

Abstract
For decades, cytogenetic studies have demonstrated that somatically acquired structural rearrangements of the genome are a common feature of most classes of human cancer. However, the characteristics of these rearrangements at sequence-level resolution have thus far been subject to very limited description. One process that is dependent upon somatic genome rearrangement is gene amplification, a mechanism often exploited by cancer cells to increase copy number and hence expression of dominantly acting cancer genes. The mechanisms underlying gene amplification are complex but must involve chromosome breakage and rejoining. We sequenced 133 different genomic rearrangements identified within four cancer amplicons involving the frequently amplified cancer genes MYC, MYCN, and ERBB2. The observed architectures of rearrangement were diverse and highly distinctive, with evidence for sister chromatid breakage–fusion–bridge cycles, formation and reinsertion of double minutes, and the presence of bizarre clusters of small genomic fragments. There were characteristic features of sequences at the breakage–fusion junctions, indicating roles for nonhomologous end joining and homologous recombination-mediated repair mechanisms together with nontemplated DNA synthesis. Evidence was also found for sequence-dependent variation in susceptibility of the genome to somatic rearrangement. The results therefore provide insights into the DNA breakage and repair processes operative in somatic genome rearrangement and illustrate how the evolutionary histories of individual cancers can be reconstructed from large-scale cancer genome sequencing.
Gene amplification may be defined as a somatically acquired increase in copy number of a restricted genomic region and is often found in cancer cells as a mechanism of increasing the transcript and therefore protein levels of dominantly acting cancer genes (Schwab 1999; Savelyeva and Schwab 2001; Myllykangas and Knuutila 2006). Several cancer genes (for example, ERBB2, MYC, MYCN, MYCL1, EGFR, and AKT2) are involved in cancer development exclusively or predominantly through gene amplification (Futreal et al. 2004; http://www.sanger.ac.uk/genetics/CGP/Census/). Targeting of anticancer therapies to the proteins encoded by amplified cancer genes has proven effective in the case of Trastuzumab, an antibody directed against ERBB2, in breast cancer (Nahta and Esteva 2006).
The processes underlying the development of gene amplification are incompletely understood. One approach to understanding the genesis of amplicons in cancer cells is to characterize their genomic structure. At the level of resolution of light microscopy, amplified regions may exist as extrachromosomal DNA (double minutes) or as large contiguous stretches of amplified DNA (homogenously staining regions, HSRs) (Schwab 1999; Savelyeva and Schwab 2001). There is evidence, including that derived from model systems (Selvarajah et al. 2006), that conversion between these structural forms may occur (Corvi et al. 1995; Coquelle et al. 1998). Fluorescent in situ hybridization (FISH) and other studies have shown that amplicons may be composed of DNA from multiple different parts of the genome (Guan et al. 1994; Muleris et al. 1995; Volik et al. 2003, 2006; Lim et al. 2005; Van Roy et al. 2006). Where the amplified DNA is from the same genomic region, both inverted orientation of amplified repeat units (Hellman et al. 2002; Herrick et al. 2005) and noninverted (tandem) orientation (Amler et al. 1992; Kuwahara et al. 2004; Vogt et al. 2004; Herrick et al. 2005) have been reported. Each of these patterns may reflect different pathogenetic processes.
Studies of amplicon structure have thus far been conducted at the microscopic level of analysis using FISH and thus have limited resolution. Volik et al. (2003, 2006) reported an approach by which the structure of genomic rearrangements, including those within amplicons, can be determined at the sequence level of resolution. This depends upon formation of a genomic library from the cancer genome, end sequencing of clones in order to identify those with potential rearrangements, and subsequent shotgun sequencing of rearranged clones. In an analysis of the breast cancer cell line, MCF7, end sequencing revealed many rearranged bacterial artificial chromosomes (BACs) connecting multiple amplified genomic regions (Volik et al. 2003, 2006). Complete sequencing of a single BAC revealed a patchwork of genomic DNA fragments (Volik et al. 2003). Using this strategy, we have now conducted a detailed investigation of the structure at sequence-level resolution of somatic rearrangements in multiple amplified regions in cancer. We have shown strikingly different patterns of rearrangement that provide insights into the processes of genomic breakage and repair underlying DNA amplification in cancer cells.
Results
Cell lines
HCC1954 is an immortal cell line derived from an invasive ductal carcinoma of the breast diagnosed in a 61-yr-old woman; NCI-H2171 is an immortal cell line derived from a metastatic small cell lung cancer in a pleural effusion from a 50-yr-old male Caucasian smoker; and NCI-H1770 is a lung neuroendocrine neoplasm arising in a 57-yr-old male Caucasian non-smoker.
Copy number analysis
Copy number analysis was performed using Affymetrix 10K SNP arrays and qPCR. Regions of amplification >2.5-fold diploid copy number in each of these cancers are shown in Table 1. NCI-H1770 exhibited a single region of amplification on chromosome 2 involving MYCN. HCC1954 showed multiple amplified regions on chromosomes 5, 8, and 17, including ERBB2 and MYC on chromosomes 17q and 8q, respectively. NCI-H2171 also showed multiple amplified regions on chromosomes 8, 11, 12, and 14, including one on chromosome 8q that harbors MYC. The BAC end-sequence data can also be used to assess copy number, and plots of the density of BAC ends across each amplicon are presented in Supplemental Figure 1.
Table 1.
Amplified genomic regions in NCI-H2171, HCC1954, and NCI-H1770

Amplicon size and mean copy number ratio are derived from the Affymetrix 10K array comparative genomic hybridization. BAC end density and proportion of recombinant BACs are from the BAC end-sequencing screen (data not shown).
aThe amplification level of ERBB2 in the 17q21.1 amplicon of HCC1954 was calculated by qPCR, as this was not detected by the Affymetrix array.
Spectral karyotype and FISH analysis
The spectral karyotype of HCC1954 was pseudotetraploid (http://www.path.cam.ac.uk/~pawefish; M. Grigorova, unpubl.). FISH using BACs from the amplified region on chromosome 17q that includes ERBB2 showed that this amplicon was constituted as two extended HSRs. Multicolor FISH using BACs from the chromosome 5p15.33, 5q35.2-q35.3, and 8q24.21-q24.22 (MYC) regions of amplification revealed two chimeric amplicons where all three signals co-localized. Multicolor FISH further demonstrated that these chimeric amplicons were physically separate from the chromosome 17q HSRs that include ERBB2 (Supplemental Fig. 2).
The spectral karyotype of NCI-H2171 was hypodiploid. (http://www.path.cam.ac.uk/~pawefish). Multicolor FISH showed that all the amplified regions in this cell line from chromosomes 8, 11, 12, and 14 (Table 1) mapped to one chimeric amplicon (Supplemental Fig. 2).
The spectral karyotype of NCI-H1770 was pseudotetraploid and showed a large HSR of chromosome 2 origin inserted into chromosome 12 (Grigorova et al. 2005) (Supplemental Fig. 2). The origin of the HSR and the inclusion of MYCN in the amplicon were confirmed by FISH. FISH was also used to investigate the chromosomal locations of DNA within and surrounding the amplicon (Supplemental Figs. 2, 3). BACs mapping to the 14.2- and 16.8-Mb positions on chromosome 2, and which are therefore outside the region of amplification, generated a signal on each of two apparently normal copies of chromosome 2. BACs mapping within the region of amplification highlighted the HSR but only one apparently normal copy of chromosome 2. Thus, the amplified region appears to have been excised from one of the two copies of chromosome 2 present in these cells.
End sequencing of bacterial artificial chromosome (BAC) libraries
Separate BAC libraries were constructed from HCC1954, NCI-H2171, and NCI-H1770. In total, 13,794 BACs were picked, grown, and sequenced from both ends, and both ends were mapped back to the genome. BACs from amplified regions were over-represented in each of the BAC libraries. In HCC1954, NCI-H2171, and NCI-H1770, amplified regions respectively account for ∼1.3%, ∼0.6%, and ∼0.08% of the reference human genome, while 7.5%, 3.8%, and 1.7% of BAC ends mapped back to these intervals (Table 1).
A subset of BACs from each library was not co-linear with the reference genome sequence. These putatively rearranged BACs were also over-represented in regions of amplification with 46.7%, 36.7%, and 23.2% mapping to amplicons in HCC1954, NCI-H2171, and NCI-H1770 respectively. The proportion of BACs that were rearranged in amplified regions was also elevated: 12.5%, 17.4%, and 18.0% of BACs were rearranged within the amplicons of HCC1954, NCI-H2171, and NCI-H1770 compared to 2.0%, 1.8%, and 1.3% in the whole genome (Table 1). Thus, there is a higher prevalence of genomic rearrangements in amplicons.
Sequence analysis of BACs showing evidence of rearrangement
Fifty-seven rearranged BACs were shotgun-sequenced to finished reference human genome standards: 21 from HCC1954, 28 from NCI-H2171, and 8 from NCI-H1770. A total of 170 breakage–fusion junctions (BFJs) were identified, of which 164 were confirmed as somatic events by PCR across the breakpoint in the tumor and matched normal DNAs. Four BACs from NCI-H2171 appeared to be rearranged from the BAC end-sequence data. However, when sequenced, these BACs had BFJs occurring at Sau3A restriction sites and the four BFJs could not be confirmed by PCR of genomic DNA from tumor or normal samples. They were therefore assumed to represent artefacts of BAC library construction. Two additional putative BFJs were identified in BACs 7h20 and 8j01 from NCI-H2171. These represented deletions of 256 and 7021 bp, respectively. These BFJs were shown to be present in the matched normal DNA from this line together with 50% (20/40) and 15% (6/40), respectively, of normal DNAs tested. These two BFJs therefore represent germline structural polymorphisms (Supplemental Table 1). Of the 164 confirmed somatic BFJs, 133 were unique and the remainder occurred more than once (Supplemental material Table 1, Fig. 1). Breakpoints interrupted gene sequences (Supplemental Table 2) and were located in various classes of repeat. However, there was no evidence that breakpoints occurred in genes or in repeats more frequently than expected by chance (data not shown).
Nine rearranged BACs were from the 17q amplicon in HCC1954 that includes ERBB2. In each of these BACs, only a single BFJ was present, and these joined sequences located within the 17q amplicon. One rearrangement was observed in seven independent BACs and mapped telomeric to ERBB2. The seven BACs carrying the identical rearrangement appear to be different recombinant clones as the genome–vector ligation junctions were different in each case. Two further rearrangements were each present in a single BAC. These mapped centromeric to ERBB2. Thus only three unique BFJs were identified in this amplicon (Fig. 1), and the differing frequency of the three BFJs is likely to reflect different levels of amplification of the three rearrangements. The sequences either side of each of the three BFJs were in inverted orientation with respect to each other. Moreover, in each instance the sequences to either side of each BFJ aligned to essentially the same position in the reference genome, although there were small gaps of 4567, 1556, and 1462 bp between the two ends. These rearrangements therefore are inverted duplications with small amounts of sequence missing at the BFJ.
Figure 1.

Somatic rearrangements in BACs. (A) Chromosome 17q21 amplicon in HCC1954 including ERBB2; (B) chimeric amplicon in HCC1954 including MYC; (C) chimeric amplicon in NCI-H2171 including MYC; (D) chromosome 2 amplicon in NCI-H1770 including MYCN. The color of the arrow identifies the chromosome, and the direction of the arrow indicates the orientation of DNA sequence relative to the reference genome (+ or − orientation); IVD, putative inverted duplications; black rectangles, DNA “insertions” that do not align to the reference human genome sequence with either flanking sequence. Figure parts are drawn to emphasize the types and complexity of rearrangements within the clone sequences and are therefore not drawn to scale.
Twelve rearranged BACs from the chimeric chromosome 5/8 amplicon in HCC1954 contained 57 unique BFJs. Eighteen BFJs bridged chromosomes, joining regions from chromosome 8q to regions on chromosome 5. Fifteen BFJs joined pairs of regions on chromosome 5 and 24 joined pairs of regions on chromosome 8 (Supplemental Table 1). Twenty-two intrachromosomal rearrangements were inverted, and 17 were noninverted. No inverted duplications were observed. In several BACs with multiple BFJs, the junctions were not distributed randomly throughout the BAC. Instead, the BFJs appear to cluster, with several small fragments of genome ranging from 72 bp to a few kb in length strung end to end (Supplemental material Table 1, Fig. 1).
The 24 BACs from the chimeric amplicon of NCI-H2171 contained a total of 64 different somatically acquired BFJs. Nineteen were interchromosomal, and 45 were intrachromosomal rearrangements, of which 23 were noninverted and 22 were inverted. One BAC (9g20) contained a large inverted duplication, which results in duplication of two BFJs (Supplemental material Table 1, Fig. 1). As was the case for the 5/8 amplicon in HCC1954, there were several BACS with clustering of several small fragments of genome (Supplemental material Table 1, Fig. 1).
Eight recombinant BACs from the chromosome 2 amplicon in NCI-H1770 were sequenced, yielding nine unique BFJs (Fig. 1). All were intrachromosomal and connected genomic areas within the chromosome 2 amplicon. Five were in noninverted orientation to each other, and four were inverted. No inverted duplications were observed. One BFJ (Supplemental Table 1, no. 130) was identified in two sequenced BACs as well as two additional BACs in which the junction was predicted from the BAC end-sequence data and confirmed by PCR. This BFJ was the most commonly observed and therefore the most highly amplified BFJ in this line. The structure of this BFJ bears the hallmarks expected of a double-minute chromosome (see Discussion).
DNA sequences at breakage–fusion junctions
The DNA sequences at BFJs exhibited several distinctive features. Twenty-five out of 133 (19%) rearrangements showed direct fusion of the two genomic regions, exactly as would have been predicted by simple breakage and rejoining of the reference genome sequences. However, 82/133 (62%) rearrangements included short regions of microhomology at the BFJ, i.e., identical short sequences (usually of 1–5 bp) in the reference genome at the end of each of the two fragments to be fused that are present in only single copy in the final fused sequence (Table 2; Supplemental Tables 1, 3). This pattern of microhomology is generally believed to be characteristic of non-homologous end-joining mechanisms of DNA double strand break repair (Cahill et al. 2006).
Table 2.
Patterns of sequence microhomology and putative nontemplated DNA synthesis (sequence insertions) at BFJs, for each cancer cell line

The total for each line is given.
aExtended regions of microhomology that may have resulted from nonallelic homologous recombination.
A different pattern was observed at three BFJs in HCC1954 (Supplemental Table 1, BFJs nos. 121, 71, and 74). These were characterized by relatively long microhomologies (10, 15, and 32 bp) and were within inverted Alu repeats (Fig. 2). Beyond the region of sequence identity in the microhomology, there was a much longer region of >80% sequence similarity either side of the BFJ. This extended sequence similarity was not observed at BFJs with shorter microhomologies. This pattern corresponds more closely to that expected for a double-strand break repair mechanism using nonallelic homologous recombination.
Figure 2.

Sequences at breakage–fusion junctions. (A) Example of putative homologous recombination based repair with extended microhomology; (B) example of nonhomologous end joining showing 5-bp overlapping microhomology; (C) example of nonhomologous end joining including putative nontemplated DNA synthesis. In each example, the BAC sequence is shown in the middle with the genomic sequences contributing to the rearrangement shown above and below. Regions of sequence identity are highlighted.
The remaining 26/133 (19%) of the BFJs could not be aligned to the reference genome without allowing insertion of a short DNA sequence from 1 to 64 bp in size between two putative breakpoints that did not align to the reference human genome as part of either flanking sequence or to a unique location elsewhere in the genome. Five of these insertions were longer than 20 bp. One of these gave no significant alignment to the reference human genome using BLASTN (http://www.ncbi.nlm.nih.gov/genome/seq/BlastGen/BlastGen.cgi?taxid=9606) or BLAT (no. 60 [24-bp insertion]). The remaining four (no. 20 [25-bp insertion], no. 36 [37-bp insertion], no. 5 [44-bp insertion], and no. 78 [64-bp insertion]) contained short regions that mapped to multiple locations in the genome and that did not account for the full length of the insertion. These inserted sequences may represent nontemplated DNA synthesis, clusters of very short DNA fragments, or, conceivably in some cases, unique sequences containing SNPs (see Discussion; Table 2; Supplemental Tables 1, 3).
Discussion
We have explored the underlying mechanisms of gene amplification in human cancer by identifying somatic genomic rearrangements in cancer amplicons and using the observed patterns to reconstruct the breakage and fusion processes involved. To achieve this, we end-sequenced more than 13,000 BACs and obtained finished shotgun sequences from 57, thus characterizing 133 unique somatic BFJs (derived from fusion of 266 somatic breakpoints) (Fig. 1; Supplemental Table 1), several fold more than have been previously reported in solid tumor genomes.
A total of 30 genes were affected by the somatic breakpoints identified, and in 19 BFJs the rearrangement fused part of the genomic sequences of two genes. However, none of the BFJs apposed coding sequence from the gene pair in the correct orientation while maintaining the translational frame, indicating that a functional fusion gene is unlikely to have been created. Nevertheless, the results illustrate how this strategy, particularly when coupled with the application of new sequencing technologies, could be implemented to identify fusion genes in cancer.
Three unique inverted duplications, and no other rearrangements, were observed among the nine rearranged BACs from the 17q21.1 amplicon in HCC1954 that includes ERBB2 (Fig. 1A; Table 1; Supplemental Table 1). The architecture of rearrangements in this amplicon recapitulates remarkably well the structural features predicted by a classical breakage–fusion–bridge cycle involving sister chromatids, as originally proposed by Barbara McClintock (1941) (see Supplemental Fig. 4). A double-strand chromosome break generating a free DNA end is followed by DNA synthesis and sister chromatid formation, resulting in two identical free DNA ends. The pair of sister chromatids then fuse to each other in order to eliminate the free ends, which otherwise may trigger cell death. Following chromatid separation during mitosis, an anaphase bridge is formed, resulting in a further double-strand break and re-initiation of the breakage–fusion–bridge cycle.
The model predicts that the sequences either side of each BFJ should be identical. We found, however, that there appears to have been a few kilobases of erosion leading to differences in the lengths of the sister chromatids prior to fusion. This erosion has previously been observed in an inverted duplication created as a result of sister chromatid breakage–fusion–bridge cycles during amplification of the DHFR gene in methotrexate-treated Chinese hamster cells (Okuno et al. 2004) and in cells targeted for removal of telomeric sequences (Lo et al. 2002). It may therefore be a typical feature of the repair process that allows fusion of the two sister chromatid ends. The erosion may result from single-strand exonuclease digestion of one sister chromatid in a failed attempt to allow strand invasion and repair via homologous recombination (Okuno et al. 2004).
The simplest model of the sister chromatid breakage–fusion–bridge cycle has two further predictions: (1) that the first inverted duplication will occur telomeric to the amplified cancer gene while all subsequent inverted duplications will be centromeric (see Supplemental Fig. 4); (2) that the amplified cancer gene will be present at approximately twice the copy number of the first inverted duplication, which itself will be at twice the copy number of the second BFJ. In HCC1954, the highest copy number inverted duplication in the ERBB2 amplicon (which is therefore likely to have been the first that occurred) mapped telomeric of ERBB2 and was present at about half the copy number of ERBB2 (Supplemental Table 4). The other inverted duplications were at lower copy numbers and were centromeric. Therefore, the ERBB2 amplicon in HCC1954 bears all the architectural hallmarks at the sequence level of a classical sister chromatid breakage–fusion–bridge process, the first time that this has been demonstrated in human cancer.
Rearrangements in the chromosome 2 amplicon of NCI-H1770 that includes MYCN exhibited a different pattern (Fig. 1D). Although the rearrangements were restricted to the region surrounding MYCN, there were both inverted and noninverted BFJs and inverted duplications were not observed. Four independent BACs contained an identical noninverted rearrangement (Supplemental Table 1, no. 130), which provides a clue to the processes involved in the formation of this amplicon. The particular orientation, structure, and breakpoint locations of this BFJ are compatible with looping out and excision of DNA to form a double-minute chromosome. This double minute would encompass the amplified region and bring the ends of the region, which are ∼1.9 Mb apart, together at the BFJ (see Supplemental Fig. 3). In support of this hypothesis, FISH indicates that the amplified region has been excised from one copy of chromosome 2 (Grigorova et al. 2005) (see Supplemental Figs. 2, 3). However, double minutes are not visible in the analyzed stock of NCI-H1770 cells, and the amplicon is an HSR. It is therefore likely that the double minutes have reintegrated into chromosomal DNA. Thus, analysis of amplicon architecture has enabled us to speculate about a phase of amplicon development that has now disappeared from the cell. Moreover, it has clearly distinguished the structure and development of the MYCN HSR in NCI-H1770 from the ERBB2 HSR in HCC1954.
The patterns of rearrangement found in the chimeric amplicons of HCC1954 and NCI-H2171 (Fig. 1B,C) were considerably more complex than the two previously described. Several BACs carried multiple BFJs, many of which formed interchromosomal in addition to inverted and noninverted intrachromosomal rearrangements. Notably, in many BACs from these two amplicons, the BFJs were not randomly distributed (see Supplemental material Table 1, Fig. 1). Instead, they appear to cluster, with several small fragments of genome, ranging from 20 bp to a few kilobases in length, strung end to end.
The derivation of these bizarre accretions of genomic shards is not clear. However, the small fragments almost always originate from within amplified regions. Moreover, their genomic origins are usually tightly clustered within a few kilobases of each other, often around the position of a chromosomal break (Fig. 3). We therefore propose that a cascade of small DNA fragments is sometimes generated in the vicinity of a chromosomal double strand break (either by physical or enzymatic processes). Each fragment is subsequently fused to an available free end, with a string of fragments sequentially extending during a single cell cycle, until a DNA segment with a telomere terminates the progression. This process, if true, may be similar to the capture of DNA fragments seen during double-strand break repair in model systems (Little and Chartrand 2004) and occasionally in balanced translocations (Reichel et al. 1998).
Figure 3.

Clustered genomic origins of rearranged DNA fragments. DNA segments within BAC 14g18 are shown in their reference genomic locations and orientations (see Supplemental Table 1). Fragments are shown in order 5′ to 3′ in the clone. One fragment from another BAC, 5am21, falls within the chromosome 12 interval (*). The scale shows the 120-kb region separated into 10-kb segments; the location of the expanded region is shown with respect to the amplicon for each chromosomal region. Fragments at the BAC vector insert junction are extended off-scale.
Although these unusual structures in the two chimeric amplicons bear many similarities, there are subtle differences. For example, genomic fragments from the NCI-H2171 and HCC1954 amplicons have significantly different mean lengths of 5797 and 3954 bp, respectively (P = 0.006), indicative either of distinct processes or of processes occurring at different rates. Indeed, in both amplicons the distributions of fragment lengths have significantly greater variation than a simple random breakage model (P < 0.0001), suggesting that more than one breakage process is operative in each.
The markedly different patterns of structural rearrangement found in the chromosome 17q21.1 ERBB2 amplicon and the chimeric chromosome 5/8 amplicon (Fig. 1A,B) co-exist in HCC1954 cells, albeit physically isolated from each other. The results therefore illustrate how different evolutionary paths, probably relying on different processes of breakage and repair, can be followed in the same cancer genome.
The DNA sequence at many BFJs showed overlapping microhomologies (Fig. 2; Table 2; Supplemental Tables 1, 3). These are believed to be influential in apposing fragments to be joined and are characteristic of nonhomologous end-joining mechanisms of double-strand break repair (Cahill et al. 2006). There was also, however, evidence for other classes of DNA double-strand break repair. The DNA sequences flanking most regions of microhomology do not show evidence of similarity to each other. However, three of the most extensive regions of microhomology we observed (of 10, 15, and 32 bp, all in HCC1954) showed substantially more sequence similarity in the wider region flanking the areas of sequence identity. This pattern conforms much more closely to that expected for homologous recombination based repair (Fig. 2; Supplemental Table 1). Despite analysis of a similar number of BFJs this pattern was not observed in NCI-H2171 and therefore may reflect a difference in the operative DNA repair processes between the two cancers.
There were also traces of DNA breakage and repair processes that are more cryptic to interpretation. Twenty-six BFJs showed insertion of short (1–64 bp) DNA segments between the chromosomal breakpoints that could not be aligned to the reference genome as part of either flanking sequence or to a unique location elsewhere in the genome (Supplemental Table 1). Five of these insertions were longer than 20 bp. One of these gave no significant alignment to the reference human genome, and the remaining four contained short regions that mapped to multiple locations in the genome but that did not account for the full length of the insertion. These insertions may represent clusters of such small DNA fragments that their origin in the reference genome cannot be unambiguously ascertained. Alternatively, they may be the result of nontemplate-dependent DNA synthesis or a combination of the two processes. Twenty-two such insertions were observed in NCI-H2171 and only three in HCC1954 (Fig. 1; Table 2), suggesting that the variant of breakage or repair processes that this pattern represents is more active in NCI-H2171.
The large number of breakpoints detected in this study further allows us to investigate whether there are structural features of the genome that are particularly prone to breakage or repair (Abeysinghe et al. 2003). Sequences surrounding breakpoints were GC-rich (P < 0.01). There was also a slight excess of polypurine and polypyrimidine runs (P < 0.01), which have been associated with sites of homologous recombination and formation of H-DNA (Abeysinghe et al. 2003). Additionally, several sequence motifs were significantly over-represented, including consensus sequences for deletion and frameshift hotspots, and immunoglobulin rearrangements (for a complete list of motifs analyzed, see Supplemental Table 5). These results generally indicate that the precise locations of breakage and repair processes are dictated, at least in part, by specific effects of the local sequence environment.
We recently described the variation in prevalence and pattern of somatic point mutations in human cancer genomes (Greenman et al. 2007). We have now demonstrated the diversity of rearrangement architectures and shown subtle differences between individual cancers that may reflect varying processes of breakage and repair. Because of the laborious methodology, we have only conducted a detailed analysis of four amplicons, and it is likely that further patterns remain to be discovered. The results therefore provide a vignette of the insights that will be acquired about somatic genomic rearrangements and the biological processes underlying them as large-scale sequencing of cancer genomes gathers pace.
Methods
Cell culture and copy number analysis
The HCC1954, NCI-H2171, and NCI-H1770 cell lines were obtained from the American Type Culture Collection. Copy number analysis was carried out using the Affymetrix 10K SNP array as previously described (Bignell et al. 2004; http://www.sanger.ac.uk/cgi-bin/genetics/CGP/CGH_home.cgi).
BAC library construction
DNA in PFGE agarose blocks prepared from ∼5 × 107 cells/mL was partially digested with Sau3AI and cloned into BamHI-linearized pBACe3.6 (Frengen et al. 1999). The ligation was electroporated into DH10B cells and plated on LB, and recombinant clones were picked robotically into 384-well plates containing 7.5% glycerol and grown for 20 h at 37°C.
BAC end sequencing and clone selection
BAC clones were picked, grown, and extracted in 384-well plates and end-sequenced using T7 and SP6 primers. End sequences were aligned to the reference genome sequence using SSAHA (Ning et al. 2001). The criteria for identifying potentially recombinant clones were (1) for BAC end sequences to map back onto the reference genome in the wrong orientation with respect to each other, (2) for BACs to show insert sizes >260 kb, or (3) for BAC ends to map back onto different chromosomes.
Sequencing of BACs and breakpoint analysis
Shotgun sequencing and directed finishing were carried out following established procedures (Lander et al. 2001). The final sequence was confirmed by comparison of 3–6 restriction enzyme digest patterns of the BACs to a theoretical digest of the finished BAC sequence. Rearrangements were identified by aligning the finished BAC sequence to the genome using BLAT (Kent 2002) followed by manual curation. Gaps represent DNA sequences that could not be assigned to either flanking region or that could not be confidently mapped in whole or in part to the reference genome sequence using BLAT. BFJs were investigated by PCR in both the cancer cell line and a matched normal EBV transformed cell line from the same individual, thereby confirming that the rearrangements represented somatic alterations.
Quantitative PCR
qPCR was carried out using the relative standard curve analysis in separate tubes (ABI User Bulletin #2) on the ABI 7700 sequence-detection system. Locus-specific dual-labeled probes were used for the ERBB2 and APP (endogenous control) loci, and NCI-BL2126 DNA was used as the calibrator.
Fluorescence in situ hybridization and spectral karyotyping
For detailed FISH methods, see Alsop et al. (2006). Briefly, BACs were from the 1-Mb clone set (Sanger Institute, Cambridge, UK). Purified BAC DNA was labeled by nick translation with spectrum orange-dUTP, digoxigenin-11-dUTP or biotin-16-dUTP. DOP-amplified flow-sorted chromosomes 17, 13, 5, and 8 were also labeled by nick translation with conjugated dUTPs to produce chromosome paints. Mixtures of labeled BACs and/or paints were hybridized to metaphase spreads and detected by appropriate antibody (FITC sheep anti-digoxigenin) or streptavidin (streptavidin-cy5 and biotinylated goat anti-streptavidin) conjugate.
Statistical analysis
To examine the breakpoint sequence context, 250 bp of genomic sequence surrounding each breakpoint site was compared to 100 control sequences of the same length, sampled from a 20-kb region surrounding the breakpoint but excluding the 250 bp of the breakpoint sequence itself. Overlapping breakpoint sequences were removed, so that sequence biases would not arise from multiple copies of the same sequence in the analysis set. This resulted in 219 breakpoint sequences and 21,900 control sequences matched for genomic location. Differences in GC content were assessed within ±2, ±10, and ±100 bp of breakpoints using two-tailed Fisher exact tests. Differences in polypurine/polypyrimidine and alternating purine/pyrimidine run lengths of ≥10 bp within ±10 bp and ≥25 bp within ±100 bp were assessed with one-tailed Fisher exact tests. The Bonferroni correction was applied to account for multiple testing within each analysis. To compare the two sets of genomic fragment lengths, they were first log-transformed into approximately normal distributions and then compared with a t-test.
Modeling breakages as a Poisson process will lead to shard lengths with exponential distributions. Goodness-of-fit to an exponential distribution was assessed as follows. If s represents the set of n shard lengths, the test distribution P(s) = λe−λs gives rise to likelihood L(s;λ) = λne−ns, where we have the nuisance parameter λ. If we condition by its sufficient statistic the sample mean (with distribution
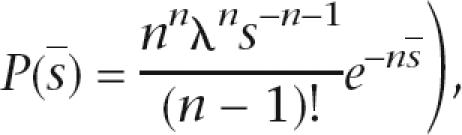 |
then the nuisance parameter is removed, giving conditional likelihood
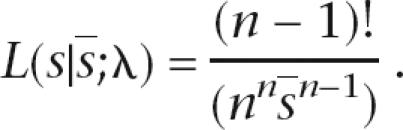 |
This is a distribution constant over the triangulation ∑s = ns. We can then simulate any test statistic using this distribution without requiring knowledge of the nuisance parameter λ. One hundred thousand simulations were used in the application. The test statistic used was the sample standard deviation, providing a goodness-of-fit test sensitive to distributions with variances either greater or smaller than that of an exponential distribution.
Acknowledgments
The studies were supported by the Wellcome Trust, Cancer Research, United Kingdom, and the Michael and Betty Kadoorie Cancer Genetics Research Programme.
Footnotes
[Supplemental material is available online at www.genome.org.]
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.6522707
References
- Abeysinghe S.S., Chuzhanova N., Krawczak M., Ball E.V., Cooper D.N., Chuzhanova N., Krawczak M., Ball E.V., Cooper D.N., Krawczak M., Ball E.V., Cooper D.N., Ball E.V., Cooper D.N., Cooper D.N. Translocation and gross deletion breakpoints in human inherited disease and cancer I: Nucleotide composition and recombination-associated motifs. Hum. Mutat. 2003;22:229–244. doi: 10.1002/humu.10254. [DOI] [PubMed] [Google Scholar]
- Alsop A.E., Teschendorff A.E., Edwards P.A.W., Teschendorff A.E., Edwards P.A.W., Edwards P.A.W. Distribution of breakpoints on chromosome 18 in breast, colorectal, and pancreatic carcinoma cell lines. Cancer Genet. Cytogenet. 2006;164:97–109. doi: 10.1016/j.cancergencyto.2005.09.011. [DOI] [PubMed] [Google Scholar]
- Amler L., Shibasaki Y., Savelyeva L., Schwab M., Shibasaki Y., Savelyeva L., Schwab M., Savelyeva L., Schwab M., Schwab M. Amplification of the N-myc gene in human neuroblastomas: Tandemly repeated amplicons within homogeneously staining regions on different chromosomes with the retention of single copy gene at the resident site. Mutat. Res. 1992;276:291–297. doi: 10.1016/0165-1110(92)90015-2. [DOI] [PubMed] [Google Scholar]
- Bignell G.R., Huang J., Greshock J., Watt S., Butler A., West S., Grigorova M., Jones K.W., Wei W., Stratton M.R., Huang J., Greshock J., Watt S., Butler A., West S., Grigorova M., Jones K.W., Wei W., Stratton M.R., Greshock J., Watt S., Butler A., West S., Grigorova M., Jones K.W., Wei W., Stratton M.R., Watt S., Butler A., West S., Grigorova M., Jones K.W., Wei W., Stratton M.R., Butler A., West S., Grigorova M., Jones K.W., Wei W., Stratton M.R., West S., Grigorova M., Jones K.W., Wei W., Stratton M.R., Grigorova M., Jones K.W., Wei W., Stratton M.R., Jones K.W., Wei W., Stratton M.R., Wei W., Stratton M.R., Stratton M.R., et al. High-resolution analysis of DNA copy number using oligonucleotide microarrays. Genome Res. 2004;14:287–295. doi: 10.1101/gr.2012304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill D., Connor B., Carney J.P., Connor B., Carney J.P., Carney J.P. Mechanisms of eukaryotic DNA double strand break repair. Front. Biosci. 2006;11:1958–1976. doi: 10.2741/1938. [DOI] [PubMed] [Google Scholar]
- Coquelle A., Toledo F., Stern S., Bieth A., Debatisse M., Toledo F., Stern S., Bieth A., Debatisse M., Stern S., Bieth A., Debatisse M., Bieth A., Debatisse M., Debatisse M. A New role for hypoxia in tumor progression: Induction of fragile site triggering genomic rearrangements and formation of complex DMs and HSRs. Mol. Cell. 1998;2:259–265. doi: 10.1016/s1097-2765(00)80137-9. [DOI] [PubMed] [Google Scholar]
- Corvi R., Savelyeva L., Amler L., Handgretinger R., Schwab M., Savelyeva L., Amler L., Handgretinger R., Schwab M., Amler L., Handgretinger R., Schwab M., Handgretinger R., Schwab M., Schwab M. Cytogenetic evolution of MYCN and MDM2 amplification in the neuroblastoma LS tumour and its cell line. Eur. J. Cancer. 1995;31A:520–523. doi: 10.1016/0959-8049(95)00031-d. [DOI] [PubMed] [Google Scholar]
- Frengen E., Weichenhan D., Zhao B., Osoegawa K., van Geel M., de Jong P.J., Weichenhan D., Zhao B., Osoegawa K., van Geel M., de Jong P.J., Zhao B., Osoegawa K., van Geel M., de Jong P.J., Osoegawa K., van Geel M., de Jong P.J., van Geel M., de Jong P.J., de Jong P.J. A modular, positive selection bacterial artificial chromosome vector with multiple cloning sites. Genomics. 1999;58:250–253. doi: 10.1006/geno.1998.5693. [DOI] [PubMed] [Google Scholar]
- Futreal P.A., Coin L., Marshall M., Down T., Hubbard T., Wooster R., Rahman N., Stratton M.R., Coin L., Marshall M., Down T., Hubbard T., Wooster R., Rahman N., Stratton M.R., Marshall M., Down T., Hubbard T., Wooster R., Rahman N., Stratton M.R., Down T., Hubbard T., Wooster R., Rahman N., Stratton M.R., Hubbard T., Wooster R., Rahman N., Stratton M.R., Wooster R., Rahman N., Stratton M.R., Rahman N., Stratton M.R., Stratton M.R. A census of human cancer genes. Nat. Rev. Cancer. 2004;4:177–183. doi: 10.1038/nrc1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenman C., Stephens P., Smith R., Dalgliesh G.L., Hunter C., Bignell G., Davies H., Teague J., Butler A., Stevens C., Stephens P., Smith R., Dalgliesh G.L., Hunter C., Bignell G., Davies H., Teague J., Butler A., Stevens C., Smith R., Dalgliesh G.L., Hunter C., Bignell G., Davies H., Teague J., Butler A., Stevens C., Dalgliesh G.L., Hunter C., Bignell G., Davies H., Teague J., Butler A., Stevens C., Hunter C., Bignell G., Davies H., Teague J., Butler A., Stevens C., Bignell G., Davies H., Teague J., Butler A., Stevens C., Davies H., Teague J., Butler A., Stevens C., Teague J., Butler A., Stevens C., Butler A., Stevens C., Stevens C., et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446:153–158. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigorova M., Lyman R.C., Caldas C., Edwards P.A.W., Lyman R.C., Caldas C., Edwards P.A.W., Caldas C., Edwards P.A.W., Edwards P.A.W. Chromosome abnormalities in 10 lung cancer cell lines of the NCI-H series analyzed with spectral karyotyping. Cancer Genet. Cytogenet. 2005;162:1–9. doi: 10.1016/j.cancergencyto.2005.03.007. [DOI] [PubMed] [Google Scholar]
- Guan X.-Y., Meltzer P.S., Dalton W.S., Trent J.M., Meltzer P.S., Dalton W.S., Trent J.M., Dalton W.S., Trent J.M., Trent J.M. Identification of cryptic sites of DNA sequence amplification in human breast cancer by chromosome microdissection. Nat. Genet. 1994;8:155–161. doi: 10.1038/ng1094-155. [DOI] [PubMed] [Google Scholar]
- Hellman A., Zlotorynski E., Scherer S.W., Cheung J., Vincent J.B., Smith D.I., Trakhtenbrot L., Kerem B., Zlotorynski E., Scherer S.W., Cheung J., Vincent J.B., Smith D.I., Trakhtenbrot L., Kerem B., Scherer S.W., Cheung J., Vincent J.B., Smith D.I., Trakhtenbrot L., Kerem B., Cheung J., Vincent J.B., Smith D.I., Trakhtenbrot L., Kerem B., Vincent J.B., Smith D.I., Trakhtenbrot L., Kerem B., Smith D.I., Trakhtenbrot L., Kerem B., Trakhtenbrot L., Kerem B., Kerem B. A role for common fragile site induction in amplification of human oncogenes. Cancer Cell. 2002;1:89–97. doi: 10.1016/s1535-6108(02)00017-x. [DOI] [PubMed] [Google Scholar]
- Herrick J., Conti C., Teissier S., Thierry F., Couturier J., Sastre-Garau X., Favre M., Orth G., Bensimon A., Conti C., Teissier S., Thierry F., Couturier J., Sastre-Garau X., Favre M., Orth G., Bensimon A., Teissier S., Thierry F., Couturier J., Sastre-Garau X., Favre M., Orth G., Bensimon A., Thierry F., Couturier J., Sastre-Garau X., Favre M., Orth G., Bensimon A., Couturier J., Sastre-Garau X., Favre M., Orth G., Bensimon A., Sastre-Garau X., Favre M., Orth G., Bensimon A., Favre M., Orth G., Bensimon A., Orth G., Bensimon A., Bensimon A. Genomic organization of amplified myc genes suggests distinct mechanisms of amplification in tumorigenesis. Cancer Res. 2005;65:1174–1179. doi: 10.1158/0008-5472.CAN-04-2802. [DOI] [PubMed] [Google Scholar]
- Kent W.J. BLAT—The BLAST-like alignment tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwahara Y., Tanabe C., Ikeuchi T., Aoyagi K., Nishigaki M., Sakamoto H., Hoshinaga K., Yoshida T., Sasaki H., Terada M., Tanabe C., Ikeuchi T., Aoyagi K., Nishigaki M., Sakamoto H., Hoshinaga K., Yoshida T., Sasaki H., Terada M., Ikeuchi T., Aoyagi K., Nishigaki M., Sakamoto H., Hoshinaga K., Yoshida T., Sasaki H., Terada M., Aoyagi K., Nishigaki M., Sakamoto H., Hoshinaga K., Yoshida T., Sasaki H., Terada M., Nishigaki M., Sakamoto H., Hoshinaga K., Yoshida T., Sasaki H., Terada M., Sakamoto H., Hoshinaga K., Yoshida T., Sasaki H., Terada M., Hoshinaga K., Yoshida T., Sasaki H., Terada M., Yoshida T., Sasaki H., Terada M., Sasaki H., Terada M., Terada M. Alternative mechanisms of gene amplification in human cancers. Genes Chromosomes Cancer. 2004;41:125–132. doi: 10.1002/gcc.20075. [DOI] [PubMed] [Google Scholar]
- Lander E., Linton L., Birren B., Nusbaum C., Zody M., Baldwin J., Devon K., Dewar K., Doyle M., FitzHugh W., Linton L., Birren B., Nusbaum C., Zody M., Baldwin J., Devon K., Dewar K., Doyle M., FitzHugh W., Birren B., Nusbaum C., Zody M., Baldwin J., Devon K., Dewar K., Doyle M., FitzHugh W., Nusbaum C., Zody M., Baldwin J., Devon K., Dewar K., Doyle M., FitzHugh W., Zody M., Baldwin J., Devon K., Dewar K., Doyle M., FitzHugh W., Baldwin J., Devon K., Dewar K., Doyle M., FitzHugh W., Devon K., Dewar K., Doyle M., FitzHugh W., Dewar K., Doyle M., FitzHugh W., Doyle M., FitzHugh W., FitzHugh W., et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Lim G., Karaskova J., Beheshti B., Vukovic B., Bayani J., Selvarajah S., Watson S., Lam W., Zielenska M., Squire J., Karaskova J., Beheshti B., Vukovic B., Bayani J., Selvarajah S., Watson S., Lam W., Zielenska M., Squire J., Beheshti B., Vukovic B., Bayani J., Selvarajah S., Watson S., Lam W., Zielenska M., Squire J., Vukovic B., Bayani J., Selvarajah S., Watson S., Lam W., Zielenska M., Squire J., Bayani J., Selvarajah S., Watson S., Lam W., Zielenska M., Squire J., Selvarajah S., Watson S., Lam W., Zielenska M., Squire J., Watson S., Lam W., Zielenska M., Squire J., Lam W., Zielenska M., Squire J., Zielenska M., Squire J., Squire J. An integrated mBAND and submegabase resolution tiling set (SMRT) CGH array analysis of focal amplification, microdeletions, and ladder structures consistent with breakage–fusion–bridge cycle events in osteosarcoma. Genes Chromosomes Cancer. 2005;42:392–403. doi: 10.1002/gcc.20157. [DOI] [PubMed] [Google Scholar]
- Little K.C.E., Chartrand P., Chartrand P. Genomic DNA is captured and amplified during double-strand break (DSB) repair in human cells. Oncogene. 2004;23:4166–4172. doi: 10.1038/sj.onc.1207570. [DOI] [PubMed] [Google Scholar]
- Lo A.W.I., Sprung C.N., Fouladi B., Pedram M., Sabatier L., Ricoul M., Reynolds G.E., Murnane J.P., Sprung C.N., Fouladi B., Pedram M., Sabatier L., Ricoul M., Reynolds G.E., Murnane J.P., Fouladi B., Pedram M., Sabatier L., Ricoul M., Reynolds G.E., Murnane J.P., Pedram M., Sabatier L., Ricoul M., Reynolds G.E., Murnane J.P., Sabatier L., Ricoul M., Reynolds G.E., Murnane J.P., Ricoul M., Reynolds G.E., Murnane J.P., Reynolds G.E., Murnane J.P., Murnane J.P. Chromosome instability as a result of double-strand breaks near telomeres in mouse embryonic stem cells. Mol. Cell. Biol. 2002;22:4836–4850. doi: 10.1128/MCB.22.13.4836-4850.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClintock B. The stability of broken ends of chromosomes in Zea mays. Genetics. 1941;26:234–282. doi: 10.1093/genetics/26.2.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muleris M., Almeida A., Gerbault-Seureau M., Malfoy B., Dutrillaux B., Almeida A., Gerbault-Seureau M., Malfoy B., Dutrillaux B., Gerbault-Seureau M., Malfoy B., Dutrillaux B., Malfoy B., Dutrillaux B., Dutrillaux B. Identification of amplified DNA sequences in breast cancer and their organization within homogeneously staining regions. Genes Chromosomes Cancer. 1995;14:155–163. doi: 10.1002/gcc.2870140302. [DOI] [PubMed] [Google Scholar]
- Myllykangas S., Knuutila S., Knuutila S. Manifestation, mechanisms and mysteries of gene amplifications. Cancer Lett. 2006;232:79–89. doi: 10.1016/j.canlet.2005.07.045. [DOI] [PubMed] [Google Scholar]
- Nahta R., Esteva F.J., Esteva F.J. Herceptin: Mechanisms of action and resistance. Cancer Lett. 2006;232:123–138. doi: 10.1016/j.canlet.2005.01.041. [DOI] [PubMed] [Google Scholar]
- Ning Z., Cox A.J., Mullikin J.C., Cox A.J., Mullikin J.C., Mullikin J.C. SSAHA: A fast search method for large DNA databases. Genome Res. 2001;11:1725–1729. doi: 10.1101/gr.194201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuno Y., Hahn P.J., Gilbert D.M., Hahn P.J., Gilbert D.M., Gilbert D.M. Structure of a palindromic amplicon junction implicates microhomology-mediated end joining as a mechanism of sister chromatid fusion during gene amplification. Nucleic Acids Res. 2004;32:749–756. doi: 10.1093/nar/gkh244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichel M., Gillert E., Nilson I., Siegler G., Greil J., Fey G., Marschalek R., Gillert E., Nilson I., Siegler G., Greil J., Fey G., Marschalek R., Nilson I., Siegler G., Greil J., Fey G., Marschalek R., Siegler G., Greil J., Fey G., Marschalek R., Greil J., Fey G., Marschalek R., Fey G., Marschalek R., Marschalek R. Fine structure of translocation breakpoints in leukemic blasts with chromosomal translocation t(4;11): The DNA damage-repair model of translocation. Oncogene. 1998;17:3035–3044. doi: 10.1038/sj.onc.1202229. [DOI] [PubMed] [Google Scholar]
- Savelyeva L., Schwab M., Schwab M. Amplification of oncogenes revisited: From expression profiling to clinical application. Cancer Lett. 2001;167:115–123. doi: 10.1016/s0304-3835(01)00472-4. [DOI] [PubMed] [Google Scholar]
- Schwab M. Oncogene amplification in solid tumors. Semin. Cancer Biol. 1999;9:319–325. doi: 10.1006/scbi.1999.0126. [DOI] [PubMed] [Google Scholar]
- Selvarajah S., Yoshimoto M., Park P.C., Maire G., Paderova J., Bayani J., Lim G., Al-Romaih K., Squire J.A., Zielenska M., Yoshimoto M., Park P.C., Maire G., Paderova J., Bayani J., Lim G., Al-Romaih K., Squire J.A., Zielenska M., Park P.C., Maire G., Paderova J., Bayani J., Lim G., Al-Romaih K., Squire J.A., Zielenska M., Maire G., Paderova J., Bayani J., Lim G., Al-Romaih K., Squire J.A., Zielenska M., Paderova J., Bayani J., Lim G., Al-Romaih K., Squire J.A., Zielenska M., Bayani J., Lim G., Al-Romaih K., Squire J.A., Zielenska M., Lim G., Al-Romaih K., Squire J.A., Zielenska M., Al-Romaih K., Squire J.A., Zielenska M., Squire J.A., Zielenska M., Zielenska M. The breakage–fusion–bridge (BFB) cycle as a mechanism for generating genetic heterogeneity in osteosarcoma. Chromosoma. 2006;V115:459–467. doi: 10.1007/s00412-006-0074-4. [DOI] [PubMed] [Google Scholar]
- Van Roy N., Vandesompele J., Menten B., Nilsson H., De Smet E., Rocchi M., De Paepe A., Pahlman S., Speleman F., Vandesompele J., Menten B., Nilsson H., De Smet E., Rocchi M., De Paepe A., Pahlman S., Speleman F., Menten B., Nilsson H., De Smet E., Rocchi M., De Paepe A., Pahlman S., Speleman F., Nilsson H., De Smet E., Rocchi M., De Paepe A., Pahlman S., Speleman F., De Smet E., Rocchi M., De Paepe A., Pahlman S., Speleman F., Rocchi M., De Paepe A., Pahlman S., Speleman F., De Paepe A., Pahlman S., Speleman F., Pahlman S., Speleman F., Speleman F. Translocation–excision–deletion–amplification mechanism leading to nonsyntenic coamplification of MYC and ATBF1. Genes Chromosomes Cancer. 2006;45:107–117. doi: 10.1002/gcc.20272. [DOI] [PubMed] [Google Scholar]
- Vogt N., Lefevre S.-H., Apiou F., Dutrillaux A.-M., Cor A., Leuraud P., Poupon M.-F., Dutrillaux B., Debatisse M., Malfoy B., Lefevre S.-H., Apiou F., Dutrillaux A.-M., Cor A., Leuraud P., Poupon M.-F., Dutrillaux B., Debatisse M., Malfoy B., Apiou F., Dutrillaux A.-M., Cor A., Leuraud P., Poupon M.-F., Dutrillaux B., Debatisse M., Malfoy B., Dutrillaux A.-M., Cor A., Leuraud P., Poupon M.-F., Dutrillaux B., Debatisse M., Malfoy B., Cor A., Leuraud P., Poupon M.-F., Dutrillaux B., Debatisse M., Malfoy B., Leuraud P., Poupon M.-F., Dutrillaux B., Debatisse M., Malfoy B., Poupon M.-F., Dutrillaux B., Debatisse M., Malfoy B., Dutrillaux B., Debatisse M., Malfoy B., Debatisse M., Malfoy B., Malfoy B. Molecular structure of double-minute chromosomes bearing amplified copies of the epidermal growth factor receptor gene in gliomas. Proc. Natl. Acad. Sci. 2004;101:11368–11373. doi: 10.1073/pnas.0402979101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volik S., Zhao S., Chin K., Brebner J.H., Herndon D.R., Tao Q., Kowbel D., Huang G., Lapuk A., Kuo W.-L., Zhao S., Chin K., Brebner J.H., Herndon D.R., Tao Q., Kowbel D., Huang G., Lapuk A., Kuo W.-L., Chin K., Brebner J.H., Herndon D.R., Tao Q., Kowbel D., Huang G., Lapuk A., Kuo W.-L., Brebner J.H., Herndon D.R., Tao Q., Kowbel D., Huang G., Lapuk A., Kuo W.-L., Herndon D.R., Tao Q., Kowbel D., Huang G., Lapuk A., Kuo W.-L., Tao Q., Kowbel D., Huang G., Lapuk A., Kuo W.-L., Kowbel D., Huang G., Lapuk A., Kuo W.-L., Huang G., Lapuk A., Kuo W.-L., Lapuk A., Kuo W.-L., Kuo W.-L., et al. End-sequence profiling: Sequence-based analysis of aberrant genomes. Proc. Natl. Acad. Sci. 2003;100:7696–7701. doi: 10.1073/pnas.1232418100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volik S., Raphael B.J., Huang G., Stratton M.R., Bignel G., Murnane J., Brebner J.H., Bajsarowicz K., Paris P.L., Tao Q., Raphael B.J., Huang G., Stratton M.R., Bignel G., Murnane J., Brebner J.H., Bajsarowicz K., Paris P.L., Tao Q., Huang G., Stratton M.R., Bignel G., Murnane J., Brebner J.H., Bajsarowicz K., Paris P.L., Tao Q., Stratton M.R., Bignel G., Murnane J., Brebner J.H., Bajsarowicz K., Paris P.L., Tao Q., Bignel G., Murnane J., Brebner J.H., Bajsarowicz K., Paris P.L., Tao Q., Murnane J., Brebner J.H., Bajsarowicz K., Paris P.L., Tao Q., Brebner J.H., Bajsarowicz K., Paris P.L., Tao Q., Bajsarowicz K., Paris P.L., Tao Q., Paris P.L., Tao Q., Tao Q., et al. Decoding the fine-scale structure of a breast cancer genome and transcriptome. Genome Res. 2006;16:394–404. doi: 10.1101/gr.4247306. [DOI] [PMC free article] [PubMed] [Google Scholar]