

Abstract
Alzheimer disease (AD) is the most common cause of dementia. We conducted a genome screen of 103 patients with late-onset AD who were ascertained as part of the Genetic Research in Isolated Populations (GRIP) program that is conducted in a recently isolated population from the southwestern area of The Netherlands. All patients and their 170 closely related relatives were genotyped using 402 microsatellite markers. Extensive genealogy information was collected, which resulted in an extremely large and complex pedigree of 4,645 members. The pedigree was split into 35 subpedigrees, to reduce the computational burden of linkage analysis. Simulations aiming to evaluate the effect of pedigree splitting on false-positive probabilities showed that a LOD score of 3.64 corresponds to 5% genomewide type I error. Multipoint analysis revealed four significant and one suggestive linkage peaks. The strongest evidence of linkage was found for chromosome 1q21 (heterogeneity LOD [HLOD]=5.20 at marker D1S498). Approximately 30 cM upstream of this locus, we found another peak at 1q25 (HLOD=4.0 at marker D1S218). These two loci are in a previously established linkage region. We also confirmed the AD locus at 10q22-24 (HLOD=4.15 at marker D10S185). There was significant evidence of linkage of AD to chromosome 3q22-24 (HLOD=4.44 at marker D3S1569). For chromosome 11q24-25, there was suggestive evidence of linkage (HLOD=3.29 at marker D11S1320). We next tested for association between cognitive function and 4,173 single-nucleotide polymorphisms in the linked regions in an independent sample consisting of 197 individuals from the GRIP region. After adjusting for multiple testing, we were able to detect significant associations for cognitive function in four of five AD-linked regions, including the new region on chromosome 3q22-24 and regions 1q25, 10q22-24, and 11q25. With use of cognitive function as an endophenotype of AD, our study indicates the that the RGSL2, RALGPS2, and C1orf49 genes are the potential disease-causing genes at 1q25. Our analysis of chromosome 10q22-24 points to the HTR7, MPHOSPH1, and CYP2C cluster. This is the first genomewide screen that showed significant linkage to chromosome 3q23 markers. For this region, our analysis identified the NMNAT3 and CLSTN2 genes. Our findings confirm linkage to chromosome 11q25. We were unable to confirm SORL1; instead, our analysis points to the OPCML and HNT genes.
Alzheimer disease (AD) is a progressive neurodegenerative disorder that accounts for the vast majority of dementia. The population prevalence of the disease rises steeply with age from <2% at age 65 years to >35% after age 90 years.1,2 Family history is an important indicator of risk of AD, and, in a large number of families, the disease segregates as an autosomal dominant trait. The heritability for AD was recently estimated to be 79%.3 Several dominant mutations have been identified, including mutations in the presenilin 1 (PSEN1 [MIM 104311]),4 presenilin 2 (PSEN2 [MIM 600759]),5,6 and amyloid precursor protein (APP [MIM 104760]) genes.7 A common polymorphism (ɛ4) in the gene encoding apolipoprotein E (APOE [MIM 107741]) increases susceptibility to both early- and late-onset AD.8,9 These four genes together explain less than a quarter of the disease prevalence, indicating that additional genetic risk factors remain to be identified.10,11 In addition to APOE, various candidate genes were reported to be associated with late-onset AD. In most cases, findings have not been consistently replicated.12,13 A large meta-analysis of all genes studied so far pinpointed 13 potential AD-susceptibility genes: angiotensin I converting enzyme (ACE [MIM 106180]); cholinergic receptor, nicotinic, beta 2 (CHRNB2 [MIM 118507]); cystatin C (CST3 [MIM 604312]); estrogen receptor 1 (ESR1 [MIM 133430]); glyceraldehyde-3-phosphate dehydrogenase, spermatogenic (GAPDHS [MIM 609169]); insulin-degrading enzyme (IDE [MIM 146680]); 5,10-methylenetetrahydrofolate reductase (MTHFR [MIM 607093]); nicastrin (NCSTN [MIM 605254]); prion protein (PRNP [MIM 176640]); PSEN1; transferrin (TF [MIM 190000]); transcription factor A, mitochondrial (TFAM [MIM 600438]); and tumor necrosis factor (TNF [MIM 191160]).14 Furthermore, genome screens targeting AD loci have been conducted. As reviewed online by the Alzheimer Research Forum, the replicated regions from previous genome screens include 1p36, 1q21-31, 2p23-24, 4q35, 5p13-15, 6p21, 6q15-16, 6q25-27, 9p21-22, 10q21-22, 10q25, 12p11-12, 19q13, 21q21-22, and Xp11-21.9,15–29 Several genes have been suggested to explain the linkage to chromosome 9, 10, 12, and 19, but, so far, these genes also remain to be confirmed. Finally, there is evidence of linkage to chromosome 11,22 which was explained recently by the identification of SORL1 (MIM 602005).30
Each of the established loci for AD (APP, PSEN1, PSEN2, and APOE) was initially localized by linkage analyses. However, pedigrees suitable for localizing genes have become scarce, particularly for late-onset forms of AD. Genetically isolated populations provide opportunities for linkage analysis. With use of genealogical records, extended pedigrees can be constructed. Furthermore, the complexity of disease may be reduced in terms of number of genes involved, particularly for rare Mendelian forms.31,32 Linkage analysis of complex traits has been used successfully in Iceland for complex diseases such as type 2 diabetes and stroke,33,34 whereas, for AD, genome screens have been conducted successfully with Caribbean Hispanics.35 We have followed this approach in a genetically isolated community from the southwestern area of The Netherlands, as part of the Genetic Research in Isolated Populations (GRIP) program.36 A total of 103 patients with late-onset AD were ascertained and were included in a large pedigree on the basis of genealogical records. In this study, we present a genomewide screen of these families. The linkage analysis was followed by an association study of cognitive function in a series of 197 unrelated and nondemented people from the GRIP region who were extensively characterized by a cognitive battery. To further investigate the evidence of linkage, the regions identified in the linkage study were characterized with a dense panel of SNPs. Decline in cognitive function, particularly mild cognitive impairment, is an early predictor of AD,37–39 and the heritability of cognitive function is as high as 56%, which suggests that cognition is a valuable endophenotype.40–42 Further, memory function was found to be an endophenotype for families multiply affected with AD.43
Material and Methods
Population and Genealogy
This study was performed within the framework of the previously described GRIP program.32,44,45 The Medical Ethics Committee of the Erasmus Medical Center approved study protocol. The GRIP population is a genetically isolated community in the southwestern area of The Netherlands. Fewer than 400 individuals were present in the region in the middle of the 18th century. Considerable population growth occurred in 1850–1900, as was the case in many European populations. An estimated 20,000 descendants of the population are now scattered over eight adjacent communities. There was minimal immigration. The genealogical database currently contains information about 107,091 people spanning 23 generations. Residents in the GRIP area are generally related via multiple lines of descent and are inbred via multiple consanguineous loops.45
Patients with AD were traced through general practitioners, neurologists, and nursing-home physicians. Data relevant for the diagnosis of AD were collected by a research physician, and the diagnosis of AD was verified by two independent neurologists with criteria of the National Institute of Neurological and Communicative Diseases and Stroke/Alzheimer's Disease and Related Disorders Association.46 Data about the presence of AD, parkinsonism, essential tremor, and dementia were collected for first-, second-, and third-degree relatives by means of a family-history questionnaire. First-degree relatives also underwent a brief neurological examination. All patients and their relatives who were invited to participate in our study provided informed consent. A total of 112 probable patients with late-onset AD (age at onset ⩾65 years, mean age [±SD] at onset 75±5.3 years) and 170 unaffected first-degree relatives (mean age 63.5±13.1 years; range 40–102 years) were ascertained.
Tracing the genealogy of the 112 probable patients with late-onset AD, we were able to include 103 patients in a single pedigree containing 4,645 individuals in 18 generations, as depicted in figure 1. The other nine patients were singletons and therefore were not included in the linkage analysis. This large pedigree showed multiple, distant lines of descent and consanguineous loops (table 1). The average kinship coefficient among patients was 0.0018. This value is between a third cousin once removed and a fourth cousin. Using such a pedigree in linkage analysis is computationally impossible. A common approach to reduce the computational complexity is to split the large pedigree into smaller and computable units. For this purpose, we used a kinship clustering method that is similar to the maximal-cliques-partitioning method proposed by Falchi et al,47 and we added a restriction that the resulting subpedigrees have no more than 35 bits, where the bit size is twice the number of founders minus the number of nonfounders. Our software for splitting large pedigrees, PedCut, is available, free of charge, at the MGA Web site.
Figure 1. .

The entire pedigree with 4,645 members, including 103 patients with late-onset AD, from the GRIP population. Men are represented with squares and women with circles. Black dots represent marriage nodes. Affected individuals are represented in black. Unknown affection status is represented with yellow. For simplicity, unaffected relatives of the patients are not shown. This figure was drawn using Pedfiddler version 0.5.
Table 1. .
Genealogic Characteristics of 103 Patients with Late-Onset AD and Their Relatives
| Characteristic | Valuea |
| Complete genealogy: | |
| No. of family members | 4,645 |
| No. of generations | 18 |
| Average no. of consanguineous loops per patient | 71.7 (0–677) |
| Average no. of meioses in a consanguineous loop | 9.9±1.2 (0–29) |
| Mean inbreeding coefficient ×100 | .39±.73 (0–3.2) |
| Average no. of lines of descent between a pair of patients | 141.7 (0–2,673) |
| Average no. of meioses separating a pair of patients | 17.1±1.6 (0–34) |
| Mean kinship coefficient ×100 | .18±1.06 (0–26.4) |
| After clustering patients into subpedigrees: | |
| No. of subpedigrees | 35 |
| No. of founders | 564 (46.0) |
| No. of females | 630 (51.3) |
| Mean pedigree size | 29.6 (18–75) |
| Mean no. of generations | 7.5 (6–10) |
| Mean no. of genotyped individuals per pedigree | 7.8 (2–14) |
| Mean no. of patients per pedigree | 2.9 (2–6) |
Values in parentheses indicate range or percentage.
We further studied a series of 197 individuals who were not related withini 5 generations and were not related to the patients with AD. The average age of these people was 31.2±6.4 years; 51% were female. These individuals were evaluated with use of an extensive cognitive battery.48 In brief, the selection of tests included the 15-word test, the color word card of the Stroop Color Word test, part B of the Trail making test (TMTB), and the verbal fluency test. These tests were selected to target early cognitive problems related to AD. From the 15-word test, we derived three scores for further analysis—that is, learning (or working memory), delayed recall, and recognition. The verbal fluency test consists of two subdomains: semantic fluency and phonological fluency. The performance of each individual on each test was scored quantitatively. Power calculation showed that this sample has 80% power to detect a SNP explaining 4% of phenotypic variance with an α of .05.
Genotyping
For all patients and their 170 first-degree relatives, DNA was extracted from peripheral leukocytes following a standard protocol.49 Elsewhere, mutations in the APP, PSEN1, and PSEN2 genes were excluded as AD-causing genes.36 The APOE genotype was determined in all DNA samples by use of TaqMan allelic discrimination technology on an ABI Prism 7900HT Sequence Detection System with SDS version 2.1 (Applied Biosystems). Patients and their first-degree relatives underwent a full genome screen in two sequential experiments. Both screens were conducted using the same set of microsatellite markers, evenly spaced by ∼10 cM (ABI Prism Linkage Mapping Set MD-10 v. 2 and v. 2.5 [Applied Biosystems]). PCRs were performed according to the manufacturer’s specified conditions. PCR products were separately pooled and analyzed on ABI377 and ABI3100 automated sequencers (Applied Biosystems). Because the genome scan had been performed with different sequencing devices, the genetic data had to be merged. The genotypic data was pooled using Pool_STR-1.1, on the basis of the allele lengths and allele frequencies observed in each group.50 Two independent technicians read the results from the sequencers, and a third reader resolved the discordant results. Only the markers with a discordance proportion <5% were selected for further analysis (N=402). Genotyping errors leading to Mendelian inconsistencies were detected using PedCheck (Statgen).51 Unlikely double-recombination events were detected using Merlin.52 Definitive genotyping errors and unlikely genotypes were rechecked using the data from the laboratory. Regions linked to late-onset AD were later fine typed by placing 45 additional microsatellite markers between those from the initial set, at a distance of 1–5 cM.
SNPs in the linkage regions were selected from the 250K Nsp array of the GeneChip Human Mapping 500K Array Set (Affymetrix). Genomic DNA was extracted from whole-blood samples drawn at the baseline examination, with use of the salting-out method.49 The 250K Nsp array from Affymetrix was used to determine genotypes. The chips were run and analyzed according to the manufacturer’s protocols. A total of 4,173 SNPs were selected for the association test on the basis of the following criteria: (1) position within the regions that show significant or suggestive evidence of linkage after fine mapping, (2) minor-allele frequency ⩾2.5%, (3) P value for Hardy-Weinberg equilibrium test ⩾.01, and (4) call rate ⩾95%.
Statistical Analysis
In linkage analysis, we assumed a dominant model of inheritance with age-dependent penetrance. Seven liability classes were defined on the basis of age (in years): <65, 65–69, 70–74, 75–79, 80–84, 85–90, and >90. For each age group j, age-dependent population prevalence Pj was obtained from the Rotterdam Study.1 The disease-gene penetrance, fj, of the jth age group can be estimated as
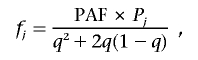 |
where PAF is the population-attributable fraction—that is, the proportion of the population prevalence that can be explained by the studied gene (10% assumed)—and q is the disease-allele frequency (1% assumed). The estimated penetrance for each defined age group is shown in table 2. Marker-allele frequencies were estimated on the basis of 144 chromosomes from unaffected elderly GRIP population members (age at last examination ⩾65 years). For small pedigrees (bits ⩾18), we used the exact calculation of multilocus likelihood, the Lander-Green algorithm implemented in GENEHUNTER 2.0.53 For larger pedigrees, we used Markov chain–Monte Carlo estimation methods implemented in SIMWALK 2.91 (Statgen). Overall LOD scores and heterogeneity LOD (HLOD) scores were computed by combining results per family with use of standard formulas, such as
where maxLR is maximized with respect to α, the proportion of the linked families, yielding maximum-likelihood estimate 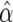 ,
,
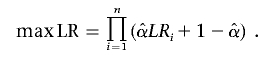 |
Table 2. .
Age-Dependent Liability Classes and Penetrances
| Liability Class |
Age (years) |
Population Prevalencea | Penetrance | No. of Patients | No. of Unaffected Relatives |
| 1 | <65 | <.02 | .00 | 0 | 129 |
| 2 | 65–69 | .02 | .09 | 4 | 6 |
| 3 | 70–74 | .05 | .23 | 22 | 11 |
| 4 | 75–79 | .09 | .46 | 32 | 14 |
| 5 | 80–84 | .23 | .99 | 30 | 8 |
| 6 | 85–89 | .35 | .99 | 24 | 1 |
| 7 | ⩾90 | >.35 | .99 | 0 | 1 |
Obtained from the Rotterdam Study.1
Haplotypes were reconstructed on the basis of the genotypes of patients, spouses of patients, and their offspring, with the Merlin package.52 These families are further expanded to depict the haplotype sharing of other patients who are relatively closely related to the patients in the families with high LOD scores but who were assigned to different families in the pedigree-splitting procedure.
Breaking pedigrees may increase the possibility of spurious linkage findings.54 Therefore, we estimated the threshold for statistical significance by use of simulations. To evaluate genomewide type I error, we simulated our genome scan 100 times. We used the complete pedigree, including all 4,645 members, for marker simulation. Unlinked markers were dropped in the complete pedigree. Number of markers, intermarker distances, and marker-allele frequencies were simulated according to the typed marker set. We performed linkage analysis using the split subpedigrees. Disease-allele frequency, liability classes, genetic model, and penetrances were the same as those we used later in the actual linkage analysis. Genotypes of untyped individuals were set to “missing.” For each genome screen, the highest HLOD score was stored. Cumulative density function of the obtained 100 maximum HLOD scores approximates the distribution of the genomewide type I error rates. Our simulations showed that an HLOD score of 3.64 corresponds to a genomewide type I error rate of 5% and that an HLOD of 2.11 corresponds to a genomewide type I error of 50% (table 3).
Table 3. .
LOD Scores and Corresponding Genomewide Type I Error Rates[Note]
| LOD | HLOD | Type I Error Rate (%) |
| 4.08 | 4.08 | 1 |
| 3.64 | 3.64 | 5 |
| 3.24 | 3.24 | 10 |
| 2.11 | 2.11 | 50 |
Note.— Values are based on 100 simulations.
We used linear regression to test for association between a single SNP with a single cognitive trait. In accordance with Affymetrix annotation, SNP genotypes were coded as 0=AA, 1=AB, and 2=BB, where A represents the allele in lower alphabetical order and B represents the other allele. Thus, in the case of a C→T change where T is the minor allele, C is coded as the A allele and T as the B allele, whereas, for a T→C change where C is the minor allele, A denotes the C allele and B denotes the T allele. In the model, we adjusted for age, sex, intelligence, and highest education. Because a causal SNP (or a SNP in linkage disequilibrium [LD] with the causal SNP) is likely to be associated with multiple cognitive domains, we used the Fisher product method for combining the findings of all cognitive tests.55 Because the SNPs that are in LD and cognitive traits are also correlated, we used a permutation method to evaluate significance level for each SNP empirically (500,000 replicates). To break the associations between the markers and traits while keeping the correlations between traits and the LD pattern between markers, we permuted the vectors of individuals’ traits (scores of cognitive tests and covariates) among individuals, without replacement. For each permutation, we tested for association between SNPs in each region and cognitive traits, and we derived corresponding Fisher products. The cumulative density function of all Fisher products for each region empirically approximates the regionwide type I error rate. Therefore, the empirical P value for each SNP can be defined as the probability of observing an equal or smaller regionwide Fisher product by chance.
Results
For linkage analysis, we clustered all patients and 170 first-degree relatives into 35 subpedigrees (fig. 2). During pedigree splitting, distant ancestors who have no phenotypic and genotypic information and do not contribute to linkage information were discarded. The resultant subpedigrees contained a total of 1,227 individuals. The characteristics of the subpedigrees are shown in table 1.
Figure 2. .



































The 35 subpedigrees obtained by applying a kinship-partitioning algorithm to the entire pedigree. Affected individuals are shown with black squares and circles. Unaffected first-degree relatives of the patients were added to the subpedigrees and are represented with white squares and circles. Ancestors of the patients and relatives are shown with gray squares and circles.
Multipoint LOD and HLOD score plots for the initial scan are shown in figure 3. A total of eight regions showed suggestive linkage (LOD or HLOD>2.11), of which the chromosome 1 region exceeded the threshold of 3.64 (LOD=4.1 at marker D1S484). These eight regions were fine typed with 45 additional markers and include chromosome 1 (14 additional markers), chromosome 3 (10 additional markers), chromosome 5 (2 additional markers), chromosome 6 (5 additional markers), chromosome 7 (3 additional markers), chromosome 10 (6 additional markers), chromosome 11 (2 additional markers), and chromosome 18 (3 additional markers).
Figure 3. .



Multipoint LOD (blue) and HLOD (pink) scores for each autosome in the genome screen of late-onset AD. Marker locations are given in Kosambi centimorgans.
Table 4 shows the regions for which the evidence of linkage remained significant or suggestive (LOD>2.11) after fine mapping. AD remained linked to three known regions, two on chromosome 1 (fig. 4A) and one on chromosome 10 (fig. 4C). The maximum HLOD at 1q21 was 5.2 at D1S498. This is the highest peak over the genome. The maximum HLOD at 1q25 was 4.0 at D1S218 and 4.2 at D10S185. References for the previously identified regions are shown in table 4. In addition to the known regions, we found genomewide significant evidence of linkage of AD to a region on chromosome 3 that spanned 18 cM from D3S3514 to D3S3626 and reached a maximum HLOD of 4.4 at D3S1569 (fig. 4B). This is the second highest peak over the genome. In table 4, we also included chromosome 11, in which a new gene (SORL1) responsible for AD was recently reported.30 There is suggestive evidence of linkage of AD to chromosome 11 (HLOD=3.3 at D11S1320) (fig. 4D), which overlaps with a region reported earlier. On chromosome 11, the HLOD at the position of SORL1 (118 cM) is 1.1.
Table 4. .
Regions with Genomewide Empirically Significant or Suggestive Linkage after Fine Mapping[Note]
| Chromosome and Marker |
Position (cM) |
LOD | HLOD | α | Region(s) Identified Elsewherea |
| 1A: | |||||
| D1S498 | 164 | 5.1 | 5.2 | .9 | A,25 D,26 F,56 and G22 |
| D1S305 | 167 | 4.5 | 4.5 | 1.0 | |
| 1B: | |||||
| D1S218 | 201 | 2.6 | 4.0 | .6 | A,25 D,26 F,56 and G22 |
| D1S366 | 208 | 2.7 | 3.5 | .6 | |
| 3: | |||||
| D3S1549 | 151 | 2.8 | 3.6 | .6 | B57 |
| D3S1569 | 158 | 4.3 | 4.4 | .8 | |
| 10: | |||||
| D10S1686 | 105 | 3.7 | 3.7 | 1.0 | C,18 E,19 F,56 G,22 H,27 and I24 |
| D10S185 | 116 | 4.2 | 4.2 | 1.0 | |
| 11b: | |||||
| D11S4151 | 127 | .3 | 2.8 | .4 | G22 |
| D11S4131 | 138 | 1.3 | 3.1 | .5 | |
| D11S1320 | 142 | 1.6 | 3.3 | .6 | |
| D11S968 | 148 | .3 | 2.0 | .5 |
Note.— Significant values are shown in bold.
Overlaps with regions reported with suggestive linkage or significant association in previous genome screens. Note that region B was screened for only two chromosomes.
Included to confirm a recent report about the SORL1 gene.30
Figure 4. .

Multipoint LOD (blue) and HLOD (pink) scores for chromosomes 1, 3, 10, and 11 in the genome screen of late-onset AD after fine typing. Marker locations are given in Kosambi centimorgans.
Haplotype analysis showed that the two linkage peaks on chromosome 1q21 and 1q25 are explained by different haplotypes segregating in different families. On chromosome 1q21, we identified a 15-cM region shared by four patients in family 1 and six other closely related patients who were assigned to different pedigrees for computational reasons in the process of pedigree splitting (fig. 5A). The 21-cM haplotype of 1q25 segregates in family 3 (four patients) and is shared by four other closely related patients who were assigned to different subpedigrees (fig. 5B). Six patients from family 9 and six closely related patients carry the haplotype of chromosome 3q23 (18 cM), as shown in figure 5C. The linkage of AD to the region on chromosome 10 was based on moderate contributions from multiple families with different haplotypes. There is not a single haplotype segregating in this region (data not shown). For chromosome 11q24, which showed suggestive linkage, we observed a single haplotype (3.4 cM) shared by four patients from family 4 and two additional closely related patients (fig. 5D).
Figure 5. .


Haplotypes of chromosomes 1, 3, and 11 segregating with AD-affected families from the GRIP population. Panels A, B, C, and D are from subpedigrees 1, 3, 9, and 4, respectively. These families are expanded to depict the haplotype sharing of other patients who are relatively closely related to the patients but were assigned to different families in the pedigree-splitting procedure. Patients with asterisks (*) were from the corresponding subpedigrees; patients without asterisks were assigned to different families. Alleles in parentheses are inferred. Pink bars depict the segment that is shared between the patients.
Next, we tested for association between cognitive function and a set of 4,173 SNPs within regions 1q21, 1q25, 3q23, 10q22-24, and 11q25, using an independent sample consisting of 197 individuals from the GRIP population (table 5). All of the linked regions except 1q21 contain at least one SNP showing significant association with use of an empirical P value of .05 (table 6). Statistically, the most significant SNP is rs7071717 at 10q23, both for the nominal P value in a single test (P=.000005, by Stroop test) and for the empirical Fisher product (P=.002), which combines the results of cognitive tests and adjust for multiple testing. This SNP, together with rs17129662 and rs11185978, is in a range of 80 kb and shows evidence of association with the Stroop test, TMTB, and semantic (except rs17129662) and phonological fluency, all of which are subdomains of executive function. These three SNPs are 2–80 kb downstream of the MPHOSPH1 gene encoding M phase phosphoprotein 1 and ∼760 kb upstream of the 5-hydroxytryptamine receptor 7 (HTR7 [MIM 182137]) gene. Another SNP, rs4110517 at 10q23, showed association with semantic (P=.00003) and phonological (P=.04) fluency (empirical P=.02). This SNP is 37.6 kb downstream of the CYP2C19 (MIM 124020) gene and 48.1 kb upstream of the CYP2C9 (MIM 601130) gene. At 1q25, the SNP rs2584820 was associated with the Stroop test (P=.0001) and phonological fluency (P=.03), with an empirical P value of .04. This SNP is in intron 4 of the regulator of G-protein signaling like 2 (RGSL2) gene. Two other SNPs in this region showed association with the TMTB (P=.0003; empirical P=.04). They are 4 kb downstream of the C1orf49 gene and 149 kb upstream of the Ral GEF with PH domain and SH3 binding motif 2 (RALGPS2) gene. At 3q23, the SNP rs952797 was associated with the Stroop test (P=.0001), the Block test (P=.0002), and learning (P=.06). When all tests were evaluated simultaneously, the association was significant (empirical P=.04). This SNP is 126 kb downstream of the gene encoding nicotinamide nucleotide adenylyltransferase 3 (NMNAT3 [MIM 608702]) and 131 kb upstream of the gene encoding calsyntein 2 (CLSTN2). SNP rs11223225 (C→T) at 11q25 showed a consistent allelic effect across key cognitive domains for AD, including learning, delayed recall, and concept shifting (Stroop and TMTB), where the minor allele of this SNP is associated with poorer performance on delayed recall (P=.0004), learning (P=.03), the Stroop test (P=.02), TMTB (P=.09), and the Block test (P=.07). When the effect of various tests was combined, the overall empirical P value was .03. This SNP is in intron 1 of the gene encoding opioid binding protein/cell adhesion molecule-like (OPCML [MIM 600632]). Four close SNPs—rs1629316, rs1547897, rs1122931, and rs11222932—at 11q25 were associated with TMTB and phonological fluency. These SNPs are in intron 1 of the gene encoding neurotrimin (HNT [MIM 607938]). The OPCML and HNT genes are <80 kb apart.
Table 5. .
Select SNPs in the Regions Linked to AD
| Start |
End |
No. of SNPs |
||||||||
| Chromosome | Marker | Marshfield (cM) |
SNP | Physical Position (bp) |
Marker | Marshfield (cM) |
SNP | Physical Position (bp) |
Initial | After QCa |
| 1q21 | D1S514 | 152.5 | rs2790308 | 141501392 | D1S2635 | 165.6 | rs16827466 | 156462773 | 828 | 585 |
| 1q25 | D1S218 | 191.5 | rs17838246 | 171261041 | D1S466 | 198.5 | rs16860717 | 179972352 | 750 | 584 |
| 3q23 | D3S1549 | 151.5 | rs7632392 | 139168654 | D3S3626 | 164.3 | rs10513332 | 149350824 | 954 | 769 |
| 10q22-24 | D10S580 | 96.7 | rs7101263 | 77715269 | D10S205 | 125.4 | rs12765878 | 105659612 | 2,518 | 2,006 |
| 11q25 | D11S4131 | 138.6 | rs1526562 | 131160829 | D11S968 | 147.8 | rs7936592 | 133620325 | 260 | 229 |
QC=quality control.
Table 6. .
SNPs Significantly Associated with Various Aspects of Cognitive Function in 197 Unrelated Individuals[Note]
| Memory |
Executive Function |
Verbal Fluency |
||||||||||||
| Linked Region and SNPa |
Marshfield (cM)b |
Physical Positionc (bp) |
Learning | Delay | Recognition | Stroop | TMTB | Block | Semantic | Phonological | All Tests Combinedd | Empirical Pe | Gene(s)f | SNP Position |
| 1q25: | ||||||||||||||
| rs17361286 | 193.9 | 176788539 | .68 | .56 | .91 | .44 | .0003 | .51 | .96 | .64 | 1.2×10−5 | .04 | C1orf49 and RALGPS2 | Intergenic |
| rs2493864 | 194.0 | 176811626 | .76 | .63 | .96 | .46 | .0003 | .49 | .99 | .62 | 1.7×10−5 | .04 | C1orf49 and RALGPS2 | Intergenic |
| rs2584820 | 197.6 | 180705709 | .20 | .19 | .76 | .0001 | .89 | .98 | .07 | .03 | 5.9×10−9 | .02 | RGSL2 | Intron 4 |
| 3q23: | ||||||||||||||
| rs952797 | 152.6 | 141005823 | .06 | .47 | .57 | .05 | .89 | .0002 | .82 | .24 | 2.8×10−8 | .04 | NMNAT3 and CLSTN2 | Intergenic |
| 10q23: | ||||||||||||||
| rs17129662 | 111.8 | 91730916 | .23 | .55 | .48 | .00003 | .00235 | .84 | .10 | .002 | 7.0×10−13 | .02 | MPHOSPH1 and HTR7 | Intergenic |
| rs11185978 | 111.8 | 91778768 | .36 | .60 | .60 | .00002 | .01 | .61 | .05 | .001 | 8.9×10−13 | .01 | MPHOSPH1 and HTR7 | Intergenic |
| rs7071717 | 111.8 | 91808267 | .30 | .73 | .67 | .000005 | .01 | .90 | .04 | .01 | 2.9×10−12 | .002 | MPHOSPH1 and HTR7 | Intergenic |
| rs4110517 | 116.8 | 96640318 | .10 | .09 | .94 | .18 | .24 | .87 | .00003 | .04 | 4.7×10−10 | .02 | CYP2C19 and CYP2C9 | Intergenic |
| 11q25: | ||||||||||||||
| rs1629316 | 139.7 | 131440484 | .07 | .21 | .41 | .94 | .0007 | .10 | .35 | .05 | 8.0×10−9 | .05 | HNT | Intron 1 |
| rs1547897 | 139.7 | 131451988 | .06 | .16 | .39 | 1.00 | .0006 | .16 | .29 | .05 | 4.8×10−9 | .04 | HNT | Intron 1 |
| rs11222931 | 139.8 | 131483366 | .07 | .15 | .33 | .87 | .0007 | .16 | .27 | .05 | 4.5×10−9 | .04 | HNT | Intron 1 |
| rs11222932 | 139.8 | 131483471 | .07 | .15 | .34 | .86 | .0006 | .18 | .30 | .05 | 4.6×10−9 | .04 | HNT | Intron 1 |
| rs11223225 | 142.7 | 132198420 | .03 | .0004 | .26 | .02 | .09 | .06 | .47 | .26 | 3.2×10−11 | .03 | OPCML | Intron 1 |
Note.— Significant values are shown in bold.
All SNPs with empirical P values ⩽.05 are shown.
According to the Marshfield map.
According to the National Center for Biotechnology Information build 35 reference map.
Fisher product over all tests.
The probability of observing an equal or smaller Fisher product by chance per region, based on permutation test of 500,000 replicates.
For intergenic SNPs, only the closest neighbor genes are listed.
Discussion
This study confirms earlier findings suggesting linkage of AD to a wide region that spans chromosome 1q21-31.25,26,56 The 1q21 region yielded the most significant evidence of linkage over the genome in our study (HLOD=5.2). This region was not replicated when testing for association with cognition in a series of 197 distantly related subjects. Although we cannot exclude the possibility of a false-positive finding, given the strength of the linkage signal and previous evidence, it is more likely that there is a rare mutation in a major gene in this region that could not be identified by association analysis in a small sample. This region contains the NCSTN gene, which binds presenilin and is required for γ-secretase activity and Aβ generation.58 Mutations in this gene have been found to be related to early-onset AD, and we have reported association in a subgroup of patients with familial early-onset AD, particularly in those who lack the APOE*4 allele.59 We have sequenced all the exons and splice sites of this gene in six patients but have not found variants. Another obvious candidate gene in this region is the gene encoding C-reactive protein (CRP [MIM 123260]), which acts as a scavenger for chromatin released by dead cells during the acute inflammatory process.60 We also sequenced the exons and splice sites of this gene in seven patients (5088, 5167, 5115, 5140, 5393, 5023, and 5394) (fig. 5A) and found that all patients except patients 5167 and 5393 carry the rare alleles of SNPs rs1130864 (C→T) and rs1417938 (T→A). The SNP rs1130864 has been reported as a tagging SNP for a haplotype associated with higher levels of CRP.61,62 We specifically tested the association of polymorphisms in CRP with cognitive function but failed to show any association (data not shown). Since CRP is a key protein involved in inflammation, a key process in life by itself, a major mutation in CRP seams unlikely for late-onset diseases, which suggests that another gene in the region may explain our high LOD score. In the 1q25 region, there was a second segregating haplotype. This region was confirmed in our association analysis by a SNP in intron 4 of RGSL2, which may be involved in the G-protein coupled receptor protein signaling pathway. Also, two SNPs upstream showed evidence of association. However, these SNPs were significant only for the TMTB and are intergenic, making it more likely that RGSL2 is the relevant gene in the 1q25 region.
The second highest linkage signal was found at chromosome 3q22-24. This region was reported earlier to be linked to AD without tau pathology in a study of a small family with four affected relatives.57 A significant LOD score of 4.1 between markers D3S1569 and D3S3554 was reported, whereas, in our study, D3S1569 is also the marker that gives the highest HLOD over chromosome 3. In the study by Poduslo et al.,57 no genomewide screen was conducted; only chromosomes 3 and 17 were screened, since the disease was expected to be related to frontotemporal dementia (FTD) and the phenotype was apparently considered to be compatible with that of FTD. Since we do not have pathology information for our patients, we cannot exclude the possibility that some of our patients also suffer from this atypical form of AD. However, all patients were carefully evaluated by a neurologist who specializes in FTD. A recent linkage-based genome scan of Caribbean Hispanic families revealed a new locus on chromosome 3q28, with a 2-point LOD score of 3.09 at marker D3S2418.35 However, this region is ∼50 cM downstream of the region we identified in our study. The linked region on chromosome 3q22-24 contains various possible candidate genes, including the TF gene, the gene encoding for butyrylcholinesterase (BCHE [MIM 177400]), the neprilysin gene (MME [MIM 120520]), and the somatostatin gene (SST [MIM 182450]). We screened these genes for mutations, but no variants were found. The SNP rs952797 at 3q23 was consistently associated with cognitive function in the 197 unrelated subjects from the GRIP population. This SNP is 126 kb downstream of the NMNAT3 gene encoding nicotinamide nucleotide adenylyltransferase 3 (NAD3). The coenzyme NAD and its derivatives are involved in hundreds of metabolic redox reactions and are used in protein ADP-ribosylation, histone deacetylation, and in some Ca2+-signaling pathways. NMNAT is a central enzyme in NAD biosynthesis, catalyzing the condensation of nicotinamide mononucleotide or nicotinic acid mononucleotide with the AMP moiety of ATP to form NAD or NaAD (Zhang et al.63); thus, the NMNAT3 gene may relevant to AD. The SNP rs952797 is 131 kb upstream of the CLSTN2 gene. It has been reported recently that SNP rs6439886, a common T→C substitution within the first intron of CLSTN2, was significantly associated with memory performance.64 Our SNP rs952797 is 160 kb upstream of the reported SNP.
Chromosome 10q22-24 is the third highest peak over the genome. This finding is consistent with previous findings about AD15,20–22,24 and plasma amyloid β42 levels.65 Our finding of linkage to late-onset AD at 10q22-24 is the first replication with use of a data set fully independent of the National Institute of Mental Health (NIMH) sample. So far, it has been difficult to identify the causal mutation(s) in this region. A series of genes has been densely genotyped, and several genes have been noted to be susceptibility genes for AD, including IDE, CH25H (MIM 604551), PLAU (MIM 191840), and LIPA (MIM 278000). In our linkage analysis, there was not a single haplotype segregating in the region, suggesting that multiple mutations in one or multiple genes may contribute to the linkage. In our association analysis, the most significant evidence of association with cognitive function was seen for this region. Three SNPs:—rs17129662, rs11185978, and rs7071717, together at 91.7 Mb—showed association with multiple cognitive domains in the 197 unrelated subjects from the GRIP population. These SNPs are intergenic SNPs of known genes. All three SNPs are <1 Mb upstream of the CH25H gene and the LIPA gene and <3 Mb downstream of the IDE gene. The genes most closely flanking these SNPs are the MPHOSPH1 gene and the HTR7 gene, which encodes 5-hydroxytryptamine receptor 7. These two genes, however, have not been extensively investigated. Another associated SNP, rs4110517 at 116.8 cM, is surrounded by four similar genes—CYP2C18 (MIM 601131), CYP2C19, CYP2C9, and CYP2C8 (MIM 601129)—in a range of <350 kb. Since no significant association was found between the CYP2C19 gene and patients with familial AD in a previous study,66 it is more likely that the SNP rs4110517 is in LD with the causal gene(s) in the region.
We also found suggestive evidence of linkage to chromosome 11q25. Blacker et al.22 previously described this region in their study of the NIMH sample, including 437 families with AD. Recent evidence suggests the SORL1 gene may be responsible.30 Our linkage peak is, however, ∼23 cM downstream of the SORL1 gene. We specifically tested the association between polymorphisms flanking SORL1 and cognitive function but failed to detect consistent associations (data not shown), suggesting that our linkage peak may be explained by other gene(s). The association for SNP rs11223225, a C→T substitution at 11q25, is one of the most promising results from our association analysis. The T allele of this SNP is consistently associated with reduced cognitive performance on multiple domains, and this SNP is an intronic SNP of the OPCML gene, which encodes the opioid-binding protein. There is evidence that the opioidergic system is affected in AD.67 Furthermore, performance on immediate memory and mental flexibility tasks has been suggestively linked to 11q25 in a recent genomewide linkage study of 260 families.68 Four other close SNPs at 11q25 also showed association with cognitive function. These SNPs are in intron 1 of the HNT gene, which encodes neurotrimin. Notably, the OPCML and HNT genes are separated by <80 kb.
In summary, we confirmed two previously well-described linkage regions for late-onset AD on chromosomes 1q21-25 and 10q22-24. With cognitive function as an endophenotype of AD, our study specifies the RGSL2, RALGPS2, and C1orf49 genes at 1q25. Our analysis of chromosome 10q22-24 points to the HTR7, MPHOSPH1, and CYP2C cluster. To our knowledge, this is the first genomewide screen that showed significant linkage to chromosome 3q23 markers. For this region, our analysis identified the NMNAT3 and CLSTN2 genes. Our findings confirm linkage to chromosome 11q25. We were unable to confirm SORL1; instead, our analysis points to the OPCML and HNT genes.
Acknowledgments
This work was supported by Netherlands Organization for Scientific Research grant 91203014, the Centre of Medical Systems Biology, Hersenstichting Nederland, Internationale Stichting Alzheimer Onderzoek, Alzheimer Association project 04516, Hersenstichting Nederland project 12F04(2).76, and the Interuniversity Attraction Poles program. P.S.-J. was supported by the post-MIR grant “Wenceslao Lopez Albo” from the Institute of the Fundación Pública Marqués de Valdecilla. All neurologists from the GRIP region are gratefully acknowledged for their support in ascertaining the patients. We thank Aaron Isaacs for valuable discussions, Leon Testers and Erik Simmons for their help in genotyping, and Petra Veraart for the collection of genealogical information. Above all, we thank the patients and their relatives from the population studied in the GRIP program and the municipality that made this work possible.
Web Resources
The URLs for data presented herein are as follows:
- AlzGene Database and Alzheimer Research Forum, http://www.alzgene.org/
- GENEHUNTER, http://linkage.rockefeller.edu/soft/gh/ (for multipoint linkage analysis with the Lander-Green algorithm)
- Merlin, http://www.sph.umich.edu/csg/abecasis/Merlin/download/ (for detecting unlikely double-recombination events and haplotype construction)
- MGA, http://mga.bionet.nsc.ru/soft/index.html (for PedCut)
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for PSEN1, PSEN2, APP, APOE, ACE, CHRNB2, CST3, ESR1, GAPDHS, IDE, MTHFR, NCSTN, PRNP, TF, TFAM, TNF, SORL1, HTR7, CYP2C19, CYP2C9, NMNAT3, OPCML, HNT, CRP, BCHE, MME, SST, CH25H, PLAU, LIPA, CYP2C18, and CYP2C8)
- Pedfiddler, http://www.medicine.mcgill.ca/statgene/software.html (v. 0.5, for drawing large pedigrees)
- Statgen, http://watson.hgen.pitt.edu/register/ (for PedCheck [for detecting Mendelian errors] and SIMWALK [for multipoint linkage analyses with the Markov chain–Monte Carlo method])
References
- 1.Ott A, Breteler MM, van Harskamp F, Claus JJ, van der Cammen TJ, Grobbee DE, Hofman A (1995) Prevalence of Alzheimer’s disease and vascular dementia: association with education. BMJ 310:970–973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rocca WA, Hofman A, Brayne C, Breteler MM, Clarke M, Copeland JR, Dartigues JF, Engedal K, Hagnell O, Heeren TJ, et al (1991) Frequency and distribution of Alzheimer’s disease in Europe: a collaborative study of 1980–1990 prevalence findings. Ann Neurol 30:381–390 10.1002/ana.410300310 [DOI] [PubMed] [Google Scholar]
- 3.Gatz M, Reynolds CA, Fratiglioni L, Johansson B, Mortimer JA, Berg S, Fiske A, Pedersen NL (2006) Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry 63:168–174 10.1001/archpsyc.63.2.168 [DOI] [PubMed] [Google Scholar]
- 4.Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, Chi H, Lin C, Li G, Holman K, et al (1995) Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 375:754–760 10.1038/375754a0 [DOI] [PubMed] [Google Scholar]
- 5.Levy-Lahad E, Wasco W, Poorkaj P, Romano DM, Oshima J, Pettingell WH, Yu CE, Jondro PD, Schmidt SD, Wang K, et al (1995) Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science 269:973–977 10.1126/science.7638622 [DOI] [PubMed] [Google Scholar]
- 6.Rogaev EI, Sherrington R, Rogaeva EA, Levesque G, Ikeda M, Liang Y, Chi H, Lin C, Holman K, Tsuda T, et al (1995) Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene. Nature 376:775–778 10.1038/376775a0 [DOI] [PubMed] [Google Scholar]
- 7.Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, Giuffra L, Haynes A, Irving N, James L, et al (1991) Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 349:704–706 10.1038/349704a0 [DOI] [PubMed] [Google Scholar]
- 8.van Duijn CM, de Knijff P, Cruts M, Wehnert A, Havekes LM, Hofman A, Van Broeckhoven C (1994) Apolipoprotein E4 allele in a population-based study of early-onset Alzheimer’s disease. Nat Genet 7:74–78 10.1038/ng0594-74 [DOI] [PubMed] [Google Scholar]
- 9.Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA (1993) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261:921–923 10.1126/science.8346443 [DOI] [PubMed] [Google Scholar]
- 10.Sleegers K, Van Duijn CM (2001) Alzheimer’s disease: genes, pathogenesis and risk prediction. Community Genet 4:197–203 10.1159/000064193 [DOI] [PubMed] [Google Scholar]
- 11.Bertram L, Tanzi RE (2005) The genetic epidemiology of neurodegenerative disease. J Clin Invest 115:1449–1457 10.1172/JCI24761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tanzi RE, Bertram L (2001) New frontiers in Alzheimer’s disease genetics. Neuron 32:181–184 10.1016/S0896-6273(01)00476-7 [DOI] [PubMed] [Google Scholar]
- 13.Myers AJ, Goate AM (2001) The genetics of late-onset Alzheimer’s disease. Curr Opin Neurol 14:433–440 10.1097/00019052-200108000-00002 [DOI] [PubMed] [Google Scholar]
- 14.Bertram L, McQueen MB, Mullin K, Blacker D, Tanzi RE (2007) Systematic meta-analyses of Alzheimer disease genetic association studies: the AlzGene database. Nat Genet 39:17–23 10.1038/ng1934 [DOI] [PubMed] [Google Scholar]
- 15.Kehoe P, Wavrant-De Vrieze F, Crook R, Wu WS, Holmans P, Fenton I, Spurlock G, Norton N, Williams H, Williams N, et al (1999) A full genome scan for late onset Alzheimer’s disease. Hum Mol Genet 8:237–245 10.1093/hmg/8.2.237 [DOI] [PubMed] [Google Scholar]
- 16.Pericak-Vance MA, Bass MP, Yamaoka LH, Gaskell PC, Scott WK, Terwedow HA, Menold MM, Conneally PM, Small GW, Vance JM, et al (1997) Complete genomic screen in late-onset familial Alzheimer disease: evidence for a new locus on chromosome 12. JAMA 278:1237–1241 10.1001/jama.278.15.1237 [DOI] [PubMed] [Google Scholar]
- 17.Pericak-Vance MA, Grubber J, Bailey LR, Hedges D, West S, Santoro L, Kemmerer B, Hall JL, Saunders AM, Roses AD, et al (2000) Identification of novel genes in late-onset Alzheimer’s disease. Exp Gerontol 35:1343–1352 10.1016/S0531-5565(00)00196-0 [DOI] [PubMed] [Google Scholar]
- 18.Curtis D, North BV, Sham PC (2001) A novel method of two-locus linkage analysis applied to a genome scan for late onset Alzheimer’s disease. Ann Hum Genet 65:473–481 10.1046/j.1469-1809.2001.6550473.x [DOI] [PubMed] [Google Scholar]
- 19.Olson JM, Goddard KA, Dudek DM (2002) A second locus for very-late-onset Alzheimer disease: a genome scan reveals linkage to 20p and epistasis between 20p and the amyloid precursor protein region. Am J Hum Genet 71:154–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Myers A, Holmans P, Marshall H, Kwon J, Meyer D, Ramic D, Shears S, Booth J, DeVrieze FW, Crook R, et al (2000) Susceptibility locus for Alzheimer’s disease on chromosome 10. Science 290:2304–2305 10.1126/science.290.5500.2304 [DOI] [PubMed] [Google Scholar]
- 21.Li Y-J, Scott WK, Hedges DJ, Zhang F, Gaskell PC, Nance MA, Watts RL, Hubble JP, Koller WC, Pahwa R, et al (2002) Age at onset in two common neurodegenerative diseases is genetically controlled. Am J Hum Genet 70:985–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blacker D, Bertram L, Saunders AJ, Moscarillo TJ, Albert MS, Wiener H, Perry RT, Collins JS, Harrell LE, Go RC, et al (2003) Results of a high-resolution genome screen of 437 Alzheimer’s disease families. Hum Mol Genet 12:23–32 10.1093/hmg/ddg007 [DOI] [PubMed] [Google Scholar]
- 23.Goddard KA, Olson JM, Payami H, van der Voet M, Kuivaniemi H, Tromp G (2004) Evidence of linkage and association on chromosome 20 for late-onset Alzheimer disease. Neurogenetics 5:121–128 10.1007/s10048-004-0174-3 [DOI] [PubMed] [Google Scholar]
- 24.Holmans P, Hamshere M, Hollingworth P, Rice F, Tunstall N, Jones S, Moore P, Wavrant DeVrieze F, Myers A, Crook R, et al (2005) Genome screen for loci influencing age at onset and rate of decline in late onset Alzheimer’s disease. Am J Med Genet B Neuropsychiatr Genet 135:24–32 [DOI] [PubMed] [Google Scholar]
- 25.Zubenko GS, Hughes HB, Stiffler JS, Hurtt MR, Kaplan BB (1998) A genome survey for novel Alzheimer disease risk loci: results at 10-cM resolution. Genomics 50:121–128 10.1006/geno.1998.5306 [DOI] [PubMed] [Google Scholar]
- 26.Hiltunen M, Mannermaa A, Thompson D, Easton D, Pirskanen M, Helisalmi S, Koivisto AM, Lehtovirta M, Ryynanen M, Soininen H (2001) Genome-wide linkage disequilibrium mapping of late-onset Alzheimer’s disease in Finland. Neurology 57:1663–1668 [DOI] [PubMed] [Google Scholar]
- 27.Farrer LA, Bowirrat A, Friedland RP, Waraska K, Korczyn AD, Baldwin CT (2003) Identification of multiple loci for Alzheimer disease in a consanguineous Israeli-Arab community. Hum Mol Genet 12:415–422 10.1093/hmg/ddg037 [DOI] [PubMed] [Google Scholar]
- 28.Wijsman EM, Daw EW, Yu C-E, Payami H, Steinbart EJ, Nochlin D, Conlon EM, Bird TD, Schellenberg GD (2004) Evidence for a novel late-onset Alzheimer disease locus on chromosome 19p13.2. Am J Hum Genet 75:398–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ashley-Koch AE, Shao Y, Rimmler JB, Gaskell PC, Welsh-Bohmer KA, Jackson CE, Scott WK, Haines JL, Pericak-Vance MA (2005) An autosomal genomic screen for dementia in an extended Amish family. Neurosci Lett 379:199–204 10.1016/j.neulet.2004.12.065 [DOI] [PubMed] [Google Scholar]
- 30.Rogaeva E, Meng Y, Lee JH, Gu Y, Kawarai T, Zou F, Katayama T, Baldwin CT, Cheng R, Hasegawa H, et al (2007) The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat Genet 39:168–177 10.1038/ng1943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Varilo T, Peltonen L (2004) Isolates and their potential use in complex gene mapping efforts. Curr Opin Genet Dev 14:316–323 10.1016/j.gde.2004.04.008 [DOI] [PubMed] [Google Scholar]
- 32.Pardo LM, MacKay I, Oostra B, van Duijn CM, Aulchenko YS (2005) The effect of genetic drift in a young genetically isolated population. Ann Hum Genet 69:288–295 [DOI] [PubMed] [Google Scholar]
- 33.Escamilla MA (2001) Population isolates: their special value for locating genes for bipolar disorder. Bipolar Disord 3:299–317 10.1034/j.1399-5618.2001.30605.x [DOI] [PubMed] [Google Scholar]
- 34.Helgason A, Yngvadottir B, Hrafnkelsson B, Gulcher J, Stefansson K (2005) An Icelandic example of the impact of population structure on association studies. Nat Genet 37:90–95 [DOI] [PubMed] [Google Scholar]
- 35.Lee JH, Cheng R, Santana V, Williamson J, Lantigua R, Medrano M, Arriaga A, Stern Y, Tycko B, Rogaeva E, et al (2006) Expanded genomewide scan implicates a novel locus at 3q28 among Caribbean Hispanics with familial Alzheimer disease. Arch Neurol 63:1591–1598 10.1001/archneur.63.11.1591 [DOI] [PubMed] [Google Scholar]
- 36.Sleegers K, Roks G, Theuns J, Aulchenko YS, Rademakers R, Cruts M, van Gool WA, Van Broeckhoven C, Heutink P, Oostra BA, et al (2004) Familial clustering and genetic risk for dementia in a genetically isolated Dutch population. Brain 127:1641–1649 10.1093/brain/awh179 [DOI] [PubMed] [Google Scholar]
- 37.Geerlings MI, Jonker C, Bouter LM, Ader HJ, Schmand B (1999) Association between memory complaints and incident Alzheimer’s disease in elderly people with normal baseline cognition. Am J Psychiatry 156:531–537 [DOI] [PubMed] [Google Scholar]
- 38.Tierney MC, Szalai JP, Snow WG, Fisher RH, Nores A, Nadon G, Dunn E, St George-Hyslop PH (1996) Prediction of probable Alzheimer’s disease in memory-impaired patients: a prospective longitudinal study. Neurology 46:661–665 [DOI] [PubMed] [Google Scholar]
- 39.Jonker C, Geerlings MI, Schmand B (2000) Are memory complaints predictive for dementia? A review of clinical and population-based studies. Int J Geriatr Psychiatry 15:983–991 [DOI] [PubMed] [Google Scholar]
- 40.Ando J, Ono Y, Wright MJ (2001) Genetic structure of spatial and verbal working memory. Behav Genet 31:615–624 10.1023/A:1013353613591 [DOI] [PubMed] [Google Scholar]
- 41.McClearn GE, Johansson B, Berg S, Pedersen NL, Ahern F, Petrill SA, Plomin R (1997) Substantial genetic influence on cognitive abilities in twins 80 or more years old. Science 276:1560–1563 10.1126/science.276.5318.1560 [DOI] [PubMed] [Google Scholar]
- 42.Swan GE, Reed T, Jack LM, Miller BL, Markee T, Wolf PA, DeCarli C, Carmelli D (1999) Differential genetic influence for components of memory in aging adult twins. Arch Neurol 56:1127–1132 10.1001/archneur.56.9.1127 [DOI] [PubMed] [Google Scholar]
- 43.Lee JH, Flaquer A, Stern Y, Tycko B, Mayeux R (2004) Genetic influences on memory performance in familial Alzheimer disease. Neurology 62:414–421 [DOI] [PubMed] [Google Scholar]
- 44.van Duijn CM, Dekker MC, Bonifati V, Galjaard RJ, Houwing-Duistermaat JJ, Snijders PJ, Testers L, Breedveld GJ, Horstink M, Sandkuijl LA, et al (2001) Park7, a novel locus for autosomal recessive early-onset parkinsonism, on chromosome 1p36. Am J Hum Genet 69:629–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Aulchenko YS, Heutink P, Mackay I, Bertoli-Avella AM, Pullen J, Vaessen N, Rademaker TA, Sandkuijl LA, Cardon L, Oostra B, et al (2004) Linkage disequilibrium in young genetically isolated Dutch population. Eur J Hum Genet 12:527–534 10.1038/sj.ejhg.5201188 [DOI] [PubMed] [Google Scholar]
- 46.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM (1984) Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 34:939–944 [DOI] [PubMed] [Google Scholar]
- 47.Falchi M, Forabosco P, Mocci E, Borlino CC, Picciau A, Virdis E, Persico I, Parracciani D, Angius A, Pirastu M (2004) A genomewide search using an original pairwise sampling approach for large genealogies identifies a new locus for total and low-density lipoprotein cholesterol in two genetically differentiated isolates of Sardinia. Am J Hum Genet 75:1015–1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sleegers K, de Koning I, Aulchenko YS, van Rijn MJ, Houben MP, Croes EA, van Swieten JC, Oostra BA, van Duijn CM (2007) Cerebrovascular risk factors do not contribute to genetic variance of cognitive function: the ERF study. Neurobiol Aging 28:735–741 10.1016/j.neurobiolaging.2006.03.012 [DOI] [PubMed] [Google Scholar]
- 49.Miller SA, Dykes DD, Polesky HF (1988) A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 16:1215 10.1093/nar/16.3.1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Aulchenko YS, Bertoli-Avella AM, van Duijn CM (2005) A method for pooling alleles from different genotyping experiments. Ann Hum Genet 69:233–238 [DOI] [PubMed] [Google Scholar]
- 51.O’Connell JR, Weeks DE (1998) PedCheck: a program for identification of genotype incompatibilities in linkage analysis. Am J Hum Genet 63:259–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Abecasis GR, Cherny SS, Cookson WO, Cardon LR (2002) Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet 30:97–101 10.1038/ng786 [DOI] [PubMed] [Google Scholar]
- 53.Kruglyak L, Daly MJ, Reeve-Daly MP, Lander ES (1996) Parametric and nonparametric linkage analysis: a unified multipoint approach. Am J Hum Genet 58:1347–1363 [PMC free article] [PubMed] [Google Scholar]
- 54.Liu F, Elefante S, van Duijn CM, Aulchenko YS (2006) Ignoring distant genealogic loops leads to false-positives in homozygosity mapping. Ann Hum Genet 70:965–970 10.1111/j.1469-1809.2006.00279.x [DOI] [PubMed] [Google Scholar]
- 55.Fisher RA (1932) Statistical methods for research workers. Oliver and Boyd, London [Google Scholar]
- 56.Myers A, Wavrant De-Vrieze F, Holmans P, Hamshere M, Crook R, Compton D, Marshall H, Meyer D, Shears S, Booth J, et al (2002) Full genome screen for Alzheimer disease: stage II analysis. Am J Med Genet 114:235–244 10.1002/ajmg.10183 [DOI] [PubMed] [Google Scholar]
- 57.Poduslo SE, Yin X, Hargis J, Brumback RA, Mastrianni JA, Schwankhaus J (1999) A familial case of Alzheimer’s disease without tau pathology may be linked with chromosome 3 markers. Hum Genet 105:32–37 10.1007/s004390051060 [DOI] [PubMed] [Google Scholar]
- 58.Yu G, Nishimura M, Arawaka S, Levitan D, Zhang L, Tandon A, Song YQ, Rogaeva E, Chen F, Kawarai T, et al (2000) Nicastrin modulates presenilin-mediated notch/glp-1 signal transduction and βAPP processing. Nature 407:48–54 10.1038/35024009 [DOI] [PubMed] [Google Scholar]
- 59.Dermaut B, Theuns J, Sleegers K, Hasegawa H, Van den Broeck M, Vennekens K, Corsmit E, St George-Hyslop P, Cruts M, van Duijn CM, et al (2002) The gene encoding nicastrin, a major γ-secretase component, modifies risk for familial early-onset Alzheimer disease in a Dutch population-based sample. Am J Hum Genet 70:1568–1574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Robey FA, Jones KD, Tanaka T, Liu TY (1984) Binding of C-reactive protein to chromatin and nucleosome core particles: a possible physiological role of C-reactive protein. J Biol Chem 259:7311–7316 [PubMed] [Google Scholar]
- 61.Despriet DD, Klaver CC, Witteman JC, Bergen AA, Kardys I, de Maat MP, Boekhoorn SS, Vingerling JR, Hofman A, Oostra BA, et al (2006) Complement factor H polymorphism, complement activators, and risk of age-related macular degeneration. JAMA 296:301–309 10.1001/jama.296.3.301 [DOI] [PubMed] [Google Scholar]
- 62.Kardys I, de Maat MP, Uitterlinden AG, Hofman A, Witteman JC (2006) C-reactive protein gene haplotypes and risk of coronary heart disease: the Rotterdam Study. Eur Heart J 27:1331–1337 10.1093/eurheartj/ehl018 [DOI] [PubMed] [Google Scholar]
- 63.Zhang X, Kurnasov OV, Karthikeyan S, Grishin NV, Osterman AL, Zhang H (2003) Structural characterization of a human cytosolic NMN/NaMN adenylyltransferase and implication in human NAD biosynthesis. J Biol Chem 278:13503–13511 10.1074/jbc.M300073200 [DOI] [PubMed] [Google Scholar]
- 64.Papassotiropoulos A, Stephan DA, Huentelman MJ, Hoerndli FJ, Craig DW, Pearson JV, Huynh KD, Brunner F, Corneveaux J, Osborne D, et al (2006) Common Kibra alleles are associated with human memory performance. Science 314:475–478 10.1126/science.1129837 [DOI] [PubMed] [Google Scholar]
- 65.Ertekin-Taner N, Graff-Radford N, Younkin LH, Eckman C, Baker M, Adamson J, Ronald J, Blangero J, Hutton M, Younkin SG (2000) Linkage of plasma Aβ42 to a quantitative locus on chromosome 10 in late-onset Alzheimer’s disease pedigrees. Science 290:2303–2304 10.1126/science.290.5500.2303 [DOI] [PubMed] [Google Scholar]
- 66.Yamada H, Dahl ML, Viitanen M, Winblad B, Sjoqvist F, Lannfelt L (1998) No association between familial Alzheimer disease and cytochrome P450 polymorphisms. Alzheimer Dis Assoc Disord 12:204–207 10.1097/00002093-199809000-00013 [DOI] [PubMed] [Google Scholar]
- 67.Mackay KB, Dewar D, McCulloch J (1994) kappa-1 Opioid receptors of the temporal cortex are preserved in Alzheimer’s disease. J Neural Transm Park Dis Dement Sect 7:73–79 10.1007/BF02252664 [DOI] [PubMed] [Google Scholar]
- 68.Buyske S, Bates ME, Gharani N, Matise TC, Tischfield JA, Manowitz P (2006) Cognitive traits link to human chromosomal regions. Behav Genet 36:65–76 10.1007/s10519-005-9008-9 [DOI] [PubMed] [Google Scholar]