

Abstract
A real-time reverse transcription-PCR method was developed to determine whether the recovery of culturability of viable but nonculturable (VBNC) Vibrio parahaemolyticus induced the expression of virulence genes coding for the thermostable direct hemolysin and for type III secretion system 2 (TTSS2). The culturability of clinical strain Vp5 of V. parahaemolyticus in artificial seawater at 4°C was monitored, and the VBNC state was obtained. One day after entry into the VBNC state, temperature upshifts to 20 and 37°C allowed the recovery of culturability. Standard curves for the relative quantification of expression of the housekeeping genes rpoS, pvsA, fur, and pvuA; the tdh2 gene; and the TTSS2 genes (VPA1354, VPA1346, and VPA1342) were established. The levels of expression of the pvsA and pvuA genes were stable and were used to normalize the levels of expression of the other genes. No transcriptional induction of the selected virulence genes under the temperature conditions used to recover the culturability of the VBNC bacteria was observed. The study results demonstrate that the recovery of culturability of VBNC cells of pathogenic V. parahaemolyticus is restricted to regrowth, without correlation with the induction of virulence gene expression. Disease induction would depend mainly on host-pathogen interactions that allow the expression of the virulence genes. This is the first time that the use of mRNA to detect viable cells was evaluated by computing the half-lives of multiple mRNA species under conditions inducing the VBNC state.
Vibrio parahaemolyticus is a marine bacterium of which some strains generate food-borne outbreaks of disease characterized by acute gastroenteritis. The thermostable direct hemolysin (TDH) and TDH-related hemolysin were previously considered to be the main factors at the origin of these enterotoxic phenomena. Recently, the genome sequencing of a clinical strain of V. parahaemolyticus, RIMD 2210633, has revealed several other factors of virulence, including genes for two type III secretion systems (TTSS), TTSS1 and TTSS2, present on chromosomes 1 and 2, respectively (18). TTSS1 has been described as a cytotoxic system, and TTSS2 has been described as an enterotoxic system (25). Under environmental stresses, such as a temperature downshift, V. parahaemolyticus appears to be fairly inactive metabolically and enters into a dormant state, namely, the viable but nonculturable (VBNC) state. The VBNC cells are able to respond to some environmental stimuli, such as a temperature upshift, and to become metabolically active and culturable. The recovery of culturability by the VBNC cells of V. cholerae, V. vulnificus, and V. parahaemolyticus has been demonstrated to cause in vivo pathogenicity (1, 6, 23). Two scenarios may explain the virulence: a high number of cells (a high infectious dose) without significant regulation of the virulence genes and/or genetic up-regulation of virulence genes under such conditions.
To determine if cells induce the expression of virulence genes after the recovery of culturability, a relative quantification of expression rates was performed with standardization by using several genes of reference. The literature shows that, in many cases, using only one gene of reference is not sufficient because the expression can vary considerably depending on experimental conditions (4, 34). The limited concentration of iron in surface seawater suggests that iron is one of the factors that affect the growth of bacteria in the oceans (32). To survive under conditions of limited iron, several genes of the iron uptake pathway are involved, including the pvsA gene required for the biosynthesis and transport of the siderophore vibrioferrin in V. parahaemolyticus (31) and the pvuA gene encoding the ferric vibrioferrin receptor (12). The gene fur, encoding a factor of transcription implicated in the siderophore-mediated iron acquisition system, was also chosen for this study (36). These genes were tested as potential control genes under our experimental conditions.
The objective was to determine (i) if V. parahaemolyticus, after the recovery of culturability under environmental conditions, could be a danger to public health by inducing the expression of virulence genes, such as tdh2 encoding TDH and VPA1354 (escU), VPA1342 (spa24), and VPA1346 (vopP) encoding cytosolic, inner membrane, and effector proteins of TTSS2, and (ii) if real-time reverse transcription (RT)-PCR could be used in a field survey to determine the potential virulence of this pathogenic bacterium. A real-time RT-PCR method was developed to quantify the relative expression ratios of the virulence genes and to detect the viable states of V. parahaemolyticus bacteria, culturable or not, by commonly used techniques based on mRNA amplification. The validity of this approach for the detection of viability was tested by calculating the half-lives of several mRNA species at a low temperature (4°C).
MATERIALS AND METHODS
Bacterial strains and culture conditions.
The clinical strain V. parahaemolyticus Vp5 was chosen for the presence of the virulence genes tdh1 and tdh2. Bacteria were grown in heart infusion (HI) broth (0.5% NaCl) at 37°C with shaking, plated onto HI agar (0.5% NaCl; Difco Laboratories, Detroit, MI), and incubated at 37°C for 24 h.
VBNC cells in artificial seawater (ASW) microcosms at 4°C were obtained by a method described previously (7). To measure the recovery of culturability, 50-ml volumes harvested from ASW microcosms 1 day after the VBNC state was reached were incubated at 20 or 37°C with shaking (70 rpm) in the dark. The CFU counts on HI (2% NaCl) agar plates after incubation at 37°C for 24 h were determined.
Strains of V. parahaemolyticus (tdh negative, tdh negative, tdh+, and/or trh+), non-O1/non-O139 V. cholerae, V. mimicus, V. alginolyticus, V. vulnificus, Aeromonas hydrophila/caviae, A. salmonicida, Pseudomonas aeruginosa, Shewanella putrefaciens, and Escherichia coli O157:H7 were selected to test the specificity of the primers used in this study.
Nucleic acid extractions.
DNA and RNA extractions were performed as previously described (7).
Probes and primers.
PCR and real-time RT-PCR were performed with the primers described in Table 1. In this study, probes and primers specific for virulence and housekeeping genes were designed based on gene sequences from 5 and 15 strains of V. parahaemolyticus, respectively. The design of the primers and probes was performed as previously described (22).
TABLE 1.
Primers used in PCR and real-time RT-PCR and probes used in slot blot hybridization for V. parahaemolyticus
| Gene or target (locus) | Primer or probe sequencea (5′ to 3′) | Primer or probe length (bases) | G+C content (%) | Melting temp (°C) | Amplicon length (bp) |
|---|---|---|---|---|---|
| rpoS (VP2553) | F-rpoS: GAC AAT GCG TCA GAG ACG | 18 | 55.6 | 60.7 | 151 |
| R2-rpoS: GAG GTG AGA AGC CAA TTT C | 19 | 47.4 | 58.3 | ||
| P2-rpoS: CGC GAG CAG TAA AAG CCT AGA CG | 23 | 56.5 | 67.1 | ||
| pvsA (VPA1658) | F2-pvsA: CTC CTT CAT CCA ACA CGA T | 19 | 47.4 | 58.3 | 104 |
| R2-pvsA: GGG CGA GAT AAT CCT TGT | 18 | 50.0 | 58.4 | ||
| fur (VP0833) | F-fur: TTG AAG AGC GCC AAC GC | 17 | 58.8 | 60.0 | 94 |
| R-fur: CAA CCA CCG TCA CTG CAT | 18 | 55.6 | 60.7 | ||
| P-fur: ACG CTA ACA AAC CAC AGC TTA TAC CTA T | 28 | 39.3 | 64.0 | ||
| pvuA (VPA1656) | F1-pvuA: CAA ACT CAC TCA GAC TCC A | 19 | 47.4 | 58.3 | 156 |
| R1-pvuA: CGA ACC GAT TCA ACA CG | 17 | 52.9 | 57.6 | ||
| tdh2 (VPA1314) | F-tdh2: CAA CTT TTA ATA CCA ATG CAC | 21 | 33.3 | 55.6 | 129 |
| R2-tdh2: GCC ATT TAG TAC CTG ACG | 18 | 50.0 | 58.4 | ||
| P3-tdh2: AGG TCT CTG ACT TTT GGA CAA ACC GTA ATG | 30 | 43.3 | 66.7 | ||
| escU (VPA1354) | F1-escU: TAA CCC GAC ACA TAT TCT GG | 20 | 45.0 | 58.4 | 163 |
| R-escU: CAT GGC TCT TGC TAA CGG | 18 | 55.6 | 60.7 | ||
| P2-escU: CAG TTT GTT ATG ACC CTA AGA TCG AAC G | 28 | 42.9 | 65.5 | ||
| vopP (VPA1346) | F2-vopP: TAG AAG TCC TCT TGA TAT GGT | 21 | 38.1 | 57.5 | 113 |
| R2-vopP: CCA CCG CTA TAC AAT GAA TG | 20 | 45.0 | 58.4 | ||
| spa24 (VPA1342) | F-spa24: TAC ACA GCA AAT CCC GCC | 18 | 55.6 | 60.7 | 166 |
| R1-spa24: TTT CGG CAT ATC GTT GTC | 18 | 44.5 | 56.1 | ||
| P1-spa24: CCA CAG ATC CAA CCA GGT ATT GAG | 24 | 50.0 | 65.4 | ||
| MS2 RNA | Oco-1: GCT CTG AGA GCG GCT CTA TTG | 21 | 57.1 | 65.3 | 69 |
| Oco-2: CGT TAT AGC GGA CCG CGT | 18 | 61.1 | 63.0 | (22) |
F, forward; R, reverse; P, probe.
PCR conditions.
Detection by PCR was performed with a 40-μl mixture containing 1× PCR buffer (10 mM Tris-HCl, 1.5 mM MgCl2, 50 mM KCl [pH 8.3]; Roche Diagnostics), 200 μM (each) deoxynucleoside triphosphates, 1 U of Taq DNA polymerase (Roche Diagnostics), 500 nM (each) primers, and 100 ng of DNA. Negative and positive controls were made with deionized water and DNA extracted from pathogenic V. parahaemolyticus Vp101, respectively. The PCR program was performed as follows: 94°C for 5 min; 35 cycles of initial denaturation at 94°C for 30 s, annealing at 60°C for 30 s, and extension at 72°C for 45 s; and a final extension at 72°C for 7 min.
Sequencing.
The PCR product obtained with each set of primers for the DNA from strain Vp5 of V. parahaemolyticus was purified using a column system (QIAquick PCR purification kit) according to the recommendations of the manufacturer (QIAGEN). The nucleotide sequence of the purified PCR product was determined as previously described (9).
Slot blot hybridization.
The slot blot hybridization approach was used to determine the specificity of the PCR amplicons obtained with newly designed primers. Slot blots were prepared using positively charged membrane (Roche Diagnostics) and processed by the method of Sambrook et al. (28). Briefly, the PCR-amplified products were denatured (100°C for 10 min) and transferred onto nylon membrane by using the slot blot apparatus BIO-DOT SF (Bio-Rad, Marnes la Coquette, France). The DNA was cross-linked to the membrane with UV light for 3 min. Oligonucleotide probes specifically designed for this assay (Table 1) were labeled using the Dig oligonucleotide tailing kit (Roche Diagnostics) and hybridized at the annealing temperature in a solution containing 5× SSC (0.75 M NaCl, 0.075 M sodium citrate [pH 7.0]), 1.0% (wt/vol) blocking reagent (Roche Diagnostics), 0.1% (wt/vol) N-lauroylsarcosine (Sigma), and 0.02% (wt/vol) sodium dodecyl sulfate (SDS; Sigma). Hybridized blots were washed twice in 2× SSC-0.1% SDS for 5 min and three times in 0.5× SSC-0.1% SDS for 5 min at the annealing temperature. The membrane was incubated with buffer containing the antidigoxigenin-alkaline phosphatase conjugate for 30 min at room temperature. The immunoblots were washed with buffer (0.1 M maleic acid, 0.15 M NaCl [pH 7.5], 0.3% Tween 20) for 15 min and developed using p-nitroblue tetrazolium chloride and BCIP (5-bromo-4-chloro-3-indolylphosphate) toluidine substrates (Roche Diagnostics) at room temperature.
cDNA synthesis.
RT was performed with the SuperScript III first-strand synthesis system for RT-PCR as described by the manufacturer (Invitrogen, Cergy-Pontoise, France). The first cDNA strand was obtained using random hexamers with 800 ng of DNase I-treated RNA. To determine the potential inhibition during the RT and/or PCR, 1 ng of MS2 RNA (Roche Diagnostics, Meylan, France) was added to each RT tube. The primers Oco-1 and Oco-2 (Table 1) were used in real-time RT-PCR to detect MS2 RNA. Each population of cDNA was also used for the construction of a standard curve specific to the corresponding targeted gene.
Real-time RT-PCR conditions.
Real-time RT-PCR was performed with viable culturable and VBNC cells (between day 0 and day 106 of the study) and VBNC samples subjected to temperature upshifts to 20 and 37°C. Detection by real-time RT-PCR was performed with the brilliant SYBR green quantitative PCR core reagent kit (Stratagene, Amsterdam, The Netherlands). Each PCR was performed with a 25-μl final volume containing 2 μl of cDNA with 1× core PCR buffer, 3.5 mM MgCl2, 200 μM (each) deoxynucleoside triphosphates, 300 nM (each) primers, 3% dimethyl sulfoxide, 0.167× SYBR green I, and 1.25 U of SureStart Taq DNA polymerase. The reactions were run on an Mx3000P quantitative PCR system (Stratagene). The following thermal cycling conditions were used: a denaturation program (95°C for 10 min), an amplification program repeated 40 times (95°C for 15 s and 60°C for 1 min), and a melting-curve program (55 to 95°C with warming of 0.2°C per s). RT and PCR positive controls (RNA and DNA, respectively) and negative controls (deionized water) were included in each run.
Construction of specific standard curves.
Specific PCR products were identified based on predicted amplicon sizes and extracted using the QIAquick gel extraction kit (QIAGEN, Courtaboeuf, France). Extracted PCR products were cloned using a TOPO TA cloning kit (pCR2.1-TOPO vector and One Shot TOP10F′ competent E. coli cells; Invitrogen). After the selection of bacterial colonies containing plasmids with the correctly sized inserts, plasmids were extracted and purified using the HiSpeed plasmid midi kit (QIAGEN). The presence of the targeted sequence was verified by PCR with specific primers. In a total volume of 40 μl, 2 μg of plasmid DNA was digested by 4 U of NotI enzyme (Roche Diagnostics) for 3 h at 37°C. For each gene, plasmid DNA dilution curves were generated and used to calculate the efficiency (E) of the real-time PCR [E = 10(−1/slope)].
Relative quantification and statistical analysis.
The relative expression ratio was calculated for each gene of interest by using a mathematical model described by Pfaffl (26):
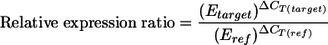 |
CT is the cycle threshold, and ΔCT(target) and ΔCT(ref) represent the CT deviation of control (untreated) minus sample (treated) of the target and reference gene transcripts, respectively. The stability of the transcription of the potential reference genes was evaluated using the freely distributed Microsoft Excel application geNorm (34). The genetic expression in culturable V. parahaemolyticus Vp5 cells, those maintained in ASW at 4°C (day 0) and those subjected to temperature upshifts to 20 and 37°C (responsible for the recovery of the culturability of VBNC cells), was quantified. Samples were taken at two different times, 2 and 6 days after the temperature upshifts. The real-time RT-PCR analysis was performed twice in triplicate. The most stable reference genes were identified, and a geometric mean was calculated and used as a normalization factor. The latter allowed the normalization of the relative expression ratio of each gene of interest. The expression ratios of the genes of interest were tested for significance by a permutation test implemented in the relative expression software tool REST which allows the user to determine if observed effects are the result of the treatment or random allocation (27).
Procedures for measurement of RNA decay.
V. parahaemolyticus Vp5 was inoculated as previously described into a final 2-liter volume of ASW in a 5-liter flask and stored at 4°C. Rifampin (Sigma Aldrich Chimie, St. Quentin Fallavier, France) was added to the ASW microcosm at a final concentration of 100 μg/ml to block transcription. After rifampin treatment, 50-ml volumes were harvested from the ASW microcosm at serial time points (0, 1, 2, 3, 4, 5, 6, and 7 days). The RNA extractions were performed as described above, and the decay of RNA was analyzed by real-time RT-PCR. Additionally, CFU were enumerated after the passage of 10-ml volumes through a 0.22-μm-pore-size nitrocellulose filter (Millipore, St. Quentin en Yvelines, France). The filters were placed on HI agar (2% NaCl) in duplicate and incubated at 37°C for 48 h.
Nucleotide sequence accession numbers.
The GenBank accession numbers for the V. parahaemolyticus Vp5 sequences obtained in this study are EF139123 (rpoS), EF139124 (pvsA), EF139126 (fur), EF139125 (pvuA), EF139129 (tdh2), EF139128 (escU), EF139127 (vopP), and EF139122 (spa24).
RESULTS
Evaluation of primers.
The specificity for V. parahaemolyticus of each primer set was tested by PCR with genomic DNA of different bacteria, i.e., V. parahaemolyticus, non-O1/non-O139 V. cholerae, V. mimicus, V. alginolyticus, V. vulnificus, A. hydrophila, A. caviae, A. salmonicida, P. aeruginosa, S. putrefaciens, and E. coli O157:H7. Only one size of amplicon for each targeted gene was obtained by PCR when DNA from V. parahaemolyticus strains was used. No amplification was observed when other species were tested (data not shown). The amplicon obtained for each gene with the strain Vp5 of V. parahaemolyticus was verified by sequencing, while slot blot hybridization was used to verify the specificity of the amplicons for all other V. parahaemolyticus strains. For example, the P2-rpoS and P3-tdh2 probes detected PCR products from all strains and only the tdh2+ strains of V. parahaemolyticus, respectively (Fig. 1). The presence of a single PCR product for each set of primers was confirmed by real-time PCR using a melting-curve analysis, which resulted in a single product-specific melting curve. Standard curves for the selected genes, rpoS, pvsA, fur, pvuA, tdh2, escU, vopP, and spa24, were developed with cDNA and plasmid DNA. The standard curves showed a highly linear relationship between the CT values and the amount of cDNA or plasmid DNA, with a significant correlation coefficient (R2) of between 0.978 and 0.999 (Table 2). With slopes between −3.22 and −3.39, the PCR efficiencies varied between 2.04 and 1.97.
FIG. 1.

Specificity of primers for V. parahaemolyticus used in real-time RT-PCR. PCR products were hybridized with digoxigenin-labeled probes P2-rpoS (A) and P3-tdh2 (B) specific for rpoS and tdh2 gene amplicons, respectively. The specificity was tested with various strains of V. parahaemolyticus, the tdh1+ tdh2+ (positions 1 to 4), tdh2+ trh+ (positions 5 and 6), tdh1 tdh2 trh negative (position 7), and trh+ (positions 8 to 14) strains, and strains of V. cholerae (position 15), V. mimicus (positions 16 and 17), V. alginolyticus (positions 18 and 19), V. vulnificus (positions 20 and 21), A. hydrophila/caviae (position 22), A. salmonicida (position 23), P. aeruginosa (position 24), S. putrefaciens (position 25), and E. coli O157:H7 (position 26). The no-template control (NTC) contained deionized water.
TABLE 2.
Standard curve evaluation of putative control and virulence genes in V. parahaemolyticus
| Gene | Plasmid DNA curve
|
cDNA curve
|
||
|---|---|---|---|---|
| Slope | R2 | Slope | R2 | |
| rpoS | −3.32 | 0.999 | −3.39 | 0.998 |
| pvsA | −3.27 | 0.999 | −3.27 | 0.984 |
| fur | −3.32 | 0.999 | −3.32 | 0.998 |
| pvuA | −3.32 | 0.999 | −3.39 | 0.978 |
| tdh2 | −3.32 | 0.999 | −3.37 | 0.996 |
| escU | −3.28 | 0.997 | −3.22 | 0.999 |
| vopP | −3.35 | 0.999 | −3.32 | 0.979 |
| spa24 | −3.32 | 0.999 | −3.32 | 0.990 |
Ranking of four putative control genes in V. parahaemolyticus and validation of the relative quantification approach.
The ranking of the four potential control genes was analyzed by geNorm software. The results of the two runs of real-time RT-PCR were used to rank the tested genes as indicated in Table 3. The stability of gene expression (M), calculated as the average pairwise variation in the expression of a gene relative to that of each of the other tested housekeeping genes, was between 0.654 and 3.017 in the first run and 0.355 and 2.840 in the second run, indicating good reproducibility of results. Under the experimental conditions described here, the two runs showed that the pvsA and pvuA genes had the most stable transcriptional expression levels. Thus, they were chosen as references to normalize the expression of the hemolysin gene tdh2 and of the TTSS2 genes escU, vopP, and spa24. M values of 3.017 and 2.840 in the two runs indicated a high level of regulation of the rpoS gene. This gene was also studied as a gene of interest, and its expression was normalized by using the normalization factor (geometric mean) calculated from the expression ratios of the pvsA and pvuA genes.
TABLE 3.
geNorm stability measurements and rankings of expression of selected housekeeping genes
| Gene | Avg stability of expression (rank)a in:
|
|
|---|---|---|
| Run 1 | Run 2 | |
| rpoS | 3.017 (3) | 2.840 (3) |
| pvsA | 0.654 (1) | 0.355 (1) |
| fur | 2.203 (2) | 2.049 (2) |
| pvuA | 0.654 (1) | 0.355 (1) |
The stability of expression is expressed as M values. Genes with the lowest M values have the most stable expression. The genes were ranked according to the stability of their expression, and the ranks are indicated in parentheses.
Regulation of rpoS, tdh2, escU, vopP, and spa24.
The expression of the genes rpoS, tdh2, escU, vopP, and spa24 after a temperature upshift to 20 or 37°C in ASW previously held at 4°C was analyzed. A permutation, or randomization, test implemented in REST version 1.9.12 was used for analyzing experimental data. No assumption concerning the distribution of the measured expression levels was made with this test. The geometric mean obtained from the expression ratios of the two control genes, pvsA and pvuA, was used each time for each temperature to normalize the raw expression levels of the genes of interest. Thus, the hemolysin gene tdh2 and the TTSS2 genes escU, vopP, and spa24 showed no significant up-regulation when the bacteria were recovering culturability after a temperature upshift to 20 or 37°C (Fig. 2A and B). Indeed, P values were close to 1 for each virulence gene. Nevertheless, the rpoS gene showed significant changes in regulation under these treatment conditions. When the temperature was increased to 20°C, this gene was up-regulated approximately 50 and 800 times at days 2 and 6, respectively (Fig. 2A). This gene was also up-regulated 170 times at day 2 when the temperature increased to 37°C (Fig. 2B). However, it was down-regulated at day 6 when the temperature increased to 37°C (Fig. 2B). After temperature upshifts to 20 and 37°C, the VBNC population recovered culturability in 2 days (Fig. 2C and D). The culturability was maintained for 18 days at 20°C (Fig. 2C) and decreased after 6 days at 37°C (Fig. 2D).
FIG. 2.

Mean relative expression ratios for five genes in ASW after temperature upshift. The temperature of ASW was increased from 4°C to 20°C (A and C) or 37°C (B and D). The levels of expression after 2 days (D2) and 6 days (D6) of temperature upshift were computed and were normalized according to the geometric mean of results obtained for two control genes, pvsA and pvuA. The data are the normalized means ± standard deviations of the results for two runs, each with three replicate samples. *, significant results (P < 0.05).
Detection of gene expression in ASW at 4°C.
The clinical strain V. parahaemolyticus Vp5 was rendered VBNC by incubation in ASW at 4°C. The VBNC state was reached at day 15, and no culturable bacteria were detected from day 15 until day 106 (data not shown). The detection of mRNA was performed nine times during the 106 days of the experiment. The housekeeping genes rpoS, pvsA, fur, and pvuA and the virulence genes tdh2, escU, vopP, and spa24 were detected in the culturable cells (day 0 to day 15) and in the VBNC bacteria (day 16 to day 106) in ASW at 4°C (data not shown). The amount of time taken for the mRNA population to decrease by half was 14.6, 22.6, 17.9, and 19.7 days for the mRNA of rpoS, pvsA, fur, and pvuA, respectively, and 13.2, 17.3, 16.7, and 16.5 days for the mRNA of tdh2, escU, vopP, and spa24, respectively (Fig. 3).
FIG. 3.

Transcriptional activity in V. parahaemolyticus Vp5 in ASW at 4°C. Quantification of the mRNA copy numbers for the rpoS, fur, pvsA, pvuA, tdh2, escU, vopP, and spa24 genes was performed by real-time RT-PCR. The number of days necessary to decrease the mRNA population by half (T½) is indicated for each gene. Sample graphs depicting the reduction in the mRNA for a housekeeping gene (rpoS; panel A) and a virulence gene (tdh2; panel B) are shown.
Half-lives of mRNA species in ASW at 4°C.
The half-lives of the various mRNA species in the Vp5 strain in ASW at 4°C were computed. After the addition of rifampin, no culturable bacteria were detected during the experiment (data not shown). The mRNA copies were quantified on each of the next 7 days, and the half-lives were determined to be 3.5, 4.3, 5.1, 3.1, 3.3, 2.1, 2.5, and 3.0 days for rpoS, fur, pvsA, pvuA, tdh2, escU, vopP, and spa24 mRNA, respectively (Fig. 4).
FIG. 4.

Half-lives of mRNA species of V. parahaemolyticus Vp5 in ASW at 4°C. The table summarizes the half-lives calculated for mRNA for eight genes, i.e., rpoS, fur, pvsA, pvuA, tdh2, escU, vopP, and spa24. Sample graphs representing the quantities of mRNA for a housekeeping gene (rpoS; panel A) and a virulence gene (tdh2; panel B) are shown. ♦, MS2 RNA was used as an internal control for detecting potential inhibitors of quantitative RT-PCR in RNA samples. •, mRNA copies were quantified on days 1, 2, 3, 4, 5, 6, and 7 after a rifampin treatment which blocked the transcriptional activity in the clinical strain V. parahaemolyticus Vp5. The data are expressed as the means ± standard deviations (SD) of results from experiments performed with three replicate samples.
DISCUSSION
The relationship of TTSS to the pathogenicity of many gram-negative bacteria is now well accepted (14). A TTSS named TTSS2, present on the small chromosome 2 of the clinical strain V. parahaemolyticus RIMD 2210633, has been described previously (18). Additionally, Park et al. (24) examined the hemolytic, cytotoxic, and enterotoxic activities of mutant strains (tdh1 negative and/or tdh2 negative) and established that the hemolytic activity should be caused exclusively by TDH and that the cytotoxic and enterotoxic activities should be caused by both TDH and other factors of virulence. The same authors confirmed these results and specified that TTSS2 is involved in enterotoxic activities of V. parahaemolyticus (25). Dziejman et al. (8) suggested that the pathogenicity of non-O1/non-O139 V. cholerae should be caused by TTSS genes related to the TTSS2 gene cluster found in the pandemic strain V. parahaemolyticus RIMD 2210633. The potential motility of the gene cluster corresponding to the TTSS and its implication in the pathogenicity of several species identifies TTSS2 as an interesting target for studying the pathogenicity of V. parahaemolyticus. We thus developed specific primers and probes to detect the expression of TTSS2 genes in V. parahaemolyticus by real-time RT-PCR.
In vitro, we observed no significant up-regulation of the virulence gene tdh2 and of the genes for TTSS2 in the bacteria after the recovery of culturability. These results are in agreement with those of Baffone et al. (1). Indeed, these authors observed that virulence characteristics disappeared after resuscitation in the mouse and were subsequently reactivated by means of two consecutive passages of V. parahaemolyticus in the rat ileal loop model. The pathogenicity of VBNC cells in vivo has been studied often. Indeed, VBNC V. cholerae cells have previously proven to maintain virulence in human volunteers after ingestion (6), VBNC V. vulnificus cells maintain virulence in a murine model after intraperitoneal injections (23), VBNC V. parahaemolyticus is virulent in a murine model after ingestion (1), and VBNC Campylobacter jejuni is virulent against embryonated eggs (5). However, these studies did not investigate if this virulence was the result of the recovery of culturability and the growth of the cells, with pathogenicity depending also on the infective dose, or if it was caused by an up-regulation of the virulence genes. Previous studies have demonstrated that VBNC bacteria maintain the expression of virulence genes, e.g., a hemolysin gene (vvhA) in V. vulnificus (9, 30, 35), cholera toxin genes (ctxAB) in V. cholerae, and a TDH gene (tdh) in V. parahaemolyticus (35). In our study, we confirmed in vitro that the virulence genes were expressed in VBNC bacteria but not induced in these cells after the recovery of culturability. However, short transitory inductions may be possible, as demonstrated with the yopH and yopE genes in Yersinia spp. after temperature upshifts (10, 17, 19, 20). Nevertheless, contact with eukaryotic cells appears to be necessary for a strong induction of the transcriptional level. This point was made previously for P. aeruginosa, in which the following environmental signals induce the TTSS: (i) the in vitro removal of calcium from the medium, (ii) in vivo contact with eukaryotic host cells, and (iii) the presence of serum (11, 33, 37). The transcriptional induction of the TTSS genes appears also to be a mechanism down-regulated by the transcriptional regulator sigma S, encoded by the rpoS gene, in enterohemorrhagic E. coli and P. aeruginosa (15, 29). Our results confirm that when the rpoS gene is highly expressed, no transcription of the TTSS genes is observed. We also observed a repression of rpoS expression before a decrease in the rate of survival of V. parahaemolyticus cells in ASW at 37°C. Thus, the RpoS factor may be involved in the maintenance of viability of the bacteria in an oligotrophic environment, as described previously for E. coli, Salmonella enterica serovar Typhimurium, and V. cholerae (21, 39).
RT-PCR, which targets short-lived mRNA molecules, has become an increasingly used molecular method for assessing the viability of bacteria, culturable or not. We developed a real-time RT-PCR for the detection of pathogenic strains of V. parahaemolyticus in a VBNC state. In this study, mRNA specific to V. parahaemolyticus was detected in ASW at 4°C and was confirmed to be the result of de novo RNA synthesis. Indeed, the amounts of time taken to decrease the quantity of mRNA by half were four to eight times higher than the mRNA half-lives, indicating transcriptional activity in V. parahaemolyticus in ASW at 4°C. In our study, we found that the rpoS mRNA half-life was 3.5 days. These results are different from previous findings, which indicated that the half-life of rpoS mRNA in V. vulnificus is less than 60 min (30). However, a number of experimental and biological variations can explain this discrepancy. To our knowledge, this is the first time that half-lives have been determined using multiple mRNA populations under conditions which induce the VBNC state. Because of the observed differences in mRNA half-lives, caution is needed when measuring the expression of more than one gene. In previous studies, the average half-life of E. coli mRNA was determined to be 1 min, with a range of 40 s to 20 min and with 80% of all mRNA half-lives computed to be between 3 and 8 min (2, 3). The mRNA turnover appears to have a large impact on transcript levels (16). However, the half-lives were not assessed under conditions which produce the VBNC state, i.e., in ASW at 4°C. Thus, the viability and the gene expression appear to be difficult to confirm because mRNA detected in the VBNC samples may result from residual mRNA and not from de novo RNA synthesis. Thus, one should take this into account when reporting the results obtained in previous studies on the expression of housekeeping (7, 13, 38) and pathogenicity (9, 30, 35) genes in VBNC populations. Indeed, the detection of mRNA in an early population of VBNC cells should be associated with a study of the mRNA half-lives under the same conditions to ensure the viability of the bacterial population.
In conclusion, a real-time RT-PCR method using virulence and housekeeping genes of V. parahaemolyticus was developed and used to study the expression of these genes. After the recovery of culturability, the VBNC cells of V. parahaemolyticus Vp5 did not significantly induce the transcription of the main virulence genes in the environment. mRNAs proved to be good viability markers, but their half-lives, a few days at 4°C, must be considered for the development of experiments and for the interpretation of real-time RT-PCR data. These first results suggest that V. parahaemolyticus needs specific conditions to induce virulence factors. In the future, verification at the translational level could be performed and the possible induction of the virulence genes in the presence of either nutrients or host cells at a favorable temperature could be investigated.
Acknowledgments
This work was financed by the French Research Institute for Exploitation of the Sea (IFREMER) and the Région Pays de la Loire.
We gratefully acknowledge Jonathan Porter (Environment Agency, Starcross, United Kingdom) and Françoise S. Le Guyader (IFREMER, Nantes, France) for critical reading of the manuscript and Jacques Le Pendu (INSERM, Nantes, France) for his advice on scientific aspects of the project.
Footnotes
Published ahead of print on 8 June 2007.
REFERENCES
- 1.Baffone, W., B. Citterio, E. Vittoria, A. Casaroli, R. Campana, L. Falzano, and G. Donelli. 2003. Retention of virulence in viable but non-culturable halophilic Vibrio spp. Int. J. Food Microbiol. 89:31-39. [DOI] [PubMed] [Google Scholar]
- 2.Baracchini, E., and H. Bremer. 1987. Determination of synthesis rate and lifetime of bacterial mRNAs. Anal. Biochem. 167:245-260. [DOI] [PubMed] [Google Scholar]
- 3.Bernstein, J. A., A. B. Khodursky, P. H. Lin, S. Lin-Chao, and S. N. Cohen. 2002. Global analysis of mRNA decay and abundance in Escherichia coli at single-gene resolution using two-color fluorescent DNA microarrays. Proc. Natl. Acad. Sci. USA 99:9697-9702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bustin, S. A. 2000. Absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assays. J. Mol. Endocrinol. 25:169-193. [DOI] [PubMed] [Google Scholar]
- 5.Cappelier, J. M., J. Minet, C. Magras, R. R. Colwell, and M. Federighi. 1999. Recovery in embryonated eggs of viable but nonculturable Campylobacter jejuni cells and maintenance of ability to adhere to HeLa cells after resuscitation. Appl. Environ. Microbiol. 65:5154-5157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Colwell, R. R., P. R. Brayton, D. Herrington, B. Tall, S. A. Huq, and M. M. Levine. 1996. Viable but non-culturable Vibrio cholerae O1 revert to a cultivable state in the human intestine. World J. Microbiol. Biotechnol. 12:28-31. [DOI] [PubMed] [Google Scholar]
- 7.Coutard, F., M. Pommepuy, S. Loaec, and D. Hervio-Heath. 2005. mRNA detection by reverse transcription-PCR for monitoring viability and potential virulence in a pathogenic strain of Vibrio parahaemolyticus in viable but nonculturable state. J. Appl. Microbiol. 98:951-961. [DOI] [PubMed] [Google Scholar]
- 8.Dziejman, M., D. Serruto, V. C. Tam, D. Sturtevant, P. Diraphat, S. M. Faruque, M. H. Rahman, J. F. Heidelberg, J. Decker, L. Li, K. T. Montgomery, G. Grills, R. Kucherlapati, and J. J. Mekalanos. 2005. Genomic characterization of non-O1, non-O139 Vibrio cholerae reveals genes for a type III secretion system. Proc. Natl. Acad. Sci. USA 102:3465-3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fischer-Le Saux, M., D. Hervio-Heath, S. Loaec, R. R. Colwell, and M. Pommepuy. 2002. Detection of cytotoxin-hemolysin mRNA in nonculturable populations of environmental and clinical Vibrio vulnificus strains in artificial seawater. Appl. Environ. Microbiol. 68:5641-5646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Forsberg, A., and H. Wolf-Watz. 1988. The virulence protein Yop5 of Yersinia pseudotuberculosis is regulated at transcriptional level by plasmid-plB1-encoded trans-acting elements controlled by temperature and calcium. Mol. Microbiol. 2:121-133. [DOI] [PubMed] [Google Scholar]
- 11.Frank, D. W. 1997. The exoenzyme S regulon of Pseudomonas aeruginosa. Mol. Microbiol. 26:621-629. [DOI] [PubMed] [Google Scholar]
- 12.Funahashi, T., K. Moriya, S. Uemura, S. I. Miyoshi, S. Shinoda, S. Narimatsu, and S. Yamamoto. 2002. Identification and characterization of pvuA, a gene encoding the ferric vibrioferrin receptor protein in Vibrio parahaemolyticus. J. Bacteriol. 184:936-946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gonzalez-Escalona, N., A. Fey, M. G. Hofle, R. T. Espejo, and C. A. Guzman. 2006. Quantitative reverse transcription polymerase chain reaction analysis of Vibrio cholerae cells entering the viable but non-culturable state and starvation in response to cold shock. Environ. Microbiol. 8:658-666. [DOI] [PubMed] [Google Scholar]
- 14.Hueck, C. J. 1998. Type III protein secretion in bacterial pathogens of animals and plants. Microbiol. Mol. Biol. Rev. 62:379-433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iyoda, S., and H. Watanabe. 2005. ClpXP protease controls expression of the type III protein secretion system through regulation of RpoS and GrlR levels in enterohemorrhagic Escherichia coli. J. Bacteriol. 187:4086-4094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kushner, S. R. 2002. mRNA decay in Escherichia coli comes of age. J. Bacteriol. 184:4658-4665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lambert de Rouvroit, C., C. Sluiters, and G. R. Cornelis. 1992. Role of the transcriptional activator, VirF, and temperature in the expression of the pYV plasmid genes of Yersinia enterocolitica. Mol. Microbiol. 6:395-409. [PubMed] [Google Scholar]
- 18.Makino, K., K. Oshima, K. Kurokawa, K. Yokoyama, T. Uda, K. Tagomori, Y. Iijima, M. Najima, M. Nakano, A. Yamashita, Y. Kubota, S. Kimura, T. Yasunaga, T. Honda, H. Shinagawa, M. Hattori, and T. Iida. 2003. Genome sequence of Vibrio parahaemolyticus: a pathogenic mechanism distinct from that of V. cholerae. Lancet 361:743-749. [DOI] [PubMed] [Google Scholar]
- 19.Michiels, T., P. Wattiau, R. Brasseur, J. M. Ruysschaert, and G. Cornelis. 1990. Secretion of Yop proteins by yersiniae. Infect. Immun. 58:2840-2849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Michiels, T., J. C. Vanooteghem, C. Lambert de Rouvroit, B. China, A. Gustin, P. Boudry, and G. R. Cornelis. 1991. Analysis of virC, an operon involved in the secretion of Yop proteins by Yersinia enterocolitica. J. Bacteriol. 173:4994-5009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Munro, P. M., G. N. Flatau, R. L. Clement, and M. J. Gauthier. 1995. Influence of the RpoS (KatF) sigma factor on maintenance of viability and culturability of Escherichia coli and Salmonella typhimurium in seawater. Appl. Environ. Microbiol. 61:1853-1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.O'Connell, K. P., J. R. Bucher, P. E. Anderson, C. J. Cao, A. S. Khan, M. V. Gostomski, and J. J. Valdes. 2006. Real-time fluorogenic reverse transcription-PCR assays for detection of bacteriophage MS2. Appl. Environ. Microbiol. 72:478-483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oliver, J. D., and R. Bockian. 1995. In vivo resuscitation, and virulence towards mice, of viable but nonculturable cells of Vibrio vulnificus. Appl. Environ. Microbiol. 61:2620-2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Park, K. S., T. Ono, M. Rokuda, M. H. Jang, K. Okada, T. Iida, and T. Honda. 2004. Cytotoxicity and enterotoxicity of the thermostable direct hemolysin-deletion mutants of Vibrio parahaemolyticus. Microbiol. Immunol. 48:313-318. [DOI] [PubMed] [Google Scholar]
- 25.Park, K. S., T. Ono, M. Rokuda, M. H. Jang, K. Okada, T. Iida, and T. Honda. 2004. Functional characterization of two type III secretion systems of Vibrio parahaemolyticus. Infect. Immun. 72:6659-6665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pfaffl, M. W. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29:2002-2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pfaffl, M. W., G. W. Horgan, and L. Dempfle. 2002. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 30:e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY.
- 29.Shen, D. K., D. Filopon, L. Kuhn, B. Polack, and B. Toussaint. 2006. PsrA is a positive transcriptional regulator of the type III secretion system in Pseudomonas aeruginosa. Infect. Immun. 74:1121-1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smith, B., and J. D. Oliver. 2006. In situ and in vitro gene expression by Vibrio vulnificus during entry into, persistence within, and resuscitation from the viable but nonculturable state. Appl. Environ. Microbiol. 72:1445-1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tanabe, T., T. Funahashi, H. Nakao, S. I. Miyoshi, S. Shinoda, and S. Yamamoto. 2003. Identification and characterization of genes required for biosynthesis and transport of siderophore vibrioferrin in Vibrio parahaemolyticus. J. Bacteriol. 185:6938-6949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tortell, P. D., M. T. Maldonado, J. Granger, and N. M. Price. 1999. Marine bacteria and biogeochemical cycling of iron in the oceans. FEMS Microbiol. Ecol. 29:1-11. [Google Scholar]
- 33.Vallis, A. J., V. Finck-Barbancon, T. L. Yahr, and D. W. Frank. 1999. Biological effects of Pseudomonas aeruginosa type III-secreted proteins on CHO cells. Infect. Immun. 67:2040-2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vandesompele, J., K. De Preter, F. Pattyn, B. Poppe, N. Von Roy, A. De Paepe, and F. Speleman. 2002. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 3:research0034.1-research0034.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vora, G. J., C. E. Meador, M. M. Bird, C. A. Bopp, J. D. Andreadis, and D. A. Stenger. 2005. Microarray-based detection of genetic heterogeneity, antimicrobial resistance, and the viable but nonculturable state in human pathogenic Vibrio spp. Proc. Natl. Acad. Sci. USA 102:19109-19114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Watnick, P. I., T. Eto, H. Takahashi, and S. B. Calderwood. 1997. Purification of Vibrio cholerae Fur and estimation of its intracellular abundance by antibody sandwich enzyme-linked immunosorbent assay. J. Bacteriol. 179:243-247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yahr, T. L., A. K. Hovey, S. M. Klich, and D. W. Frank. 1995. Transcriptional analysis of the Pseudomonas aeruginosa exoenzyme S structural gene. J. Bacteriol. 177:1169-1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yaron, S., and K. R. Matthews. 2002. A reverse transcriptase polymerase chain reaction assay for detection of viable Escherichia coli O157:H7: investigation of specific target genes. J. Appl. Microbiol. 92:633-640. [DOI] [PubMed] [Google Scholar]
- 39.Yildiz, F. H., and G. K. Schoolnik. 1998. Role of rpoS in stress survival and virulence of Vibrio cholerae. J. Bacteriol. 180:773-784. [DOI] [PMC free article] [PubMed] [Google Scholar]