome 6p harbors various other genes; noticeably, some HLA loci are <2 Mb away from the peak. Because of their known role in immune defense, the HLA loci are certainly also strong candidates.22
It is also important to consider that the genetic intermixing that has occurred in this population over the past 5 centuries has brought together new combinations of diverged genomes and therefore created a novel genetic background on which selection may have operated. In this regard, as opposed to nonadmixed groups, admixed populations offer novel and complementary information about complex interactions involving both genes and environment. Further investigations are required to reveal the targets and agents of selection that have played important roles in shaping the admixed gene pool.
Acknowledgments
We acknowledge the families and the patients for their participation. We thank Dr. Mark Shriver for contributing samples of European, African, and Native American descent, and we thank Dr. Eduardo Santiago-Delpin for providing the HLA frequencies in Puerto Ricans. This work is supported by National Institute of General Medical Sciences grants GM073059 (to H.T.) and HL078885 (to E.G.B.), American Thoracic Society “Breakthrough Opportunities in Lung Disease” Award (to S.C.), the Sandler Center for Basic Research in Asthma, and the Sandler Family Supporting Foundation.
Appendix A
Genotyping and Assessing the Data Quality
A total of 192 Puerto Ricans, 42 Europeans, 37 Africans, and 30 Native Americans were genotyped with the Affymetrix Human Mapping 100K GeneChip set,15 which comprises two arrays, each capable of genotyping >50,000 SNPs (58,960 for XbaI array and 57,244 for HindIII array). Approximately 250 ng of genomic DNA was processed for each chip, in accordance with the Affymetrix protocol.
In all analyses reported in this work, only autosomal SNPs were included. For quality control, SNPs with call rates <.85 were eliminated from the analysis. To further eliminate SNPs with possible genotyping errors, we excluded SNPs with only heterozygous calls. We chose not to use HWE as a quality-control criterion, because SNPs with large allele-frequency differences between major racial/ethnic groups would tend to deviate from HWE if there is nonrandom mating with respect to ancestry in this population.23 These exclusions left us with 112,984 autosomal SNPs for analysis. Among the ancestral samples, two Africans have allele sharing comparable to a sib pair, as do two Native Americans. In the subsequent analyses, we excluded one of the individuals from each pair.
Statistical Analysis
Marker-specific ancestry estimates were produced using the program SABER.11 We assumed a trihybrid population model and used the genotypes of the European, African, and Native American subjects to estimate the ancestral-allele frequencies, as well as the two-marker haplotype frequencies within each ancestral population. In this analysis, we excluded 400 SNPs, which were monomorphic in the entire sample. The ancestral-allele frequencies were further refined using the program FRAPPE, which estimates the ancestral-allele frequencies with use of both the ancestral and the admixed individuals.18 The starting values of genomewide average individual admixture fractions (IA), which are required input parameters in SABER, were also estimated using FRAPPE. The SABER output was used to re-estimate IA. We then reran SABER with updated IA estimates. We observed that new IA estimates from SABER did not change much in subsequent iterations. Both FRAPPE and SABER are available at the Tang Lab Web site.
Once the locus-specific ancestry was estimated, we computed the “Δ ancestry,” deficiency or excess at each locus, using the genomewide IA as a baseline. Specifically, the Δ ancestry for ancestral population k at marker m is defined as
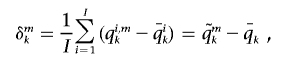 |
where qi,mk is the locus-specific ancestry of individual i at marker m, estimated from SABER; 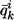 is the IA of individual i;
is the IA of individual i; 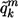 is the mean ancestry at marker m averaged over all individuals; and
is the mean ancestry at marker m averaged over all individuals; and 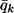 is the proportion from ancestral population k, averaged over all individuals and the entire genome.
is the proportion from ancestral population k, averaged over all individuals and the entire genome.
As shown by Long,9 genetic drift can create intermarker variation in ancestry proportions. Therefore, we performed a simulation to evaluate its potential impact on our analyses. We assumed a simple population model: at time 0, Europeans and Native Americans admix at a ratio of 0.82:0.18. The population size is kept at N=1,000 for t=5 generations. At t=5, African individuals enter the gene pool, bringing N to 1,250, achieving ancestry proportions similar to those estimated in the Puerto Ricans (i.e., .18), and reducing the European and Native American proportions to .67 and .15, respectively. This trihybrid population is then allowed to mate randomly for 10 additional generations at the constant population size. Each simulation generates the evolutionary history of one locus independently, with no linkage. The parameters of the population model were chosen on the basis of our understanding of the history of this population and to match the resulting locus-specific–ancestry distribution with the corresponding empirical distributions. Whereas 15 generations of admixing time may seem short (when the fact that Europeans arrived on the island >500 years ago is considered), we note that the effect of genetic drift diminishes as the size of the population grows, as has been the case in Puerto Rico for the past several hundred years.
We note that Kimura analytically studied the question of allele-frequency distributions over time under drift.24 He showed that the derived distribution is a complex function of t/2N, where t is the number of generations and N is the effective population size. Although his results would be directly relevant to the ancestry distribution in a dihydrid population over time, they are not directly applicable to our more complex scenario of three populations admixing at different time points, with the exception of African ancestry, which we assumed entered at a single time point 10 generations ago, when the population size was 1,250. Results with use of his analytic formulas closely mirror our simulation results.
Since the evolutionary simulation does not account for statistical sampling error and error associated with estimating ancestry, we performed a permutation test. In each individual, we randomly concatenated the ancestry estimates of 22 autosomes. This linear, long piece of permutated genome was circularized and was cut at a location chosen uniformly. A permuted sample was formed by repeating this process for each individual, with the order of concatenation and the location of linearization chosen independently in each individual. We then computed the excess/deficiency in ancestry in each permuted sample and recorded the extreme values (maximum and minimum) achieved with respect to European, African, and Native American components. When markers are evenly spaced and linkage disequilibrium between neighboring markers is similar, an advantage of this permutation procedure is that it preserves the correlation structure between markers. In our data, this assumption does not hold exactly. Further, the estimation in ancestry with use of the same set of ancestral individuals induces a correlation at each site, which is not preserved in the permutation procedure. To cope with these complexities, we further added a quantile transformation step—specifically, in each permutation, we multiplied the permuted statistics by a scale factor, so that the SD of the distribution of the permuted statistics (trimming .05 on each end) matched that in the corresponding observed distribution. In total, 20,000 permutations were performed, and the maximum absolute deviations (excess or deficiency) from these permuted genomes were treated as the null distribution, with which we computed the significance levels of the observed ancestry deviation.
One region of potential interest was the HLA region on chromosome 6p. To estimate the ancestry proportion in Puerto Ricans at the HLA loci, we used published and publicly available data. For Puerto Ricans, the gene frequencies at HLA-A, -B, -DRB, and -DQB were taken from 461 individuals who were genotyped during 1997 and 1998.16 The corresponding gene frequencies in non-Hispanic Europeans, sub-Saharan Americans, and Native Americans were compiled using the HLA Frequency database from ASHI. Population-level ancestry proportions were then estimated using maximum likelihood. For the genomewide estimate of admixture in Puerto Ricans, we used the data of Zuniga et al.,17 who studied a random collection of 15 microsatellites.
Web Resources
The URLs for data presented herein are as follows:
- ASHI, http://www.ashi-hla.org/ (for the HLA Frequency database)
- Tang Lab Web site, http://www.fhcrc.org/labs/tang (for FRAPPE and SABER software)
References
- 1.Kimura M (2003) The neutral theory of molecular evolution. Cambridge University Press, Cambridge, United Kingdom [Google Scholar]
- 2.Sabeti PC, Reich DE, Higgins JM, Levine HZ, Richter DJ, Schaffner SF, Gabriel SB, Platko JV, Patterson NJ, McDonald GJ, et al (2002) Detecting recent positive selection in the human genome from haplotype structure. Nature 419:832–837 10.1038/nature01140 [DOI] [PubMed] [Google Scholar]
- 3.Carlson CS, Thomas DJ, Eberle MA, Swanson JE, Livingston RJ, Rieder MJ, Nickerson DA (2005) Genomic regions exhibiting positive selection identified from dense genotype data. Genome Res 15:1553–1565 10.1101/gr.4326505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Voight BF, Kudaravalli S, Wen X, Pritchard JK (2006) A map of recent positive selection in the human genome. PLoS Biol 4:e72 10.1371/journal.pbio.0040072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tishkoff SA, Reed FA, Ranciaro A, Voight BF, Babbitt CC, Silverman JS, Powell K, Mortensen HM, Hirbo JB, Osman M, et al (2007) Convergent adaptation of human lactase persistence in Africa and Europe. Nat Genet 39:31–40 10.1038/ng1946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Workman PL, Blumberg BS, Cooper AJ (1963) Selection, gene migration and polymorphic stability in a US white and negro population. Am J Hum Genet 15:429–437 [PMC free article] [PubMed] [Google Scholar]
- 7.Reed TE (1969) Caucasian genes in American negroes. Science 165:762–768 10.1126/science.165.3895.762 [DOI] [PubMed] [Google Scholar]
- 8.Cavalli-Sforza LL, Bodmer WF (1971) The genetics of human populations. Freeman, San Francisco [Google Scholar]
- 9.Long J (1991) The genetic structure of admixed populations. Genetics 127:417–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Adams J, Ward RH (1973) Admixture studies and detection of section. Science 180:1137–1143 10.1126/science.180.4091.1137 [DOI] [PubMed] [Google Scholar]
- 11.Tang H, Coram M, Wang P, Zhu X, Risch N (2006) Reconstructing genetic ancestry blocks in admixed individuals. Am J Hum Genet 79:1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carrion AM (1984) Puerto Rico: a political and cultural history. Norton, New York [Google Scholar]
- 13.Burchard EG, Borrell LN, Choudhry S, Naqvi M, Tsai HJ, Rodriguez-Santana JR, Chapela R, Rogers SD, Mei R, Rodriguez-Cintron W, et al (2005) Latino populations: a unique opportunity for the study of race, genetics, and social environment in epidemiological research. Am J Public Health 95:2161–2168 10.2105/AJPH.2005.068668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burchard EG, Avila PC, Nazario S, Casal J, Torres A, Rodriguez-Santana JR, Sylvia JS, Fagan JK, Salas J, Lilly CM, et al (2004) Lower bronchodilator responsiveness in Puerto Rican than in Mexican asthmatic subjects. Am J Respir Crit Care Med 169:386–392 10.1164/rccm.200309-1293OC [DOI] [PubMed] [Google Scholar]
- 15.Kennedy GC, Matsuzaki H, Dong S, Liu WM, Huang J, Liu G, Su X, Cao M, Chen W, Zhang J, et al (2003) Large-scale genotyping of complex DNA. Nat Biotechnol 21:1233–1237 10.1038/nbt869 [DOI] [PubMed] [Google Scholar]
- 16.Santiago-Delpin EA, de Echegaray S, Rivera-Cruz F, Rodriguez-Trinidad A (2002) The histocompatibility profile of the Puerto Rican population. Transplant Proc 34:3075–3078 10.1016/S0041-1345(02)03594-7 [DOI] [PubMed] [Google Scholar]
- 17.Zuniga J, Ilzarbe M, Acunha-Alonzo V, Rosetti F, Herbert Z, Romero V, Almeciga I, Clavijo O, Stern JN, Granados J, et al (2006) Allele frequencies for 15 autosomal STR loci and admixture estimates in Puerto Rican Americans. Forensic Sci Int 164:266–270 10.1016/j.forsciint.2005.11.022 [DOI] [PubMed] [Google Scholar]
- 18.Tang H, Peng J, Wang P, Risch NJ (2005) Estimation of individual admixture: analytical and study design considerations. Genet Epidemiol 28:289–301 10.1002/gepi.20064 [DOI] [PubMed] [Google Scholar]
- 19.Lautenberger JA, Stephens JD, O’Brien SJ, Smith MW (2000) Significant admixture linkage disequilibrium across 30 cM around the FY locus in African Americans. Am J Hum Genet 66:969–978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gilad Y, Bustamante CD, Lancet D, Pääbo S (2003) Natural selection on the olfactory receptor gene family in humans and chimpanzees. Am J Hum Genet 73:489–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shimizu A, Asakawa S, Sasaki T, Yamazaki S, Yamagata H, Kudoh J, Minoshima S, Kondo I, Shimizu N (2003) A novel giant gene CSMD3 encoding a protein with CUB and sushi multiple domains: a candidate gene for benign adult familial myoclonic epilepsy on human chromosome 8q23.3-q24.1. Biochem Biophys Res Commun 309:143–154 10.1016/S0006-291X(03)01555-9 [DOI] [PubMed] [Google Scholar]
- 22.Meyer D, Single RM, Mack SJ, Erlich HA, Thomson G (2006) Signatures of demographic history and natural selection in the human major histocompatibility complex loci. Genetics 173:2121–2142 10.1534/genetics.105.052837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Choudhry S, Coyle NE, Tang H, Salari K, Lind D, Clark SL, Tsai HJ, Naqvi M, Phong A, Ung N, et al (2006) Population stratification confounds genetic association studies among Latinos. Hum Genet 118:652–664 10.1007/s00439-005-0071-3 [DOI] [PubMed] [Google Scholar]
- 24.Kimura M (1955) Solution of a process of random genetic drift with a continuous model. Proc Natl Acad Sci USA 41:144–150 10.1073/pnas.41.3.144 [DOI] [PMC free article] [PubMed] [Google Scholar]