

Abstract
Four rhesus macaques were inoculated intravenously with a cryopreserved stock of microglia obtained from a simian immunodeficiency virus (SIV)-infected rhesus macaque. Before infection, three of the four monkeys were trained and tested daily on a computerized neuropsychological test battery. After SIV infection, behavioral testing continued to monitor deficits associated with disease progression. Five additional age-matched, behaviorally trained monkeys served as controls. Neurophysiological testing for visual and auditory evoked responses was accomplished 37–52 weeks after infection in all monkeys. Subsequently, all four SIV-infected monkeys and one control subject were sacrificed, and samples of brain tissue were taken for pathological analysis. SIV-infected monkeys demonstrated abnormal responses in both auditory and visual evoked responses. In addition, around the time of electrophysiological recording, all three SIV-infected, behaviorally trained monkeys exhibited significant decreases in progressive-ratio performance, reflecting a reduction in reinforcer efficacy. One subject also demonstrated impairments in shifting of attentional set and motor ability at that time. Neuropathological evaluation revealed that all four SIV-infected monkeys exhibited numerous perivascular and parenchymal infiltrating T cells. These findings document that SIV causes electrophysiological, behavioral, and neuropathological sequelae similar to what has been observed in the human neuroAIDS syndrome. Our observations further validate the simian model for the investigation of the pathogenesis of AIDS dementia and for the investigation of drugs with potential therapeutic benefits.
Keywords: evoked potentials, behavioral performance, pathogenesis
HIV infects the central nervous system (CNS) early in the time course of the infection (1). Although the virus resides and can be detected in the CNS, and in spite of the fact that neuronal cell loss has been reported, neurons do not seem to become directly infected with virus (2). However, neuronal pathology may result from toxicity elicited indirectly by viral products derived from the HIV envelope or from a host-derived immune response to infection (3, 4, 5). Moreover, several neurophysiological variables are impaired in infected patients; for example, sleep patterns, visual, auditory, and somatosensory evoked potentials, as well as event-related potentials, are reported to be altered after HIV-1 infection (6, 7, 8, 9, 10). A dementia associated with AIDS affects 15–20% of patients and consists of a clinical triad, including progressive cognitive decline, motor dysfunction, and behavioral abnormalities (4, 11, 12). More generally, neurological disorders are found in 30–60% of those patients with advanced HIV disease (13). Associations have been reported between neurological and neuropsychological abnormalities and between a more rapid progression of cognitive deterioration and death (14).
Simian immunodeficiency virus (SIV) is one member of a family of primate lentiviruses, the closest relatives of which are both HIV-1 and HIV-2 (15, 16). Experimental infection of SIV into rhesus monkeys produces a progressive immunodeficiency syndrome that is remarkably similar to AIDS (15, 16, 17). Once inoculated, SIV readily infects CD4+ T cells and tissue macrophages, as HIV does in humans. SIV invades the brain soon after the experimental intravenous infection (18, 19, 20, 21). Infectious virus and a virus-specific host immune response can be detected in the cerebrospinal fluid as early as 1 week after the infection (21, 22). In addition, systematic studies seeking to characterize the neuropathological changes caused by SIV have established remarkable similarities with the alterations produced by HIV-1 in human brains (23). To improve our understanding of the pathogenesis of the AIDS-associated dementia caused by HIV in humans, we have investigated electrophysiological parameters in the SIV-infected rhesus monkey as an animal model that closely reproduces several clinical/neurological manifestations of the human disease.
MATERIALS AND METHODS
Subjects.
Nine adult male rhesus monkeys free of type D simian retroviruses and herpes B virus were obtained from an isolated primate colony (Key Lois, Charles River Breeding Laboratories). Experimental animals (n = 4) were infected with a cryopreserved stock of microglia from an SIV-infected rhesus monkey that constituted the second round, in vivo, of serial microglia passage. We have reported that this serial passage results in a neuropathogenic strain of SIV (derived from the SIVmac251 strain) (24, 25).
Infection and bleeding were performed under ketamine anesthesia (10 mg/kg, i.m.). Peripheral blood mononuclear cells were separated from plasma from heparinized blood samples using Histopaque (Sigma). Peripheral blood mononuclear cells were stored in a 10% dimethyl sulfoxide solution in liquid nitrogen, and plasma samples were stored at −70°C until analysis. The presence of plasma SIV p27 gag antigen was detected using a commercially available ELISA (Coulter). Virus was recovered from peripheral blood mononuclear cell samples using phytohemagglutinin stimulation as described (24).
Electrophysiological Analysis.
Cortical visual evoked potentials and brainstem cortical, as well as auditory, evoked potentials were evaluated in all nine monkeys. Electrophysiological studies were accomplished during anesthesia (ketamine, 20 mg/kg, i.m.), between 37 and 52 weeks after infection with SIV. Anesthetized monkeys were aseptically fitted with subcutaneous monopolar needle electrodes (TECA Intropak DMG-50, Vickers Medical, Pleasantville, NY). One active electrode was placed at the cranial vertex. One electrode placed s.c. in the nose over the greater alar cartilage was used as reference. An electrical ground electrode was inserted into the musculature of the neck. To minimize superficial dermal penetrations in the infected monkeys, the same s.c. electrode array was used for both auditory and visual modalities.
Recording.
Raw electroencephalographic signals were amplified by a Grass 12A5 ac amplifier (Grass Instruments, Quincy, MA) and filtered within a bandwidth of 30–3000 Hz for brainstem auditory evoked potentials (BSAEP) and 1–100 Hz for visual (VEP) and cortical auditory evoked potentials (AEP). A computer program written using National Instruments Software (labview) for the Macintosh II microcomputer was used to generate the stimuli and to average, on-line, the electroencephalographic signals. BSAEPs were obtained by averaging 1024 samples of electroencephalograms obtained during the first 10 msec after the stimulus delivery. For VEP and AEP, samples were obtained during the first 200 msec after stimuli delivery, and a total of 100 samples were averaged.
Auditory stimulation.
Binaural condensation stimulation, produced by clicks generated by 0.10-msec square waves and delivered at a 70-dB sound pressure level at a repetition rate of 10 clicks/sec, was used to generate BSAEP. A frequency of 1/sec was used to generate AEPs. Clicks were amplified by a Grass Instruments Audio Amplifier. Environmental white noise was at a 40-dB sound pressure level.
Visual stimulation.
For binocular stimulation, a Grass clinical photostimulator was used to deliver flashes (0.1 msec duration) with the photostimulator intensity set at 16. The flash lamp was placed 30 cm from the nose of the monkey. Ambient lights remained on.
Analysis.
Several waves were determined by visual inspection in all evoked potential modalities. The latency of these waves from the stimulus artifact to the apex of each selected wave, as well as peak-to-trough amplitude, was calculated off-line by using custom-made labview software. Statistical differences between groups were determined by using a Student’s t test.
Behavioral Testing.
Three of four infected monkeys were trained on a computerized test battery (Cambridge Neuropsychological Test Automated Battery, Paul Fray, United Kingdom) adapted for use with monkeys from a similar battery that has been used to test HIV+ patient populations (26). Baseline data were collected before inoculation with SIV-infected microglia. Shifting of attentional set was measured the week after electrophysiological recording (monkey 193) using an intradimensional/extradimensional shift task consisting of four stages (simple visual discrimination, compound discrimination, intra- and extradimensional shifts, and reversals; ref. 27). Reinforcer efficacy was probed at the end of each week using a progressive-ratio schedule, in which the number of responses required to obtain a food pellet was increased progressively throughout the session. The progressive-ratio task was run using two slightly different schedules. In one case, the ratio requirement increased by one response per reinforcer for the first eight reinforcers, then increased by two responses per reinforcer for the next eight reinforcers, and so on, incrementing by 4, 8, 16, etc., for each eight reinforcers (monkeys 231 and 193). In the second case, each ratio was simply increased by two throughout the session (monkey 187). Motor coordination was monitored with a bimanual motor task daily after computerized testing. In this task, the monkey was timed for its ability to use both hands to retrieve raisins from a holeboard mounted perpendicular to the door of the transport cage. Behavioral testing was suspended the day of electrophysiological testing.
Histology.
For necropsy, the animals were maintained at a near lethal level of anesthesia and then transcardially perfused with cold PBS. To examine the integrity of the blood–brain barrier, Evans blue dye (3 ml/kg of a 2% solution) was infused aseptically into the saphenous vein of one anesthetized monkey 30 min before necropsy. The brain was removed and divided sagittally, with one-half sectioned at 5-mm intervals, immersion-fixed in 10% neutral-buffered formalin, and embedded in paraffin. Sections (6 μm) were stained with hematoxylin and eosin for histopathological analysis, and indirect immunocytochemical staining was performed after trypsin digestion using a rabbit polyclonal antibody directed against CD3 (Dako) to identify T cells, and a mouse monoclonal antibody against the leukocyte antigen L1 (MAC387, Dako) to identify reactive macrophages.
RESULTS
Electrophysiological Responses.
Sensory evoked responses were obtained in all nine monkeys at the time corresponding to 37–52 weeks after infection. Except for one (monkey 231), SIV subjects were tested at least twice. No significant inter-test variability was observed. Data presented in Figs. 1, 2, 3 are averaged values taken from 40 or 44 weeks after infection. Five waves were identified in the short latency BSAEP complex by visual inspection, P1–P5. The BSAEP from SIV-infected monkeys showed no latency changes in the first three waves (P1–P3; Fig. 1), but showed a significant delay in the latency of waves P4 and P5. The intervals P1–P4 and P1–P5 were significantly longer in the infected monkeys (P < 0.05). Amplitude of the BSAEP complex was, in general, smaller in SIV-infected monkeys than in control monkeys; however, this change did not reach levels of statistical significance.
Figure 1.

BSAEPs. (a) Average of control (n = 5) versus SIV-infected (n = 4) monkeys. Significant delay of the waves P4 and P5 from SIV-infected monkeys is depicted in these traces. (b) Actual values, in milliseconds, of the latencies of the different waves measured from the stimulus artifact (time = 0). (c) Interpeak latencies. Note that waves P1–P4 and P1–P5 show significant delays in the SIV-infected monkeys. ∗, P < 0.05.
Figure 2.
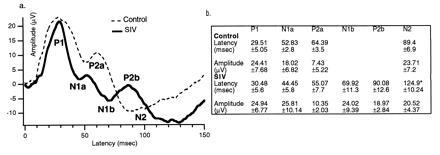
AEPs. (a) AEPs obtained from control (n = 5) and SIV-infected (n = 4) monkeys. The AEPs from SIV-infected monkeys demonstrate a more complex trace. This trace depicts the appearance of more waves and a delay in the latency of N2. (b) Actual values for the latencies of each wave in milliseconds, measured from the stimulus artifact (time = 0). ∗, P < 0.05.
Figure 3.
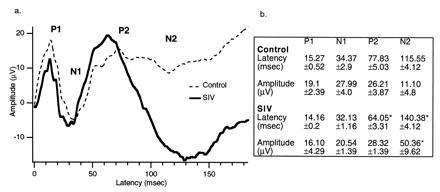
VEPs. (a) VEPs from SIV-infected monkeys (n = 4) compared with controls (n = 5). The latencies obtained from SIV-infected monkeys are shorter than the VEPs from the control group. This figure also illustrates a large amplitude N2 wave uniquely appearing in the trace of the SIV monkeys. (b) Values of the latencies of both groups measured from the stimulus artifact (time = 0). ∗, P < 0.05.
The long-latency cortical AEPs from SIV-infected monkeys demonstrated waveforms dissimilar to the waves obtained from the control group (Fig. 2). Two positive (P1 and P2) and two negative (N1 and N2) salient peaks were identified in the AEPs’ complex from the control monkeys. However, in the complex obtained from SIV monkeys, the second positive peak (P2) appeared to be split into two components: one with shorter and one with longer latency than the P2 in the control monkeys AEPs. This difference resulted in the N2 wave exhibiting a prolonged latency. The amplitude of the waves was similar between infected and control groups. Regarding the long-latency cortical VEP, two positive (P1 and P2) and two negative (N1 and N2) peaks were identified. VEPs from SIV-infected monkeys exhibited a P2 wave with shorter latency compared with the control group (Fig. 3). In addition, wave N2 appeared with a significantly higher amplitude and prolonged latency in the SIV-infected monkeys (Fig. 3). Table 1 illustrates the relationship between these electrophysiological findings and concurrent behavioral and subsequent pathological assessment described below.
Table 1.
Assessment of neurophysiology, coincident behavioral impairment, clinical condition at sacrifice, and pathology for four SIV-infected subjects
| Monkey | Neurophysiology | Behavioral impairment | Clinical condition at sacrifice | Neuropathology |
|---|---|---|---|---|
| 187 | 44, 46 weeks | Reinforcer efficacy 37 → | 46; no disease | Infiltrating T cells |
| 193 | 44, 52 weeks | Reinforcer efficacy 44, 50 → | 71; progressive gingivitis | Infiltrating T cells |
| Attentional shifting 45 Motor 44, 57–60 | Perivascular Mø Disrupted BBB | |||
| 231 | 40 weeks | Reinforcer efficacy 36 → | 41; weight loss, colitis, pneumonia | Infiltrating T cells |
| Infiltrating Mø | ||||
| 232 | 40, 52 weeks | Not assessed | 67; lymphoma of the left orbit and lung | Infiltrating T cells |
Numbers indicate the weeks after infection for each measure, and arrows indicate sustained impairment until the termination of behavioral testing. Cohort control monkeys (not shown) 225–228 were neurophysiologically tested with SIV-infected monkeys on week 40 after infection. An additional control monkey (monkey 178) was neurophysiologically tested with SIV-infected monkeys on week 44 after infection. Neuropathological assessment of monkey 178 revealed no significant pathological changes. BBB, blood–brain barrier; Mø, macrophage.
Behavioral Testing.
Performance in the behavioral tasks was evaluated for the period surrounding electrophysiological recording (Table 1). A full description of the behavioral profile throughout the course of SIV infection will be described in a subsequent paper. SIV-infected monkeys exhibited significant behavioral impairment measured in the progressive-ratio task during the week of electrophysiological testing. In all three monkeys, performance, measured either by last ratio completed or number of reinforcers earned, had declined to a level below 2 SDs of preinfection performance in this task. In one case (monkey 193), deficits in shifting of attentional set and motor coordination were also observed for the week coincident with or following electrophysiological recording.
Pathological Findings.
All four experimental animals became productively infected by SIV after intravenous injection of cryopreserved microglia. Plasma viremia (SIV p27 gag antigen) was first detected 10 days after infection in all animals and was undetectable from 4 weeks after infection through sacrifice in the three behaviorally trained animals (monkeys 187, 193, and 231). Monkey 232 was viremic at 10 days after infection and cleared the viremia by 4 weeks; however, this animal showed intermittent evidence of antigen through the course of infection (at 17, 31, 44, and 67 weeks after infection). In addition, virus could be recovered from the peripheral blood mononuclear cells of all four infected monkeys at all times tested after infection. One of the animals (monkey 231) was sacrificed at 41 weeks after infection (8 days after the recording) due to progressive weight loss (10% of body weight) secondary to Pneumocystis carinii pneumonia and Balantidium coli colitis. This was the only monkey with an AIDS illness near the time of study. At 46 weeks after infection, another monkey (monkey 187) was sacrificed, but showed no symptoms of SIV-induced disease. A third monkey (monkey 232) was sacrificed at 67 weeks after infection due to an extranodal lymphoma, involving the left orbit and lung, but not the CNS. The last SIV-infected monkey (monkey 193) was sacrificed at 71 weeks after infection due to a progressive gingivitis.
The neuropathological examination showed that all four SIV-infected monkeys showed perivascular and parenchymal infiltrating T cells in the brain, as evidenced by immunocytochemical staining for the CD3 antigen (Fig. 4). Additionally, monkeys 193 and 231 had appreciable numbers of perivascular macrophages, with infiltrating macrophages noted only in monkey 231. Neither the T cell nor macrophage infiltrates showed any consistent pattern of involvement of specific brain regions. Finally, the brain of monkey 193, the only animal examined by the Evans blue technique for leakage of the blood–brain barrier, showed one discrete area of a compromised blood–brain barrier in the temporal lobe, which histopathologically did not differ from other regions of the brain. One control monkey brain (monkey 178) was similarly examined for neuropathology and was observed to be pathologically unremarkable. Table 1 illustrates the relationship of the time course of measured dependent variables in the three components of this study.
Figure 4.

Photomicrograph of immunocytochemical staining for T cells in the brainstem of SIV-infected monkey 232. Lymphocytes immunoreactive for CD3 stain darkly, and can be found both perivascularly and infiltrating the parenchyma.
DISCUSSION
This study provides further evidence that SIV invades the CNS after intravenous infection and produces changes in brain physiology, behavioral performance, and CNS tissue integrity. The occurrence of parallel behavioral and physiological alterations in animals infected with microglia-passaged SIV further support the ability of this virus to induce SIV-related CNS disease (See Table 1).
Human neuropsychological symptoms of HIV infection include forgetfulness, loss of concentration, mental inflexibility, and mental and psychomotor slowing (28, 29). One advantage of the Cambridge Neuropsychological Test Automated Battery is that data collected in monkeys can be directly compared with data collected in human patients. A study of HIV-infected patients (symptomatic and asymptomatic) tested using this battery (26) revealed impairments in the ability to shift attentional set similar to that observed in the monkeys after SIV infection. These results reflect an inability to redirect attention toward a dimension of the stimuli that was previously uncorrelated with reward and that has been shown to involve frontal lobe function (27, 30). In addition, infected monkeys also exhibited significant reductions in progressive-ratio responding over the course of SIV infection, and one (monkey 193) exhibited decreased bimanual motor coordination at the time of electrophysiological testing. Disruptions in progressive-ratio performance measured in short probe sessions result from decreases in overall rates of responding and can be interpreted to reflect both motor slowing and a reduction in motivation to work for food. Cognitive and motor impairments have also been reported in monkeys infected with a different strain of SIV (31). A convergence of evidence from electrophysiological and neuropsychological measurements will help to fully characterize the neurological changes associated with SIV infection.
The electrophysiological findings we observed are comparable to those described in humans. For example, the BSAEP from SIV monkeys show a delay in waves P4 and P5, as reported in HIV+ subjects (6, 8, 9, 10). SIV-infected monkeys also exhibited generally smaller-amplitude evoked potentials compared with controls. There is evidence indicating that waves P4 and P5 are generated by the upper brainstem in monkeys as well as in humans (32, 33, 34). Therefore, this finding suggests a functional abnormality of the upper level of the auditory pathway. Interestingly, we have also observed similar alterations in BSAEP waves P4 and P5 in another animal model of AIDS, the feline immunodeficiency virus-infected cat (35). This abnormality indicates a common pattern in which virus infection leads to a functional pathophysiology of the brainstem.
Regarding the long-latency cortical AEPs, these waveforms showed more complexity in the SIV-infected monkeys than in the normal controls. Cortical VEPs exhibited a P2 wave occurring with significantly shorter latency in the SIV-infected monkeys compared with controls. The visually evoked N2 wave developed a significantly larger amplitude and was delayed compared with controls, similar to previously recognized so-called giant potentials observed in varied human clinical disease states (36). For example, the appearance of more waves in the entire evoked complex, shortening of latency, or giant potentials has been observed in progressive myoclonic, photosensitive, and petit mal epilepsy, as well as in subacute encephalopathies, including Creutzfeldt–Jakob disease and Lafora body disease (36, 37, 38). All these diseases have in common a theme of central hyperexcitability. Indeed, in related animal studies, topical application of bicuculline [a γ-aminobutyric acid (GABA) antagonist] or the excitatory amino acid transmitter glutamate directly to the visual cortex of cats or rats produces similar shortened latencies of the VEPs and can induce comparable giant potentials (39, 40). These data suggest that the changes observed in the AEP and VEP of the SIV-infected rhesus may be due to a viral-induced cortical hyperexcitability that may have specific neuropharmacological correlates.
As the neuropathogenesis in neuroAIDS has also been associated with overfunction of excitatory amino acids such as glutamate (41), SIV may be similarly pathogenic. Although our evoked potentials were obtained under anesthesia, a state that potentially could mask the hyperactivity of the cortex, the observation of multiple-wave AEPs and shorter-latency VEPs in the infected group suggests that anesthesia is not a confounding factor. Moreover, impairment in other neurochemical systems as well, such as GABAergic (40) and cholinergic (42) systems, can also produce the effects we observed.
Unlike BSAEP and VEPs, AEPs have not, to our knowledge, been recorded in HIV+ subjects; therefore, it is not possible to compare our observations with human findings. However, our results suggest that AEPs may be a sensitive measure for detecting pathophysiological changes induced by HIV in humans. Hence, these findings would strongly encourage the analysis of AEPs in HIV+ and AIDS patients.
Our experimental observations do not establish how early in the time course of the disease these electrophysiological changes take place. Our studies focused on monkeys about 11 months after SIV infection, but the abnormalities may be present much earlier. Indeed, feline immunodeficiency virus-infected cats display abnormal AEPs as early as 1 month after infection with a feline-specific microglia-derived virus stock (S.J.H., unpublished results), and at 3 months after infection with a molecularly cloned feline immunodeficiency virus inoculate (43).
Taken together, these data indicate that SIV infection produces neurological abnormalities detectable by sensitive neuropsychological and electrophysiological tests. Although the neuropathological assessment did not indicate consistent or substantial damage in brain areas predicted by these tests (e.g., the cerebral cortex and upper brainstem), it does not rule out a functional abnormality. We have identified SIV-specific cytotoxic T lymphocytes in the CNS of SIV-infected monkeys (22). These T cells were present in the cerebrospinal fluid as early as 1 week after infection and could be found in the brain at autopsy. In the animals studied here, although cytotoxic T lymphocyte assays were not performed, the presence of T cells in the brains was a consistent pathological finding. T cells and their products can directly or indirectly damage neurons, and may contribute to the functional deficits noted in this study.
Future experiments will disclose how early in the disease the electrophysiological changes can be detected. The early detection of these abnormalities will provide a unique opportunity for assaying drugs to prevent or at least delay neurological damage.
Acknowledgments
We thank Amanda Ormsby for technical assistance. This work was supported by National Institutes of Health Grants P50-MH47680 to F.E.B. and MH55836 to H.S.F. This is publication No. 10222-NP from The Scripps Research Institute.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: CNS, central nervous system; SIV, simian immunodeficiency virus; BSAEP, brainstem auditory evoked potential; VEP, visual evoked potential; AEP, auditory evoked potential.
References
- 1.Davis L E, Hjelle B L, Miller V E, Palmer D L, Llewellyn A L, Merlin T L, Young S A, Mills R G, Wachsman W, Wiley C A. Neurology. 1992;42:1736–1739. doi: 10.1212/wnl.42.9.1736. [DOI] [PubMed] [Google Scholar]
- 2.Everall I P, Luthert P, Lantos P. J Neuropathol Exp Neurol. 1993;52:561–566. doi: 10.1097/00005072-199311000-00002. [DOI] [PubMed] [Google Scholar]
- 3.Glass J D, Wesselingh S L, Selnes O A, McArthur J C. Neurology. 1993;43:2230–2237. doi: 10.1212/wnl.43.11.2230. [DOI] [PubMed] [Google Scholar]
- 4.Power C, Johnson R T. Can J Neurol Sci. 1995;22:92–100. doi: 10.1017/s0317167100040154. [DOI] [PubMed] [Google Scholar]
- 5.Brew B J, Rosenblum M, Cronin K, Price R W. Ann Neurol. 1995;38:563–570. doi: 10.1002/ana.410380404. [DOI] [PubMed] [Google Scholar]
- 6.Bankaitis A E. Ear Hear. 1995;16:321–324. doi: 10.1097/00003446-199506000-00009. [DOI] [PubMed] [Google Scholar]
- 7.Darko D F, Mitler M M, Henriksen S J. Adv Neuroimmunol. 1995;5:55–77. doi: 10.1016/0960-5428(94)00044-o. [DOI] [PubMed] [Google Scholar]
- 8.Hausler R, Vibert D, Koralnik I J, Hirschel B. Acta Otolaryngol. 1991;481:515–521. doi: 10.3109/00016489109131461. [DOI] [PubMed] [Google Scholar]
- 9.Pagano M A, Cahn P E, Grarau M L, Mangone C A, Figini H A, Yorio A A, Dellepiane M C, Amores M G, Perez H M, Casiro A D. Arch Neurol. 1992;49:166–169. doi: 10.1001/archneur.1992.00530260068022. [DOI] [PubMed] [Google Scholar]
- 10.Pierelli F, Soldati G, Zambardi P, Garrubba C, Spadaro M, Tilia G, Pauri F, Morocutti C. Acta Neurol Belg. 1993;93:78–87. [PubMed] [Google Scholar]
- 11.Price R W. In: HIV, AIDS and the Brain. Price R W, Perry S W, editors. New York: Raven; 1994. pp. 1–45. [Google Scholar]
- 12.Everall I P. J Neurol Neurosurg Psychiatry. 1995;58:399–402. doi: 10.1136/jnnp.58.4.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McArthur J C, Selnes O A, Glass J D, Hoover D R, Bacellar H. In: HIV, AIDS and Brain. Price R W, Perry S W, editors. Vol. 72. New York: Raven; 1994. pp. 251–272. [Google Scholar]
- 14.Stern Y, Liu X, Marder K, Todak G, Sano M, Ehrhardt A, Gorman J. Neurology. 1995;45:467–472. doi: 10.1212/wnl.45.3.467. [DOI] [PubMed] [Google Scholar]
- 15.Desrosiers R. Annu Rev Immunol. 1990;8:557–578. doi: 10.1146/annurev.iy.08.040190.003013. [DOI] [PubMed] [Google Scholar]
- 16.Kestler H, Kodama T, Ringler D, Marthas M, Pedersen N, Lackner A, Regier D, Sehgal P, Daniel M, King N, Desrosiers R. Science. 1990;248:1109–1112. doi: 10.1126/science.2160735. [DOI] [PubMed] [Google Scholar]
- 17.Kindt T, Hirsch V, Johnson P, Sawasdikosol S. Adv Immunol. 1992;52:425–473. doi: 10.1016/s0065-2776(08)60880-9. [DOI] [PubMed] [Google Scholar]
- 18.Chakrabarti L, Hurtrel M, Maire M A, Vazeux R, Dormont D, Montagnier L, Hurtrel B. Am J Pathol. 1991;139:1273–1280. [PMC free article] [PubMed] [Google Scholar]
- 19.Sharer L R, Michaels J, Murphey-Corb M, Hu F S, Kuebler D J, Martin L N, Baskin G B. J Med Primatol. 1991;20:211–217. [PubMed] [Google Scholar]
- 20.Lackner A A, Smith M O, Munn R J, Martfeld D J, Gardner M B, Marx P A, Dandekar S. Am J Pathol. 1991;139:609–621. [PMC free article] [PubMed] [Google Scholar]
- 21.Smith M O, Heyes M P, Lacker A A. Lab Invest. 1995;72:547–555. [PubMed] [Google Scholar]
- 22.von-Herrath M, Oldstone M B A, Fox H S. J Immunol. 1995;154:5582–5589. [PubMed] [Google Scholar]
- 23.Simon M A, Chalifoux L V, Ringler D J. AIDS Res Hum Retroviruses. 1992;8:327–337. doi: 10.1089/aid.1992.8.327. [DOI] [PubMed] [Google Scholar]
- 24.Watry D, Lane T E, Steb M, Fox H S. Am J Pathol. 1995;146:914–923. [PMC free article] [PubMed] [Google Scholar]
- 25.Lane T E, Buchmeier M J, Watry D D, Jakubowski D B, Fox H S. Virology. 1995;212:458–465. doi: 10.1006/viro.1995.1503. [DOI] [PubMed] [Google Scholar]
- 26.Sahakian B J, Elliot R, Low N, Mehta M, Clark R T, Pozniak A L. Psychol Med. 1995;25:1233–1246. doi: 10.1017/s0033291700033201. [DOI] [PubMed] [Google Scholar]
- 27.Roberts A C, Sahakian B J. In: Behavioral Neuroscience: A Practical Approach. Sahgal A, editor. New York: Oxford Univ. Press; 1993. pp. 165–184. [Google Scholar]
- 28.Navia B A, Jordan B D, Price R W. Ann Neurol. 1986;19:517–524. doi: 10.1002/ana.410190602. [DOI] [PubMed] [Google Scholar]
- 29.Robertson K, Hall C. Semin Neurol. 1992;12:18–27. doi: 10.1055/s-2008-1041153. [DOI] [PubMed] [Google Scholar]
- 30.Dias R, Robbins T W, Roberts A C. Nature (London) 1996;380:69–72. doi: 10.1038/380069a0. [DOI] [PubMed] [Google Scholar]
- 31.Murry E, Rausch D, Lendvay J, Sharer L, Eiden L. Science. 1992;255:1246–1249. doi: 10.1126/science.1546323. [DOI] [PubMed] [Google Scholar]
- 32.Hashimoto I, Ihiyama Y, Yoshimoto T, Nemoto S. Brain. 1981;104:841–859. doi: 10.1093/brain/104.4.841. [DOI] [PubMed] [Google Scholar]
- 33.Legatt A D, Arezzo J C, Hebert G, Vaughan J. Electroencephalogr Clin Neurophysiol. 1986;64:53–73. doi: 10.1016/0013-4694(86)90043-x. [DOI] [PubMed] [Google Scholar]
- 34.Møller A R, Burgess J. Electroencephalogr Clin Neurophysiol. 1986;65:361–372. doi: 10.1016/0168-5597(86)90015-8. [DOI] [PubMed] [Google Scholar]
- 35.Phillips T R, Prospéro-García O, Puaoi D L, Lerner D L, Fox H S, Olmsted R A, Bloom F E, Henriksen S J, Elder J H. J Gen Virol. 1994;75:979–987. doi: 10.1099/0022-1317-75-5-979. [DOI] [PubMed] [Google Scholar]
- 36.Mauguiere F, Holder G E, Luxon L M, Pottinger R C. In: Clinical Neurophysiology. Binnie C D, Cooper R, Fowler C J, Mauguiere F, Prior P F, editors. Cambridge, U.K.: The University Press; 1995. pp. 431–481. [Google Scholar]
- 37.Shibasaki H, Yamashita Y. In: Evoked Potentials. Cracco R, Bodis-Wollner I, editors. New York: Liss; 1986. pp. 402–408. [Google Scholar]
- 38.Shibasaki H, Yamashita Y, Neshige R, Tobimatsu S, Fukui R. Brain. 1985;108:225–240. doi: 10.1093/brain/108.1.225. [DOI] [PubMed] [Google Scholar]
- 39.Cohn R, Xu S, Wagner H G, Joo F, Klatzo I. Neurol Res. 1992;14:248–251. doi: 10.1080/01616412.1992.11740063. [DOI] [PubMed] [Google Scholar]
- 40.Zemon V, Kaplan E, Ratliff F. In: Evoked Potentials. Cracco R, Bodis-Wollner I, editors. New York: Liss; 1986. pp. 287–295. [Google Scholar]
- 41.Lipton S A. In: HIV, AIDS and Brain. Price R W, Perry S W, editors. New York: Raven; 1994. pp. 183–202. [Google Scholar]
- 42.Kirby A W, Wiley R W, Harding T H. In: Evoked Potentials. Cracco R, Bodis-Wollner I, editors. New York: Liss; 1986. pp. 296–306. [Google Scholar]
- 43.Phillips, T. R., Prospéro-García, O., Wheeler, D. W., Wagaman, P. C., Lerner, D. L., Fox, H. S., Whalen, L. R., Bloom, F. E., Elder, J. H. & Henriksen, S. J. (1996) J. NeuroVirology, in press. [DOI] [PubMed]