

Abstract
The homotypic cell adhesion molecule PECAM-1 is a major participant in the migration of leukocytes across endothelium. We examined the ability of a chimeric soluble sPECAM-1 fused to human IgG-Fc to impair leukocyte entry through the blood-brain barrier and reduce CNS autoimmunity. sPECAM-Fc impaired migration of lymphocytes across brain endothelial monolayers and diminished the severity of EAE, an experimental model of MS, when administered at the onset of symptoms. However, in mice transgenic for sPECAM-Fc, the chronically elevated levels of sPECAM-Fc hastened onset of EAE disease without significantly changing clinical score severity. Our data suggests that short-term treatment of diseases like MS with sPECAM-Fc has therapeutic potential.
Keywords: PECAM-1, EAE, sPECAM-Fc, MS, cellular migration
1. Introduction
Platelet endothelial cell adhesion molecule (PECAM)-1 (CD31) is a cell adhesion molecule found on leukocytes and vascular endothelium. Homophilic interactions between PECAM-1 on both leukocyte and endothelium is necessary for the migration of leukocytes through the vasculature from blood to tissue (Muller et al., 1993). In CNS autoimmunity, leukocytes migrate through the blood-brain barrier, a unique vascular structure, to enter the brain and spinal cord. Soluble (s)PECAM-1 has the potential to compete with and interfere with the PECAM-PECAM interactions between the brain microvascular endothelial cells and the entering leukocytes and may reduce the number of invading cells and improve clinical outcome.
Several in vitro and in vivo studies have examined the effects of PECAM-1 inhibition on cellular migration across endothelial cells or vessels. In vitro, sPECAM-1 and anti-PECAM-1 antibodies inhibit the passage of immune cells across layers of endothelial cells (Goldberger et al., 1994; Liao et al., 1995; Liao et al., 1997; Muller et al., 1993; Wong et al., 1995). In vivo in several species, treatment with sPECAM-1 blocks leukocyte entry into the peritoneum in response to thioglycolate (Bogen et al., 1994; Liao et al., 1999; Schenkel et al., 2004), lungs in response to immune complexes (Vaporciyan et al., 1993) and muscle in response to ischemia/reperfusion injury (Farooq et al., 2001; Gumina et al., 1996; Murohara et al., 1996). Additionally, T cell accumulation in the cerebral spinal fluid in response to infused antigen is impaired in the presence of exogenous sPECAM-Fc or anti-PECAM-1 antibody (Qing et al., 2001). These studies promote the hypothesis that sPECAM-1 may ameliorate MS pathology.
The role of PECAM-1 in experimental autoimmune encephalomyelitis (EAE) has partially been examined. The use of anti-PECAM antibody had no effect on EAE in rats, although this antibody was not shown to be effective at blocking leukocyte migration (Williams et al., 1996). Furthermore, PECAM-1 deficient animals had an earlier onset of EAE clinical signs compared to wild-type animals (Graesser et al., 2002). This effect was attributed to increased vascular permeability, particularly of the blood-brain barrier, in the PECAM-1 deficient mice. Similar results were seen in studies of PECAM-1 deficient mice in collagen induced arthritis models (Tada et al., 2003; Wong et al., 2005). Both of these studies were carried out in the C57Bl/6 strain of mice, which are unique in that they do not respond to PECAM blockade in several inflammatory models (Schenkel et al., 2004). However, these studies suggest that PECAM-1 interactions could play an important role in autoimmune responses.
We examined the therapeutic potential of a chimeric soluble PECAM-1 fused human IgG-Fc in EAE. We found that sPECAM-Fc was able to impair migration of lymphocytes across brain endothelium in vitro and was able to reduce the severity of clinical symptoms in SJL mice treated at the onset of disease. To examine the effect of continued sPECAM-Fc therapy, we examined EAE symptoms in transgenic mice secreting serum sPECAM-Fc. Interestingly, animals with long-term elevated levels of sPECAM-Fc experienced earlier onset of symptoms. Our data suggest that sPECAM-Fc may be an efficacious acute, but not extended, therapy for multiple sclerosis (MS).
2. Materials and Methods
2.1 Mice
SJL/J mice were purchased from Jackson Laboratories (Bar Harbor, ME).Mice transgenic for a chimeric, soluble, murine PECAM-1 fused to the Fc region of human IgG1 (Liao et al., 1999) were backcrossed six generations from the FVB/n to the SJL/J background. Expression of the sPECAM-Fc was monitored by serum ELISA using goat anti-human IgG- Fcγ fragment specific antibodies (Jackson ImmunoResearch, West Grove, PA) at Weill College of Medicine (New York, NY). Transgene expression in the colony was heterogeneous, producing offspring of varying levels of transgene expression. “High” expression of sPECAM-Fc in serum was defined as 9-12.5 μg/ml. “Low” expression was defined as 2-9 μg/ml. Mice used in EAE experiments were 7-13 week old females. The University of Wisconsin-Madison School of Medicine and Public Heath Institutional Animal Care and Use Committee approved all experimental protocols.
2.2 Migration assay
Brain endothelial cells were harvested as described (Deli et al., 2003; Deli and Joo, 1996). In brief, brain cortexes of adult female SJL/J mice were isolated and processed. Brain tissue microvessels were plated on to 3μm Transwells® (Corning, Acton, MA) precoated with fibronectin and collagen IV (Sigma-Aldrich) in 20% fetal bovine serum in DMEM supplemented with 2mM L-glutamine, 1ng/ml basic fibroblast growth factor (Roche Applied Science, Roche Diagnostics Corporation, Indianapolis, IN) and antibiotics. Cells were incubated in the presence of 4 μg/ml puromycin (Sigma-Aldrich) for two days to eliminate contaminating pericytes. Two days prior to use, cell monolayers were cultured in serum-free DMEM-HAM's-F12 media containing L-glutamine and antibiotics and supplemented with 550 nM hydrocortisone (Sigma-Aldrich) (Weidenfeller et al., 2005). Transendothelial resistance of endothelial monolayers was measured prior to use to ensure integrity of the layer. Splenocytes were isolated according to as previously described (Fee et al., 2003; Qing et al., 2001; Qing et al., 2000; Fabry et al., 1993; Fabry et al., 1990) and incubated in a plastic dish for 30 min to deplete adherent cells. Non-adherent cells were washed and counted. Immediately prior to use, the chambers were washed and the media replaced with 2% FBS DMEM-HAM's-F12 media supplemented with hydrocortisone. 2×105 cells were put into the upper chambers with 20 μg/ml sPECAM-Fc (Liao et al., 1999) or purified Fc fragment of human IgG (Bethyl Laboratories, Inc., Montgomery, TX). Cells were allowed to migrate at 37°C for four hours after which the abluminal face of the Transwell was gently rinsed into the lower chamber media. Cells were then collected and counted. Experiments were repeated two times using three or four separate migration chambers.
2.3 EAE induction and evaluation
Proteolipid protein (PLP 139-151) synthesized by Annovis (Ashton, PA) was dissolved at 1 mg/ml in sterile PBS and mixed by sonication in equal volumes with complete Freunds adjuvant (CFA), supplemented with Mycobacterium tuberculosis H37Ra (Difco, Detroit, MI) to 5 mg/ml. 100 μl of the homogenized solution was injected subcutaneously between the shoulder blades of the mouse. Animals were coded and clinical scores were monitored on a blind manner daily beginning 5-7 days after EAE induction. Clinical scoring was as follows: 0, no motor defect; 1, tail weakness; 2, partial hind limb paralysis; 3, complete hind limb paralysis; 4, complete fore limb and hind limb paralysis; 5, moribund or dead. Intermediate scores were assigned for intermediate symptoms.
2.4 Acute treatment of SJL mice with EAE
EAE was induced in SJL mice, and 200 ng of Pertussis toxin (List Biological Laboratories, Inc. Campbell, CA) was administered intraperitoneally on the day of induction and two days later. Mice were injected with 100μg purified sPECAM-Fc or 100μg human Ig-Fc intraperitoneally in 100μl on the day of EAE symptom onset, then every other day for a total of 5 injections. The half-life of sPECAM-Fc is ∼24 hrs (Liao et al., 1999). Statistical comparison of clinical scores included calculating the average clinical score for each mouse after onset and performing a Wilcox analysis (Fleming et al., 2005). p values were calculated using the statistical package R version 1.12 (http://www.R-project.org) (R-Development-Core-Team, 2005)
2.5 Treatment of transgenic and wildtype mice
40μg purified sPECAM-Fc, 40μg human IgG-Fc, or PBS was injected intravenously in 100μl into “high” sPECAM-Fc expressing, “low” sPECAM-Fc expressing, or wildtype mice, respectively; every other day from day 5-13. Average clinical score was calculated for each mouse for 15 days after onset and a Kruskal-Wallis analysis was performed (Fleming et al., 2005). Onset comparisons were performed using survival analysis
2.6 Histology
At 40 days post EAE induction, spinal cords were removed from the spinal column by liquid expulsion, fixed in 10% formalin and embedded in paraffin. 10μm sections were cut and stained with hematotoxylin and eosin (H&E) or luxol fast blue (LFB) and examined with an Olympus BX40 microscope (Olympus America Inc., Melville, N.Y.) with an Olympus Qcolor 3 camera (Olympus America Inc., Melville, N.Y. Images were adjusted in Adobe Photoshop (Adobe Systems, Inc.)
3. Results
3.1 Migration of lymphocytes across brain endothelium in vitro in the presence of sPECAM-Fc
sPECAM-Fc is capable of blocking migration of monocytes and neutrophils across human umbilical vein endothelial cells (Liao et al., 1997), and anti-PECAM antibody is capable of reducing T cell migration across human brain microvessel endothelial cells (Wong et al., 1999). We investigated whether sPECAM-Fc would also block migration of lymphocytes across a primary murine brain endothelial layer. Isolated murine SJL/J brain microvessels were grown to confluence on 3μm Transwell® membranes and hydrocortisone was used to induce blood-brain barrier–like characteristics (Weidenfeller et al., 2005). Splenocytes harvested from a naïve SJL/J were enriched for lymphocytes by plastic adherence. Purity of lymphocytes reached approximately 90%. 2×105 cells were applied to the luminal face of the endothelial monolayer in media with either purified sPECAM-Fc or human IgG-Fc. Cells were allowed to migrate for four hours, then collected and counted. In the presence of sPECAM-Fc, 2.4 times fewer lymphocytes migrated across the endothelial cells into the lower chamber. This supports previous findings and established that in vitro, sPECAM-Fc blocks lymphocyte migration across brain endothelium.
3.2 Treatment of EAE with sPECAM-Fc
In order to analyze the effect of sPECAM-Fc in an autoimmune disease of the CNS in vivo, we tested the effect of injected sPECAM-Fc in EAE. We induced EAE in SJL/J mice using PLP139-151 peptide and injected 100μg sPECAM-Fc or human IgG-Fc intraperitoneally on the day when clinical symptoms were detected and then every other day for a total of five injections throughout of the disease. As we demonstrate in figures 2B and C, the proportion of mice without clinical symptoms or with mild EAE (clinical score 0.5-2) was higher in the sPECAM-Fc treated groups. This also corresponded with the lower proportion of mice experiencing severe EAE (clinical score higher than 2) in this treated group. Although the initial peak of disease was similar in the sPECAm-Fc and control Fc injected groups, improvement of clinical signs in the sPECAM-Fc treated group was evident in the relapse phase of the disease, after the injections had ended (Figure 2A). These data indicate that the inhibition by sPECAM-Fc is capable of providing a lasting clinical improvement of ongoing EAE disease in vivo.
Figure 2.
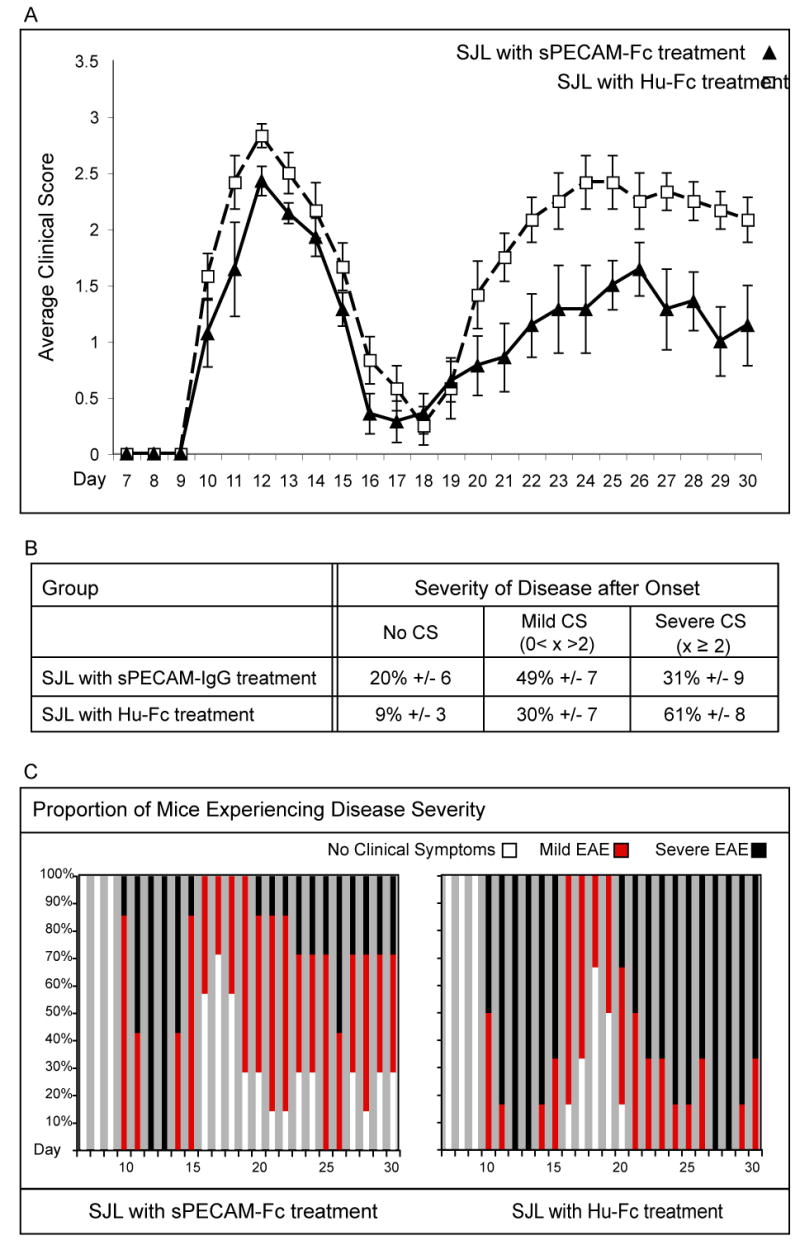
SJL mice treated with sPECAM-Fc at symptom onset have improved clinical symptom severity. A. Daily mean clinical score for each group is plotted. Error bars represent the standard error of the mean. p = 0.02, n= 7 PECAM-IgG treated, 6 human IgG-Fc, combined from 2 independent experiments. B. The average percentage of time after onset that mice in each group experienced no clinical symptoms, mild EAE with clinical symptoms greater than zero and less than 2, and severe EAE with clinical symptoms of 2 and above is shown. C. Graphical representation shows the proportion of mice in the group on each day experiencing no onset or no clinical symptoms (white portion of bar), mild EAE (red portion of bar), or severe EAE (black portion of bar).
3.3 EAE in transgenic mice with elevated levels of sPECAM-Fc
In the above experiments, soluble PECAM levels in vivo were acutely increased by injection of sPECAM-Fc. To test whether sPECAM-Fc would be more effective with extended sPECAM-Fc exposure, we utilized transgenic mice expressing serum sPECAM-Fc (Liao et al., 1999) bred onto the SJL/J background. These mice allowed us to avoid complications from an accumulation of potentially blocking antibodies to the exogenous human portion of the protein chimera that might develop with many injections in a wildtype animal and kept stress to the animals at a minimum. To avoid possible complications from parental strain influences, wildtype littermates were used as controls. Mice from the colony were divided into three groups based on serum level of sPECAM-Fc. High sPECAM-Fc expressing mice were defined as having serum levels of sPECAM-Fc ranging from 9-12.5 μg/ml, and low expressors defined as having levels ranging from 2-9 μg/ml. Wildtype mice had no detectable sPECAM-Fc. Initial experiments with low sPECAM-Fc expressing and wildtype mice indicated that sPECAM-Fc at a concentration in serum between 2-9 μg/ml is not sufficient to significantly alter the disease course from that seen in wildtype mice (Figure 3).
Figure 3.
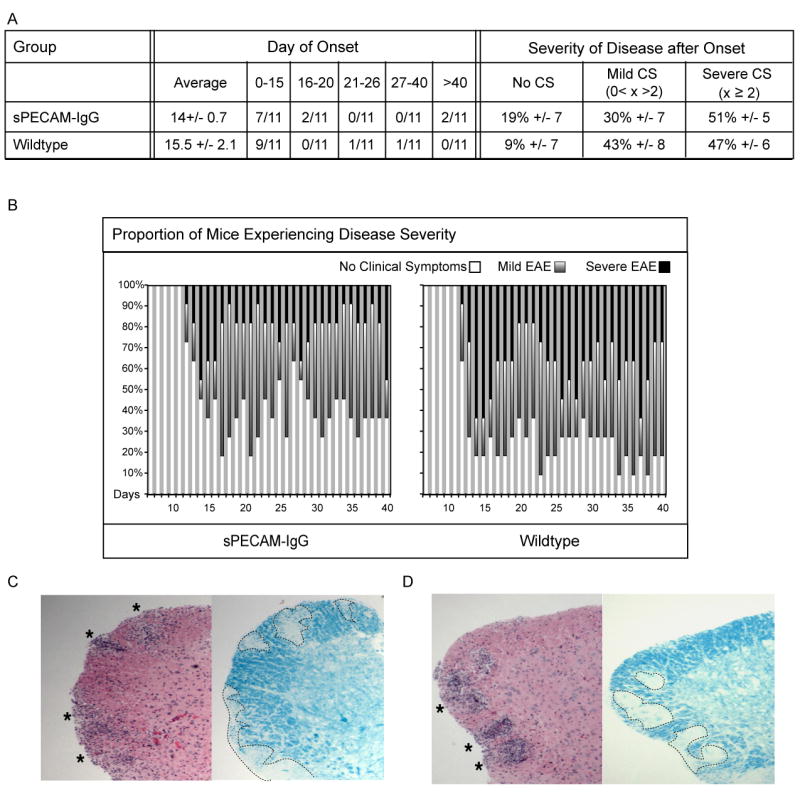
sPECAM-Fc mice with 2-9 μg/ml in serum do not have different onset or severity of EAE symptoms than wildtype mice. A. Shown is average day onset, plus or minus standard error of the mean, and the number of mice per group to experience onset within the given time frames. The average percentage of time during the fifteen days after onset that mice in each group experienced no clinical symptoms, mild EAE with clinical symptoms greater than zero and less than 2, and severe EAE with clinical symptoms of 2 and above is shown. B. Graphical representation shows the proportion of mice in the group on each day experiencing no onset or no clinical symptoms (white portion of bar), mild EAE (gray portion of bar), or severe EAE (black portion of bar). The protection afforded by the low levels of sPECAM-Fc did not reach statistical significance. (C) Wildtype and (D) sPECAM-Fc mice. Spinal cords were removed 40 days post EAE induction. Paraffin-embedded sections were stained with H&E or LFB to detect infiltrating mononuclear cells and demyelinated areas, respectively. Both wildtype and sPECAM-Fc transgenic mice showed visible signs of infiltration and demyelination in the spinal cord.
To increase the likelihood of protection, we supplemented the serum level of sPECAM-Fc in high producing mice to raise it closer to the level found previously to be effective in blocking transmigration after thioglycollate challenge (Laio et al., 1999). Every other day from day 5-13, mice received an intravenous injection of 40μg sPECAM-Fc into high expressors, 40μg of human IgG-Fc into low expressors, or PBS into wildtype. The mice were monitored for clinical symptoms until day 40. Onset of EAE clinical symptoms was on average 2-3 days earlier in the high expressing mice treated with additional sPECAM-Fc (Figure 4), indicating that in contrast to the results with acute treatment after disease onset, chronically elevated levels of sPECAM-Fc were not protective. However, the severity of disease post-onset was unaffected by the presence of increased sPECAM-Fc. On average, within the first fifteen days after onset, all three groups experienced similar numbers of days with severe symptoms (clinical scores of two or above); mild symptoms (clinical scores less than two, but more than zero); or without discernable disability. This indicates that chronically high levels of sPECAM-Fc were no longer an effective inhibitor of clinical symptoms in EAE.
Figure 4.
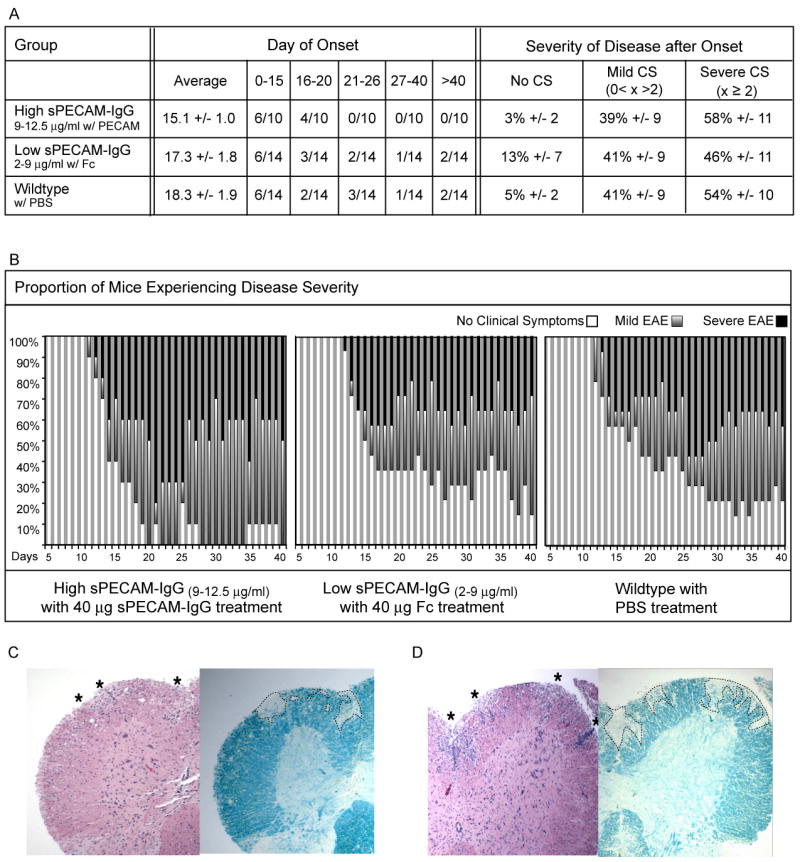
sPECAM-Fc treatment of high sPECAM-Fc expressing mice increases disease onset but not disease severity as compared to wildtype mice. High sPECAM-Fc expressors, low sPECAM-Fc expressors, and wildtype mice were treated 5,7,9,11,13 days after induction of EAE. High expressors received 40μg of purified sPECAM-Fc; low expressors received 40μg of human IgG-Fc; wildtype mice received PBS alone. A. Shown is average day onset, plus or minus standard error of the mean, and the number of mice per group to experience onset within the given time frames for all three groups. For wildtype to high comparison, p= 0.06, for all other comparisons, p > 0.1 The average percentage of time during the fifteen days after onset that mice in each group experienced no clinical symptoms, mild EAE with clinical symptoms greater than zero and less than 2, and severe EAE with clinical symptoms of 2 and above is shown. B. Graphical representation shows the proportion of mice in the group on each day experiencing no onset or no clinical symptoms (white portion of bar), mild EAE (red portion of bar), or severe EAE (black portion of bar). (C) Low sPECAM-Fc expressor and (D) high sPECAM-Fc expressor. Spinal cords were removed 40 days post EAE induction. Paraffin-embedded sections were stained with H&E or LFB to detect infiltrating mononuclear cells and demyelinated areas, respectively. Both low and high expressors showed visible signs of infiltration and demyelination in the spinal cord.
4. Discussion
Multiple lines of in vitro and in vivo evidence suggest that blocking PECAM-PECAM interactions between leukocytes and endothelium either through anti-PECAM-1 antibodies or through the use of recombinant sPECAM-1 constructs diminishes migration of leukocytes across endothelium (Liao et al., 1995; Liao et al.,1997; Muller et al., 1993; Wakelin et al., 1996; Wong et al., 1999). We investigated the hypothesis that blocking PECAM-PECAM interactions could improve clinical outcome in EAE.
We first addressed whether our sPECAM-Fc would block lymphocyte migration across brain endothelial cells in vitro using a Transwell® assay. We determined that passage of murine lymphocytes across murine brain endothelium was diminished in the presence of sPECAM-Fc, similar to PECAM blocking experiments using monocytes and neutrophils on human umbilical vein endothelial cells (Liao et al., 1995; Liao et al., 1997) and T cells on human brain endothelium (Wong et al., 1999). Thus, we had additional support for the hypothesis that sPECAM-Fc might prevent mice from developing EAE or at least diminish the severity of the disease by impairing cell entry into the CNS.
We then investigated whether the blocking effect of sPECAM-Fc seen in vitro would be sufficient to change the course of infiltration and destruction seen in CNS autoimmunity using EAE in SJL mice. We treated animals with a regimen of five injections of sPECAM-Fc over nine days beginning at the onset of symptoms. We found that sPECAM-Fc was effective in vivo and capable of reducing the severity of clinical disease, even after the treatment was no longer being administered. This data shows that acutely administered sPECAM-Fc is a novel treatment with potential as a therapeutic agent for MS.
Evaluation of sPECAM-Fc as a possible long-term therapy was evaluated using sPECAM-Fc transgenic mice. Unexpectedly, sPECAM-Fc did not show a protective effect in the transgenic animals, but instead allowed an earlier onset of disease onset without changing the disease severity. Earlier onset of EAE without an overall increase in disease severity was also described in the PECAM-1 deficient strain (Graesser et al., 2002). In their paper, the authors conclude that the accelerated onset of EAE in the absence of PECAM-1 is due to brain microvessel hyperpermeability in response to inflammatory stimuli. In previous studies, sPECAM-Fc transgenic mice had reduced migration into the peritoneum in response to acute inflammatory challenge (Liao et al., 1999), indicating that sPECAM-Fc transgenicity does not lead to vascular hyperpermability. Our data suggest, therefore, that long-term sPECAM-Fc exposure can contribute to the onset of the inflammatory process.
Interestingly, a number of pro-inflammatory effects have been shown when cross-linked anti-PECAM antibodies were used to stimulate PECAM-1 outside-in signaling. For example, when macrophages or neutrophils were treated with anti-PECAM antibodies an increased CD11b/CD18 activity (Berman and Muller, 1995; Elias et al., 1998) and pro-matrix metalloproteinase-9 production were observed (Nelissen et al., 2002), in parallel with elevated superoxide and TNF-α (Elias et al., 1998). Activation of naïve T cells can also be potentiated using anti-PECAM-1 antibodies (Elias et al., 1998). We measured T cell activation markers in sPECAM-Fc transgenic and wildtype mice (data not shown). Brains and spleens from EAE mice were harvested 40 days after EAE induction. Expression of IFN-γ and high LFA-1 were measured as indicators of activation. The proportion of LFA-1 high, CD4+ cells in the spleen was unchanged among groups. Similarly, in brain we did not observe changes in the PLP139-151-induced IFN-γ + CD4+ populations in the transgenic mice (data not shown). It seems unlikely then, that sPECAM-Fc has an effect on T cell activation once the disease has been initiated. We cannot exclude that with extended exposure, sPECAM-Fc may be signaling through PECAM-1 on leukocytes to contribute to initial inflammatory responses in mice chronically producing sPECAM-Fc in vivo.
Accumulation of sPECAM-1 in blood is a characteristic of diseases effecting endothelium. An increase of serum levels of sPECAM-1 is well documented in MS (Losy et al., 1999; Jimenez et al., 2005; Minagar et al., 2001; Kuenz et al., 2005.), although studies have not been able to link genetic changes in PECAM-1 with MS (Sciacca et al., 2000; Nelissen et al. 2000.) In addition to MS, increased levels of sPECAM-1 are detected in serum after acute myocardial infarction (Serebruany et al.,1999b; Soeki et al., 2003), congestive heart failure (Serebruany et al., 1999a), stroke (Zaremba and Losy, 2002), and umbilical placental vascular disease (Wang et al., 2002). This indicates that elevated sPECAM-1 is naturally associated with disease states. It is possible that chronic elevation of sPECAM-1 can be an inflammatory mediator and not merely a marker of chronic inflammation, although the mechanisms for this remain to be identified.
Our data together suggest sPECAM-1 acts both to enhance and curb inflammatory processes. sPECAM-Fc is a new and exciting possible treatment for MS during acute attack, but should not be used chronically or in periods of remission as it may also have long-term proinflammatory effects.
Figure 1.
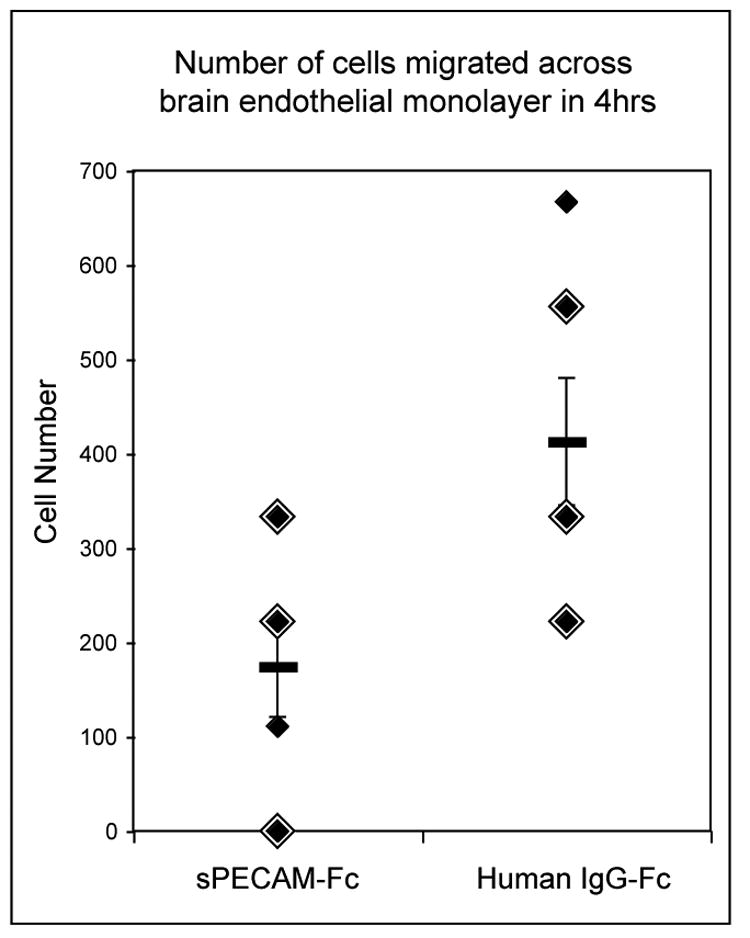
sPECAM-Fc blocks migration of lymphocytes across an in vitro murine brain endothelial cell monolayer. Lymphocytes were incubated for four hours in QCM™ 24-well Invasion Assay chambers with endothelial cell monolayers in the presence of 20 μg/ml sPECAM-Fc or human IgG-Fc. Migrated cells were collected, washed, and counted as described in Material and Methods. In the presence of sPECAM-Fc, on average, 2.4 times fewer cells migrated across the monolayers. Error bars represent the standard deviation. p < 0.02 Double size diamonds indicate a second measurement of the same magnitude.
Acknowledgments
We thank Tina Chew and Alana DeRoche (Weill Cornell Medical Center) for performing serum ELISAs, Khen Macvilay for flow cytometry, and Dr. Laura Hogan for critical reading of the manuscript. This work was supported by the National Institutes of Health grant RO1- NS 37570 01 to Z. Fabry and HL046849 to WAM.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Berman ME, Muller WA. Ligation of platelet/endothelial cell adhesion molecule 1 (PECAM-1/CD31) on monocytes and neutrophils increases binding capacity of leukocyte CR3 (CD11b/CD18) J Immunol. 1995;154:299–307. [PubMed] [Google Scholar]
- Bogen S, Pak J, Garifallou M, Deng X, Muller WA. Monoclonal antibody to murine PECAM-1 (CD31) blocks acute inflammation in vivo. J Exp Med. 1994;179:1059–1064. doi: 10.1084/jem.179.3.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deli MA, Abraham CS, Niwa M, Falus A. N,N-diethyl-2-[4-(phenylmethyl)phenoxy]ethanamine increases the permeability of primary mouse cerebral endothelial cell monolayers. Inflamm Res. 2003;52 1:S39–40. doi: 10.1007/s000110300045. [DOI] [PubMed] [Google Scholar]
- Deli MA, Joo F. Cultured vascular endothelial cells of the brain. Keio J Med. 1996;45:183–198. doi: 10.2302/kjm.45.183. discussion 198-189. [DOI] [PubMed] [Google Scholar]
- Elias CG, 3rd, Spellberg JP, Karan-Tamir B, Lin CH, Wang YJ, McKenna PJ, Muller WA, Zukowski MM, Andrew DP. Ligation of CD31/PECAM-1 modulates the function of lymphocytes, monocytes and neutrophils. Eur J Immunol. 1998;28:1948–1958. doi: 10.1002/(SICI)1521-4141(199806)28:06<1948::AID-IMMU1948>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Fabry Z, Waldschmidt MM, Van Dyk L, Moore SA, Hart MN. Activation of CD4+ lymphocytes by syngeneic brain microvascular smooth muscle cells. J Immunol. 1990;145:1099–1104. [PubMed] [Google Scholar]
- Fabry Z, Sandor M, Gajewski TF, Herlein JA, Waldschmidt MM, Lynch RG, Hart MN. Differential activation of Th1 and Th2 CD4+ cells by murine brain microvessel endothelial cells and smooth muscle/pericytes. J Immunol. 1993;151:38–47. [PubMed] [Google Scholar]
- Farooq MM, Serra A, Newman PJ, Cambria RA, Seabrook GR, Towne JB, Freischlag JA. PECAM-1/IgG attenuates peroxynitrite-mediated extremity reperfusion injury. J Vasc Surg. 2001;34:555–558. doi: 10.1067/mva.2001.115813. [DOI] [PubMed] [Google Scholar]
- Fee D, Crumbaugh A, Jacques T, Herdrich B, Sewell D, Auerbach D, Piaskowski S, Hart MN, Sandor M, Fabry Z. Activated/effector CD4(+) T cells exacerbate acute damage in the central nervous system following traumatic injury. J Neuroimmunol. 2003;136:54–66. doi: 10.1016/s0165-5728(03)00008-0. [DOI] [PubMed] [Google Scholar]
- Fleming KK, Bovaird JA, Mosier MC, Emerson MR, LeVine SM, Marquis JG. Statistical analysis of data from studies on experimental autoimmune encephalomyelitis. J Neuroimmunol. 2005;170:71–84. doi: 10.1016/j.jneuroim.2005.08.020. [DOI] [PubMed] [Google Scholar]
- Goldberger A, Middleton KA, Oliver JA, Paddock C, Yan HC, DeLisser HM, Albelda SM, Newman PJ. Biosynthesis and processing of the cell adhesion molecule PECAM-1 includes production of a soluble form. J Biol Chem. 1994;269:17183–17191. [PubMed] [Google Scholar]
- Graesser D, Solowiej A, Bruckner M, Osterweil E, Juedes A, Davis S, Ruddle NH, Engelhardt B, Madri JA. Altered vascular permeability and early onset of experimental autoimmune encephalomyelitis in PECAM-1-deficient mice. J Clin Invest. 2002;109:383–392. doi: 10.1172/JCI13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumina RJ, el Schultz J, Yao Z, Kenny D, Warltier DC, Newman PJ, Gross GJ. Antibody to platelet/endothelial cell adhesion molecule-1 reduces myocardial infarct size in a rat model of ischemia-reperfusion injury. Circulation. 1996;94:3327–3333. doi: 10.1161/01.cir.94.12.3327. [DOI] [PubMed] [Google Scholar]
- Jimenez J, Jy W, Mauro LM, Horstman LL, Ahn ER, Ahn YS, Minagar A. Elevated endothelial microparticle-monocyte complexes induced by multiple sclerosis plasma and the inhibitory effects of interferon-beta 1b on release of endothelial microparticles, formation and transendothelial migration of monocyte-endothelial microparticle complexes. Mult Scler. 2005;11:310–5. doi: 10.1191/1352458505ms1184oa. [DOI] [PubMed] [Google Scholar]
- Kuenz B, Lutterotti A, Khalil M, Ehling R, Gneiss C, Deisenhammer F, Reindl M, Berger T. Plasma levels of soluble adhesion molecules sPECAM-1, sP-selectin and sE-selectin are associated with relapsing-remitting disease course of multiple sclerosis. J Neuroimmunol. 2005;167:143–9. doi: 10.1016/j.jneuroim.2005.06.019. [DOI] [PubMed] [Google Scholar]
- Liao F, Ali J, Greene T, Muller WA. Soluble domain 1 of platelet-endothelial cell adhesion molecule (PECAM) is sufficient to block transendothelial migration in vitro and in vivo. J Exp Med. 1997;185:1349–1357. doi: 10.1084/jem.185.7.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao F, Huynh HK, Eiroa A, Greene T, Polizzi E, Muller WA. Migration of monocytes across endothelium and passage through extracellular matrix involve separate molecular domains of PECAM-1. J Exp Med. 1995;182:1337–1343. doi: 10.1084/jem.182.5.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao F, Schenkel AR, Muller WA. Transgenic mice expressing different levels of soluble platelet/endothelial cell adhesion molecule-IgG display distinct inflammatory phenotypes. J Immunol. 1999;163:5640–5648. [PubMed] [Google Scholar]
- Losy J, Niezgoda A, Wender M. Increased serum levels of soluble PECAM-1 in multiple sclerosis patients with brain gadolinium-enhancing lesions. J Neuroimmunol. 1999;99:169–72. doi: 10.1016/s0165-5728(99)00092-2. [DOI] [PubMed] [Google Scholar]
- Minagar A, Jy W, Jimenez JJ, Sheremata WA, Mauro LM, Mao WW, Horstman LL, Ahn YS. Elevated plasma endothelial microparticles in multiple sclerosis. Neurology. 2001;56:1319–24. doi: 10.1212/wnl.56.10.1319. [DOI] [PubMed] [Google Scholar]
- Muller WA, Weigl SA, Deng X, Phillips DM. PECAM-1 is required for transendothelial migration of leukocytes. J Exp Med. 1993;178:449–460. doi: 10.1084/jem.178.2.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murohara T, Delyani JA, Albelda SM, Lefer AM. Blockade of platelet endothelial cell adhesion molecule-1 protects against myocardial ischemia and reperfusion injury in cats. J Immunol. 1996;156:3550–3557. [PubMed] [Google Scholar]
- Nelissen I, Ronsse I, Van Damme J, Opdenakker G. Regulation of gelatinase B in human monocytic and endothelial cells by PECAM-1 ligation and its modulation by interferon-beta. J Leukoc Biol. 2002;71:89–98. [PubMed] [Google Scholar]
- Nelissen I, Fiten P, Vandenbroeck K, Hillert J, Olsson T, Marrosu MG, Opdenakker G. PECAM1, MPO and PRKAR1A at chromosome 17q21-q24 and susceptibility for multiple sclerosis in Sweden and Sardinia. J Neuroimmunol. 2000;108:153–159. doi: 10.1016/s0165-5728(00)00293-9. [DOI] [PubMed] [Google Scholar]
- Qing Z, Sewell D, Sandor M, Fabry Z. Antigen-specific T cell trafficking into the central nervous system. J Neuroimmunol. 2000;105:169–178. doi: 10.1016/s0165-5728(99)00265-9. [DOI] [PubMed] [Google Scholar]
- Qing Z, Sandor M, Radvany Z, Sewell D, Falus A, Potthoff D, Muller WA, Fabry Z. Inhibition of antigen-specific T cell trafficking into the central nervous system via blocking PECAM1/CD31 molecule. J Neuropathol Exp Neurol. 2001;60:798–807. doi: 10.1093/jnen/60.8.798. [DOI] [PubMed] [Google Scholar]
- R-Development-Core-Team. R: A Language and Environment for Statistical Computing. Vienna, Austria: 2005. [Google Scholar]
- Schenkel AR, Chew TW, Muller WA. Platelet endothelial cell adhesion molecule deficiency or blockade significantly reduces leukocyte emigration in a majority of mouse strains. J Immunol. 2004;173:6403–6408. doi: 10.4049/jimmunol.173.10.6403. [DOI] [PubMed] [Google Scholar]
- Sciacca FL, Ferri C, D'Alfonso S, Bolognesi E, Martinelli Boneschi F, Cuzzilla B, Colombo B, Comi G, Canal N, Grimaldi LM. Association study of a new polymorphism in the PECAM-1 gene in multiple sclerosis. J Neuroimmunol. 2000;104:174–178. doi: 10.1016/s0165-5728(99)00274-x. [DOI] [PubMed] [Google Scholar]
- Serebruany VL, Murugesan SR, Pothula A, Atar D, Lowry DR, O'Connor CM, Gurbel PA. Increased soluble platelet/endothelial cellular adhesion molecule-1 and osteonectin levels in patients with severe congestive heart failure. Independence of disease etiology, and antecedent aspirin therapy. Eur J Heart Fail. 1999a;1:243–249. doi: 10.1016/s1388-9842(99)00029-x. [DOI] [PubMed] [Google Scholar]
- Serebruany VL, Murugesan SR, Pothula A, Semaan H, Gurbel PA. Soluble PECAM-1, but not P-selectin, nor osteonectin identify acute myocardial infarction in patients presenting with chest pain. Cardiology. 1999b;91:50–55. doi: 10.1159/000006876. [DOI] [PubMed] [Google Scholar]
- Soeki T, Tamura Y, Shinohara H, Sakabe K, Onose Y, Fukuda N. Increased soluble platelet/endothelial cell adhesion molecule-1 in the early stages of acute coronary syndromes. Int J Cardiol. 2003;90:261–268. doi: 10.1016/s0167-5273(02)00564-8. [DOI] [PubMed] [Google Scholar]
- Tada Y, Koarada S, Morito F, Ushiyama O, Haruta Y, Kanegae F, Ohta A, Ho A, Mak TW, Nagasawa K. Acceleration of the onset of collagen-induced arthritis by a deficiency of platelet endothelial cell adhesion molecule 1. Arthritis Rheum. 2003;48:3280–3290. doi: 10.1002/art.11268. [DOI] [PubMed] [Google Scholar]
- Vaporciyan AA, DeLisser HM, Yan HC, Mendiguren II, Thom SR, Jones ML, Ward PA, Albelda SM. Involvement of platelet-endothelial cell adhesion molecule-1 in neutrophil recruitment in vivo. Science. 1993;262:1580–1582. doi: 10.1126/science.8248808. [DOI] [PubMed] [Google Scholar]
- Wakelin MW, Sanz MJ, Dewar A, Albelda SM, Larkin SW, Boughton-Smith N, Williams TJ, Nourshargh S. An anti-platelet-endothelial cell adhesion molecule-1 antibody inhibits leukocyte extravasation from mesenteric microvessels in vivo by blocking the passage through the basement membrane. J Exp Med. 1996;184:229–239. doi: 10.1084/jem.184.1.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Athayde N, Trudinger B. Endothelial cell expression of adhesion molecules is induced by fetal plasma from pregnancies with umbilical placental vascular disease. Bjog. 2002;109:770–777. doi: 10.1111/j.1471-0528.2002.01240.x. [DOI] [PubMed] [Google Scholar]
- Weidenfeller C, Schrot S, Zozulya A, Galla HJ. Murine brain capillary endothelial cells exhibit improved barrier properties under the influence of hydrocortisone. Brain Res. 2005;1053:162–174. doi: 10.1016/j.brainres.2005.06.049. [DOI] [PubMed] [Google Scholar]
- Williams KC, Zhao RW, Ueno K, Hickey WF. PECAM-1 (CD31) expression in the central nervous system and its role in experimental allergic encephalomyelitis in the rat. J Neurosci Res. 1996;45:747–757. doi: 10.1002/(SICI)1097-4547(19960915)45:6<747::AID-JNR11>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Wong D, Prameya R, Dorovini-Zis K. In vitro adhesion and migration of T lymphocytes across monolayers of human brain microvessel endothelial cells: regulation by ICAM-1, VCAM-1, E-selectin and PECAM-1. J Neuropathol Exp Neurol. 1999;58:138–152. doi: 10.1097/00005072-199902000-00004. [DOI] [PubMed] [Google Scholar]
- Wong MX, Hayball JD, Hogarth PM, Jackson DE. The inhibitory co-receptor, PECAM-1 provides a protective effect in suppression of collagen-induced arthritis. J Clin Immunol. 2005;25:19–28. doi: 10.1007/s10875-005-0354-7. [DOI] [PubMed] [Google Scholar]
- Zaremba J, Losy J. sPECAM-1 in serum and CSF of acute ischaemic stroke patients. Acta Neurol Scand. 2002;106:292–298. doi: 10.1034/j.1600-0404.2002.01339.x. [DOI] [PubMed] [Google Scholar]