

Abstract
Mitochondrial DNA (mtDNA) mutations are a common cause of human disease and accumulate as part of normal ageing and in common neurodegenerative disorders. Cells express a biochemical defect only when the proportion of mutated mtDNA exceeds a critical threshold, but it is not clear whether the actual cause of this defect is a loss of wild-type mtDNA, an excess of mutated mtDNA, or a combination of the two. Here, we show that segments of human skeletal muscle fibers harboring two pathogenic mtDNA mutations retain normal cytochrome c oxidase (COX) activity by maintaining a minimum amount of wild-type mtDNA. For these mutations, direct measurements of mutated and wild-type mtDNA molecules within the same skeletal muscle fiber are consistent with the “maintenance of wild type” hypothesis, which predicts that there is nonselective proliferation of mutated and wild-type mtDNA in response to the molecular defect. However, for the m.3243A→G mutation, a superabundance of wild-type mtDNA was found in many muscle-fiber sections with negligible COX activity, indicating that the pathogenic mechanism for this particular mutation involves interference with the function of the wild-type mtDNA or wild-type gene products.
Pathogenic mtDNA mutations affect the quality or quantity of respiratory-chain proteins.1,2 Point mutations affecting tRNA genes and large deletions that remove multiple tRNA genes lead to a global decrease in mitochondrial protein synthesis and defects affecting a number of different respiratory chain complexes, which often involve complex IV (cytochrome c oxidase [COX]). Most patients with multisystem mtDNA disease harbor a mixture of mutated and wild-type mtDNA (heteroplasmy). Single-cell3,4 and transmitochondrial cybrid5–8 studies have shown that the proportion of mutated mtDNA exceeds a critical threshold in cells expressing a biochemical defect. The precise threshold differs from mutation to mutation and also between cell types. For some mtDNA mutations, the threshold appears to be sharp and clearly defined,3 but, for others, the transition is gradual.9 Understanding the threshold effect is the key to understanding the mitochondrial defect at the cellular level that underpins organ dysfunction and human disease.
The threshold could be the result of the functional complementation of mutated mtDNA by excess wild-type genomes,10 possibly through increased rates of transcription.8,11 However, there is emerging evidence that the amount of mtDNA within cells is carefully regulated, and most, if not all, mtDNA molecules are required to maintain optimal mitochondrial function, including mtDNA transcription and translation.12,13 In keeping with this, compensatory mitochondrial proliferation in response to “sick mitochondria”14 can occur in the absence of an overt biochemical defect, forming COX-positive ragged-red skeletal muscle fibers.3,4
Adjacent cells within the same organ often contain different proportions of mutated and wild-type mtDNA,3,4,15,16 limiting the value of data collected from tissue homogenates. To provide a definitive understanding of the threshold effect, the absolute amount of wild-type and mutated mtDNA must be studied at the cellular level. Skeletal muscle is ideal for this purpose, because different segments of the same multinucleate giant cell can have normal or abnormal COX activity.17 Our preliminary data revealed considerable variation in the amount of mtDNA between healthy muscle fibers, at least partly due to differences in fiber volume. This could obscure a clear relationship between the biochemical defect and the absolute amount of mutated and wild-type mtDNA. Subsequent work was therefore performed on serial sections through the same muscle fiber.
Left-quadricep-muscle biopsies were performed under local anesthetic, and 3–5-mm3 blocks of fresh skeletal muscle were transversely orientated and were mounted on filter paper before being snap frozen in supercooled isopentane cooled (to −150°C) in liquid nitrogen. The entire tissue block was sectioned (into 20-μm sections) onto glass-membrane slides for laser microdissection. Muscle histochemical analysis was performed on all the sections from each biopsy by staining for COX and succinate dehydrogenase (SDH) sequentially. Entirely synthesized from nuclear gene transcripts, SDH (complex II) is an effective counterstain used in the identification of COX-deficient muscle fibers. The sections were digitally photographed. Each fiber was given a numerical code and was followed up for the entire depth of the biopsy (42–49 sections). Each muscle-fiber section was classified as COX positive (i.e., showed brown staining), COX negative (i.e., showed the blue SDH counterstaining), or intermediate, as described elsewhere.18 Specific fibers were chosen for further analysis on the basis of the following criteria: (1) the same fiber showed a clear transition from COX positive to COX negative, and (2) the cross-sectional area of the fiber remained constant. Individual fibers were photographed, were laser captured (Leica LMD), and were subjected to cell lysis and DNA extraction.
Before investigating the patients with mtDNA mutations, we studied the effect of different lysis techniques and enzyme histochemistry on the amount of mtDNA within single muscle fibers from healthy control subjects. Serial sections from the same control muscle fiber were stained for COX in isolation, were stained sequentially with COX and SDH, and were left unstained. Individual fiber sections were laser captured and were lysed using different techniques (tables 1 and 2). We observed no effect on the quality of the data obtained from any of the combinations of histochemical assays. The most consistent and greatest amount of mtDNA was obtained from cells lysed for 16 h in 50 mM Tris-HCl (pH 8.5), with 0.5% Tween 20 and 100 μg Proteinase K at 55°C, followed by heat inactivation at 95°C for 10 min. Similar experiments showed that a more reliable estimate of cellular mtDNA content was obtained by splitting the lysate into 2-μl fractions and taking a minimum of four measurements, rather than a single measurement from 20 μl (data not shown). Each muscle-fiber section was lysed in 20 μl, which was split into two aliquots: 10 μl for real-time PCR quantification (4× 2-μl replicates) of the total amount of mtDNA within each fiber section (mtDNA copy number) and 10 μl for quantifying heteroplasmy. mtDNA copy number was measured by real-time PCR by use of iQ Sybr Green on the BioRad ICycler (BioRad) with a target template spanning from nt 3459 to nt 3568 of MTND1 (MIM 516000) (with forward primer 5′-ACGCCATAAAACTCTTCACCAAAG-3′ and reverse primer 5′-GGGTTCATAGTAGAAGAGCGATGG-3′). Absolute quantification was performed by the standard-curve method by use of a purified PCR product quantified by UV absorbance at 260 nm, with each measurement made in quadruplicate, as described elsewhere.18
Table 1. .
Amount of mtDNA Measured in Type 1 Skeletal Muscle Fibers Dissected from 20-μm Cryostat Sections of Control Muscle[Note]
| No. of mtDNA Molecules with Method |
||||
| Measurement | Standard Lysis Buffer | Lysis Buffer without EDTA | PCR Buffer | Lysis and Purification |
| 1 | 32,200 | 19,800 | 761 | 8,300 |
| 2 | 27,800 | 13,400 | 9,430 | 6,030 |
| 3 | 1,950 | 11,500 | 2,420 | 2,740 |
| 4 | 2,050 | 15,400 | 2,990 | 7,910 |
| 5 | 42,800 | 12,900 | 3,630 | 7,640 |
| 6 | 12,200 | 10,100 | 3,200 | 4,230 |
| 7 | 1,300 | 13,200 | 15,800 | 17,300 |
| 8 | 5,010 | 12,500 | 4,210 | 2,740 |
| 9 | 957 | 13,300 | 11,500 | 16,700 |
| 10 | 42,100 | 14,800 | 1,530 | 40,500 |
| 11 | 8,350 | 45,200 | … | 7,520 |
| 12 | 33,800 | 11,100 | … | 64,600 |
| Mean | 17,543 | 16,100 | 5,547 | 15,518 |
| SD | 16,835 | 9,494 | 4,966 | 18,630 |
Note.— Fibers were lysed using different methods. “Standard lysis buffer” was 50 mM Tris-HCl (pH 8.5), with 1 mM EDTA (pH 8), 0.5% Tween 20, and 100 μg Proteinase K. “Lysis buffer without EDTA” was 50 mM Tris-HCl (pH 8.5), 0.5% Tween 20, and 100 μg Proteinase K. “PCR buffer” was ABI GeneAmp PCR Buffer (1×), 0.5% Tween 20, and 100 μg Proteinase K. “Lysis and purification” was the same as for standard lysis buffer, followed by column purification (Montage PCR filter units [Millipore]) in accordance with the manufacturer's instructions. Lysis was performed at 55°C for 2 h, followed by heat inactivation at 95°C for 10 min before immediate real-time PCR to measure mtDNA copy number (see text). The inclusion of EDTA in the buffer increases the variability, whereas a post-lysis purification markedly decreases the copy number measurement. Lysis buffer without EDTA was used for subsequent experiments. Each value in the table was obtained from a different muscle fiber section.
Table 2. .
Effect of Lysis Temperature and Lysis Incubation Time on mtDNA Copy Number in Control Muscle[Note]
| No. of mtDNA Molecules with |
||||
| 2-h Incubation at |
16-h Incubation at |
|||
| Measurement | 37°C | 55°C | 37°C | 55°C |
| 1 | 15,200 | 18,500 | 17,800 | 23,000 |
| 2 | 7,640 | 19,500 | 20,700 | 20,400 |
| 3 | 22,100 | 16,300 | 12,400 | 18,000 |
| 4 | 15,400 | 15,400 | 17,100 | 37,200 |
| 5 | 20,700 | 16,800 | 27,200 | 29,800 |
| 6 | 15,400 | 24,400 | 15,900 | 19,500 |
| 7 | 21,400 | 19,800 | 51,200 | 22,400 |
| 8 | 15,300 | 15,400 | 19,800 | 24,300 |
| 9 | 15,600 | 14,900 | 23,200 | 25,100 |
| 10 | 15,600 | 22,200 | 22,500 | 27,900 |
| 11 | 17,800 | 16,900 | 14,900 | 26,000 |
| 12 | 16,600 | 18,500 | 21,600 | 31,300 |
| Mean | 16,562 | 18,217 | 22,025 | 25,408 |
| SD | 3,818 | 2,903 | 10,043 | 5,483 |
Note.— Measurements from control skeletal muscle fibers were taken from 20-μm cryostat sections, by use of lysis buffer without EDTA and real-time PCR to measure total mtDNA (see text) in control muscle type 1 fibers. Consistently higher values were obtained by incubation at 55°C for 16 h. Each value in the table was obtained from a different muscle fiber section.
For the 4.9-kb deletion, the proportion of mutated mtDNA was determined by real-time PCR (with forward primer 5′-ACCTTGGCTATCATCACCCGAT-3′ and reverse primer 5′-AGTGCGATGAGTAGGGGAAGG-3′) by use of iQ Sybr Green on the BioRad ICycler (BioRad) with a target template spanning from nt 11144 to nt 11250 of MTND4 (MIM 516003), which was deleted in the patient.19 Each assay was performed in quadruplicate. The proportion of mutated mtDNA was then calculated from the data for MTND4 (wild-type molecules) and MTND1 (total mtDNA). For the mtDNA point mutations, the proportion of mutated mtDNA was determined by last-cycle fluorescent PCR with use of a labeled primer to generate mutated and wild-type fragments, which were sized on a fluorescent analyzer (Beckman Coulter CEQ 8000) as described elsewhere20,21 (by an RFLP-based method for m.3243A→G and m.10010T→C), enabling a direct calculation of the amount of mutated and wild-type mtDNA within each fiber.
A total of 400 muscle fibers were examined through 49 serial 20-μM sections from a patient harboring the 4.9-kb “common deletion” of mtDNA (for patient details, see appendix A). COX-SDH histochemistry was used to identify seven transitions from regions of normal COX activity (COX-positive regions) to regions of COX deficiency (COX-negative regions) (n=41 fiber sections) (fig. 1a). COX-negative sections contained more mtDNA molecules than did the COX-positive sections (fig. 1a) (mean increase 2.94-fold, SD=1.50; paired t test P=.004) and had a higher percentage of mutated mtDNA (96.02% vs. 55.15%; SD=24.97; P=7.17×10-5), consistent with published data,15,16 and a lower percentage of wild-type mtDNA (mean decrease 5.15-fold, SD=5.10; P<.001). Between these regions, fiber sections contained intermediate values, demonstrating the gradual transition from COX-positive to COX-negative sections (fig. 1a and fig. 2a).
Figure 1. .

Relationship between wild-type and mutated mtDNA and COX deficiency for three pathogenic mtDNA mutations. a, 4.9-kb mtDNA deletion. b, m.10010T→C MTTG point mutation. c, m.3243A→G MTTL1 point mutation. Serial 20-μm sections through the same left-quadricep skeletal muscle fiber show the absolute amount of mutated mtDNA (red bars), the absolute amount of wild-type mtDNA (green bars), and the percentage of mutated mtDNA (blackened squares). Adjacent sections are linked by a solid line, and nonadjacent sections are linked by a dashed line. Error bars correspond to the SEM for the three measurements made on each tissue section. A photomicrograph of the corresponding muscle-fiber section before lysis is shown below, stained histochemically for COX activity (brown) and SDH activity (blue). Strong brown staining indicates normal COX activity (Positive), whereas strong blue staining indicates COX deficiency (Negative). Brown-blue staining indicates an intermediate level of COX activity (Transition), corresponding to the transition region between normal and abnormal COX activity. Individual fibers were stained, photographed, captured by laser microdissection, and lysed before molecular genetic analysis (see text).
Figure 2. .



Relationship between wild-type and mutated mtDNA and COX deficiency for three pathogenic mtDNA mutations. a, 4.9-kb mtDNA deletion. b, m.10010T→C MTTG point mutation. c, m.3243A→G MTTL1 point mutation. Serial 20-μm sections through the same left-quadricep skeletal muscle fiber show the absolute amount of mutated mtDNA (left panels) subdivided into the absolute amount of mutated and wild-type mtDNA (CN = copy number in each muscle-fiber section). Right panels show the corresponding percentage of mutated mtDNA (% MUTATION). POSITIVE = COX positive; NEGATIVE = COX negative; TRANSITION = region of intermediate COX activity (see text).
Plotting the data from all 41 segments on one graph revealed a clear relationship between COX activity and the amount of wild-type mtDNA (fig. 3a). COX-positive sections contained a varied amount of mutated mtDNA, but the number of wild-type molecules was relatively constant (SD=0.15). By contrast, high percentage levels of mutated mtDNA were associated with a dramatic reduction in wild-type levels and COX deficiency. These observations correspond with predictions made by an in silico model based on the “maintenance of wild type” hypothesis, which assumes that healthy fiber segments contain an optimal amount of mtDNA required to maintain COX activity22 (for an overview of the modeling methods, see appendix B). When the mtDNA are mutated and compromise expression of the 13 mtDNA-encoded proteins, a segment of the multinucleated cell responds by nonselectively proliferating its entire mtDNA content, to restore wild-type mtDNA to its optimal level. This mechanism will maintain wild-type mtDNA at near-optimal levels, but only up to a specific point. Beyond this point, further proliferation leads to the loss of wild-type genomes at the expense of mutated genomes, which are in excess, with the consequent loss of COX activity.22
Figure 3. .

Relationship between the amount of wild-type mtDNA and the percentage of mutated mtDNA. The amount of wild-type mtDNA is shown relative to an estimation of the optimal amount of wild-type mtDNA based on the average amount of wild-type mtDNA in adjacent COX-positive regions from the same muscle fiber. Each data point represents measurements made on a single muscle-fiber segment. Error bars correspond to the SEM for three independent assays. a, 4.9-kb mtDNA deletion (n=41). b, m.10010T→C MTTG point mutation (n=37). c, m.3243A→G MTTL1 point mutation (n=33). The histochemical phenotype is color coded: red circles indicate COX-positive regions, blue triangles indicate COX-negative regions, and gray squares indicate transition regions. The superimposed curve describes predictions made by the in silico model of mtDNA replication and heteroplasmy in nondividing cells that is based on the “maintenance of wild type” hypothesis (see text and appendix B). The model results are calculated using equation (1) with α=16. This value is derived from independent experimental data measuring the maximum amount of mitochondrial proliferation seen in skeletal muscle harboring pathogenic mtDNA mutations. (See work by Chinnery and Samuels22 and Capps et al.23 for a detailed explanation with citations of the original data that underpin the model.)
To determine whether this effect was specific to the 4.9-kb “common deletion,” we performed similar analysis on six transitions from COX-positive to COX-negative regions (n=37 fiber sections) (figs. 1b and 2b) for an mtDNA tRNA gene point mutation also known to globally impair intramitochondrial protein synthesis: m.10010T→C MTTG21 (MIM 590035) (for patient details, see appendix A). As with the 4.9-kb deletion, the COX-negative fiber sections contained a greater number of mtDNA molecules (mean increase 4.77-fold, SD=2.23; P=.037), a greater percentage of mutated mtDNA (98.56% vs. 91.67%, SD=3.69; P=4.45×10-6), and a lower number of wild-type mtDNA (mean decrease 1.76-fold, SD=1.10; P=.203) than did the COX-positive sections, although the last observation did not reach statistical significance. There was a similar relationship between the amount of wild-type mtDNA and the percentage level of mutated mtDNA (fig. 3b).
Here, we provide the first direct evidence supporting the in silico model.22 This accurately describes events in single muscle-fiber segments in vivo and provides an alternative explanation for previously published data. Proliferation of the entire mtDNA pool in response to a decrease in the amount of wild-type mtDNA gives the impression of the preferential replication of mutated molecules, when the signal to increase replication is not selective. The threshold effect is a direct consequence of this process, rather than being the result of the loss of reserve capacity, and is due to an inability to maintain wild-type mtDNA at an optimal level.
The m.3243A→G MTTL1 (MIM 590050) point mutation has distinct histopathological features, including COX-positive ragged-red fibers. Since it is the most common inherited heteroplasmic mtDNA mutation, we also studied eight transitions in five muscle fibers from a patient with this mutation (n=33 fiber sections) (figs. 1c and 2c) (for patient details, see appendix A). As expected, COX-negative fiber sections contained a greater number of mtDNA molecules (mean increase 9.95-fold, SD=4.99; P=.006) and a higher percentage of mutated mtDNA (91.35% vs. 58.55%, SD=22.15; P=2.23×10-4) than did the COX-positive sections. Again, each COX-positive segment contained a similar amount of wild-type mtDNA, despite each segment containing different percentage levels of mutated mtDNA (fig. 3c). However, unlike for the other mutations, regions of intermediate COX activity contained the same amount of wild-type mtDNA as did the COX-positive regions, with the amount of wild-type mtDNA up to sixfold greater in COX-negative segments (mean increase 5.86-fold, SD=1.84; P=.013). In other words, supranormal levels of wild-type mtDNA could not maintain COX-activity, raising the possibility that the m.3243A→G MTTL1 mutation suppresses the normal function of the wild-type tRNALeu(UUR) in human skeletal muscle fibers.
All three of these mtDNA mutations impair intramitochondrial protein synthesis, but there is a poor correlation between the translational and biochemical defects for m.3243A→G,24,25 indicating an additional pathogenic mechanism other than decreased steady-state levels of aminoacylated tRNA.26 A primary defect in 5-taurinomethyluridine modification of the first (wobble) position of the anticodon leads to impaired UUG decoding in m.3243A→G and to a qualitative defect in translation.26,27 Although similar defects were observed with other tRNA mutations,26 they are not a universal finding,26 indicating a fundamental difference in disease mechanism between different tRNA mutations that may correlate with the clinical phenotype.27
How could a mutated tRNA interfere with the function of wild-type mtDNA molecules? Specific MTTL1 mutations have been shown to disrupt the aminoacylation of wild-type tRNA, although this does not appear to be the case for m.3243A→G.28 Alternatively, since m.3243A→G is located within the binding site of the mitochondrial transcription termination factor, this could lead to reduced transcription termination at the 16S/tRNALeu(UUR) boundary, and the high levels of steady-state 16S transcripts observed in myoblasts.29 Associated high levels of the 19S RNA in m.3243A→G cybrids (an RNA intermediate incorporating the 16S RNA, tRNALeu(UUR), and ND1 transcripts) incorporated into mitochondrial polyribosomes could lead to ribosomal stalling.30 mtDNA proliferation and increased rates of transcription would exacerbate this effect, generating a positive-feedback mechanism and further inhibition of ribosomal function. In some ways, this is similar to the “dominant negative effect” seen in some Mendelian traits. However, this term must be used with caution, and comparing mitochondrial and Mendelian genetics is not straightforward. For example, mtDNA deletions have been described as having both “dominant”31 and “recessive”32 properties, on the basis of measurement of different aspects of the associated molecular biology. Analysis of the tRNA levels in each segment could help to resolve this complex issue for specific mtDNA mutations, including m.3243A→G.
It is intriguing that we consistently observed a decrease in the total amount of mtDNA in leukocytes from patients with the m.3243A→G MTTL1 mutation.20 Unlike skeletal muscle, which is postmitotic, the leukocyte population is continuously recycled in the peripheral circulation. The low levels of mtDNA seen in m.3243A→G leukocytes may reflect this fundamental difference between postmitotic and dividing cells, although other mechanisms have been proposed involving mitochondrial fragility.33
Our current model does not account for the structural organization of mitochondria and mtDNA within skeletal muscle fibers but assumes that the mtDNA molecules are freely mixing.22 Some degree of compartmentalization clearly must occur to produce defined segments of COX deficiency,17 but the correspondence between observed values measured in skeletal muscle and the predictions made by the model indicate that this is less important over short distances (such as the 20-μm section thickness studied here). Incorporating some form of compartmentalization is likely to be important for studying the long-term spread of a COX-deficient segment along the muscle fiber length.
These observations add to the growing body of evidence against the concept of a surplus of mitochondrial genomes within healthy human cells and favor the close correlation between transcription rates and mtDNA copy number. This supports the development of treatment strategies aimed at increasing the amount of mtDNA in patients with mtDNA disease. A similar approach would also prevent somatic mtDNA mutations that contribute to aging and neurodegeneration. However, this might not be appropriate for every mtDNA defect, because some may lead to high levels of mtDNA proliferation, which correlates with apoptosis.34 For these mutations, new therapeutic strategies should be aimed at increasing specifically the amount of wild-type mtDNA, not at increasing the total amount of mtDNA.
Acknowledgments
P.F.C. is a Wellcome Trust Senior Fellow in Clinical Science. This study was funded by a Wellcome Trust project grant (awarded to P.F.C. and D.C.S.). P.F.C. also receives funding from the United Mitochondrial Diseases Foundation, and the European Union Framework Programme EUmitocombat and MITOCIRCLE. We are grateful to Rob Taylor, for his help in obtaining muscle blocks, and to two anonymous reviewers, for helpful comments.
Appendix A: Clinical Details of the Patients with mtDNA Myopathy
Patient 1 is a 37-year-old man with chronic progressive external ophthalmoplegia who presented in his 20s with bilateral ptosis and ophthalmoplegia and who later developed a mild proximal myopathy and dysphagia. His serum creatine kinase was 215 IU/liter (normal <170 IU/liter), his fasting blood lactate was 1.1 mM/liter, and his fasting cerebrospinal fluid lactate was 1.6 mM/liter. A left-quadricep-muscle biopsy, which was described in detail elsewhere,17 revealed 15% ragged-red skeletal muscle fibers and 20% COX-negative fibers. A Southern blot showed that 20% of the muscle mtDNA had the 4.9-kb “common” mtDNA deletion.
Patient 2 is a 43-year-old woman with short stature who was described elsewhere21 and who presented with an encephalopathy at age 20 years. She went on to develop cognitive impairment, bilateral optic atrophy, sensorineural deafness, ataxia, dystonia, and a spastic paraparesis. She had glucose intolerance. Her serum creatine kinase ranged between normal and 1,800 IU/liter (normal <170 IU/liter), and her serum lactate was 5.3 mM/liter. Her diagnostic muscle biopsy revealed 30% ragged-red fibers and 21% COX-negative fibers. Complete sequencing of muscle mtDNA revealed two heteroplasmic substitutions: m.10010T→C MTTG, thought to be the primary pathogenic mutation in this case, and the m.5656A→G noncoding substitution, which has been described in a large number of healthy control subjects (mtDB Web site).21
Patient 3 is a 30-year-old woman with longstanding bilateral sensorineural deafness and short stature, who presented at age 30 years with recurrent seizures and an encephalopathy. She later developed migraine, diabetes, recurrent vomiting, gastrointestinal pseudo-obstruction, cardiac failure, optic atrophy, and a proximal myopathy. Her serum creatine kinase was 302 IU/liter (normal <170 IU/liter), and her fasting blood lactate ranged from 3.3 to 5.5 mM/liter. A left-quadricep-muscle biopsy revealed 30% ragged-red fibers and 2% COX-negative fibers. The m.3243A→G MTTL1 mutation was detected in blood (49% mutated mtDNA) and muscle (52% mutated mtDNA).
Appendix B: In Silico Modeling
Our simulation and mathematical model of mtDNA replication is explained elsewhere.22,23 For loss-of-function mutations (where the total amount of wild-type mtDNA is the primary determinant of mitochondrial function), this model predicts that the wild-type copy number W in a cell will be a simple function of the mutation level m, given by
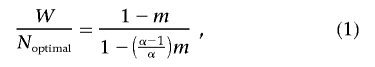 |
where Noptimal is the number of mtDNA molecules in a normal cell with no mutant mtDNA and α is a parameter defining the maximum proliferation in the cell.
Web Resources
The URLs for data presented herein are as follows:
- mtDB, http://www.genpat.uu.se/mtDB/
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for MTND1, MTND4, MTTG, and MTTL1)
References
- 1.DiMauro S, Schon EA (2003) Mitochondrial respiratory-chain diseases. N Engl J Med 348:2656–2668 10.1056/NEJMra022567 [DOI] [PubMed] [Google Scholar]
- 2.Taylor RW, Turnbull DM (2005) Mitochondrial DNA mutations in human disease. Nat Rev Genet 6:389–402 10.1038/nrg1606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boulet L, Karpati G, Shoubridge EA (1992) Distribution and threshold expression of the tRNALys mutation in skeletal muscle of patients with myoclonic epilepsy and ragged-red fibers (MERRF). Am J Hum Genet 51:1187–1200 [PMC free article] [PubMed] [Google Scholar]
- 4.Tokunaga M, Mita S, Murakami T, Kumamoto T, Uchino M, Nonaka I, Ando M (1994) Single muscle fiber analysis of mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS). Ann Neurol 35:413–419 10.1002/ana.410350407 [DOI] [PubMed] [Google Scholar]
- 5.Koga Y, Davidson M, Schon EA, King MP (1995) Analysis of cybrids harboring MELAS mutations in the mitochondrial tRNALeu(UUR) gene. Muscle Nerve 3:S119–S123 10.1002/mus.880181424 [DOI] [PubMed] [Google Scholar]
- 6.Dunbar DR, Moonie PA, Zeviani M, Holt IJ (1996) Complex I deficiency is associated with 3243G:C mitochondrial DNA in osteosarcome cell cybrids. Hum Mol Genet 5:123–129 10.1093/hmg/5.1.123 [DOI] [PubMed] [Google Scholar]
- 7.Chomyn A, Martinuzzi A, Yoneda M, Daga A, Hurko O, Johns D, Lai ST, Nonaka I, Angelini C, Attardi G (1992) MELAS mutation in mtDNA binding site for transcription termination factor causes defects in protein synthesis and in respiration but no change in levels of upstream and downstream mature transcripts. Proc Natl Acad Sci USA 89:4221–4225 10.1073/pnas.89.10.4221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yoneda M, Miyatake T, Attardi G (1994) Complementation of mutant and wild-type human mitochondrial DNAs coexisting since the mutation event and lack of complementation of DNAs introduced separately into a cell within distinct organelles. Mol Cell Biol 14:2699–2712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clark KM, Taylor RW, Johnson MA, Chinnery PF, Chrzanowska-Lightowlers ZMA, Andrews RM, Nelson IP, Wood NW, Lamont PJ, Hanna MG, et al (1999) An mtDNA mutation in the initiation codon of cytochondrome c oxidase subunit II results in lower levels of the protein and a mitochondrial encephalomyopathy. Am J Hum Genet 64:1330–1339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hammans SR, Sweeney MG, Holt IJ, Cooper JM, Toscano A, Clark JB, Morgan-Hughes JA, Harding AE (1992) Evidence for intramitochondrial complementation between deleted and normal mitochondrial DNA in some patients with mitochondrial myopathy. J Neurol Sci 107:87–92 10.1016/0022-510X(92)90213-5 [DOI] [PubMed] [Google Scholar]
- 11.Attardi G, Yoneda M, Chomyn A (1995) Complementation and segregation behavior of disease causing mitochondrial DNA mutations in cellular model systems. Biochim Biophys Acta 1271:241–248 [DOI] [PubMed] [Google Scholar]
- 12.Dubeau F, De Stefano N, Zifkin BG, Arnold DL, Shoubridge EA (2000) Oxidative phosphorylation defect in the brains of carriers of the tRNAleu(UUR) A3243G mutation in a MELAS pedigree. Ann Neurol 47:179–185 [DOI] [PubMed] [Google Scholar]
- 13.Villani G, Attardi G (1997) In vivo control of respiration by cytochrome c oxidase in wild-type and mitochondrial DNA mutation-carrying human cells. Proc Natl Acad Sci USA 94:1166–1171 10.1073/pnas.94.4.1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yoneda M, Chomyn A, Martinuzzi A, Hurko O, Attardi G (1992) Marked replicative advantage of human mtDNA carrying a point mutation that causes the MELAS encephalomyopathy. Proc Natl Acad Sci USA 89:11164–11168 10.1073/pnas.89.23.11164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sciacco M, Bonilla E, Schon EA, DiMauro S, Moraes CT (1994) Distribution of wild-type and common deletion forms of mtDNA in normal and respiration-deficient muscle fibers from patients with mitochondrial myopathy. Hum Mol Genet 3:13–19 [erratum 3:687] 10.1093/hmg/3.1.13 [DOI] [PubMed] [Google Scholar]
- 16.Oldfors A, Larsson NG, Holme E, Tulinius M, Kadenbach B, Droste M (1992) Mitochondrial DNA deletions and cytochrome c oxidase deficiency in muscle fibres. J Neurol Sci 110:169–177 10.1016/0022-510X(92)90025-G [DOI] [PubMed] [Google Scholar]
- 17.Elson JL, Samuels DC, Johnson MA, Turnbull DM, Chinnery PF (2002) The length of cytochrome c oxidase-negative segments in muscle fibres in patients with mtDNA myopathy. Neuromuscul Disord 12:858–864 10.1016/S0960-8966(02)00047-0 [DOI] [PubMed] [Google Scholar]
- 18.Durham SE, Bonilla E, Samuels DC, DiMauro S, Chinnery PF (2005) Mitochondrial DNA copy number threshold in mtDNA depletion myopathy. Neurology 65:453–455 10.1212/01.wnl.0000171861.30277.88 [DOI] [PubMed] [Google Scholar]
- 19.He L, Chinnery PF, Durham SE, Blakely EL, Wardell TM, Borthwick GM, Taylor RW, Turnbull DM (2002) Detection and quantification of mitochondrial DNA deletions in individual cells by real-time PCR. Nucleic Acids Res 30:e68 10.1093/nar/gnf067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pyle A, Taylor RW, Durham SE, Deschauer M, Schaefer AM, Samuels DC, Chinnery PF (2007) Depletion of mitochondrial DNA in leucocytes harbouring the 3243A→G mtDNA mutation. J Med Genet 44:69–74 10.1136/jmg.2006.043109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bidooki SK, Johnson MA, Chrzanowska-Lightowlers Z, Bindoff LA, Lightowlers RN (1997) Intracellular mitochondrial triplasmy in a patient with two heteroplasmic base changes. Am J Hum Genet 60:1430–1438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chinnery PF, Samuels DC (1999) Relaxed replication of mtDNA: a model with implications for the expression of disease. Am J Hum Genet 64:1158–1165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Capps GJ, Samuels DC, Chinnery PF (2003) A model of the nuclear control of mitochondrial DNA replication. J Theor Biol 221:565–583 10.1006/jtbi.2003.3207 [DOI] [PubMed] [Google Scholar]
- 24.Florentz C, Sohm B, Tryoen-Toth P, Putz J, Sissler M (2003) Human mitochondrial tRNAs in health and disease. Cell Mol Life Sci 60:1356–1375 10.1007/s00018-003-2343-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hayashi J, Ohta S, Takai D, Miyabayashi S, Sakuta R, Goto Y, Nonaka I (1993) Accumulation of mtDNA with a mutation at position 3271 in tRNALeu(UUR) gene introduced from a MELAS patient to HeLa cells lacking mtDNA results in progressive inhibition of mitochondrial respiratory function. Biochem Biophys Res Commun 197:1049–1055 10.1006/bbrc.1993.2584 [DOI] [PubMed] [Google Scholar]
- 26.Yasukawa T, Kirino Y, Ishii N, Holt IJ, Jacobs HT, Makifuchi T, Fukuhara N, Ohta S, Suzuki T, Watanabe K (2005) Wobble modification deficiency in mutant tRNAs in patients with mitochondrial diseases. FEBS Lett 579:2948–2952 10.1016/j.febslet.2005.04.038 [DOI] [PubMed] [Google Scholar]
- 27.Kirino Y, Yasukawa T, Marjavaara SK, Jacobs HT, Holt IJ, Watanabe K, Suzuki T (2006) Acquisition of the wobble modification in mitochondrial tRNALeu(CUN) bearing the G12300A mutation suppresses the MELAS molecular defect. Hum Mol Genet 15:897–904 10.1093/hmg/ddl007 [DOI] [PubMed] [Google Scholar]
- 28.Hao R, Yao YN, Zheng YG, Xu MG, Wang ED (2004) Reduction of mitochondrial tRNALeu(UUR) aminoacylation by some MELAS-associated mutations. FEBS Lett 578:135–139 10.1016/j.febslet.2004.11.004 [DOI] [PubMed] [Google Scholar]
- 29.Suomalainen A, Majander A, Pihko H, Peltonen L, Syvanen AC (1993) Quantification of tRNA3243Leu point mutation of mitochondrial DNA in MELAS patients and its effects on mitochondrial transcription. Hum Mol Genet 2:525–534 10.1093/hmg/2.5.525 [DOI] [PubMed] [Google Scholar]
- 30.King MP, Koga Y, Davidson M, Schon EA (1992) Defects in mitochondrial protein synthesis and respiratory chain activity segregate with the tRNALeu(UUR) mutation associated with mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes. Mol Cell Biol 12:480–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shoubridge EA, Karpati G, Hastings KE (1990) Deletion mutants are functionally dominant over wild-type mitochondrial genomes in skeletal muscle fiber segments in mitochondrial disease. Cell 62:43–49 10.1016/0092-8674(90)90238-A [DOI] [PubMed] [Google Scholar]
- 32.Mita S, Schmidt B, Schon EA, DiMauro S, Bonilla E (1989) Detection of “deleted” mitochondrial genomes in cytochrome-c oxidase-deficient muscle fibers of a patient with Kearns-Sayre syndrome. Proc Natl Acad Sci USA 86:9509–9513 10.1073/pnas.86.23.9509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim JY, Hwang J-M, Ko HS, Seong M-W, Park SS (2005) Mitochondrial DNA content is decreased in autosomal dominant optic atrophy. Neurology 64:966–972 [DOI] [PubMed] [Google Scholar]
- 34.Aure K, Fayet G, Leroy JP, Lacene E, Romero NB, Lombes A (2006) Apoptosis in mitochondrial myopathies is linked to mitochondrial proliferation. Brain 129:1249–1259 10.1093/brain/awl061 [DOI] [PubMed] [Google Scholar]