

Abstract
Bdellovibrio bacteriovorus is a species of unique obligate predatory bacteria that utilize gram-negative bacteria as prey. Their life cycle alternates between a motile extracellular phase and a growth phase within the prey cell periplasm. The mechanism of prey cell invasion and the genetic networks and regulation during the life cycle have not been elucidated. The obligate predatory nature of the B. bacteriovorus life cycle suggests the use of this bacterium in potential applications involving pathogen control but adds complexity to the development of practical genetic systems that can be used to determine gene function. This work reports the development of a genetic technique for allelic exchange or gene inactivation by construction of in-frame markerless deletion mutants including the use of a counterselectable marker in B. bacteriovorus. A suicide plasmid carrying the sacB gene for counterselection was used to inactivate the strB gene in B. bacteriovorus HD100 by an in-frame deletion. Despite the inactivation of the strB gene, B. bacteriovorus was found to retain resistance to high concentrations of streptomycin. The stability of a plasmid for use in complementation experiments was also investigated, and it was determined that pMMB206 replicates autonomously in B. bacteriovorus. Development of this practical genetic system now facilitates the study of B. bacteriovorus at the molecular level and will aid in understanding the regulatory networks and gene function in this fascinating predatory bacterium.
Bdellovibrio is a small, motile gram-negative bacterium with an obligate predatory life cycle that utilizes other gram-negative bacteria in the predation process. The Bdellovibrio life cycle consists of two phases: a free-swimming extracellular attack phase and an intraperiplasmic phase in which the bacteria replicate by filamentous fragmentation in the periplasm of the prey cell. The growth phase ends with lysis of the prey cell by the Bdellovibrio progeny, which enter the free-swimming phase ready to invade other prey cells. Bdellovibrio is different from other predatory bacteria in that it enters the prey cell but remains in the periplasm. After the Bdellovibrio bacterium penetrates the prey cell, the prey cell remains intact but becomes rounded in shape. This prey cell, containing the growing Bdellovibrio bacterium, has been termed a bdelloplast. Interestingly, some strains of Bdellovibrio can generate mutant cells that are able to grow axenically and are referred to as host independent (1, 5). Bdellovibrio preys on a variety of other gram-negative bacteria, some of which are pathogens and thus have potential applications in either environmental or medical settings (19, 25, 29). The development of practical molecular genetic techniques that can be used for research involving gene function may lead to the discovery of novel antibacterial enzymes in this fascinating bacterium.
Despite the growing interest in this unique bacterium for potential applications, the mechanism of prey cell invasion and the genetic networks and regulation during the life cycle, as well as the mechanism underlying the phenotype change to host independence, are not understood. This lack of understanding of almost all aspects of Bdellovibrio is due in large part to the scarcity of work at the molecular level that has been reported. The obligate predatory nature of Bdellovibrio adds some complexity to the development of a feasible system for genetic manipulation to determine gene function in these bacteria. However, given the possible applications involving use of either live Bdellovibrio bacteria or enzymes encoded by the Bdellovibrio genome, it is important to continue to expand the repertoire of useful genetic tools for studying this bacterium at the molecular level.
Genetic work with Bdellovibrio was pioneered in 1992 by Cotter and Thomashow, who transferred plasmids to Bdellovibrio bacteriovorus strain 109J by using conjugal matings and determined that IncQ plasmids are autonomously replicated in B. bacteriovorus but that some IncP plasmids, including pVK100, are not (5). They then proceeded to clone an open reading frame (ORF) from B. bacteriovorus 109J into pVK100, which they then integrated by homologous recombination into the chromosomal DNA of the host-independent form of the B. bacteriovorus 109J strain harboring a mutation in that ORF (6). Phenotype characterization was carried out using the merodiploid, and no attempt to select for excisants was reported. Despite the integration of the wild-type gene in these merodiploids, they never returned to the fully wild-type phenotype that would be expected if an allelic exchange method had been available for use instead. It was over 10 years later, in 2003, that further molecular genetic work with Bdellovibrio was reported. Building on the earlier results showing that the IncP plasmid pVK100 cannot autonomously replicate in B. bacteriovorus and thus can be used as a suicide vector, Lambert and coworkers used the plasmid pSET151, which also has the IncP origin of replication, as a suicide vector (14). The plasmid pSET151 was used to independently inactivate the mcp2 and mviN genes by insertion of alleles disrupted with a kanamycin (Km) resistance cassette on the chromosome of B. bacteriovorus strain 109J. Knockout mutants were obtained by screening for loss of the wild-type gene without the use of counterselection. This same method was used later to inactivate flagellin genes in B. bacteriovorus 109J (13).
Recently, the complete genome sequence of B. bacteriovorus strain HD100 was reported (19). With this genome sequence now available, a combination of bioinformatics and transcriptional analysis during different times in the Bdellovibrio life cycle can suggest specific ORFs that might be associated with a particular function of interest. Reverse genetic approaches can be used to determine gene function, and furthermore, in studying genes associated with the invasion process, development of genetic methods that can be used with Bdellovibrio to determine gene essentiality will become important. Thus far, chromosomal knockouts have been limited to insertion of a Km resistance cassette, and these mutations are not reported to be in-frame; therefore, downstream polar effects might have some effect on phenotype analysis. Furthermore, validation of phenotype change due solely to mutations in specific genes by complementation is an important issue to be addressed for Bdellovibrio. As mentioned, Cotter and Thomashow demonstrated that several IncQ plasmids replicate autonomously in B. bacteriovorus (5). This result suggests that these plasmids can be used for complementation experiments. In fact, more recent work by others made use of this information, whereupon pMMB206 (15), a plasmid with an IncQ origin of replication, was found to replicate autonomously in B. bacteriovorus and was used in RNA interference experiments to reduce expression of the MotA protein in B. bacteriovorus 109J (10). Despite another recent report that there is no suitable vector for complementation in B. bacteriovorus (13), we have been successful in maintaining pMMB206 extrachromosomally in both the HD100 and the B610 strains of B. bacteriovorus for over 4 months. The apparent discrepancy among reports pertaining to the stability of pMMB206 in B. bacteriovorus may be due to the low copy number of the plasmid.
Although gene inactivation by antibiotic cassette insertion was an important step forward, if a deeper understanding of the Bdellovibrio life cycle at the molecular level is to be gained, a genetic system in which markerless in-frame chromosomal deletion mutants can be constructed and complemented must be developed. In addition, for some experiments it may be desirable to replace the wild-type allele with a mutant allele rather than completely knock out a gene; thus, a means of allelic exchange rather than insertion inactivation is important. The evaluation of possible counterselectable markers that can be successfully used in Bdellovibrio bacteria has not been reported. One method of counterselection widely used with gram-negative bacteria is detection of sucrose sensitivity in the presence of the Bacillus subtilis sacB gene coding for levansucrase (20). This method of counterselection has been found to be useful for several other environmental bacteria, including Geobacter (4), Desulfovibrio (11), and Myxococcus (28) bacteria. However, Leptospira, another environmental bacterium, was found to be sensitive to sucrose; thus, counterselection using the sacB gene was not possible (18).
In the present work, we describe a practical technique for allelic exchange or gene inactivation by in-frame deletion and the use of a counterselectable marker in B. bacteriovorus. In choosing a gene to inactivate, only genes known to be nonessential for cell viability were considered. The strB gene, which is known to confer streptomycin (Sm) resistance in other bacteria (27), seems a logical choice since it is not essential for survival unless bacterial cultures are grown in Sm. Use of the sacB gene was found to be a valuable method for selecting double recombinants in B. bacteriovorus. A suicide plasmid, pSSK10, which is derived from pDS132 (17) by addition of a Km resistance cassette and which carries the sacB gene for counterselection and the RK6 origin of replication, was constructed. Using our constructed suicide plasmid, we have inactivated the strB gene in B. bacteriovorus strain HD100 by an in-frame deletion, showing for the first time in Bdellovibrio the ability to make markerless deletion mutants and to use a counterselectable marker. In addition, the stability of a plasmid for use in complementation experiments was investigated, and it was determined that pMMB206 replicates autonomously in B. bacteriovorus.
MATERIALS AND METHODS
Bacterial strains and plasmids.
Experiments reported in this work used B. bacteriovorus strain HD100 (gi 42494925) and strain B610 (7). The Escherichia coli strains and plasmids used are listed in Table 1. E. coli DH5αλpir or S17-1λpir was used for cloning manipulations involving the suicide plasmids, and either E. coli SM10λpir or E. coli S17-1λpir was used for conjugal matings since the plasmids used are based on the R6K origin of replication, thus requiring the product of the λpir gene for replication.
TABLE 1.
Escherichia coli strains and plasmids used in this study
| Strain or plasmid | Relevant features | Reference or source |
|---|---|---|
| E. coli strains | ||
| DH5α | F− ϕ80lacZΔM15 Δ(lacZYA-argF)U169 deoR recA1 endA1 hsdR17(rk− mk+) phoA supE44 λ−thi-1 gyrA96 relA1 | Gibco-BRL |
| ML35 | lacI lacY | 21 |
| DH5αλpir | DH5α with λpir lysogen | M. Donnenberg |
| SM10λpir | thiL thrL leuB6 supE44 tonA21 lacY1 recA::RP4-2-Tc::Mu Kmr | 23 |
| S17-1λpir | pro recA thi hsdR Hfr RP4-2 (Tc::Mu) (Km::Tn7) Smr Tpr λpir lysogen | 24 |
| Plasmids | ||
| pDS132 | R6K ori mobRP4 cat sacB | 17 |
| pCR2.1-TOPO | Source of Kmr | Invitrogen |
| pSSK10 | Derived from pDS132; insertion of Kmr along with NdeI, MluI, and XhoI cloning sites | This work |
| pSSK20 | ΔstrB cloned into pSSK10 | This work |
| pMMB206 | IncQ lacIq Ptaclac lacZα rrnB cat mobRP4 | 15 |
| pMMB0307 | pMMB206 with strB cloned | This work |
| pACYC184 | Cmr | New England Biolabs |
Media, growth conditions, and conjugation.
B. bacteriovorus HD100 was grown in liquid cocultures along with E. coli prey by using dilute nutrient broth (DNB) consisting of 0.8 g/liter nutrient broth (Difco) supplemented with 2 mM CaCl2 and 3 mM MgCl2. Wild-type HD100 was typically maintained using E. coli ML35, and Km-resistant HD100 was grown in broth supplemented with Km (25 μg ml−1), with E. coli SM10λpir as prey. To obtain isolated plaques, HD100 was plated using the double overlay method (14). Liquid cocultures were shaken and grown at room temperature, and plates were incubated at 30°C. Km (50 μg ml−1) was added to both the top and the bottom agar for selection of merodiploid HD100. DNB medium prepared without the additional salts and supplemented with 5% sucrose was used for selection of chromosomal deletion mutants. All E. coli strains were grown at 37°C in Luria-Bertani (LB) media supplemented with Km (50 μg ml−1), Sm (50 μg ml−1), or chloramphenicol (Cm; 30 μg ml−1) when appropriate. The suicide plasmid and the plasmid pMMB206 were transferred by conjugation to HD100 by using E. coli S17-1λpir and SM10λpir donor cells, respectively, according to the conjugal mating procedure previously described for B. bacteriovorus (5). Briefly, 12 ml of donor cells grown to the top of log phase was pelleted and resuspended in 100 μl DNB. Cocultures grown in 50 ml DNB were filtered and pelleted as described above, and the B. bacteriovorus cells were resuspended in 100 μl DNB and then spread on a nitrocellulose filter placed on a peptone yeast agar plate. The donor cells were then spread on the B. bacteriovorus cells, and the plate was incubated at 30°C for 22 h. After the mating, the filter was vortexed in 2 ml DNB and appropriate dilutions were plated in DNB supplemented with Km (50 μg ml−1) by using the double overlay method described above.
Cloning, PCR, and RT-PCR.
Pfu Turbo DNA polymerase (Stratagene) was used for all cloning and sequencing PCRs. A QIAprep spin mini prep kit (QIAGEN) or a plasmid midi kit (QIAGEN) was used for plasmid extractions. Bacterial pellets for both HD100 RNA and genomic DNA extractions were prepared free of contaminating E. coli prey cells by first filtering the cocultures through a 0.8-μm syringe filter and then two times through 0.45-μm syringe filters, which allow the smaller B. bacteriovorus cells but not the E. coli prey cells to pass. The absence of prey cells in the filtrate, as well as the absence of donor cells after experiments involving conjugation, was confirmed by incubation of an aliquot of the filtrate on LB media overnight. DNA was extracted from B. bacteriovorus pellets by using a DNeasy tissue kit (QIAGEN). All sequences were obtained with primers indicated in Table 2. Specifically, the strB gene was sequenced from genomic HD100 DNA by using primers Bd0307F and Bd0307R, and the rpsL gene was sequenced using primers Bd2981F and Bd2981R. All sequencing reactions were carried out using a BigDye Terminator cycle sequencing kit (Applied Biosystems) and an ABI 3730xl automatic sequencer. For PCR screening, reaction mixtures of B. bacteriovorus in cocultures with puRe Taq ready-to-go PCR beads (Amersham Biosciences) were used. For these reaction mixtures, 1.6 ml of clear coculture was pelleted in a microcentrifuge tube at maximum speed for 30 min at 4°C. The pellet was resuspended in 100 μl H2O, and the cell suspension was boiled for 5 min, after which 10 μl was used in a 25-μl-total-volume PCR mixture. With all sets of primers used, the absence of a PCR product for only E. coli prey cells confirms amplification of only B. bacteriovorus DNA in the pellets prepared from the cocultures. RNA was stabilized in the filtered B. bacteriovorus cultures before pellet preparation using RNAprotect bacterial reagent (QIAGEN), and RNA was extracted from the pellets using an RNeasy kit (QIAGEN). Possible contaminating DNA was removed by RQ1 RNase-free DNase (Promega) treatment followed by a cleanup with the RNeasy kit. Absence of DNA was determined by lack of PCR product by using universal primers for the 16S rRNA gene and HD100 DNA as a positive control. Two primers for the strB gene, RTStrBF (outside the deleted region in the ΔstrB mutant) and RTStrBR (within the deleted region in the ΔstrB mutant), were designed, along with two primers for the rpsL gene, rpsLF2 and rpsLR, as a load control. These primer sets were tested using both HD100 and E. coli ML35 DNA to show specificity for HD100 by the absence of a PCR product for the E. coli DNA. Reverse transcription (RT)-PCR was performed with 200 ng of total RNA extracted from wild-type HD100 and the HD100ΔstrB mutant by using a OneStep RT-PCR kit (QIAGEN), along with E. coli ML35 RNA as a control for further confirmation of the absence of prey cell RNA contributing to the RT-PCR product from the extracted HD100 RNA.
TABLE 2.
Primers used in this work
| Primer | Sequence (5′ to 3′)a |
|---|---|
| Bd0307F | GCCAATCTGGATCAAGGTC |
| Bd0307R | GCTGTTTTCTGTCAGTGGTC |
| Bd2981F | GAAATGTAAGTTCCAGCGTC |
| Bd2981R | AACTTAGCAATTACTAGATCCTTG |
| PTPKRF | ATAAGAGCTCCTCGAGACGCGTCATATGGGACAAGGGAAAACGCAAG |
| PTPKRR | ACTAGAGCTCCGACACGGAAATGTTGAATACTC |
| PDS132F | CTGTTGCATGGGCATAAAG |
| PDS132R | AGGAACACTTAACGGCTGAC |
| STRBF1 | ATAGTAACATATGGCAAAGTGGCGGAGCTGAC |
| STRBR1 | AATAGCATGCCGTCAGATCCCAGCGCAG |
| STRBF2 | TGATGCATGCTCTGCAGCCTGGAGCGTG |
| STRBR2 | ATACTCGAGAGTAACGGTACTGGGGCGTC |
| SBF | TATTAACCTTTACTACCGCACTG |
| SBR | GTCCTTGTTCAAGGATGCTG |
| P206F | ATGATAGCCTACGAGACAGCAC |
| P206R | GCTGCGCTAGGCTACACAC |
| P206R2 | TTGAGAATATGTTTTTCGTCTCAG |
| SBcomR | TATCCCGGGATCCATTGAGCGTTGGTG |
| SBcomF | TATAAGCTTCCAGAGGATTTTCATTCAAC |
| PB206F | CGTATGTTGTGTGGAATTGTGAG |
| PB206R | GATTAAGTTGGGTAACGCCAG |
| RTStrBF | ATTTTCCCGCACCGTTC |
| RTStrBR | GCAAGAAGTCACGCACATG |
| rpsLF2 | CATCAAAAGTGAGCGTACTG |
| rpsLR | CGCAGACGACCGTTTACAC |
Restriction enzyme sites are underlined.
Construction of the suicide plasmid pSSK10.
A suicide vector for making deletion mutants of B. bacteriovorus was constructed by insertion of a Km resistance cassette along with additional unique restriction enzyme sites into pDS132, a suicide vector with the R6K origin of replication that carries the cat gene for selection of integrants and the sacB gene for counterselection of excisants (17). The Km resistance cassette including the promoter was amplified from pCR2.1-TOPO by using primers PTPKRF and PTPKRR, which include SacI sites. Primer PTPKRF also includes unique sites NdeI, MluI, and XhoI, which can be used for cloning. The PCR-amplified fragment containing the Km resistance cassette was blunt end ligated into the unique SacI site in pDS132, resulting in the plasmid pSSK10. The orientation of the Km resistance cassette was determined by digestion and confirmed by sequencing using primers PDS132F and PDS132R.
Construction of HD100 in-frame strB deletion mutant.
The strB gene (locus tag Bd0307), which is known to confer Sm resistance in other bacteria (27), was knocked out by allelic exchange using an in-frame deletion cloned on the suicide plasmid pSSK10. Construction of the strB knockout suicide vector pSSK20 was accomplished by triple ligation of the two flanking regions with the pSSK10 plasmid digested with NdeI and XhoI. One set of primers, STRBF1 and STRBR1, was designed to include the first 40 amino acids of the StrB protein, and a second set of primers, STRBF2 and STRBR2, was used to produce a PCR product including the 28 C-terminal amino acids. These two PCR products were each ∼700 bp and had an SphI site included on the primers within the strB gene that was used for the triple ligation. The correct construction was confirmed by sequencing. E. coli S17-1λpir harboring pSSK20 was used as a donor for conjugation into the recipient HD100. The exconjugates were plated with E. coli SM10λpir prey on plates containing no antibiotic, in addition to the plates containing Km (50 μg ml−1), to determine the frequency of integration of the suicide plasmid. Eight of the plaques grown under Km selection were picked and transferred to liquid cocultures in DNB supplemented with Km (25 μg ml−1). Upon clearance, the presence of merodiploid HD100 in the cocultures was confirmed by PCR using the same outer flanking primers that had been used for cloning. Further corroboration of the integration of the suicide plasmid was obtained by PCR amplification of the sacB gene using primers SBF and SBR. An aliquot of a merodiploid culture was pelleted, the pellet was washed with DNB, and then the cells were added to fresh DNB with no antibiotic and E. coli ML35 prey. This coculture was grown for 48 h to allow for excision of the suicide plasmid by a second homologous recombination event. An aliquot of this coculture was transferred to DNB broth containing 5% sucrose and fresh prey cells to select for excisants. After clearing of the cocultures, dilutions were plated and 40 plaques were picked into DNB broth with fresh prey. These cocultures were then screened by PCR for knockout mutants using the outer flanking primers STRBF1 and STRBR2. The DNA was extracted from one of the B. bacteriovorus strB knockout mutants for confirmation of the correct in-frame deletion of the strB gene on the chromosome by sequencing.
Confirmation of the stability of pMMB206 in B. bacteriovorus.
Two B. bacteriovorus strains, HD100 and B610, were used for experiments to confirm the autonomous replication of pMMB206 in B. bacteriovorus. The plasmid pMMB206 was transferred to HD100 and B610 by conjugation using E. coli SM10λpir donor cells as described earlier (5). E. coli SM10λpir made Cm resistant by transformation with pACYC184 was used as prey for plating the exconjugates. A single plaque of exconjugate HD100 or B610 was picked and grown in liquid media supplemented with Cm (10 μg ml−1). DNA was extracted from the Cm-resistant B. bacteriovorus after filtration to remove the prey and analyzed by PCR for the presence of the plasmid pMMB206. Two sets of primers that give PCR product sizes of 514 bp and 1,323 bp with pMMB206 as a template but do not result in PCR products from the plasmid pACYC184 were used. These primer sets consisted of one forward primer, P206F, paired with two different reverse primers, P206R and P206R2. Wild-type HD100 or B610 DNA was used as a negative control. In order to definitively confirm that the B. bacteriovorus bacteria were carrying pMMB206 extrachromosomally, the extracted B610 DNA was used for transformation into E. coli DH5α. The HD100 strain harboring pMMB206 was transferred weekly and filtered numerous times for 4 months in DNB containing Cm (10 μg ml−1). To rule out the possibility that any donor E. coli SM10 λpir cells were still present in the cocultures even after the filtering process, the HD100 strain carrying pMMB206 was transferred twice in DNB media containing ampicillin (Amp; 100 μg ml−1) in addition to the Cm. DNA was then extracted from the B. bacteriovorus bacteria and used for transformation into E. coli DH5α. Following each of the transformations, a colony was grown and the plasmid was extracted. The size of the extracted plasmid was compared to that of pMMB206 by gel electrophoresis, and the extracted plasmid was employed as a template for PCR, using the primers described above that are specific for the pMMB206 plasmid.
Cloning of the strB gene on pMMB206.
The strB gene, along with 236 bp upstream, was amplified from the wild-type HD100 genomic DNA by PCR using primers SBcomF and SBcomR, which included SmaI and HindIII restriction sites for directional cloning into the plasmid pMMB206, to create pMMB0307. This construction was transformed into E. coli DH5α, and after confirmation of the correct sequence using primers PB206F and PB206R, located on either side of the cloning site, a clone was tested for Sm resistance. The plasmid pMMB0307 carries the cat gene and the IPTG (isopropyl-β-d-thiogalactopyranoside)-inducible Ptaclac promoter upstream of the cloned fragment. Therefore, the HD100 StrB protein function in the E. coli DH5αpMMB0307 clone was tested on LB plates supplemented with Cm, 0.1 mM IPTG (Amresco), and Sm at either 0, 5, 10, or 50 μg ml−1.
RESULTS
Construction of the knockout suicide plasmid pSSK20.
Several clones resulting from the use of blunt-end ligation of the Km resistance cassette into pDS132 to derive pSSK10 were screened for the orientation of the inserted fragment. All clones had the same orientation, which is shown in Fig. 1; thus, this plasmid construction was used to clone for the strB knockout. The shortage of restriction enzyme sites available for cloning on pDS132 was at least partly alleviated by insertion of the additional NdeI, MluI, and XhoI sites cloned with the Km resistance cassette into pSSK10. Two of these sites were subsequently used for cloning the flanking regions for the in-frame deletion. The construction results in a deletion of 231 amino acids from the 299-amino-acid StrB protein.
FIG. 1.

Construction of the suicide plasmid pSSK10. The plasmid pSSK10 was derived from pDS132 by addition of the Kmr cassette from pCR2.1-TOPO and unique restriction enzyme sites NdeI, MluI, and XhoI. The PCR product containing the Kmr cassette was obtained with primers having terminal SacI sites and was ligated into pDS132 at the unique SacI site.
Construction of HD100 in-frame strB deletion mutant.
A comparison of the numbers of plaques obtained from exconjugates plated with and without Km selection showed that integration of the suicide plasmid took place at a frequency of about 10−6. Since it was desired to use the sacB gene for counterselection of excisants, wild-type HD100 was tested for sucrose sensitivity and then for the best concentration of sucrose to use for the counterselection. Concentrations of 2%, 5%, and 10% sucrose (wt/vol) were screened. Although B. bacteriovorus bacteria were found to be viable in 10% sucrose, the coculture takes significantly longer to clear. Counterselection was, therefore, attempted using 5% sucrose plates but resulted in plaques corresponding to predominantly merodiploids. As a result, selection of excisants was accomplished in this work using liquid 5% sucrose media rather than sucrose plates. With this method, the cocultures were observed to clear in 4 days (typically, cocultures of HD100 clear in 2 days when transferred using DNB media with no sucrose). Of the 40 plaques screened via PCR after sucrose selection, only one merodiploid was found, thus showing that sucrose counterselection is an efficient method for use with B. bacteriovorus. Further substantiating this method of counterselection for B. bacteriovorus, during the selection process in constructing a deletion mutant of a different gene, over 120 plaques were screened for the second homologous crossover event and all were found to be excisants (S. R. Steyert and S. A. Pineiro, unpublished results). In the present work, 26 of the 40 plaques screened by PCR were found to contain wild-type revertant B. bacteriovorus and 13 corresponded to strB knockout mutants. An agarose gel representative of the results of the PCR screening for 19 of the plaques is shown in Fig. 2.
FIG. 2.

PCR screening of exconjugate HD100. PCR products obtained using primers STRBF1 and STRBR2 (outer primers used for cloning the flanking regions of the strB gene deletion into the suicide vector) were visualized in agarose gels. This gel, one of several, shows 19 of the 40 plaques screened and includes 13 wild-type revertants, 1 merodiploid (lane 13), and 5 knockout mutants (lanes 1, 4, 6, 14, and 18). Lane 11 contains a molecular size marker.
Confirmation of the strB gene in-frame deletion.
DNA from one of the HD100ΔstrB mutants was extracted and sequenced to confirm that the deletion was in-frame as constructed. As further confirmation of the deletion, RT-PCR products can be seen for the strB gene during the free-swimming extracellular stage in the life cycle in wild-type HD100 but not in the HD100ΔstrB mutant. Figure 3 shows the presence of a transcription product in the wild-type HD100 isolate (Fig. 3, lane 2), in contrast to the mutant for which there is no product (Fig. 3, lane 3). In addition, the HD100ΔstrB mutant was tested for Sm sensitivity but was found to retain resistance to high concentrations of Sm (>2,000 μg ml−1). Since the strB gene was shown by RT-PCR to be transcribed in wild-type HD100, however, we questioned the functionality of the StrB protein. The DNA sequence of the strB gene in the HD100 isolate from our culture collection (EF493834) was compared to the strB gene sequence of the published HD100 genome (19) and was found to have 12 nucleotide differences. Some of these base pair changes are silent mutations, but the protein sequence does have four amino acid changes. Presumably, these mutations have occurred during propagation of HD100 in the laboratory under nonselective conditions. It is worth noting that the 16S rRNA gene sequence, as well as sequences of other loci from this strain, shows 100% identity with the GenBank sequences (data not shown). The functionality of the StrB protein in our HD100 strain was tested with E. coli DH5α carrying the plasmid pMMB0307, which contains the HD100 strB gene and promoter region. It was found to be sensitive to Sm concentrations of 5 μg ml−1; therefore, the strB gene did not appear to confer Sm resistance in E. coli and thus presumably not in B. bacteriovorus either. The high level of Sm resistance that the strB deletion mutant was found to retain suggested the possibility of a mutation in the rpsL gene in our HD100 isolate. Mutations in the rpsL gene encoding the ribosomal protein S12 have been shown to confer Sm resistance in many bacteria (3). Therefore, the rpsL gene in our HD100 isolate was sequenced (EF493835) and this sequence compared to the published rpsL gene sequence (19). A BLASTp (http://www.ncbi.nlm.nih.gov/blast/) search of the DNA sequences reveals point mutation K42R (E. coli and Bdellovibrio numbering followed), which is an rpsL mutation commonly found to result in high levels of Sm resistance (3).
FIG. 3.
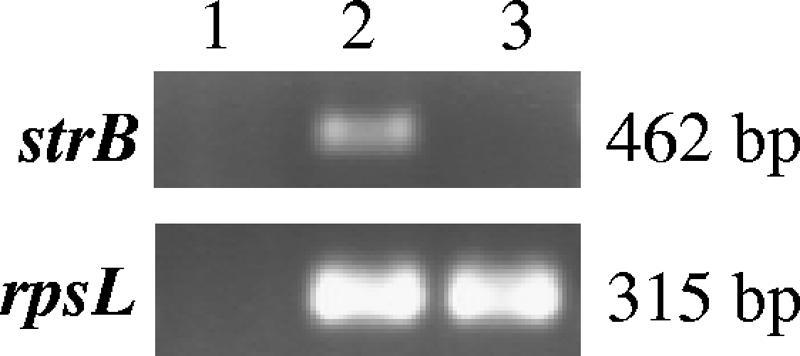
RT-PCR analysis of wild-type HD100 and the strB mutant. Total RNA was extracted from E. coli ML35 (lane 1), wild-type HD100 (lane 2), and HD100ΔstrB (lane 3) and used in RT-PCRs with primers specific for the strB gene. The rpsL gene was used as a load control for B. bacteriovorus RNA. The absence of an RT-PCR product in lane 1 indicates that the product seen for both the strB and the rpsL genes corresponds to B. bacteriovorus RNA rather than any possible contaminating E. coli RNA.
Stability of pMMB206 in B. bacteriovorus.
The stability of the plasmid pMMB206 for use in complementation experiments was tested with the two B. bacteriovorus strains HD100 and B610. The plasmid pMMB206 was found by PCR to be present in the extracted DNA from both the HD100 and the B610 B. bacteriovorus strains, showing that this plasmid was successfully transferred into both strains. The extracted plasmids recovered from transformation of the DNA into E. coli DH5α were found in both cases to be the same size as pMMB206, and the two expected PCR products were obtained (data not shown). This confirms that pMMB206 is stably maintained extrachromosomally by autonomous replication in B. bacteriovorus and can be used for complementation experiments.
DISCUSSION
From the time of the sequencing of the B. bacteriovorus HD100 genome (19), there has been a renewed interest in the investigation of this predatory bacterium at the molecular level. Various aspects of the growth and development of B. bacteriovorus during its unique life cycle have been investigated biochemically and microscopically in the past, with a few more-recent molecular investigations, as is outlined in a recent review (12). The annotation of the B. bacteriovorus genome reveals the existence of many hydrolytic enzymes (19), some of which have no homology with known enzymes and may be involved in the unique predatory nature of the Bdellovibrio life cycle. In order to make further advances in determining gene function or elucidating the regulatory networks in Bdellovibrio, tools for more-efficient genetic manipulation are necessary.
The first suicide plasmid used in B. bacteriovorus was a cosmid, pVK100, belonging to the IncP incompatibility group, which does not replicate in B. bacteriovorus (6) and which carries antibiotic resistance cassettes for Km and tetracycline (Tc) as markers for selection. The large size of this plasmid makes it difficult to work with. More-recent genetic work with B. bacteriovorus has made use of another IncP plasmid, pSET151, as a suicide plasmid (13, 14). This plasmid has a more manageable size of 6.2 kb and a reasonable selection of cloning sites but lacks a useful counterselectable marker (2). An additional drawback to the use of this plasmid is that the marker for selection of plasmid integration after the first homologous recombination is Amp resistance. We have found B. bacteriovorus HD100 to be resistant to Amp at concentrations up to at least 100 μg ml−1. In agreement, Cotter and Thomashow were unable to use either Tc or Amp to select for conjugal transfer of plasmids but were able to use Cm or Km (5). Use of pSET151 as a suicide plasmid was still possible because gene inactivation was carried out by insertion of a Km resistance cassette which could be used for selection of integrants. The problem of no counterselection was circumvented by resorting to the less efficient method of relying solely on screening by Southern blotting.
An alternative to using a plasmid with the IncP origin of replication as a suicide plasmid in Bdellovibrio is to use a plasmid with the RK6 origin of replication. Plasmids with the RK6 origin of replication are dependent on the π protein supplied in trans by the product of the λpir gene for replication (8, 17). The plasmid pCVD442, which has been used successfully as a suicide vector in other gram-negative bacteria, is based on the RK6 origin of replication and carries the sacB gene for counterselection (8). However, pCVD442 uses Amp resistance as a marker for integration and thus cannot be used without modification for work with B. bacteriovorus. A derivative of pCVD442 carrying the cat gene for Cm resistance selection in place of Amp resistance as well as having the insertion sequences found in pCVD442 removed has been reported (17). This plasmid, pDS132, was used to construct the suicide vector used in this work. Our initial work using Cm to select for B. bacteriovorus containing the chromosomally integrated plasmid was unsatisfactory due to the fact that low Cm concentrations were necessary to maintain viability and reasonable times for coculture clearing, and this would lead to the appearance of some plaques on plates of exconjugates due to the wild-type HD100 strain. The other antibiotic to which B. bacteriovorus bacteria are sensitive and that has been used successfully is Km; thus, we decided to use Km selection by addition of a Km resistance cassette to pDS132. We also included three unique restriction enzyme sites that can be used to clone knockout constructions directly into the suicide vector.
Counterselection based on the sacB gene was found to be an efficient method for B. bacteriovorus, provided that the selection is done in liquid culture. Because of the added complexity of growing the prey bacteria with the Bdellovibrio bacteria, ideal concentrations of antibiotics to use for selection can be different depending on whether plates or liquid culture are used. Interestingly, attempts to pick merodiploid plaques into DNB broth with fresh prey supplemented with Km at the concentration of 50 μg ml−1 resulted in either no viable B. bacteriovorus bacteria or the need for an unreasonably long period of time for the coculture to clear even though plaques were observed to start growing in the plates within 2 days. Therefore, plaques were picked into DNB containing Km at a concentration of 25 μg ml−1, which was determined to be sufficient to maintain the B. bacteriovorus bacteria as merodiploids. It appears that the lawn of prey growing in the soft agar has a protective effect on the Bdellovibrio bacteria possibly because it can immediately prey on the E. coli bacteria rather than swim to find the prey as it does in liquid coculture.
The strB gene codes for an aminoglycoside phosphotransferase, APH(6), usually linked upstream with another aminoglycoside kinase gene, strA, which confers Sm resistance. The linked strA-strB genes are found within the transposon Tn5393 on large conjugative plasmids in bacterial plant isolates or on small nonconjugative broad-host-range plasmids in human clinical isolates (27). B. bacteriovorus is unusual in that it possesses no strA gene, but rather, a gene coding for carboxypeptidase and oriented antisense to strB is upstream of strB. Just downstream of the strB gene is an ORF annotated as a hypothetical protein. No ORF with homology to the strA gene is reported to be in the genome sequence of HD100 (19). Since we were successful in creating an in-frame deletion in the strB gene, we proceeded to test our deletion mutant to determine the level of Sm resistance that the gene confers in B. bacteriovorus.
The HD100ΔstrB mutant retains a high level of Sm resistance. Sequencing of the rpsL gene in our wild-type HD100 strain showed that this high level of Sm resistance is most likely due to a common point mutation, K42R, known to confer Sm resistance at >2,000 μg ml−1 in other bacteria (3). This mechanism of Sm resistance is consistent with the observation that resistance was retained in the ΔstrB mutant. Also, it cannot be ruled out that the lack of permeability of the B. bacteriovorus outer membrane might account for some of the Sm resistance. We reasoned that if the StrB protein contributes significantly to the level of resistance to Sm in the wild-type HD100 strain, then observing a phenotype change in the deletion mutant followed by complementation would still be possible. The strB gene, along with an upstream sequence, had been cloned into pMMB206 for complementation. In order to determine whether the StrB protein is functional and, if so, what level of Sm resistance it confers, we tested the sensitivity to Sm of E. coli DH5α harboring this plasmid. We have determined by RT-PCR that the strB gene is transcribed, but it appears that the StrB protein is either not expressed or not functional, since E. coli DH5α bacteria harboring this plasmid were not viable in Sm concentrations as low as 5 μg ml−1. This result is not completely surprising, given that there is certainly no selective pressure to keep a functional allele under normal conditions in which HD100 is maintained in the laboratory, and especially in our strain with an rpsL mutation.
We have shown that the plasmid pMMB206 autonomously replicates in two different B. bacteriovorus strains. This is evidenced both by the ability to continuously grow and transfer these B. bacteriovorus bacteria harboring pMMB206 in media supplemented with Cm at a concentration of 10 μg ml−1 (in which wild-type HD100 and B610 are not viable) and by the recovery of pMMB206 from the extracted DNA. This result is consistent with the report that not only was green fluorescent protein expression detected in B. bacteriovorus strain 109J harboring pMMB206 carrying the gfp gene, but a phenotype difference in life cycle duration as well as altered motility was observed in 109J harboring pMMB206 with antisense motA cloned (10). Therefore, complementation using the plasmid pMMB206 in B. bacteriovorus appears feasible. Since a phenotype change in the HD100ΔstrB mutant could not be detected, the mutant was not complemented.
Bdellovibrio is an obligate predatory bacterium; thus, any study attempting to determine at the genetic level how the Bdellovibrio bacterium invades the prey cell and how its life cycle is regulated will presumably involve investigation of genes that are essential for the viability of the Bdellovibrio bacterium. Therefore, there is a possibility of attempts to construct deletion mutants of essential genes. Different variations of gene essentiality experiments with other organisms have been reported (9, 16, 22, 26). One involves introducing a wild-type copy of the gene to be deleted on an autonomously replicating plasmid carrying a counterselection marker different from that used on the suicide plasmid into the strain merodiploid for the knockout. The wild-type rpsL gene acts in a dominant-negative manner to confer Sm sensitivity to the cell and has been used as a counterselection marker (20). Therefore, the HD100 rpsL ΔstrB mutant that we have constructed would be ideal for use in gene essentiality studies using a wild-type copy of the rpsL gene cloned into pMMB206 as a counterselection marker.
Now that we have developed a system for allelic exchange or construction of in-frame markerless deletion mutants of B. bacteriovorus HD100, this bacterium can be genetically manipulated to test gene function. Exciting new investigations can be undertaken to study many aspects of Bdellovibrio, from chemotaxis in the free-living stage of the life cycle to metabolic changes within the bdelloplast as well as functional characterization of hydrolytic enzymes. Furthermore, the genetic system described for constructing mutants in the host-dependent Bdellovibrio bacterium can be easily modified for purposes of genetic manipulation in the host-independent phenotype. Importantly, application of the methods described in this work coupled with transcriptional analysis will aid in understanding the regulatory networks in this unique prokaryotic predator bacteria.
Acknowledgments
We thank Dominique Schneider for pDS132 and Michael Donnenberg for the E. coli strains DH5αλpir, SM10λpir, and S17-1λpir.
This work was supported by departmental start-up funds.
Footnotes
Published ahead of print on 8 June 2007.
REFERENCES
- 1.Barel, G., and E. Jurkevitch. 2001. Analysis of phenotypic diversity among host-independent mutants of Bdellovibrio bacteriovorus 109J. Arch. Microbiol. 176:211-216. [DOI] [PubMed] [Google Scholar]
- 2.Bierman, M., R. Logan, K. O'Brien, E. T. Seno, R. N. Rao, and B. E. Schoner. 1992. Plasmid cloning vectors for the conjugal transfer of DNA from Escherichia coli to Streptomyces spp. Gene 116:43-49. [DOI] [PubMed] [Google Scholar]
- 3.Bottger, E. C., B. Springer, T. Prammananan, Y. Kidan, and P. Sander. 2001. Structural basis for selectivity and toxicity of ribosomal antibiotics. EMBO Rep. 2:318-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Coppi, M. V., C. Leang, S. J. Sandler, and D. R. Lovely. 2001. Development of a genetic system for Geobacter sulfurreducens. Appl. Environ. Microbiol. 67:3180-3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cotter, T. W., and M. F. Thomashow. 1992. A conjugation procedure for Bdellovibrio bacteriovorus and its use to identify DNA sequences that enhance the plaque-forming ability of a spontaneous host-independent mutant. J. Bacteriol. 174:6011-6017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cotter, T. W., and M. F. Thomashow. 1992. Identification of a Bdellovibrio bacteriovorus genetic locus, hit, associated with the host-independent phenotype. J. Bacteriol. 174:6018-6024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davidov, Y., and E. Jurkevitch. 2004. Diversity and evolution of Bdellovibrio-and-like organisms (BALOs), reclassification of Bacteriovorax starrii as Peredibacter starrii gen. nov., comb. nov., and description of the Bacteriovorax-Peredibacter clade as Bacteriovoracaceae fam. nov. Int. J. Syst. Evol. Microbiol. 54:1439-1452. [DOI] [PubMed] [Google Scholar]
- 8.Donnenberg, M. S., and J. B. Kaper. 1991. Construction of an eae deletion mutant of enteropathogenic Escherichia coli by using a positive-selection suicide vector. Infect. Immun. 59:4310-4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dziadek, J., S. A. Rutherford, M. V. Madiraju, M. A. L. Atkinson, and M. Rajagopalan. 2003. Conditional expression of Mycobacterium smegmatis ftsZ, an essential cell division gene. Microbiology 149:1593-1603. [DOI] [PubMed] [Google Scholar]
- 10.Flannagan, R. S., M. A. Valvano, and S. F. Koval. 2004. Downregulation of the motA gene delays the escape of the obligate predator Bdellovibrio bacteriovorus 109J from bdelloplasts of bacterial prey cells. Microbiology 150:649-656. [DOI] [PubMed] [Google Scholar]
- 11.Fu, R., and G. Voordouw. 1997. Targeted gene-replacement mutagenesis of dcrA, encoding an oxygen sensor of the sulfate-reducing bacterium Desulfovibrio vulgaris Hildenborough. Microbiology 143:1815-1826. [DOI] [PubMed] [Google Scholar]
- 12.Lambert, C., K. A. Morehouse, C. Y. Chang, and R. E. Sockett. 2006. Bdellovibrio: growth and development during the predatory cycle. Curr. Opin. Microbiol. 9:1-6. [DOI] [PubMed] [Google Scholar]
- 13.Lambert, C., K. J. Evans, R. Till, L. Hobley, M. Capeness, S. Rendulic, S. C. Schuster, S.-I. Aizawa, and R. E. Sockett. 2006. Characterizing the flagellar filament and the role of motility in bacterial prey-penetration by Bdellovibrio bacteriovorus. Mol. Microbiol. 60:274-286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lambert, C., M. C. M. Smith, and R. E. Sockett. 2003. A novel assay to monitor predator-prey interactions for Bdellovibrio bacteriovorus 109J reveals a role for methyl-accepting chemotaxis proteins in predation. Environ. Microbiol. 5:127-132. [DOI] [PubMed] [Google Scholar]
- 15.Morales, V. M., A. Backman, and M. Bagdasarian. 1991. A series of wide-host-range low-copy-number vectors that allow direct screening for recombinants. Gene 97:39-47. [DOI] [PubMed] [Google Scholar]
- 16.Murphy, C. K., E. J. Stewart, and J. A. Beckwit. 1995. Double counter-selection system for the study of null alleles of essential genes in Escherichia coli. Gene 155:1-7. [DOI] [PubMed] [Google Scholar]
- 17.Philippe, N., J. P. Alcaraz, E. Coursange, J. Geiselmann, and D. Schneider. 2004. Improvement of pCVD442, a suicide plasmid for gene allele exchange in bacteria. Plasmid 51:246-255. [DOI] [PubMed] [Google Scholar]
- 18.Picardeau, M., A. Brenot, and I. S. Girons. 2001. First evidence for gene replacement in Leptospira spp. Inactivation of L. biflexa flab results in non-motile mutants deficient in endoflagella. Mol. Microbiol. 40:189-199. [DOI] [PubMed] [Google Scholar]
- 19.Rendulic, S., P. Jagtap, A. Rosinus, M. Eppinger, C. Baar, C. Lanz, H. Keller, C. Lambert, K. Evans, A. Goesmann, F. Meyer, R. E. Sockett, and S. C. Schuster. 2004. A predator unmasked: life cycle of Bdellovibrio bacteriovorus from a genomic perspective. Science 303:689-692. [DOI] [PubMed] [Google Scholar]
- 20.Reyrat, J.-M., V. Pelicic, B. Gicquel, and R. Rappuloi. 1998. Counterselectable markers: untapped tools for bacterial genetics and pathogenesis. Infect. Immun. 66:4011-4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Romo, A. J., E. G. Ruby, and M. H. Saier, Jr. 1992. Effect of Bdellovibrio bacteriovorus infection on the phosphoenolpyruvate: sugar phosphotransferase system in Escherichia coli: evidence for activation of cytoplasmic proteolysis. Res. Microbiol. 143:5-14. [DOI] [PubMed] [Google Scholar]
- 22.Salama, N. R., B. Shepherd, and S. Falkow. 2004. Global transposon mutagenesis and essential gene analysis of Helicobacter pylori. J. Bacteriol. 186:7926-7935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Simon, R., U. Priefer, and A. Puhler. 1983. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in Gram negative bacteria. Bio/Technology 1:784-791. [Google Scholar]
- 24.Simons, R. W., F. Houman, and N. Kleckner. 1987. Improved single and multicopy lac-based cloning vectors for protein and operon fusions. Gene 53:85-96. [DOI] [PubMed] [Google Scholar]
- 25.Sockett, R. E., and C. Lambert. 2004. Bdellovibrio as therapeutic agents: a predatory renaissance? Nat. Rev. Microbiol. 2:669-675. [DOI] [PubMed] [Google Scholar]
- 26.Sun, J., L. Zheng, C. Landwehr, J. Yang, and Y. Ji. 2005. Identification of a novel essential two-component signal transduction system, YhcSR, in Staphylococcus aureus. J. Bacteriol. 187:7876-7880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sundin, G. W. 2002. Distinct recent lineages of the strA-strB streptomycin-resistance genes in clinical and environmental bacteria. Curr. Microbiol. 45:63-69. [DOI] [PubMed] [Google Scholar]
- 28.Wu, S. S., and D. Kaiser. 1996. Markerless deletions of pil genes in Myxococcus xanthus generated by counterselection with the Bacillus subtilis sacB gene. J. Bacteriol. 178:5817-5821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yair, S., D. Yaacov, S. Koval, and E. Jurkevitch. 2003. Small eats big: ecology and diversity of Bdellovibrio and like organisms, and their dynamics in predator-prey interactions. Agronomie 23:433-439. [Google Scholar]