

Abstract
Immunoliposomes (antibody-directed liposomes) were used in the present study for delivery of the antineoplastic agent daunomycin to the rat brain. A coupling procedure was introduced, which allows conjugation of a thiolated antibody to maleimide-grafted 85-nm liposomes sterically stabilized with PEG. Antibody was thereby coupled to the terminal end of a PEG-conjugated linker lipid. No brain uptake of PEG-conjugated liposomes carrying [3H]daunomycin was observed. However, brain targeting of immunoliposomes carrying [3H]daunomycin was mediated by the OX26 monoclonal antibody to the rat transferrin receptor, which is selectively enriched at the brain microvascular endothelium that comprises the blood–brain barrier in vivo. Coupling of 30 OX26 antibodies per liposome resulted in optimal brain delivery. Saturation of delivery was observed at higher antibody densities. Determination of brain levels of immunoliposomes over 24 h revealed that immunoliposomes accumulate in brain tissue. Brain targeting of immunoliposomes was not observed in immunoliposomes conjugated with a mouse IgG2a isotype control. In addition, coinjection of free OX26 saturated plasma clearance of immunoliposomes. Since a single liposome may carry ≥10,000 drug molecules, the use of PEG-conjugated immunoliposomes increases the drug carrying capacity of the monoclonal antibody by up to 4 logarithmic orders in magnitude. In summary, specific OX26-mediated targeting of daunomycin to the rat brain was achieved by the use of an immunoliposome-based drug delivery system.
Keywords: PEG conjugation, blood–brain barrier, drug targeting, daunomycin
Immunoliposomes (antibody-directed liposomes) have been recognized as a promising tool for the site-specific delivery of drugs and diagnostic agents. However, the in vivo use of classical immunoliposomes is hampered by the very rapid clearance of immunoliposomes from the circulation by the reticuloendothelial system (1, 2). Avoidance of this obstacle is possible if gangliosides (3) or PEG-derivatized lipids (4) are inserted within the bilayer of conventional liposomes, as these modifications prolong considerably the liposome half-life in the circulation. Liposomes coated with the inert and biocompatible polymer PEG are widely used and are often referred to as “sterically stabilized” or “stealth liposomes” (5). PEG coating is believed to prevent recognition of liposomes by macrophages due to reduced binding of plasma proteins (4, 6). Unfortunately, it has been difficult to combine steric stabilization of liposomes with efficient immunotargeting. PEG coating of liposomes can create steric hindrances for antibody–target interaction (7, 8). It has therefore been proposed to attach a cell-specific ligand to the distal end of a few lipid-conjugated PEG molecules rather than conjugate the ligand to a lipid headgroup on the surface of a PEG-conjugated liposome. This has been done recently with folic acid (9) and monoclonal antibodies (10, 11, 12, 13) to target liposomes to cells in tissue culture and organs in vivo.
The application of PEG-conjugated immunoliposomes to in vivo brain targeting of drugs has not been attempted thus far. Conventional liposomes are not delivered to brain in vivo (14, 15, 16), because these agents are not transported through the brain capillary endothelial wall, which makes up the blood–brain barrier (BBB) in vivo. However, certain receptor specific monoclonal antibodies (mAbs) undergo receptor-mediated transcytosis through the BBB (17), and mAb–gold conjugates are transcytosed through the BBB (18) in vivo. Therefore, the present studies were designed to achieve the following goals. First, PEG-conjugated immunoliposomes were synthesized using thiolated mAb and a bifunctional 2000-Da PEG (PEG2000) that contains a lipid at one end and a maleimide at the other end. Second, the pharmacokinetics and brain uptake of [3H]daunomycin was examined following intravenous administration of the drug in free form, as a conventional liposome, as a PEG-conjugated liposome, and as a PEG-conjugated immunoliposome. The mAb used in these studies is the OX26 mAb to the rat transferrin receptor (19), which is abundant on brain microvascular endothelium (20, 21).
MATERIALS AND METHODS
Materials.
Cholesterol was from Sigma. Distearoylphosphatidylcholine and distearoylphosphatidylethanolamine (DSPE) were from Avanti Polar Lipids. DSPE-PEG was purchased from Shearwater Polymers (Huntsville, AL). DSPE-PEG-maleimide was custom-synthesized by Shearwater Polymers. DSPE-PEG-maleimide was prepared by derivatization of DSPE-PEG. The efficiency of this reaction was 84%, as determined by NMR analysis. PEG2000 was used in all PEG-containing compounds. [3H]Daunomycin was from DuPont/NEN. 2-Iminothiolane (Traut’s reagent) and the bicinchoninic acid protein assay kit (used with bovine serum albumin as a standard) were obtained from Pierce. Sephadex G-25, Sepharose CL-4B, and protein G Sepharose were from Pharmacia. Mouse myeloma ascites IgG2a (κ) was from Cappel. Centriprep-30 (molecular weight cut-off: 30,000) concentrators were from Amicon. Male Sprague–Dawley rats (weighing 260–290 g) were obtained from Harlan–Sprague–Dawley. All other chemicals were of analytical grade.
Purification and Thiolation of Antibodies.
The anti-rat transferrin receptor OX26 mAb (19) was harvested from cell culture supernatants of the OX26 hybridoma cell line as described (22). OX26 as well as the control IgG2a antibody were purified by protein G Sepharose affinity chromatography (23). OX26 mAb was iodinated to a specific activity of 10 μCi/μg (1 Ci = 37 GBq) using [125I]iodine and chloramine T, as described previously (24). The radiolabeled protein was purified by Sephadex G-25 gel filtration chromatography and was >96% TCA-precipitable. [125I]OX26 or IgG2a were thiolated using 2-iminothiolane (Traut’s reagent; ref. 25). mAb was dissolved in 0.15 M Na-borate buffer/0.1 mM EDTA, pH 8.5 followed by the addition of Traut’s reagent. After incubation for 60 min at room temperature, mAb solutions were concentrated and the buffer exchanged with 0.1 M Na-phosphate (pH 8.0) using a Centriprep-30 concentrator (Amicon). Thiolated mAb was immediately used for conjugation with liposomes (see below). Ellmann’s reagent (26) was used to determine the number of sulfhydryl groups added by thiolation to mAb. Using a mAb/Traut’s ratio of 1:40 (mol/mol), an average of one primary amine per mAb was thiolated.
Liposome Preparation.
Distearoylphosphatidylcholine (5.2 μmol), cholesterol (4.5 μmol), DSPE (0.3 μmol), and, for the preparation of immunoliposomes, linker lipid (DSPE-PEG-maleimide, 0.015 μmol) were dissolved in chloroform/methanol [2:1 (vol/vol)]. For the synthesis of PEG-liposomes and immunoliposomes, DSPE was substituted by DSPE-PEG. A lipid film was prepared by vacuum evaporation using a Speedvac concentrator (Savant) for 70 min. Dried lipid films were hydrated at 65°C in 0.3 M citrate (pH 4.0), such that a final lipid concentration of 10 mM was achieved. Lipids were subjected to five freeze-thaw cycles followed by extrusion (4 times) at room temperature through a 100-nm pore size polycarbonate membrane employing an extruder (Avestin, Ottawa, Canada). Extrusion was repeated 9 times using a 50-nm polycarbonate membrane. Mean vesicle diameters were determined by quasielastic light scattering using a Microtrac Ultrafine Particle Analyzer (Leeds–Northrup, St. Petersburg, FL).
Doxorubicin Loading and Antibody Conjugation.
Loading of liposomes with [3H]daunomycin was done via a pH gradient (27). By addition of NaOH to liposomes (10 μmol of lipid), the pH of the external buffer was raised to pH 7.8. [3H]Daunomycin was added and the incubation mix was incubated for 10 min at 60°C. Loaded liposomes were either used for coupling with mAb or external buffer was exchanged by passing the liposomes over a Sephadex G-25 column and eluting with 0.001 M PBS (0.001 M Na-phosphate/0.15 M NaCl, pH 7.4). For antibody conjugation, thiolated antibody was incubated with liposomes containing DSPE-PEG-maleimide overnight at room temperature. Buffer was exchanged by applying the reaction mix to a 1.6 × 18 cm Sephadex CL-4B column and eluting with 0.001 M PBS. Aliquots of column eluates were analyzed by scintillation counting. Efficiency of entrapment of daunomycin and efficiency of OX26 coupling was determined by analysis of the column elution profiles. In the case where no 125I-labeled antibody was available (control IgG2a mAb), the amount of liposome-conjugated protein was determined by a protein assay as described (28). The number (n) of OX26 molecules attached per liposome are designated as OX26n.
Pharmacokinetics and Brain Delivery of Immunoliposomes.
Pharmacokinetic experiments were performed as described (29). In brief, rats were anesthetized with 100 mg of ketamine and 2 mg of xylazine per kg of body weight intraperitoneally. The left femoral vein was cannulated and injected with 0.001 M PBS containing 4 μCi of free [3H]daunomycin, [3H]daunomycin-loaded liposomes, or immunoliposomes. The injected dose of mAb and lipid was always ≤50 μg per rat and ≤1 μmol per rat, respectively. Blood samples were collected via a cannula implanted in the left femoral artery at 0.25, 1, 2, 5, 15, 30, and 60 min after intravenous injection of the isotope. The blood volume was replaced with an equal volume of saline. After 60 min, the animals were decapitated for removal of the brain. For some experiments, animals were sacrificed 6 h or 24 h after intravenous injection. In this case, animals were allowed to recover after surgery and terminal blood only was sampled. The plasma and organ samples were solubilized with Soluene-350 (Packard) and neutralized with glacial acetic acid before liquid scintillation counting. Pharmacokinetic parameters were calculated by fitting plasma radioactivity data to a biexponential equation—i.e, A(t) = A1 e−k1t + A2 e−k2t, where A(t) = %ID per ml of plasma 3H radioactivity (%ID, percent injected dose). The biexponential equation was fit to plasma data using a nonlinear regression analysis. The pharmacokinetic parameters, such as plasma clearance (Cl), the initial plasma volume (VC), steady-state volume of distribution (Vss) and steady-state area under the plasma concentration curve (AUC0t) were determined from A1, A2, k1, and k2. The brain volume of distribution (VD) of the [3H]sample at a given timepoint after i.v. injection was determined from the ratio of disintegrations/min (dpm) per g of tissue divided by the dpm/μl of terminal plasma. The brain permeability surface area (PS) product was determined as follows:
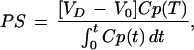 |
where Cp(T) = the terminal plasma concentration and V0 = the brain volume distribution of a plasma volume marker. The brain volume of distribution of PEG-liposomes of 7.48 ± 0.34 μl/g (n = 3) was lower than the corresponding value of the commonly used plasma volume marker [125I]mouse IgG2a of 11 μl/g (23). The brain VD of the PEG-liposomes was therefore used as a plasma volume marker (V0) for the calculation of brain PS products. Brain uptake was expressed as %ID per g of brain and was calculated from: %ID/g(t) = PS × AUC0t.
Daunomycin Octanol/Aqueous Partition Coefficient.
The 1-octanol/buffer partition coefficent (P) was determined for [3H]daunomycin as described previously (17) and was 0.40 ± 0.04 (mean ± SEM, n = 3), which yielded a logarithmic P = −0.40.
RESULTS
Synthesis of Immunoliposomes.
Liposomes were prepared by repeated extrusion through polycarbonate filter membranes with pore sizes of 100 and 50 nm, respectively. By this method, an average diameter of the liposomes of 85 nm was obtained with a sharp distribution of size (80% of the liposomes had a size between 65 and 115 nm; Fig. 1B). Using a pH-shift method, loading of liposomes with [3H]daunomycin was achieved with high and reproducible yield (89.9 ± 1.1%, value is mean ± SEM, n = 7). Loading was not affected by lipid composition of the vesicles or the lipid to daunomycin molar ratio (range of ratios tested was from 16:1 to 2500:1). For the synthesis of immunoliposomes, the linker lipid DSPE-PEG-maleimide was incorporated in liposomes. Daunomycin-loaded vesicles were allowed to react with thiolated antibody (Fig. 1A). mAb OX26 was thiolated using 2-iminothiolane (Traut’s reagent). The ratio of Traut’s reagent and mAb was adjusted to yield an average of one thiolated primary amine per mAb. It has been shown previously (30) that thiolation of OX26 does not interfere with its target recognition. Since the maleimide group slowly hydrolyses when in contact with water, it was essential to proceed for the synthesis of immunoliposomes without unnecessary delay. The efficiency of coupling was not affected by mAb concentration using molar ratios of phospholipid/mAb from 100:1 to 400:1. An average of 10% of the antibody could be attached by a thioether bond to the liposome in most cases. Immunoliposomes were separated from unincorporated [3H]daunomycin and free mAb and the external buffer was exchanged for 0.001 M PBS by a Sephadex CL-4B gel filtration chromatography (Fig. 1C). [125I]mAb OX26 eluted in two baseline-separated peaks at 12 and 29 ml, corresponding to liposome-conjugated mAb overlapping with the peak of [3H]daunomycin containing liposomes and unbound mAb, respectively. The salt volume of the column was 35 ml. Analysis of elution profiles allowed the determination of efficiency of [3H]daunomycin loading and conjugation of mAb. The conversion of μg of IgG per μmol of phospholipid to the number of mAb molecules conjugated per liposome was based on the assumption that a 100-nm liposome contains ≈100,000 molecules of phospholipid (11).
Figure 1.
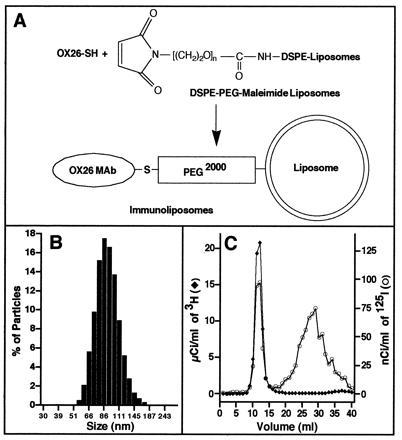
Preparation of PEG-conjugated immunoliposomes. (A) Schematic diagram of coupling of the thiolated monoclonal antibody OX26 (OX26-SH) with sterically stabilized liposomes containing DSPE-PEG-maleimide. (B) Size distribution of sterically stabilized liposomes prepared by rapid extrusion. (C) Elution profile of the separation of [3H]daunomycin-loaded immunoliposomes from unencapsulated [3H]daunomycin and unbound 125I-labeled mAb OX26 by gel filtration.
Comparison of Free Daunomycin, Liposomes, Sterically Stabilized Liposomes, and Immunoliposomes.
The disappearance of free daunomycin, liposomes, immunoliposomes, and sterically stabilized PEG-liposomes from the plasma compartment occurred in a biexponential manner (Fig. 2A). From the data in Fig. 2A, the distribution volume at steady state and the plasma clearance were calculated. There was a pronounced difference between these compounds. Free daunomycin and not PEG-conjugated liposomes containing daunomycin disappear rapidly from the circulation, with plasma clearances of 45 ± 7 and 13 ± 6 ml/min per kg, respectively (Table 1). The plasma clearance of the liposome was reduced 66-fold by PEG conjugation (Table 1). The 235-fold difference in plasma clearance between free daunomycin and daunomycin encapsulated within PEG-liposomes is indicative of adequate retention of the drug within liposomes. Coupling of 29 OX26 mAbs per PEG-liposome partially reversed the effect of PEG conjugation and resulted in a 5-fold increase in plasma clearance (Table 1). Analysis of brain tissue revealed a permeability surface area (PS) product of 1.6 μl/min per g of free daunomycin (Fig. 3), a value comparable to morphine-6-glucuronide (31). However, the area under the plasma curve (AUC) of daunomycin is very small, resulting in poor brain tissue accumulation (reflected by its low %ID/g value) of this substance (Fig. 3). The BBB PS product of the [3H]daunomycin-containing liposomes was decreased 8-fold, whereas the plasma AUC was increased 4-fold compared with free daunomycin, and these offsetting effects resulted in no change in brain drug delivery (Fig. 3). The use of PEG-conjugated liposomes reduced the BBB PS value to zero: therefore, no brain uptake of the PEG-liposomes was observed, despite the marked increase in plasma AUC (Fig. 3). Conversely, the use of PEG-conjugated OX26 immunoliposomes increased the BBB PS product, relative to PEG-liposomes, and a brain uptake of 0.03 %ID/g at 60 min was observed (Fig. 3).
Figure 2.
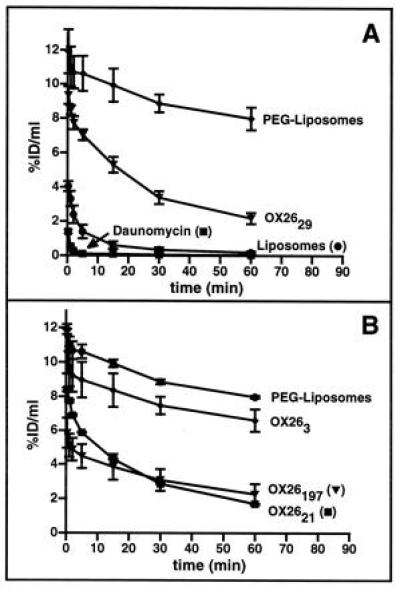
%ID/ml of plasma of daunomycin, liposomes, and immunoliposomes is plotted versus various times after intravenous injection. (A) Comparison of free daunomycin, liposomes, sterically stabilized liposomes (PEG-liposomes), and PEG-conjugated immunoliposomes (OX2629). (B) Titration of OX26 bound to PEG-conjugated liposomes. The liposomes designated as PEG-liposomes have no mAb attached. Data are means ± SEM of n = 3 rats/point.
Table 1.
Systemic volume of distribution and plasma clearance of daunomycin, liposomes, and immunoliposomes
| Systemic volume of distribution,* ml/kg | Plasma clearance,* ml/min/kg | |
|---|---|---|
| Daunomycin | 1400 ± 253 | 44.7 ± 6.8 |
| Liposomes | 290 ± 69 | 12.6 ± 6.3 |
| PEG-liposomes | 34 ± 4 | 0.19 ± 0.01 |
| OX263 | 30 ± 3 | 0.22 ± 0.03 |
| OX2621 | 55 ± 2 | 1.20 ± 0.06 |
| OX2629 | 58 ± 9 | 0.91 ± 0.11 |
| OX26197 | 59 ± 3 | 0.67 ± 0.05 |
| OX26197 plus 1 mg of OX26 | 50 ± 4 | 0.39 ± 0.04† |
| 20 Mouse IgG2a | 39 ± 4‡ | 0.37 ± 0.04‡ |
Values represent means ± SEM of n = 3 experiments. All immunoliposomes are PEG-conjugated.
Statistically significant difference by Student’s t test (P = 0.012) when compared to corresponding value of OX26197 immunoliposomes.
Statistically significant difference by Student’s t test (P < 0.015) when compared to corresponding value of OX2621 immunoliposomes.
Figure 3.
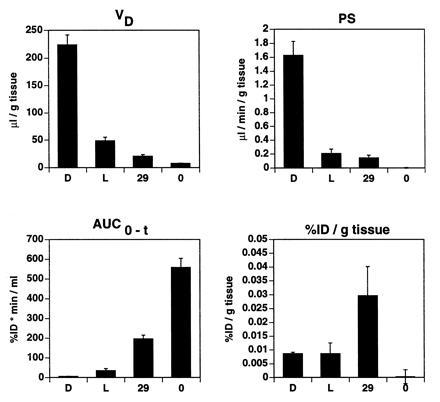
Pharmacokinetic parameters and brain delivery of free daunomycin (column D), not PEG-conjugated liposomes (column L), PEG-conjugated OX2629 immunoliposomes (column 29), and PEG-conjugated liposomes without mAb (column 0) at 60 min after intravenous injection. Data represent means ± SEM (n = 3).
Titration of OX26.
Titration of the amount of OX26 conjugated per liposome revealed an increase in plasma clearance and a decrease in the systemic volume of distribution of immunoliposomes at higher OX26 concentrations (Fig. 2B and Table 1). The PEG-liposomes are designated as OX260 immunoliposomes in Fig. 4. Highest PS product values and brain tissue accumulation were observed for OX2629 immunoliposomes (Fig. 4). At higher OX26 densities on the liposome, a saturation effect was observed resulting in reduced VD, PS product, and brain tissue accumulation of OX26197 immunoliposomes.
Figure 4.
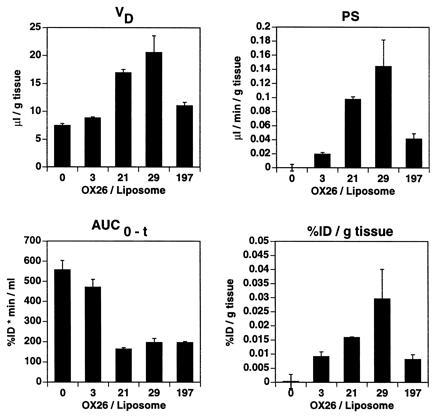
Pharmacokinetic parameters and brain delivery of sterically stabilized liposomes (PEG-liposomes) and immunoliposomes at 60 min after intravenous injection. All liposomes are PEG-conjugated. Data represent means ± SEM (n = 3).
Twenty-four-Hour Pharmacokinetics and Tissue Distribution.
To determine if immunoliposomes accumulate in brain tissue over time, 1-h, 6-h, and 24-h brain uptake experiments were performed using OX2630 immunoliposomes, and the brain VD was observed to increase over time (Fig. 5). This result indicates that immunoliposomes directed to the brain are trapped in the brain. Similar results were obtained from experiments using OX26197 immunoliposomes (data not shown).
Figure 5.
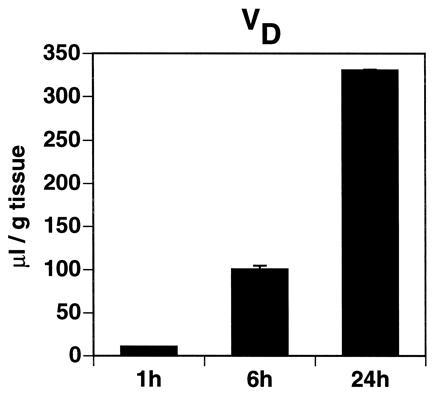
Brain VD of PEG-conjugated immunoliposomes (OX2630). Brain tissue was analyzed 1 h, 6 h, or 24 h after intravenous injection of immunoliposomes packaged with [3H]daunomycin. Data represent means ± SEM (n = 3).
Coinjection of OX26 and Use of Nonspecific Control IgG Immunoliposomes.
To determine if brain delivery of immunoliposomes was mediated by the mAb OX26, two series of control experiments were performed. First, OX26197 immunoliposomes were coinjected with 1 mg of OX26. Comparison of plasma pharmacokinetics revealed a statistically significant difference (Student’s t test, P < 0.012) between OX26197 and coinjection of 1 mg of OX26 with OX26197 immunoliposomes with respect to plasma clearance (Table 1) and steady-state area under the plasma concentration curve (197.2 ± 5.1 versus 460.5 ± 27.8 %ID·min/ml, means ± SEM, n = 3). The coinjection of free OX26 reduced the plasma clearance of the PEG-conjugated OX26 immunoliposome to a value equal to the plasma clearance of the PEG-conjugated IgG2a immunoliposome (Table 1). Second, mouse IgG2a (i.e., the OX26 isotype) was coupled to PEG-liposomes (20 mouse IgGs per liposome), and pharmacokinetic parameters and tissue distribution were compared with PEG-conjugated OX2621 immunoliposomes. The plasma clearance of the PEG-conjugated IgG2a immunoliposomes was reduced 70% to a value identical to the clearance of the OX26 immunoliposome under saturating conditions (Table 1). There was no brain uptake of the PEG-conjugated IgG2a immunoliposomes, as the brain VD, 7.4 ± 0.5 μl/g (n = 3), was not different from the brain VD of PEG-liposomes, 7.5 ± 0.3 μl/g (n = 3).
DISCUSSION
By introducing a novel antibody–liposome coupling procedure, immunoliposomes were synthesized that were subsequently used to target an encapsulated drug, daunomycin, to the rat brain in vivo. The antibody used for this study was the OX26 mAb. This antibody is able to traverse the BBB by a mechanism that normally serves to mediate BBB transcytosis of circulating transferrin (32, 33). OX26 was thiolated and conjugated by a thioether bond to sterically stabilized liposomes. As a linker, a PEG-conjugated lipid was used carrying at its distal end a maleimide group. The reaction between maleimide and thiol groups is a rapid and efficient reaction that is widely used in bioconjugate chemistry (34, 35). However, direct conjugation of maleimide to lipids without the use of a spacer may considerably interfere with subsequent doxorubicin loading of liposomes (11). In addition, surface-bound maleimide groups are poorly accessible when used in combination with PEG-lipids, which results in very poor coupling efficiency with thiolated ligands (13). In our experimental system, these problems were not observed, since the maleimide group was attached at the distal end of PEG-conjugated lipids. Thus a long, flexible PEG spacer was provided which separated the maleimide group from the liposome surface. Consequently, reproducible and efficient loading of liposomes with daunomycin and subsequent coupling of thiolated mAb OX26 was possible.
Plasma pharmacokinetics and brain delivery of free daunomycin, conventional liposomes, sterically stabilized liposomes, and immunoliposomes were determined after single intravenous injection in the rat. The brain delivery of a given substance (expressed as %ID/g of tissue) may thereby be described as the product of its permeability surface area (PS) product and its plasma area under the concentration curve (AUC) at a given time after injection (17). Thus, brain delivery of free daunomycin or conventional liposomes is poor due to their rapid clearance from the circulation, resulting in a low plasma AUC. The residual brain uptake of daunomycin packaged within conventional liposomes (Fig. 3) may arise from brain uptake of free daunomycin released to the blood following dissolution of these liposomes in peripheral tissues. Daunomycin has a log P (refer to Materials and Methods) comparable to morphine (17), which accounts for the measurable brain uptake of free daunomycin (Fig. 3). The plasma AUC is greatly increased with the use of PEG-liposomes. However, the brain uptake of PEG-liposomes is zero (Fig. 3). Therefore, the use of sterically stabilized liposomes confers no advantage in brain drug delivery. In contrast, the use of PEG-conjugated OX26 immunoliposomes results in a measurable PS product with a moderate plasma clearance (Figs. 3 and 4). Consequently, brain delivery of immunoliposomes is greater than brain delivery of free daunomycin, conventional liposomes, or sterically stabilized liposomes. Plasma pharmacokinetic and brain delivery studies over 24 h indicate that immunoliposomes, once delivered to the brain, are sequestered in brain tissue. The VD of OX2630 immunoliposomes increases over time (Fig. 5). The observed properties of immunoliposomes were specifically mediated by the OX26 mAb. Brain targeting of immunoliposomes was eliminated by substitution of OX26 by a mouse IgG2a isotype control.
OX2630 immunoliposomes had the most favorable properties of the immunoliposome preparations tested. Coupling of higher amounts of OX26 to immunoliposomes (e.g., OX26197 immunoliposomes) results in relatively poor brain targeting and brain accumulation of immunoliposomes. These results are similar to findings in lung using PEG-conjugated 34A immunoliposomes. The 34A mAb targets pulmonary endothelial cells, and the attachment of >30 34A mAb molecules per 110-nm liposome confers no additional advantage in targeting PEG-liposomes to the lung (12).
Using immunoliposomes, an average brain delivery of 0.03 %ID/g brain tissue is possible at 60 min after injection (Fig. 3). This value is about 3- to 4-fold lower than the values that can be attained using biotinylated drugs individually attached to conjugates of OX26 and avidin analogues (29, 30). However, only 2–3 small molecules can be attached to a given OX26–avidin conjugate. In contrast, >10,000 small molecules may be entrapped in a single 100-nm liposome. For example, lipid/drug ratios of 3.2 are achieved with 100-nm liposomes (36). Given ≈100,000 lipid molecules on the liposome surface (11), ≈28,000 daunomycin molecules are packaged within a single liposome. Therefore, the conjugation of PEG-liposomes greatly increases the carrying capacity of the OX26 mAb molecule by up to 4 logarithmic orders in magnitude.
In conclusion, the present studies demonstrate BBB drug delivery vectors such as the OX26 mAb may be successfully conjugated to the tips of PEG-liposomes for the purpose of greatly expanding the loading of drugs per individual OX26 molecule. With this approach, it is feasible for vector-mediated drug delivery systems, which have heretofore been used for peptides or peptide nucleic acids (17), to be applied to small molecule drug delivery to the brain. It is generally necessary to achieve micromolar concentrations in the brain for many small molecules to be pharmacologically active. Micromolar drug concentrations in brain cannot be achieved using BBB drug delivery vectors unless the carrying capacity of the vectors is increased by logarithmic orders. This is possible with the use of PEG-conjugated immunoliposomes and mAbs that target the brain and BBB receptors.
Acknowledgments
We would like to thank Jing Yang for technical assistance. We thank Dr. K. Shelly for assisting with dynamic light scattering measurements. Dr. J. Huwyler is recipient of a research grant from the Swiss National Science Foundation (32-42179.94).
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: BBB, blood–brain barrier; DSPE, distearoylphosphatidylethanolamine; %ID, percent injected dose; PEG2000, 2000-Da PEG.
References
- 1.Aragnol D, Leserman L D. Proc Natl Acad Sci USA. 1986;83:2699–2703. doi: 10.1073/pnas.83.8.2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Derksen J T, Morselt H W, Scherphof G L. Biochim Biophys Acta. 1988;971:127–136. doi: 10.1016/0167-4889(88)90184-x. [DOI] [PubMed] [Google Scholar]
- 3.Gabizon A, Papahadjopoulos D. Biochim Biophys Acta. 1992;1103:94–100. doi: 10.1016/0005-2736(92)90061-p. [DOI] [PubMed] [Google Scholar]
- 4.Papahadjopoulos D, Allen T M, Gabizon A, Mayhew E, Matthay K, Huang S K, Lee K D, Woodle M C, Lasic D D, Redemann C, Martin F J. Proc Natl Acad Sci USA. 1991;88:11460–11464. doi: 10.1073/pnas.88.24.11460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Allen T M. Trends Pharmacol Sci. 1994;15:215–220. doi: 10.1016/0165-6147(94)90314-x. [DOI] [PubMed] [Google Scholar]
- 6.Moghimi S M, Patel H M. Biochim Biophys Acta. 1992;1135:269–274. doi: 10.1016/0167-4889(92)90230-9. [DOI] [PubMed] [Google Scholar]
- 7.Klibanov A L, Maruyama K, Beckerleg A M, Torchilin V P, Huang L. Biochim Biophys Acta. 1991;1062:142–148. doi: 10.1016/0005-2736(91)90385-l. [DOI] [PubMed] [Google Scholar]
- 8.Torchilin V P. ImmunoMethods. 1994;4:244–258. doi: 10.1006/immu.1994.1027. [DOI] [PubMed] [Google Scholar]
- 9.Lee R J, Low P S. J Biol Chem. 1994;269:3198–3204. [PubMed] [Google Scholar]
- 10.Allen T M, Brandeis E, Hansen C B, Kao G Y, Zalipsky S. Biochim Biophys Acta. 1995;1237:99–108. doi: 10.1016/0005-2736(95)00085-h. [DOI] [PubMed] [Google Scholar]
- 11.Hansen C B, Kao G Y, Moase E H, Zalipsky S, Allen T M. Biochim Biophys Acta. 1995;1239:133–144. doi: 10.1016/0005-2736(95)00138-s. [DOI] [PubMed] [Google Scholar]
- 12.Maruyama K, Takizawa T, Yuda T, Kennel S J, Huang L, Iwatsuru M. Biochim Biophys Acta. 1995;1234:74–80. doi: 10.1016/0005-2736(94)00263-o. [DOI] [PubMed] [Google Scholar]
- 13.Shahinian S, Silvius J R. Biochim Biophys Acta. 1995;1239:157–167. doi: 10.1016/0005-2736(95)00145-s. [DOI] [PubMed] [Google Scholar]
- 14.Schackert G, Fan D, Nayar R, Fidler I J. Sel Cancer Ther. 1989;5:73–79. doi: 10.1089/sct.1989.5.73. [DOI] [PubMed] [Google Scholar]
- 15.Gennuso R, Spigelman M K, Chinol M, Zappulla R A, Nieves J, Vallabhajosula S, Alberto P P, Goldsmith S J, Holland J F. Cancer Invest. 1993;11:118–128. doi: 10.3109/07357909309024829. [DOI] [PubMed] [Google Scholar]
- 16.Sakamoto A, Ido T. Brain Res. 1993;629:171–175. doi: 10.1016/0006-8993(93)90499-d. [DOI] [PubMed] [Google Scholar]
- 17.Pardridge W M. Adv Drug Delivery Rev. 1995;15:109–146. [PubMed] [Google Scholar]
- 18.Bickel U, Kang Y S, Yoshikawa T, Pardridge W M. J Histochem Cytochem. 1994;42:1493–1497. doi: 10.1177/42.11.7930531. [DOI] [PubMed] [Google Scholar]
- 19.Jefferies W A, Brandon M R, Williams A F, Hunt S V. Immunology. 1985;54:333–341. [PMC free article] [PubMed] [Google Scholar]
- 20.Jefferies W A, Brandon M R, Hunt S V, Williams A F, Gatter K C, Mason D Y. Nature (London) 1984;312:162–163. doi: 10.1038/312162a0. [DOI] [PubMed] [Google Scholar]
- 21.Pardridge W M, Eisenberg J, Yang J. Metabolism. 1987;36:892–895. doi: 10.1016/0026-0495(87)90099-0. [DOI] [PubMed] [Google Scholar]
- 22.Kang Y S, Pardridge W M. J Pharmacol Exp Ther. 1994;269:344–350. [PubMed] [Google Scholar]
- 23.Yoshikawa T, Pardridge W M. J Pharmacol Exp Ther. 1992;263:897–903. [PubMed] [Google Scholar]
- 24.Triguero D, Buciak J B, Yang J, Pardridge W M. Proc Natl Acad Sci USA. 1989;86:4761–4765. doi: 10.1073/pnas.86.12.4761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marsh J W. J Biol Chem. 1988;263:15993–15999. [PubMed] [Google Scholar]
- 26.Riddles P W, Blakeley R L, Zerner B. Anal Biochem. 1979;94:75–81. doi: 10.1016/0003-2697(79)90792-9. [DOI] [PubMed] [Google Scholar]
- 27.Mayer L D, Bally M B, Cullis P R. Biochim Biophys Acta. 1986;857:123–126. doi: 10.1016/0005-2736(86)90105-7. [DOI] [PubMed] [Google Scholar]
- 28.Heath T D, Macher B A, Papahadjopoulos D. Biochim Biophys Acta. 1981;640:66–81. doi: 10.1016/0005-2736(81)90532-0. [DOI] [PubMed] [Google Scholar]
- 29.Kang Y S, Bickel U, Pardridge W M. Drug Metab Dispos. 1994;22:99–105. [PubMed] [Google Scholar]
- 30.Pardridge W M, Boado R J, Kang Y S. Proc Natl Acad Sci USA. 1995;92:5592–5596. doi: 10.1073/pnas.92.12.5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bickel U, Schumacher O, Kang Y-S, Voigt K. J Pharmacol Exp Ther. 1996;278:107–113. [PubMed] [Google Scholar]
- 32.Pardridge W M, Buciak J L, Friden P M. J Pharmacol Exp Ther. 1991;259:66–70. [PubMed] [Google Scholar]
- 33.Skarlatos S, Yoshikawa T, Pardridge W M. Brain Res. 1995;683:164–171. doi: 10.1016/0006-8993(95)00363-u. [DOI] [PubMed] [Google Scholar]
- 34.Martin F J, Papahadjopoulos D. J Biol Chem. 1982;257:286–288. [PubMed] [Google Scholar]
- 35.Heath T D, Martin F J. Chem Phys Lipids. 1986;40:347–358. doi: 10.1016/0009-3084(86)90078-2. [DOI] [PubMed] [Google Scholar]
- 36.Mayer L D, Tai L C, Ko D S, Masin D, Ginsberg R S, Cullis P R, Bally M B. Cancer Res. 1989;49:5922–5930. [PubMed] [Google Scholar]