

Abstract
Histone acetylation, regulated by the opposing actions of histone acetyltransferases (HATs) and deacetylases, is an important epigenetic mechanism in eukaryotic transcription. Although an acetyltransferase (PfGCN5) has been shown to preferentially acetylate histone H3 at K9 and K14 in Plasmodium falciparum, the scale of histone acetylation in the parasite genome and its role in transcriptional activation are essentially unknown. Using chromatin immunoprecipitation (ChIP) and DNA microarray, we mapped the global distribution of PfGCN5, histone H3K9 acetylation (H3K9ac) and trimethylation (H3K9m3) in the P. falciparum genome. While the chromosomal distributions of H3K9ac and PfGCN5 were similar, they are radically different from that of H3K9m3. In addition, there was a positive, though weak correlation between relative occupancy of H3K9ac on individual genes and the levels of gene expression, which was inversely proportional to the distance of array elements from the putative translational start codons. In contrast, H3K9m3 was negatively correlated with gene expression. Furthermore, detailed mapping of H3K9ac for selected genes using ChIP and real-time PCR in three erythrocytic stages detected stage-specific peak H3K9ac enrichment at the putative transcriptional initiation sites, corresponding to stage-specific expression of these genes. These data are consistent with H3K9ac and H3K9m3 as epigenetic markers of active and silent genes, respectively. We also showed that treatment with a PfGCN5 inhibitor led to reduced promoter H3K9ac and gene expression. Collectively, these results suggest that PfGCN5 is recruited to the promoter regions of genes to mediate histone acetylation and activate gene expression in P. falciparum.
Plasmodium falciparum, the causative agent of malignant tertian malaria, is responsible for more than 300 million cases and 1 to 3 million deaths per year (51). Extensive research efforts have led to the deciphering of its genome, transcriptome, and proteome, which has greatly advanced our understanding of the fundamental biology of the parasite (18, 22). The parasite life cycle encompasses several morphologically distinct stages while alternating between a vertebrate and an invertebrate host. Recent microarray studies revealed that tightly regulated gene expression in the parasite is in the form of a continuous cascade, with the induction of most genes occurring only once, when their products are needed (3, 39). In addition, antigenic variation of the highly polymorphic P. falciparum erythrocyte membrane protein 1 (PfEMP1), a major virulence factor, through monoallelic expression of ∼60 var genes, is primarily regulated at the transcription level (33). Although how such a global and localized transcription regulation is achieved is poorly understood, recent extensive studies of var gene regulation indicate that epigenetic control plays an important role in these processes (41, 45).
Compared to transcription in prokaryotes, there is an added layer of complexity in eukaryotes arising from packaging of genomes with histones into chromatin. The dynamic structure of chromatin serves as the scaffold on which the basic cellular processes of DNA replication, repair, and transcription take place. The N-terminal tails of histones are subject to various posttranslational modifications, including phosphorylation, acetylation, methylation, ubiquitination, and sumoylation. Different modifications affect the histone-DNA interactions and act sequentially or in combination to create a “histone code” (30, 53) that serves as signals read by regulatory molecules containing specific interacting domains such as the bromodomain, chromodomain, and SANT domain (12). These modifications play pivotal roles in regulating gene expression, and aberrance in modifications is associated with an array of disorders in humans. Among the histone modifications, acetylation is the most extensively studied and has formed the basis for an evolving model on how covalent histone modifications regulate transcription.
Similar to other eukaryotes, the 14 P. falciparum chromosomes have a typical nucleosomal organization involving core and variant histones that are dynamically acetylated at a number of lysine residues during the asexual erythrocytic cycle (6, 42). Although how histone acetylation regulates parasite genes is still poorly understood, the recent discovery of its role in regulating monoallelic expression of var gene family sheds new light on its significance (13, 21). In particular, these studies have demonstrated that the P. falciparum homologue of the yeast Sir2 (for silent information regulator 2) deacetylase appears to play a similar role in establishing subtelomeric heterochromatin structure, thus repressing the expression of a subset of subtelomeric var genes. While var gene expression is controlled exclusively at the level of transcription without the feedback of the PfEMP1 protein (14), transfection experiments have begun to elucidate the roles of var promoters and introns in mediating silencing (15, 19). Along this line, a number of recent studies implicated the role of cooperative interactions in cis between var upstream sequences and var intron in silencing (5, 11, 20), whereas Voss et al. (58) showed that the var promoter is sufficient to infiltrate a transgene into the allelic exclusion program of var gene expression with the var promoter mediating nucleation and spreading of stably inherited heterochromatin (58). Despite this discrepancy regarding the role of the var intron, epigenetics is likely the major regulator of monoallelic var gene expression. Furthermore, this epigenetic memory is probably inherited through covalent histone modifications such as H3K9 trimethylation (H3K9m3), which can persist in parasite lines for generations (7). In accordance with these notions, a glimpse of the parasite genome has revealed a full assembly of proteins regulating the organization and structure of chromatin (1). These include histone acetyltransferases (HATs) and histone deacetylases that catalyze the reversible histone lysine acetylation. In particular, a GNAT (GCN5-related N-acetyltransferase) family member, PfGCN5, has been characterized that preferentially acetylates H3 at K9 and K14 (16). The significance of PfGCN5 is further underscored by the finding that PfGCN5 is the center of an extensive interacting network of proteins, many of which are potentially involved in transcription (17, 35). Moreover, inhibition of PfGCN5 activity in vivo by a polyphenolic compound curcumin leads to hypoacetylation of histones, which may partially contribute to the toxicity of curcumin (10). This further testifies to the potential of enzymes regulating covalent modifications of histones as targets for novel chemotherapy of parasitic diseases (55).
To elucidate transcriptional regulatory networks and roles of relative occupancy and modification states of histones in controlling global transcription, many studies have used chromatin immunoprecipitation (ChIP) coupled with DNA microarray analysis (chip), a powerful genome-wide analysis technique known as ChIP-chip (24); however, the extreme AT-rich genome of P. falciparum has hampered application of this tool to study its gene regulatory circuitry despite the availability of the genome information and DNA microarrays. To dissect the complex network of transcription regulation in P. falciparum, we addressed the function of PfGCN5-mediated histone H3 acetylation by developing a genome map of histone acetylation and PfGCN5 localization using a preexisting DNA microarray. Here we report the first application of the ChIP-chip technique to map the global distribution of histone H3K9 acetylation (H3K9ac), trimethylation, and the coactivator PfGCN5 in the P. falciparum genome. By combining this analysis with gene expression profiling, we were able to associate H3 acetylation with gene expression and H3K9m3 with silent genes. Using conventional ChIP and real-time quantitative PCR analysis, we have shown that promoters of active genes are enriched with H3K9ac and deficient in H3K9m3, suggesting that PfGCN5-mediated H3 acetylation is likely involved in regulating gene expression in the malaria parasite.
MATERIALS AND METHODS
Parasite culture.
The P. falciparum 3D7 clone was cultured at 5% hematocrit in type O+ human red blood cells in complete medium in an atmosphere of 5% CO2, 3% O2, and 92% N2 (56). The parasites were synchronized twice by 5% sorbitol treatments (36). To obtain parasite materials, infected erythrocytes were lysed by 0.1% saponin treatment, and parasite pellets were collected by centrifugation and washed twice with cold phosphate-buffered saline (PBS). For stage-specific gene expression, synchronized parasites were harvested at 12, 20, 30, and 42 h to represent ring, early trophozoite, late trophozoite, and schizont stages, respectively.
ChIP-chip.
ChIP was performed as described previously with modifications (46). Briefly, 3 × 109 ∼28-h-old synchronized trophozoites were harvested in 10 ml of cold PBS and cross-linked with 1% formaldehyde at room temperature for 30 min. After the cross-linker was quenched with glycine, parasites were washed twice with cold PBS and resuspended in 1.5 ml of immunoprecipitation (IP) buffer (50 mM HEPES [pH 7.5], 140 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate) with a protease inhibitor cocktail (Roche, Indianapolis, IN). Sonication was performed on ice and monitored to ensure that the average size of sonicated DNA was 0.5 to 1 kb to suit the preexisting expression array. IP was performed at 4°C overnight with a 1:100 dilution of anti-H3K9ac and anti-H3K9m3 (Millipore, Billerica, MA) and a 1:50 dilution of anti-PfGCN5 (17). Antibodies for both H3K9ac and H3K9m3 are specific for the corresponding modified histones (16, 54). The DNA-protein complexes were eluted with an elution buffer (50 mM Tris-HCl [pH 8.0], 10 mM EDTA, 1% sodium dodecyl sulfate) and incubated at 65°C to reverse the cross-linking. DNA was isolated by using proteinase K digestion and phenol-chloroform extraction and then dissolved in TE buffer (10 mM Tris [pH 8.0], 5 mM EDTA). Labeling of immunoprecipitated and input DNA with Cy5 or Cy3 was performed by using the randomly primed PCR amplification method (40, 49). The labeled products were purified by using a MinElute PCR purification kit (QIAGEN, Valencia, CA). Labeling or real-time PCR analysis was repeated with immunoprecipitated DNA from at least three IP experiments.
Spotted microarrays used in the present study consisted of 7,462 individual 70-mer oligonucleotides representing 4,488 of the 5,409 open reading frames (ORFs) annotated by the malaria genome sequencing consortium (3). Cy5- and Cy3-labeled probes were mixed and hybridized to DNA microarrays using the MAUI hybridization system (BioMicro Systems, Salt Lake City, UT). Slides were scanned with a GenePix 4000B array scanner (Axon Instruments, Union City, CA). After scanning, the quality of the spots was verified individually by visual inspection. The signals from each array spot were calculated by the median pixel value of the spot minus that of background and normalized so that the median Cy5/Cy3 ratio was 1.0. Spots with signals less than the median pixel value of background plus twice the standard deviation of background pixel values were removed. The median value of individual spots from all experiments was used as the final data set. The data were transformed into the log2(Cy5:Cy3) ratio for statistical analysis. Pearson correlation between data sets was performed by using Minitab Software 13.0 (Minitab, Inc., State College, PA).
Quantitative analysis of ChIP enrichment by real-time PCR.
To verify the results from the microarray analysis and define the distribution of H3K9ac and H3K9m3 in individual genes, we first chose the MAL7P1.37 gene locus, which was highly enriched in both H3K9ac and PfGCN5 ChIP-chip analyses, for more detailed mapping using conventional ChIP and real-time PCR. The MAL7P1.37-38 locus spans ∼7 kb, with the two genes being arranged head to head and an intergenic region of ∼2.5 kb. MAL7P1.37 encodes a Sin3-associated p18-like protein, a component of the Sin3 complex that is responsible for the repression of transcription via histone deacetylation. MAL7P1.38 is a homologue of the regulator of chromatin condensation that regulates nuclear import and export. We designed nine pairs of primers spanning the entire MAL7P1.37-38 locus (ORFs and intergenic region) and assessed H3K9ac and H3K9m3 enrichment in the trophozoite stage (30 h) by ChIP and real-time PCR analysis. Stage-specific expression of these two genes was examined by real-time reverse transcriptase PCR (RT-PCR) analysis with the constitutively expressed seryl-tRNA synthetase as the reference gene.
To further correlate stage-specific H3K9ac and corresponding gene activation, we selected eight stage-specific or constitutively expressed genes for detailed mapping in ring, trophozoite, and schizont stage. These genes were selected largely due to their previously established expression patterns and/or transcription initiation sites, which include DNA polymerase δ (PF10_0165) and glycophorin-binding protein 130 (GBP130, PF10_0159) expressed in trophozoites (26, 37), MAL7P1.170 and heat shock protein 101 (HSP101, PF11_0175) expressed in rings (52), erythrocyte-binding protein 175 (EBA175, MAL7P1.176) and merozoite surface protein 1 (MSP1, PFI1475w) expressed in schizonts, and constitutively expressed actin-related gene (PF14_0218) and calmodulin (PF14_0323) (9). For PF14_0218, its constitutive expression was verified by real-time RT-PCR using four different stages of synchronized parasites (42). ChIP was performed with 9 × 109 parasites at the ring stage, and 3 × 109 parasites at the trophozoite and schizont stages (12, 28, and 42 h), respectively. Sets of primers were designed to amplify 150- to 300-bp fragments covering both the promoter and the coding regions (see Table S1 in the supplemental material). For real-time PCR analysis, the final concentration of the templates was adjusted to 1 ng/μl. Real-time PCR was performed as previously described (42) using the FastStart SYBR green master mix (Roche). Relative fold enrichment at different sites was calculated by using the 2ΔCT method, and the standard deviation was calculated from at least three IP experiments (42). For reproducibility of the data, IP experiments were done using chromatin preparations from two different cultures and representative data were shown.
Inhibition of PfGCN5 activity and gene expression.
To determine whether inhibition of PfGCN5 leads to reduced H3K9ac and gene expression, synchronized 3D7 cells at 18 h were treated with the PfGCN5 inhibitor curcumin (5 and 20 μM) for 12 h, which was shown to cause no obvious changes of parasite morphology or growth (10). Parasites from both control and treated groups were harvested and used for total RNA extraction and ChIP. RNA was isolated from aliquots of the same parasite populations using TRIzol reagent and treated with RNase-free DNase I (Promega, Madison, WI) to remove contaminating genomic DNA. cDNA was synthesized from 5 μg of total RNA using SuperScript III RT (Invitrogen, Carlsbad, CA) with oligo(dT) primer. A reference gene, PF07_0047, encoding a putative ATPase, whose expression was not significantly affected by curcumin treatment (unpublished microarray data), two trophozoite-specific genes, and two constitutively expressed genes mentioned above were evaluated by real-time RT-PCR analysis using pretested primer sets (see Table S1 in the supplemental material). The amplification efficiency of individual genes was normalized with the expression level of the reference gene, seryl-tRNA synthetase. Relative expression levels of tested genes were determined by using the 2−ΔΔCT method with ring stage being set as the reference stage (42). To determine whether the effect of the curcumin on gene expression was the result of the reduced acetylation in the promoter regions of the genes, ChIP was performed with anti-H3K9ac using equal numbers of parasites, and real-time PCR was performed using upstream regions of peak H3K9ac as described above.
RESULTS
Validation of the ChIP-chip method.
While a whole-genome tiling array is ideal for global localization study, here we used a preexisting DNA array and sought to develop a reproducible protocol for the ChIP-chip procedure that is suitable for the highly AT-rich P. falciparum genome. We first tested a rabbit preimmune serum and anti-H3K9ac antibodies for ChIP experiments on synchronized late-trophozoite-stage parasites. This stage was chosen because of the ease of procuring sufficient parasite materials for ChIP-chip analysis. We compared the feasibility of two modified labeling methods involving either ligation-mediated or randomly primed PCR amplification (46, 49) for fluorescent labeling of the ChIP products and input DNA with Cy5 or Cy3. The labeled products were hybridized to the spotted P. falciparum DNA microarrays, and the hybridization signals of each spot were obtained. To validate these methods, each experiment was repeated at least three times with parasites from different cultures; data from all experiments were analyzed statistically. We found that the latter labeling method yielded the most reproducible results, with high Pearson correlation efficiency between experiments (R > 0.90, P < 0.001).
Genome-wide localization of H3K9ac.
Using this modified labeling method, we first explored the genome-wide occupancy of H3K9ac during the late trophozoite stage (∼28 h). Hybridization signals from each spot were transformed into the log2(IP/input) values, and the data from three replicates of each antiserum were averaged and combined into bins with an increment of 0.075 for histogram analysis. Histograms showed that ChIP-chip with the control serum generated a normal distribution of the log2(IP/input) data, with 74% of the array elements within the range of −1 to +1 (Fig. 1A). In comparison, distribution of the anti-H3K9ac data formed a second peak at approximately the 2.2 to 2.8 log2(IP/input) points, which reproducibly identified 258 array elements (225 genes) with more than fourfold enrichment, representing 4.9% of the 4,488 genes on the microarray (Fig. 1B). With this low-density array, we found that elements enriched for H3K9ac were distributed on all 14 chromosomes (see Fig. S1A in the supplemental material). Classification of the highly enriched genes using the gene ontology (GO) terms grouped them into nine categories (see Fig. S2 in the supplemental material). Except for the largest category of genes with unknown functions (mostly encoding hypothetical proteins), proteins with binding activities (17.3%) and catalytic functions (16%) were among the largest categories. Interestingly, five genes in the binding category are expressed in merozoites and involved in erythrocyte invasion, suggesting that H3K9ac might also serve as a marker for genes that are poised for activation.
FIG. 1.

Histograms showing the distribution of enrichment of array elements from ChIP-chip experiments. Hybridizations were performed using synchronized P. falciparum late trophozoites (∼28 h) with a preimmune serum (A) and antibodies to H3K9ac (B). For each antibody, ChIP was performed in triplicate, and the averages of three experiments were used to construct the histograms. Each bar indicates the number of array elements with similar log2(IP/input) values binned together.
Genome-wide occupancy of PfGCN5 and its correlation with that of H3K9ac.
The GCN5 HAT in model organisms preferentially acetylates histone H3 at K9 and K14 (31, 57). To investigate whether the observed global H3K9ac distribution was the result of GCN5 HAT activity, we performed ChIP-chip analysis of late trophozoites (∼28 h) with anti-PfGCN5. Genome-wide distribution of PfGCN5 occupancy had a very similar pattern to that of H3K9ac (data not shown), with 207 genes having ≥4-fold enrichment. Not only were the H3K9ac and PfGCN5 data sets highly correlated (Pearson R = 0.88, P < 0.001) (Fig. 2A), but the highly enriched genes also had extensive overlap, with 136 genes being highly enriched in both data sets (Fig. 2B). Upon inspection of the expression profiles of these genes using the available microarray data (3, 39), 63% (86 of 136) were found expressed in trophozoites, suggesting that PfGCN5-mediated acetylation at least partially regulates the expression of these genes.
FIG. 2.

Genome-wide H3K9ac and PfGC5 occupancy profiles are highly correlated but different from that of H3K9m3 occupancy. (A) Correlation between the ChIP-chip datasets. R indicates Pearson correlation values. (B) Venn diagram comparing genes highly enriched (≥4-fold) for H3K9ac, PfGCN5, and H3K9m3.
Genome-wide occupancy of H3K9m3.
To further demonstrate that the significant correlation between the H3K9ac and PfGCN5 data sets was of functional importance but not the result of an accidental association, ChIP-chip was performed with parasites of the same developmental stage using antibodies for the silent chromatin marker H3K9m3. The results showed that H3K9m3 distribution was dramatically different from that of either H3K9ac or PfGCN5 (Fig. S1B), and the H3K9m3 data set was only weakly correlated with that of H3K9ac or PfGCN5 (R = 0.23 and R = 0.13, respectively) (Fig. 2A). H3K9m3 ChIP-chip analysis identified 205 genes with greater than fourfold enrichment. Consistent with H3K9m3 being a heterochromatin marker and associated with silent genes, microarray profiling data indicated that 153 of 205 (75%) were silent in the trophozoite stage (3). Furthermore, Of the 205 genes highly enriched for H3K9m3, only 5 and 11 genes overlapped with the highly enriched genes from H3K9ac and PfGCN5 analyses, respectively (Fig. 2B). These results further validated the consistency of the ChIP-chip experimental procedure in P. falciparum and indicated that H3K9ac and H3K9m3 were opposing epigenetic markers associated with different sets of genes.
Association of H3K9ac and H3K9m3 with transcription.
In model organisms, histone acetylation at the promoters is associated with gene activation. To determine whether H3K9ac is associated with transcriptionally active genes in P. falciparum, we compared the log2(IP/input) data set from H3K9ac ChIP-chip analysis with that from gene expression profiling at the same late trophozoite stage (3). The results showed that the two data sets were weakly, yet positively correlated by Pearson correlation analysis (R = 0.285, P < 0.102). This was expected given that many oligonucleotides in the microarray were located far downstream of the promoters. To evaluate this position effect, we divided the oligonucleotides into three groups according to their distances from the putative ATG start codon. Pearson correlation analysis demonstrated a stronger correlation between H3K9ac enrichment and gene expression for array elements within 500 bp of the putative ATG codon (Fig. 3). In contrast, when a similar analysis was performed with the H3K9m3 data, a negative correlation with the gene expression was observed, especially for array elements within 500 bp of the putative ATG codon (Fig. 3).
FIG. 3.
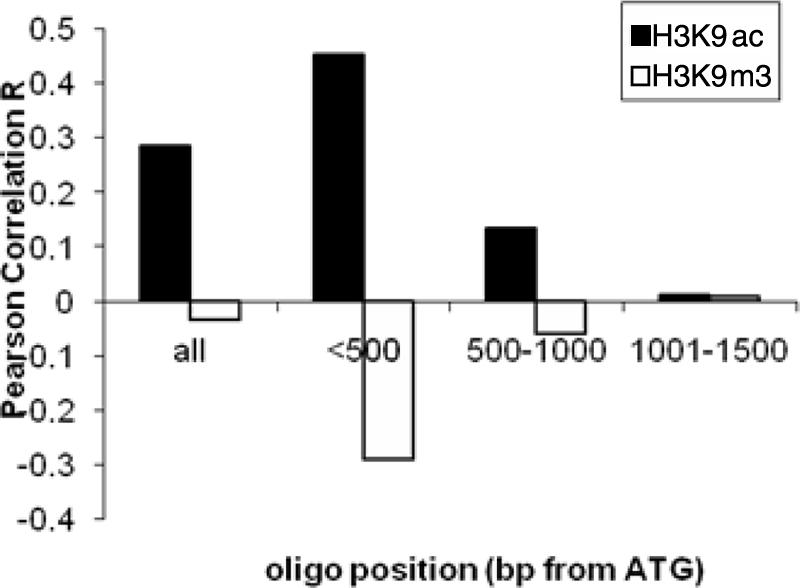
Correlation of H3K9ac and H3K9m3 enrichment with gene expression divided on the positions of the array elements (bp from the putative ATG). For the four groups of elements, the correlations (R) between H3K9ac and gene expression were 0.285 (P < 0.102), 0.453 (P < 0.002), 0.135 (P < 0.236), and 0.011 (P < 0.468), whereas the correlations (R) between H3K9m3 and gene expression were −0.035 (P < 0.019), −0.29 (P < 0.001), −0.061 (P < 0.135), and 0.009 (P < 0.553).
Association of H3K9ac with 5′ regions of active genes.
In general, H3K9ac is highly enriched in the promoters of active genes in model eukaryotes (43). To determine whether this principle also applies to P. falciparum, we first tested the MAL7P1.37-38 locus. Real-time RT-PCR analysis showed that MAL7P1.37 is most active in late trophozoites, whereas MAL7P1.38 is active in late schizonts (Fig. 4). ChIP and real-time PCR mapping of this region at the trophozoite stage showed that the intergenic region was enriched for H3K9ac, with the peak H3K9ac being skewed toward the active MAL7P1.37 gene (Fig. 4). In contrast, mapping with H3K9m3 showed that the distribution pattern of H3K9m3 was opposite of that for H3K9ac, with the peak H3K9m3 being located closer to the putative ATG codon of the inactive MAL7P1.38 gene. This observation is consistent with H3K9ac and H3K9m3 as opposing epigenetic markers of active and silent genes, respectively.
FIG. 4.

Mapping of H3K9ac and H3K9m3 at the MAL7P1.37-38 locus in trophozoites (28 h). The array element for MAL7P1.37 was highly enriched in the H3K9ac and PfGCN5 ChIP-chip data sets. (Top) RT-PCR analysis of MAL7P1.37 and MAL7P1.38 expression in asexual erythrocytic stages. R, rings; ET, early trophozoites; LT, late trophozoites; S, schizonts. Cycles indicate RT-PCR cycles for the respective genes, and numbers above the gels indicate average 2−ΔΔCT values by real-time RT-PCR with ring stage as the reference stage (42). (Middle) Schematic drawing to show the ORF of the active (filled arrow) and inactive (open arrow) genes in the trophozoite stage and their intergenic region (line). Primers were designed to amplify nine regions of this locus. Primer pairs 2 and 8 were used in RT-PCR analysis. (Bottom) ChIP enrichment (IP/input) at different regions of the locus to show that H3K9ac was higher in the 5′ region of MAL7P1.37, whereas H3K9m3 was highly enriched near the inactive MAL7P1.38 gene.
To further extend this observation to gene expression during different stages of intraerythrocytic development, we selected four sets of genes representing constitutively expressed and stage-specific genes and mapped their H3K9ac. The constitutive expression of the actin-related gene shown from microarray analysis was confirmed by real-time RT-PCR analysis (Fig. 5). To study the stage specificity of H3K9ac enrichment, ChIP and real-time PCR analysis were performed on synchronized parasites from the ring, trophozoite, and schizont stages. For stage-specific genes, the levels of H3K9ac enrichment were generally higher in the developmental stages when these genes were expressed, whereas the constitutively expressed genes had relatively steady levels of H3K9ac enrichment throughout the erythrocytic development of the parasite (Fig. 5). Mapping of H3K9m3 by ChIP and real-time PCR at the DNA polymerase δ and GBP130 loci in trophozoites again confirmed the opposing trends between H3K9ac and H3K9m3 (see Fig. S3 in the supplemental material). In addition to the correlation between H3K9ac enrichment levels and developmental activation of the genes, H3K9ac peaked in the 5′ regions of the genes and was likely associated with the promoter regions. For DNA polymerase δ, GPB130, and calmodulin genes, peak H3K9ac enrichment was mapped near the previously determined transcriptional initiation sites (9, 37, 44).
FIG. 5.

Mapping of H3K9ac to the promoters of active genes using ChIP and real-time PCR analysis. Selected genes represent stage-specific and constitutively expressed genes (marked on the right). (A and B) Expressed in trophozoite stage (DNA pol δ and GBP130); (C and D) expressed in ring stage (MAL7P1.170 and HSP101); (E and F) expressed in schizont stage (EBA175 and MSP1); (G and H) expressed in all stages (actin-related and calmodulin). ChIP was performed with synchronized parasites at the ring, trophozoite, and schizont stages. Real-time PCR was done using input DNA and DNA enriched by ChIP with anti-H3K9ac. For the gene schemes, filled and open block arrows indicate the orientation of expressed and silent genes, respectively. Intergenic regions are shown as thin lines. The positions of previously determined transcription initiation sites for DNA pol δ, GBP130, and calmodulin are indicated as arrows. The relative positions of PCR fragments (A to F) are indicated under the gene schemes. In each graph, the x axis indicates the position of the PCR fragments, and the y axis shows fold enrichment (IP/input) determined by real-time PCR. The inset in panel G shows the expression of the PF14_0218 gene in rings (R), early trophozoites (ET), late trophozoites (LT), and schizonts (S) by real-time RT-PCR compared to constitutive expression of seryl-tRNA synthetase in Fig. 4. Numbers below the gel indicate the average 2−ΔΔCT values with the ring stage as the reference stage (42).
Curcumin treatment, histone acetylation, and gene expression.
With the establishment of H3K9ac association with the promoters of active genes, we further sought to test the significance of PfGCN5-mediated histone acetylation in parasite development. Unable to genetically knock out PfGCN5 (L. Cui et al., unpublished), we sought to use the inhibitor curcumin to downregulate PfGCN5 activity and determine its effect on gene activity and acetylation. Treatment of early trophozoites with sub-50% inhibitory concentration doses of curcumin (5 and 20 μM) for 12 h, which had no obvious effect on parasite gross morphology or growth, did not alter the expression of the reference gene PF07_0047, whereas it reduced the expression of other four genes in a dose-dependent manner (Fig. 6A). In addition, ChIP and real-time PCR analysis detected dramatic reduction of H3K9ac in the upstream regions of these genes compared to insignificant change in the 5′ region of the PF07_0047 gene (Fig. 6B). These results suggest that disturbing PfGCN5 activity might be responsible for reduced acetylation in the promoters and reduced expression of certain tested genes, which may also explain the long-term effect of curcumin on parasites (10).
FIG. 6.
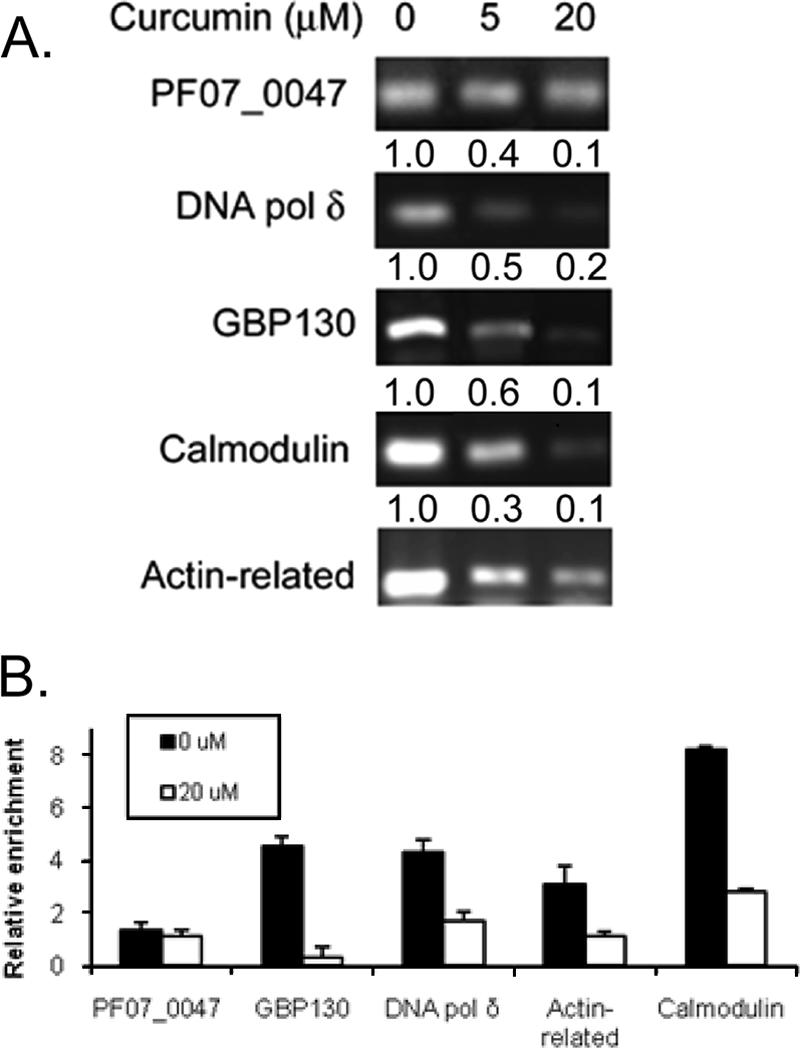
Curcumin treatment is associated with reduced gene expression and promoter acetylation. (A) Curcumin treatment (5 and 20 μM) of early trophozoites (18 h) for 12 h resulted in the reduced expression of DNA pol δ, GBP130, actin-related, and calmodulin genes. Shown here are gel pictures of semiquantitative RT-PCR for the PF07_0047 gene (20 cycles) and four other genes (25 cycles). Numbers above each panel indicate average 2−ΔΔCT values of the genes compared to that of PF07_0047 gene by real-time PCR with untreated parasites as the reference (42). (B) Reduced acetylation in the upstream regions of genes. Fragments used for real-time PCR analysis are DNA pol δ (c), GBP130 (d), actin-related (c), and calmodulin (b) as shown in Fig. 4. The region of the PF07_0047 gene was selected from a mapping experiment and the primers are shown in Table S1 in the supplemental material.
DISCUSSION
Genome-wide expression analysis of P. falciparum intraerythrocytic development reveals an interesting cascade-like pattern of gene expression (3, 39). Although how the parasite achieves such transcription regulation is not understood, recent advances in functional genomics of the parasite demonstrated the potential significance of epigenetic mechanisms in gene regulation (41, 45). Understanding these mechanisms will benefit greatly from the global view of dynamics of chromatin modifications in the parasite genome. With the availability of the parasite genome sequence and microarrays, we have demonstrated the applicability of the ChIP-chip technique for genome-wide localization analysis of transcriptional factors and chromatin modifications in P. falciparum through successful analysis of genome-wide localization of H3K9ac, H3K9m3, and PfGCN5 in late trophozoites.
In P. falciparum, the presence of basal RNA polymerase II transcription machinery and many components of chromatin remodeling complexes indicates evolutionary conservation of similar transcriptional activation pathways (1, 8). The GCN5-containing HAT complexes have been documented in yeast, humans, and fruit flies (4, 23, 32). That PfGCN5 forms an interaction network with a number of transcription-associated proteins suggests that transcription in Plasmodium may also be regulated by PfGCN5-containing protein complexes (17, 35). In yeast, deletion of GCN5 only affects the expression of 5 to 10% of genes, and a subset of these genes is stress related (25, 29, 38). The small set of genes affected by GCN5 in yeast may reflect its partial redundancy with other HAT proteins (27, 38). Our analysis revealed that in the late trophozoite stage of the P. falciparum intraerythrocytic development cycle, ∼5% of the genes were enriched with H3K9ac, 60% of which were also enriched with PfGCN5. Although it is not clear how the H3K9ac mark is delivered to the rest of genes, we can speculate that it could be the result of other HAT activities (e.g., the elongation factor protein 3 homologue PFL1345c) or that not all chromatin-associated PfGCN5 is measurable by the ChIP procedure due to its limited sensitivity. Similar chromosomal distributions of H3K9ac and PfGCN5 are congruent with the notion that GCN5 complexes are recruited to mediate the acetylation of H3K9, which may, in turn, enhance recruitment of PfGCN5 to these sites through the interaction of its bromodomain with H3K9ac (28). Although the present study suggests that PfGCN5 may be similarly involved in regulating ∼5% of genes, the rest of the genome may be regulated by other transcription complexes such as TFIID (29). Future studies using high-density arrays and different developmental stages will identify the full spectrum of genes regulated by PfGCN5. Regardless of the seemingly small pool of genes as potential PfGCN5 targets, our studies suggest that PfGCN5 has a more profound effect on parasite gene expression. Whereas yeast GCN5 is dispensable for growth in rich media, PfGCN5 appears essential, and its inhibition leads to developmental arrest of the parasite (10). Our data indicate that the cytotoxic effect of a PfGCN5 inhibitor could be attributed to its disturbance of the histone mark H3K9ac in the upstream sequences of certain genes, leading to their aberrant expression.
H3K9 is subject to both acetylation and methylation. In general, H3K9 acetylation is associated with active transcription, whereas methylation is associated with transcriptional repression (34). With the lack of many canonical eukaryotic transcriptional factors in P. falciparum, we wonder whether these markers can also be used to distinguish between active and silent genes in the parasite. Our analysis revealed a positive, albeit weak, correlation between PfGCN5, H3K9ac, and transcription activity; This correlation became stronger if the analysis used array elements within 500 bp of the putative translation start sites of genes, suggesting that PfGCN5 and H3K9ac occupancies were higher in the 5′ regions of active genes. In contrast, H3K9m3 had a distinct occupancy pattern and had little correlation with the H3K9ac or PfGCN5 data set. This result suggests that acetylation and methylation of H3K9 (mutually exclusive) occupy different domains on P. falciparum chromosomes and that their switching may contribute to regulating gene activity. Our data are consistent with the association of H3K9ac with active genes found in yeast and higher eukaryotes (2, 43, 48). In addition, detailed mapping of selected genes indicated that relative levels of H3K9ac at the promoters are dynamically regulated, coincidental with gene activation and silencing through various developmental stages. Our analysis suggests that, as seen in yeast, HAT proteins are recruited to the predicted transcription start sites to activate genes in P. falciparum (43, 47). Although H3K9m3 appears to be an epigenetic mark associated with long-term memory of the var genes (7), its association with promoters of silent genes suggests that it may also serve as a dynamic mark controlling the on-off status of the genes. Noteworthy, the recent identification of a large group of evolutionarily conserved histone lysine demethylases has revealed the reversible and highly dynamic nature of histone lysine methylation marks and their involvements in regulating transcription (50).
Collectively, our study has demonstrated the suitability of ChIP-chip technique for genome-wide association studies on the AT-rich genome of the malaria parasite. We have determined low-density maps of global occupancies of histone acetylation, methylation, and a transcription coactivator in the late trophozoite stage. This study has established a solid foundation for future detailed global localization analysis of the histone code in this parasite at different stages using high-density arrays. We have demonstrated that, as in model eukaryotes, PfGCN5-mediated H3 acetylation is also involved in general gene regulation of the malaria parasite and that these epigenetic pathways constitute viable targets for designing novel chemotherapy.
Supplementary Material
Acknowledgments
This study was supported by a NIH grant to L.C. (AI064553) and the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health, to X.S.
We thank NIAID intramural editor Brenda Rae Marshall for assistance.
Footnotes
Published ahead of print on 20 April 2007.
Supplemental material for this article may be found at http://ec.asm.org/.
REFERENCES
- 1.Aravind, L., L. M. Iyer, T. E. Wellems, and L. H. Miller. 2003. Plasmodium biology: genomic gleanings. Cell 115:771-785. [DOI] [PubMed] [Google Scholar]
- 2.Bernstein, B. E., M. Kamal, K. Lindblad-Toh, S. Bekiranov, D. K. Bailey, D. J. Huebert, S. McMahon, E. K. Karlsson, E. J. Kulbokas III, T. R. Gingeras, S. L. Schreiber, and E. S. Lander. 2005. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell 120:169-181. [DOI] [PubMed] [Google Scholar]
- 3.Bozdech, Z., M. Llinas, B. L. Pulliam, E. D. Wong, J. Zhu, and J. L. DeRisi. 2003. The transcriptome of the intraerythrocytic developmental cycle of Plasmodium falciparum. PLOS Biol. 1:E5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brown, C. E., T. Lechner, L. Howe, and J. L. Workman. 2000. The many HATs of transcription coactivators. Trends Biochem. Sci. 25:15-19. [DOI] [PubMed] [Google Scholar]
- 5.Calderwood, M. S., L. Gannoun-Zaki, T. E. Wellems, and K. W. Deitsch. 2003. Plasmodium falciparum var genes are regulated by two regions with separate promoters, one upstream of the coding region and a second within the intron. J. Biol. Chem. 278:34125-34132. [DOI] [PubMed] [Google Scholar]
- 6.Cary, C., D. Lamont, J. P. Dalton, and C. Doerig. 1994. Plasmodium falciparum chromatin: nucleosomal organisation and histone-like proteins. Parasitol. Res. 80:255-258. [DOI] [PubMed] [Google Scholar]
- 7.Chookajorn, T., R. Dzikowski, M. Frank, F. Li, A. Z. Jiwani, D. L. Hartl, and K. Deitsch. 2007. Epigenetic memory at malaria virulence genes. Proc. Natl. Acad. Sci. USA 104:899-902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coulson, R. M., N. Hall, and C. A. Ouzounis. 2004. Comparative genomics of transcriptional control in the human malaria parasite Plasmodium falciparum. Genome Res. 14:1548-1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crabb, B. S., and A. F. Cowman. 1996. Characterization of promoters and stable transfection by homologous and nonhomologous recombination in Plasmodium falciparum. Proc. Natl. Acad. Sci. USA 93:7289-7294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cui, L., J. Miao, and L. Cui. 2007. Cytotoxic effect of curcumin on malaria parasite Plasmodium falciparum: inhibition of histone acetylation and generation of reactive oxygen species. Antimicrob. Agents Chemother. 51:488-494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deitsch, K. W., M. S. Calderwood, and T. E. Wellems. 2001. Malaria. Cooperative silencing elements in var genes. Nature 412:875-878. [DOI] [PubMed] [Google Scholar]
- 12.De la Cruz, X., S. Lois, S. Sanchez-Molina, and M. A. Martinez-Balbas. 2005. Do protein motifs read the histone code? Bioessays 27:164-175. [DOI] [PubMed] [Google Scholar]
- 13.Duraisingh, M. T., T. S. Voss, A. J. Marty, M. F. Duffy, R. T. Good, J. K. Thompson, L. H. Freitas-Junior, A. Scherf, B. S. Crabb, and A. F. Cowman. 2005. Heterochromatin silencing and locus repositioning linked to regulation of virulence genes in Plasmodium falciparum. Cell 121:13-24. [DOI] [PubMed] [Google Scholar]
- 14.Dzikowski, R., M. Frank, and K. Deitsch. 2006. Mutually exclusive expression of virulence genes by malaria parasites is regulated independently of antigen production. PLoS Pathogens 2:184-194 (e22). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dzikowski, R., T. J. Templeton, and K. Deitsch. 2006. Variant antigen gene expression in malaria. Cell. Microbiol. 8:1371-1381. [DOI] [PubMed] [Google Scholar]
- 16.Fan, Q., L. An, and L. Cui. 2004. Plasmodium falciparum histone acetyltransferase, a yeast GCN5 homologue involved in chromatin remodeling. Eukaryot. Cell 3:264-276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fan, Q., L. An, and L. Cui. 2004. PfADA2, a Plasmodium falciparum homologue of the transcriptional coactivator ADA2 and its in vivo association with the histone acetyltransferase PfGCN5. Gene 336:252-261. [DOI] [PubMed] [Google Scholar]
- 18.Florens, L., M. P. Washburn, J. D. Raine, R. M. Anthony, M. Grainger, J. D. Haynes, J. K. Moch, N. Muster, J. B. Sacci, D. L. Tabb, A. A. Witney, D. Wolters, Y. Wu, M. J. Gardner, A. A. Holder, R. E. Sinden, J. R. Yates, and D. J. Carucci. 2002. A proteomic view of the Plasmodium falciparum life cycle. Nature 419:520-526. [DOI] [PubMed] [Google Scholar]
- 19.Frank, M., and K. Deitsch. 2006. Activation, silencing and mutually exclusive expression within the var gene family of Plasmodium falciparum. Int. J. Parasitol. 36:975-985. [DOI] [PubMed] [Google Scholar]
- 20.Frank, M., R. Dzikowski, D. Costantini, B. Amulic, E. Berdougo, and K. Deitsch. 2006. Strict pairing of var promoters and introns is required for var gene silencing in the malaria parasite Plasmodium falciparum. J. Biol. Chem. 281:9942-9952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Freitas-Junior, L., R. Hernandez-Rivas, S. A. Ralph, D. Montiel-Condado, O. K. Ruvalcaba-Salazar, A. P. Rojas-Meza, L. Mancio-Silva, R. J. Leal-Silvestre, A. M. Gontijo, S. Shorte, and A. Scherf. 2005. Telomeric heterochromatin propagation and histone acetylation control mutually exclusive expression of antigenic variation genes in malaria parasites. Cell 121:25-36. [DOI] [PubMed] [Google Scholar]
- 22.Gardner, M. J., N. Hall, E. Fung, et al. 2002. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature 419:498-511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grant, P. A., D. E. Sterner, L. J. Duggan, J. L. Workman, and S. L. Berger. 1998. The SAGA unfolds: convergence of transcription regulators in chromatin-modifying complexes. Trends Cell Biol. 8:193-197. [DOI] [PubMed] [Google Scholar]
- 24.Hanlon, S. E., and J. D. Lieb. 2004. Progress and challenge in profiling the dynamics of chromatin and transcription factor binding with DNA microarrays. Curr. Opin. Genet. Dev. 14:697-705. [DOI] [PubMed] [Google Scholar]
- 25.Holstege, F. C., E. G. Jennings, J. J. Wyrick, T. I. Lee, C. J. Hengartner, M. R. Green, T. R. Golub, E. S. Lander, and R. A. Young. 1998. Dissecting the regulatory circuitry of a eukaryotic genome. Cell 95:717-728. [DOI] [PubMed] [Google Scholar]
- 26.Horrocks, P., M. Jackson, S. Cheesman, J. H. White, and B. J. Kilbey. 1996. Stage specific expression of proliferating cell nuclear antigen and DNA polymerase δ from Plasmodium falciparum. Mol. Biochem. Parasitol. 79:177-182. [DOI] [PubMed] [Google Scholar]
- 27.Howe, L., D. Auston, P. Grant, S. John, R. G. Cook, J. L. Workman, and L. Pillus. 2001. Histone H3 specific acetyltransferases are essential for cell cycle progression. Genes Dev. 15:3144-3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hudson, B. P., M. A. Martinez-Yamout, H. J. Dyson, and P. E. Wright. 2000. Solution structure and acetyl-lysine binding activity of the GCN5 bromodomain. J. Mol. Biol. 304:355-370. [DOI] [PubMed] [Google Scholar]
- 29.Huisinga, K. L., and B. F. Pugh. 2004. A genome-wide housekeeping role for TFIID and a highly regulated stress-related role for SAGA in Saccharomyces cerevisiae. Mol. Cell 13:573-585. [DOI] [PubMed] [Google Scholar]
- 30.Jenuwein, T., and C. D. Allis. 2001. Translating the histone code. Science 293:1074-1080. [DOI] [PubMed] [Google Scholar]
- 31.Kuo, M.-H., J. Brownell, R. E. Sobel, T. A. Ranalli, D. G. Edmonson, S. Y. Roth, and C. D. Allis. 1996. Transcription-linked acetylation by Gcn5p of histone H3 and H4 at specific lysines. Nature 383:269-272. [DOI] [PubMed] [Google Scholar]
- 32.Kusch, T., S. Guelman, S. M. Abmayr, and J. L. Workman. 2003. Two Drosophila Ada2 homologues function in different multiprotein complexes. Mol. Cell. Biol. 23:3305-3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kyes, S., P. Horrocks, and C. Newbold. 2001. Antigenic variation at the infected red cell surface in malaria. Annu. Rev. Microbiol. 55:673-707. [DOI] [PubMed] [Google Scholar]
- 34.Lachner, M., R. J. O'Sullivan, and T. Jenuwein. 2003. An epigenetic road map for histone lysine methylation. J. Cell Sci. 116:2117-2124. [DOI] [PubMed] [Google Scholar]
- 35.LaCount, D. J., M. Vignali, R. Chettier, A. Phansalker, R. Bell, J. R. Hesselberth, L. W. Schoenfeld, I. Ota, S. Sahasrabuhde, C. Kurschner, S. Fields, and R. Hughes. 2004. A protein interaction network of the malaria parasite Plasmodium falciparum. Nature 438:103-107. [DOI] [PubMed] [Google Scholar]
- 36.Lambros, C., and J. P. Vanderberg. 1979. Synchronization of Plasmodium falciparum erythrocyte stages in culture. J. Parasitol. 65:418-420. [PubMed] [Google Scholar]
- 37.Lanzer, M., D. de Bruin, and J. V. Ravetch. 1992. Transcription mapping of a 100 kb locus of Plasmodium falciparum identifies an intergenic region in which transcription terminates and reinitiates. EMBO J. 11:1949-1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee, T. I., H. C. Causton, F. C. P. Holstege, W. C. Shen, N. Hannett, E. G. Jannings, F. Winston, M. R. Green, and R. A. Young. 2000. Redundant roles for the TFIID and SAGA complexes in global transcription. Nature 405:701-704. [DOI] [PubMed] [Google Scholar]
- 39.Le Roch, K. G., Y. Zhou, P. L. Blair, M. Grainger, J. K. Moch, J. D. Haynes, P. De la Vega, A. A. Holder, S. Batalov, D. J. Carucci, and E. A. Winzeler. 2003. Discovery of gene function by expression profiling of the malaria parasite life cycle. Science 301:1503-1508. [DOI] [PubMed] [Google Scholar]
- 40.Lieb, J. D., X. Liu, D. Botstein, and P. O. Brown. 2001. Promoter-specific binding of Rap1 revealed by genome-wide maps of protein-DNA association. Nat. Genet. 28:327-334. [DOI] [PubMed] [Google Scholar]
- 41.Merrick, C. J., and M. T. Duraisingh. 2006. Heterochromatin-mediated control of virulence gene expression. Mol. Microbiol. 62:612-620. [DOI] [PubMed] [Google Scholar]
- 42.Miao, J., Q. Fan, L. Cui, J. Li, J. Li, and L. Cui. 2006. The malaria parasite Plasmodium falciparum histones: organization, expression, and acetylation. Gene 369:53-65. [DOI] [PubMed] [Google Scholar]
- 43.Pokholok, D. K., C. T. Harbison, S. Levine, M. Cole, N. M. Hannett, T. I. Lee, G. W. Bell, K. Walker, P. A. Rolfe, E. Herbolsheimer, J. Zeitlinger, F. Lewitter, D. K. Gifford, and R. A. Young. 2005. Genome-wide map of nucleosome acetylation and methylation in yeast. Cell 122:517-527. [DOI] [PubMed] [Google Scholar]
- 44.Porter, M. E. 2001. The DNA polymerase delta promoter from Plasmodium falciparum contains an unusually long 5′ untranslated region and intrinsic DNA curvature. Mol. Biochem. Parasitol. 114:249-255. [DOI] [PubMed] [Google Scholar]
- 45.Ralph, S. A., and A. Scherf. 2005. The epigenetic control of antigenic variation in Plasmodium falciparum. Curr. Opin. Microbiol. 8:434-440. [DOI] [PubMed] [Google Scholar]
- 46.Ren, B., F. Robert, J. J. Wyrick, O. Aparicio, E. G. Jennings, I. Simon, J. Zeitlinger, J. Schreiber, N. Hannett, E. Kanin, T. L. Volkert, C. J. Wilson, S. P. Bell, and R. A. Young. 2000. Genome-wide location and function of DNA binding proteins. Science 290:2306-2309. [DOI] [PubMed] [Google Scholar]
- 47.Robert, F., D. K. Pokholok, N. M. Hannett, N. J. Rinaldi, M. Chandy, A. Rolfe, J. L. Workman, D. K. Gifford, and R. A. Young. 2004. Global position and recruitment of HATs and HDACs in the yeast genome. Mol. Cell 16:199-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schübeler, D., D. M. MacAlpine, D. Scalzo, C. Wirbelauer, C. Kooperberg, F. van Leeuwen, D. E. Gottschling, L. P. O'Neill, B. M. Turner, J. Delrow, S. P. Bell, and M. Groudine. 2004. The histone modification pattern of active genes revealed through genome-wide chromatin analysis of a higher eukaryote. Genes Dev. 18:1263-1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schübeler, D., D. Scalzo, C. K. Kooperberg, B. Van Steensel, J. Delrow, and M. Groudine. 2002. Genome-wide DNA replication profile for Drosophila melanogaster: a link between transcription and replication timing. Nat. Genet. 32:438-442. [DOI] [PubMed] [Google Scholar]
- 50.Shi, Y., and J. R. Whetstine. 2007. Dynamic regulation of histone lysine methylation by demethylases. Mol. Cell 25:1-14. [DOI] [PubMed] [Google Scholar]
- 51.Snow, R. W., C. A. Guerra, A. M. Noor, H. Y. Myint, and S. I. Hay. 2005. The global distribution of clinical episodes of Plasmodium falciparum malaria. Nature 434:214-217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Spielman, T., and H. P. Beck. 2000. Analysis of stage-specific transcription in Plasmodium falciparum reveals a set of genes exclusively transcribed in ring stage parasites. Mol. Biochem. Parasitol. 111:453-458. [DOI] [PubMed] [Google Scholar]
- 53.Strahl, B. D., and C. D. Allis. 2000. The language of covalent histone modifications. Science 403:41-45. [DOI] [PubMed] [Google Scholar]
- 54.Suka, N., Y. Suka, A. A. Carmen, J. Wu, and M. Grunstein. 2001. Highly specific antibodies determine histone acetylation site usage in yeast heterochromatin and euchromatin. Mol. Cell 8:473-479. [DOI] [PubMed] [Google Scholar]
- 55.Sullivan, W. J., Jr., A. Naguleswaran, and S. O. Angel. 2006. Histones and histone modifications in protozoan parasites. Cell. Microbiol. 8:1850-1861. [DOI] [PubMed] [Google Scholar]
- 56.Trager, W., and J. B. Jensen. 1976. Human malaria parasites in continuous culture. Science 193:673-675. [DOI] [PubMed] [Google Scholar]
- 57.Vogelauer, M., J. Wu, N. Suka, and M. Grunstein. 2000. Global histone acetylation and deacetylation in yeast. Nature 408:495-498. [DOI] [PubMed] [Google Scholar]
- 58.Voss, T. S., J. Healer, A. J. Marty, M. F. Duffy, J. K. Thompson, J. G. Beeson, J. C. Reeder, B. S. Crabb, and A. F. Cowman. 2006. A var gene promoter controls allelic exclusion of virulence genes in Plasmodium falciparum malaria. Nature 439:1004-1008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.