

Abstract
Transcription of the Pseudomonas aeruginosa type III secretion system (T3SS) is induced under Ca2+-limiting growth conditions or following the contact of the bacteria with host cells. The regulatory response to low Ca2+ levels is initiated by the T3SS-mediated secretion of ExsE, a negative regulatory protein that prevents T3SS gene transcription. In the present study, we demonstrated that ExsE plays an analogous role in transcriptional induction following host cell contact. By using a flow cytometry assay, the host contact-dependent induction of T3SS gene expression was found to be dependent upon the presence of functional type III translocation machinery. Using three independent assays, we demonstrated that ExsE was translocated into Chinese hamster ovary cells in a T3SS-dependent manner. Deletion mapping experiments indicated that the amino terminus of ExsE is required both for secretion under Ca2+-limiting growth conditions and for translocation into host cells. A P. aeruginosa mutant expressing an exsE allele lacking codons 3 through 20 was deficient in ExsE secretion and translocation and showed constitutive repression of T3SS gene expression under Ca2+-limiting growth conditions. The mutant also failed to induce T3SS gene expression following host cell contact and demonstrated a significant reduction in T3SS-dependent cytotoxicity towards Chinese hamster ovary cells, indicating that the translocation of ExsE is required for the host contact-dependent induction of T3SS gene expression.
Infections resulting from the gram-negative opportunistic pathogen Pseudomonas aeruginosa are usually restricted to patients with underlying immunodeficiency, cystic fibrosis, severe burns, or ulcerations (22, 23). P. aeruginosa utilizes a type III secretion system (T3SS) to evade phagocytosis and intoxicate eukaryotic cells (2, 25). The T3SS consists of ∼40 coordinately expressed genes that encode structural components of the secretion and translocation machinery, translocated effector proteins, and regulatory factors (6).
Although the physiological induction signal for T3SS gene expression appears to be the intimate contact of P. aeruginosa with a target eukaryotic cell (30), the mechanism involved in sensing host cell contact is poorly understood. Fortunately, T3SS gene expression can be induced experimentally by growing P. aeruginosa in Ca2+-limiting medium (14), and most of the details of T3SS gene regulation have been deduced from previous studies using this artificial condition. Ca2+ limitation indirectly activates T3SS gene expression by converting the type III secretion machinery from a secretion-incompetent to a secretion-competent state through an unknown mechanism (17). The central regulator of T3SS gene transcription is ExsA, a member of the AraC/XylS family of transcriptional activators (13, 34, 35). ExsA binds DNA elements within T3SS promoters to activate transcription. The transcriptional activity of ExsA is intimately coupled with secretion competency by a regulatory cascade consisting of ExsD, ExsC, and ExsE (37). ExsD is a negative regulator (antiactivator) of T3SS gene expression and functions by directly binding ExsA to inhibit transcription (17). ExsC functions as an anti-antiactivator by directly binding to and inhibiting the negative regulatory activity of ExsD (5, 38). Finally, ExsE antagonizes ExsC activity through a direct binding interaction (24, 29, 38). Under high-Ca2+-level conditions, ExsE sequesters ExsC, allowing ExsD to inhibit ExsA-dependent transcription. In response to Ca2+ limitation, however, ExsE is secreted via the T3SS. Under this condition, ExsC sequesters ExsD and ExsA-dependent transcription ensues. In the present study, we tested the hypothesis that the ExsADCE regulatory cascade involved in the response to Ca2+ limitation also functions for the more physiologically relevant host cell contact induction signal.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
The bacterial strains used in this study are given in Table 1. P. aeruginosa strains were maintained on Vogel-Bonner minimal (VBM) medium (31). For the expression of the T3SS, strains were grown at 30°C in TSB (Trypticase soy broth supplemented with 1% glycerol and 100 mM monosodium glutamate) either with or without 2 mM EGTA as indicated below. Strains carrying plasmids were grown in the presence of 300 μg of carbenicillin/ml and 100 μg of gentamicin/ml where indicated.
TABLE 1.
Bacterial strains and plasmids
| Strain or plasmid | Relevant characteristic(s)b | Source or reference |
|---|---|---|
| Strainsa | ||
| PA103 | P. aeruginosa wild-type parent | 7 |
| exsA::Ω mutant | Ω element inactivation of exsA | 7 |
| ΔexsC mutant | Nonpolar deletion of exsC | 5 |
| ΔexsD mutant | Nonpolar deletion of exsD | 17 |
| exsE107ΔN mutant | Nonpolar deletion of exsE codons 3-20 | This study |
| ΔpcrV mutant | Nonpolar deletion of pcrV | 26 |
| popD::Ω mutant | Ω element inactivation of popD | 36 |
| ΔpscC mutant | Nonpolar deletion of pscC | This study |
| ΔexoUT::Ω mutant | Deletion of exoU; Ω element inactivation of exoT | 36 |
| Plasmids | ||
| mini-CTX1 | Cloning vector | 11 |
| pPROBE-NT | Carries promoterless gfp gene | 19 |
| pNMD60 | Source of B. pertussis cyaA gene | N. Carbonetti |
| pJN105 | Cloning vector; carries arabinose-inducible promoter; Gmr | 20 |
| pUCPexsC | ExsC overexpression plasmid; Cbr | 5 |
| pJNE05 | Carries transcriptional fusion of P. aeruginosa strain 388 exoS promoter region to gfp gene; Gmr | This study |
| pUY44wt | Arabinose-inducible expression of ExsE-GSK; Gmr | This study |
| pUY30 | Arabinose-inducible expression of ExsE-CyaA; Gmr | This study |
| pUY36ΔN5 | Deletion of exsE codons 2-5 in pUY30; Gmr | This study |
| pUY36ΔN10 | Deletion of exsE codons 2-10 in pUY30; Gmr | This study |
| pUY34ΔN20 | Deletion of exsE codons 3-20 in pUY30; Gmr | This study |
All mutant strains are derivatives of P. aeruginosa strain PA103.
Cbr, carbenicillin resistance; Gmr, gentamicin resistance.
To construct the exsE107ΔN strain, a deletion plasmid based on vector pEX18Gm (10) and containing DNA flanking the exsE gene from P. aerugnosa strain PA103 was constructed by overlap PCR using internal exsE primers that contained a deletion of exsE codons 3 through 20. The mutated allele was transferred into the chromosome of wild-type PA103 by homologous recombination with the removal of plasmid vector sequences as previously described (17). The resulting mutant, the exsE107ΔN strain, was verified by PCR and by DNA sequencing of the entire region involved in the construction. The ΔpscC, ΔexsC, and ΔexoT mutants were similarly constructed in the PA103 genetic background by using the following plasmids: pEX18GmΔpscC, resulting in an in-frame deletion of pscC codons 10 to 567 (33); pEX18TcΔexsC, resulting in an in-frame deletion of exsC codons 8 to 141 (5); and pEX18GmΔexoT (this study), resulting in an in-frame deletion of exoT codons 4 to 444.
Plasmids.
The plasmids used in this study are given in Table 1. Plasmid pJNE05 was constructed by PCR amplification of the promoter region of the exoS gene from reporter plasmid pMini-PexoS-lacZ (17) and cloning of the resulting HindIII-EcoRI fragment in front of the coding region of the green fluorescent protein (GFP) gene on plasmid pPROBE-NT. The exoS-gfp transcriptional fusion was then PCR amplified as a SacI-KpnI fragment, cloned into plasmid miniCTX1, and finally transferred as an XbaI fragment to plasmid pJN105, which had been modified by a SmaI-DraI deletion of araC and the PBAD promoter.
Plasmid pUY30 was constructed by first PCR amplifying codons 2 through 399 of the Bordetella pertussis cyaA gene carried on plasmid pNMD60 by using XbaI- and SacI-containing primers and cloning the resulting ∼1,200-bp XbaI-SacI restriction fragment into vector pJN105. Subsequently, the exsE gene from P. aeruginosa strain PA103 was PCR amplified using an upstream primer incorporating an EcoRI site and the ribosome binding region from the exsC gene fused to the start codon of exsE via an NdeI restriction site (5′AAGAATTCAGGAGGCGCCCATATGAAAATCGAATCGATTCCGCCGGTGC); the downstream primer changed the exsE stop codon to a serine codon and introduced an XbaI restriction site. The resulting EcoRI-XbaI fragment was cloned in front of the cyaA gene, resulting in a translational fusion of exsE to cyaA. Plasmids pUY36ΔN5, pUY36ΔN10, and pUY34ΔN20 were constructed by replacing the NdeI-XbaI restriction fragment in plasmid pUY30 with analogous PCR-generated fragments having deletions of exsE codons 2 through 5, 2 through 10, and 3 through 20, respectively. Plasmid pUY44wt was constructed by replacing the XbaI-SacI cyaA fragment on pUY30 with a 55-bp XbaI-SacI fragment encoding the 15-amino-acid (LQMSGRPRTTSFAES) glycogen synthase kinase 3β (GSK) tag (8), resulting in an exsE-gsk translational fusion.
Flow cytometry.
Chinese hamster ovary (CHO) cells (ATCC CCL-61) were maintained and grown as previously described (4) in Ham's F-12 nutrient medium containing glutamine (Invitrogen, Carlsbad, CA). CHO cells were seeded into 24-well tissue culture plates (2 × 105 cells/well), grown overnight, and then washed with 1 ml of prewarmed Ham's F-12 tissue culture medium. Bacteria grown overnight on VBM agar containing gentamicin were suspended in prewarmed tissue culture medium (106 cells/ml), and 1-ml aliquots were added to CHO cells. Plate contents were centrifuged (500 × g for 5 min at 25°C), and then the plates were incubated at 37°C for 4 h. After incubation, the growth medium was aspirated and the CHO cells were washed three times with phosphate-buffered saline (PBS), and then 1 ml of a mixture of PBS and 0.1% Triton X-100 was added to each well. To facilitate the lysis of the CHO cells and the dispersion of the bacterial cells, the plate was subjected to nutation (5 min at 25°C) and the contents of each well were then transferred into a 1.5-ml tube and sonicated in a bath sonicator (5 min at 25°C). As an uninduced control, a sample of each bacterial strain in 1 ml of Ham's F-12 medium was incubated for 4 h in an empty well on the same plate and then centrifuged, and the pellet was resuspended in 1 ml of PBS-0.1% Triton X-100. To further control for the effects of CHO cell debris on fluorescence, this resuspended pellet solution was added to a well of PBS-washed CHO cells and the mixture was subjected to nutation and sonication as described above. This control sample reflected the level of uninduced PexoS-gfp expression during the 4-h incubation in Ham's F-12 medium in the absence of CHO cell contact. For the flow cytometry analysis of P. aeruginosa alone, cells were grown at 30°C in TSB in the absence or presence of EGTA to an optical density at 540 nm of 1 and suspended (107 CFU/ml) in PBS. All samples were assayed for fluorescence by counting 50,000 bacteria per sample using a Becton Dickson FACScan at the University of Iowa Flow Cytometry Facility.
Translocation assays.
The GSK tag translocation assay was based on a previously reported method (8). CHO cells were seeded into six-well tissue culture plates at 106 cells/well, grown overnight, and then washed twice with 2.5 ml of tissue culture medium. Bacterial strains grown overnight on VBM agar (containing gentamicin and carbenicillin) were suspended in 2.5 ml of prewarmed tissue culture medium containing 0.4% arabinose and carbenicillin. The suspension (108 cells) was added to the washed CHO cells, the plate contents were centrifuged (500 × g for 5 min at 25°C), and the cocultures were incubated at 37°C. After 2 h, the growth medium was aspirated and the monolayers were lysed by the addition of 200 μl of sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) loading buffer. Lysates were subjected to SDS-PAGE, and duplicate gels were immunoblotted using either a monoclonal antibody specific for the phosphorylated form of ExsE-GSK or a monoclonal antibody that recognizes both the phosphorylated and nonphosphorylated forms (antibody no. 9336 and no. 9332, respectively; Cell Signaling Technology, Danvers, MA).
The differential solubility translocation assay was based on a previously reported method (15), except that 0.1% Triton X-100 was used in place of digitonin. Briefly, CHO cells were cultured with P. aeruginosa strains (multiplicity of infection [MOI], 40) as in the GSK tag assay described above and incubated at 37°C. After 2 h, the growth medium was aspirated and the CHO cells were washed twice with tissue culture medium. One milliliter of sterile PBS-0.1% Triton X-100 was added to each well, and the cell monolayer was scraped from the well with a rubber policeman. Thirty-microliter aliquots were removed and used to determine counts of viable bacteria; the remaining sample was incubated for 20 min at 25°C with intermittent vortexing and then centrifuged (20,000 × g for 15 min at 25°C). The supernatants were transferred into clean tubes (soluble fraction) and the pellets were resuspended in 1 ml of PBS-0.1% Triton X-100 and sonicated (pellet fraction). Detergent was then removed, and the protein was precipitated using a methanol-chloroform-H2O mixture as described previously (32). Protein samples were subjected to SDS-PAGE, and duplicate gels were immunoblotted using anti-ExsE and anti-ExsA antisera.
The CyaA translocation assay was based on a previously reported method (28). CHO cells were seeded into six-well tissue culture plates (4 × 105 cells/well), grown overnight, and then washed twice with 1 ml of tissue culture medium. Bacterial strains grown overnight on VBM agar containing gentamicin and carbenicillin were resuspended in prewarmed tissue culture medium (1.6 × 107 cells/ml) containing 0.4% arabinose and carbenicillin, and 1 ml of the suspension was added to washed CHO cells. Cultures were centrifuged (500 × g for 5 min at 25°C) and incubated at 37°C. After 4 h, the medium was aspirated and the monolayers were washed twice with 1 ml of tissue culture medium. Two hundred fifty microliters of 0.1 M HCl was added, and the plates were incubated for 20 min at 25°C to lyse the cells. The lysed cells were scraped from the wells and centrifuged, and the supernatants were assayed for cyclic AMP (cAMP) levels by using an enzyme-linked immunoassay according to the instructions of the manufacturer (Cayman Chemical Co., Ann Arbor, MI).
β-galactosidase and secretion assays.
Cells were grown in TSB at 30°C to an optical density at 540 nm of 1. Levels of β-galactosidase activity were determined in triplicate as described previously (5). An aliquot of cell culture (109 cells) was centrifuged, and the cell pellet and trichloroacetic acid-precipitated supernatant proteins were subjected to immunoblotting as described previously (17).
Cytotoxicity assay.
P. aeruginosa-mediated cytotoxicity to CHO cells was assayed as described previously (4). Briefly, adherent CHO cell monolayers grown in 24-well plates (2.5 × 105 cells/well) were coinfected with P. aeruginosa strains (MOI, 10) for 1 to 4 h at 37°C. The plate contents were centrifuged, and the degree of CHO cell lysis was determined by assaying the supernatant for lactate dehydrogenase (LDH) activity using the Cytotox 96 system according to the instructions of the manufacturer (Promega, Madison, WI). Statistical analyses were performed using analysis of variance followed by Tukey's test for multiple comparisons with the program Prism (GraphPad Software, San Diego, CA).
RESULTS AND DISCUSSION
CHO cell-mediated induction of T3SS gene transcription requires a functional T3SS translocation apparatus.
A flow cytometry-based assay was developed to examine T3SS gene transcription following the contact of P. aeruginosa with CHO cells. The flow cytometry assay relied upon a transcriptional reporter plasmid (pJNE05) consisting of a T3SS promoter (PexoS) cloned upstream of the gene encoding green fluorescent protein (gfp). To verify the activity of the PexoS-gfp reporter, wild-type P. aeruginosa strain PA103 and T3SS mutants were transformed with pJNE05 and assayed for GFP fluorescence under Ca2+-replete and -limiting growth conditions. Consistent with the findings of previous studies (12, 17, 36), both wild-type PA103 and the secretion-competent but translocation-defective popD mutant displayed populations of cells with increased expression of the PexoS-gfp reporter under Ca2+-limiting growth conditions (Fig. 1A). Mutants lacking either exsA (encoding the transcriptional activator of the T3SS) or pscC (encoding a major structural component of the type III secretory apparatus), however, were defective in the induction of the PexoS-gfp reporter under the same conditions.
FIG. 1.

CHO cell contact-mediated induction of T3SS gene expression requires a functional secretion/translocation channel. (A) P. aeruginosa strains carrying pJNE05 encoding the exoS-gfp transcriptional fusion were grown under Ca2+-replete (−EGTA) or -limiting (+EGTA) conditions, and GFP fluorescence was examined by flow cytometry. wt, wild type. (B) P. aeruginosa strains carrying pJNE05 were cultured either in the presence (+ CHO cells) or in the absence (− CHO cells) of CHO cells as indicated, and then GFP fluorescence was examined by flow cytometry. The curves plot the relative number of bacterial cells (y axis) at each relative level of GFP fluorescence (x axis).
To measure the host contact-dependent transcription of the PexoS-gfp reporter, CHO cell monolayers were cultured with bacterial strains (MOI, 5) carrying the GFP reporter plasmid at 37°C and then washed to remove unattached bacteria. CHO cell-associated bacteria were then released from the monolayer by selectively solubilizing the CHO cells with a mild detergent treatment, and GFP fluorescence was measured by flow cytometry. As a negative control, samples of the same bacterial strains were incubated for 4 h in tissue culture medium in the absence of CHO cells and then added to CHO cells in the presence of detergent prior to examination by flow cytometry. These control samples reflected the level of PexoS-gfp expression in the absence of CHO cells during the 4-h incubation in tissue culture medium and further controlled for any effects of solubilized CHO cell debris on fluorescence.
The initial coculture experiments were performed with wild-type strain PA103, which is highly cytotoxic and lyses >80% of the CHO cells within 1 h. Under these conditions, the expression of the PexoS-gfp reporter was undetectable at all time points tested (0.5 to 4 h). For this reason, we found it necessary to use a noncytotoxic mutant (the ΔexoUT mutant) lacking the ExoU and ExoT effector proteins to prevent the premature destruction of the CHO cells during the coculture experiments. When tested in the flow cytometry assay under Ca2+-replete and -limiting growth conditions, the induction of GFP fluorescence in the ΔexoUT mutant carrying the pJNE05 reporter plasmid was similar to that in wild-type PA103 (Fig. 1A).
Coculture of the ΔexoUT strain carrying pJNE05 with CHO cells resulted in a population of bacteria having a significant increase in GFP fluorescence compared to that of the same strain incubated in the absence of CHO cells (Fig. 1B). In contrast, mutants lacking either ExsA or PscC demonstrated no increase in GFP fluorescence in the presence of CHO cells. Furthermore, the secretion-competent but translocation-defective popD mutant (26) also failed to induce GFP fluorescence in the presence of CHO cells. We concluded that the CHO cell contact-mediated induction of PexoS-gfp expression required a functional type III secretion and translocation apparatus. The need for the latter suggests that the translocation of a protein substrate may be required for the transcriptional induction of T3SS gene expression.
ExsE is translocated into CHO cells.
Previous studies identified ExsE as a negative regulator of T3SS gene transcription (24, 29). ExsE secretion by P. aeruginosa under calcium-limiting growth conditions allows the induction of T3SS gene expression. We hypothesized that the translocation of ExsE into eukaryotic host cells may play a similar role in initiating T3SS gene transcription following the contact of P. aeruginosa with host cells. Three different assays were used to determine whether ExsE is translocated into host cells.
In the first translocation assay, cells were engineered to express ExsE tagged at the carboxy terminus with a 15-amino-acid peptide derived from eukaryotic GSK. The GSK tag is a substrate for a eukaryotic serine/threonine kinase and is not phosphorylated in the bacterial cytoplasm (8). Phosphorylation of the tag, therefore, should occur only following the translocation of the ExsE-GSK fusion protein into the host cell cytoplasm. The exsE-gsk gene fusion was placed under the transcriptional control of the arabinose-inducible PBAD promoter. The resulting plasmid (pUY44wt) was used to transform the ΔexoUT and popD mutants, both of which also carried a second plasmid (pUCPexsC) that overexpresses ExsC. The inclusion of the ExsC expression plasmid was necessary for two reasons. First, the negative regulatory activity of ExsE-GSK is similar to that of wild-type ExsE and the overexpression of ExsE (29) or ExsE-GSK (data not shown) results in the superrepression of T3SS gene expression; this superrepression is relieved upon concomitant ExsC overexpression (29). Second, ExsC also functions as a chaperone and is necessary for ExsE secretion (29).
Transformants expressing ExsE-GSK and ExsC were cultured with CHO cell monolayers for 2 h. Monolayers were then lysed using SDS, and whole-cell extracts were subjected to immunoblot analyses using either a monoclonal antibody specific for the phosphorylated form of ExsE-GSK or a monoclonal antibody that recognizes phosphorylated and nonphosphorylated forms (8). Both the ΔexoUT and popD mutants yielded strong signals when cell lysates were probed with the antibody recognizing both forms of ExsE-GSK (Fig. 2A). When the lysates were probed with the antibody specific for the phosphorylated GSK tag, however, a signal was detected in lysates from the ΔexoUT strain coculture but not in lysates from CHO cells cultured with the popD translocation mutant.
FIG. 2.

Translocation of ExsE into CHO cells. For each of the assays, CHO cells were cocultured with P. aeruginosa strains expressing the indicated ExsE fusion proteins. (A) ExsE-GSK translocation assay. Following coculture, CHO cell lysate samples were processed for SDS-PAGE and duplicate gels were immunoblotted using antibody specific for the phosphorylated GSK tag (anti-GSK*P) or antibody recognizing both phosphorylated and nonphosphorylated GSK tags (anti-GSK). The phosphorylated (GSK*P) and nonphosphorylated (GSK) forms of endogenous CHO cell glycogen synthase kinase are indicated. Duplicate cocultures were performed for each strain. (B) Selective lysis translocation assay. Following coculture incubation, CHO cells were selectively lysed with 0.1% Triton X-100 and the lysate was separated by centrifugation into fractions consisting of pelleted intact bacteria (pel) and soluble CHO cell cytoplasm (sol). The fractions were processed for SDS-PAGE, and duplicate gels were immunoblotted using anti-ExsE antiserum or anti-ExsA antibody. (C) ExsE-CyaA translocation assay. Bacterial strains expressing either the full-length ExsE-CyaA fusion protein from pUY30 (wild type [wt]) or an amino-terminal deletion derivative from pUY34ΔN20 (ΔN20) were cocultured with CHO cells for 4 h. Monolayers were then washed, lysed, and assayed for calmodulin-dependent adenylate cyclase activity. The cAMP levels have been normalized to the value obtained for the ΔexoUT strain carrying pUY30 (8.6 ± 5.6 pmol/μg of CHO cell lysate). The standard errors are indicated by vertical lines.
A second translocation assay based upon the differential detergent solubilities of eukaryotic and prokaryotic cells (15) was used to confirm the GSK tag results. CHO cell monolayers were infected as described above, except that a different translocation-defective mutant, the ΔpcrV strain (16), was used as the negative control. Following a 2-h coculture, the CHO cells were washed and selectively lysed with 0.1% Triton X-100 to release the cytoplasmic contents. Lysates were fractionated into supernatant and pellet fractions by centrifugation and examined by immunoblotting using anti-ExsE antibody. In this assay, the supernatant fraction reflected the cytoplasmic contents of the CHO cells whereas the pellet fraction included the intact bacterial cells. The presence of ExsE in the supernatant fraction should occur only if the protein has been translocated into the CHO cell cytoplasm. Consistent with the results from the GSK experiments, ExsE was present in the soluble cell fraction from CHO cells cultured with the ΔexoUT mutant but absent in the fraction from cells cultured with the ΔpcrV translocation mutant (Fig. 2B). Unfortunately, our ExsE antiserum lacked the sensitivity to detect ExsE present in the low number of adherent bacterial cells. To confirm that Triton X-100 selectively lysed the CHO cells, the fractions were also analyzed for ExsA, a bacterial cytoplasmic protein that is not translocated, by immunoblotting. As expected, ExsA was found exclusively in the pellet fraction for both the ΔexoUT and ΔpcrV mutants (Fig. 2B).
Finally, a biochemical translocation assay was used as further confirmation of ExsE translocation into CHO cells. For this assay, we constructed a plasmid consisting of exsE fused to DNA encoding a truncated form of the calmodulin-activated adenylate cyclase (CyaA) from B. pertussis (28). The enzymatic activity of CyaA requires calmodulin, a protein present in CHO cells but not in P. aeruginosa; thus, the formation of cAMP by the ExsA-CyaA fusion protein should occur only following the translocation of the fusion protein into the CHO cell cytoplasm (28). Importantly, ExsE-CyaA secretion required the presence of EGTA in the culture medium (low Ca2+ level), indicating T3SS-dependent secretion of the fusion protein (Fig. 3A). The ΔexoUT and ΔpcrV mutants carrying pUCPexsC were transformed with the ExsE-CyaA overexpression plasmid pUY30 as before, and the double transformants were cultured with CHO cells as described above (MOI, 40). Following a 4-h coculture, the growth medium was removed and the CHO cells were washed, lysed, and processed for the determination of cAMP levels by using an enzyme-linked immunoassay. As shown in Fig. 2C, coculture of CHO cells with the ΔexoUT mutant resulted in a >100-fold increase in cAMP levels compared to the levels seen with the uninfected control and with the translocation-defective ΔpcrV mutant. Based on the results of these three independent translocation assays, we concluded that the coculture of P. aeruginosa with CHO cells results in the T3SS-dependent translocation of ExsE into the CHO cell cytoplasm.
FIG. 3.

Requirements for ExsE secretion. (A) P. aeruginosa strains (exsD and exsD exsC mutants) carrying expression plasmid pUY44wt (ExsE-GSK) or pUY30 (ExsE-CyaA) were grown under Ca2+-replete (EGTA −) or -limiting (EGTA +) conditions. Cultures were centrifuged, and the growth medium (supernatant) and cell pellet (lysate [lys]) fractions were analyzed by immunoblotting using either anti-ExsE or anti-CyaA antibody. (B) The exsD mutant was transformed with plasmids encoding either the full-length ExsE-CyaA fusion protein (wild type [wt]) or amino-terminal deletion derivatives (those lacking amino acids corresponding to codons 2 through 5 [ΔN5], 2 through 10 [ΔN10], and 3 through 20 [ΔN20]). Cultures were grown under Ca2+-limiting conditions, and supernatant and lysate fractions were prepared as described above and immunoblotted using anti-CyaA antibody.
The ExsE amino terminus is required for secretion and translocation.
The signal required for most proteins secreted by the T3SS resides in the amino-terminal region of the protein (27, 28). To examine the requirements for ExsE secretion, we constructed derivatives of the exsE-cyaA gene fusion lacking codons 2 through 5 (pUY36ΔN5), 2 through 10 (pUY36ΔN10), and 3 through 20 (pUY34ΔN20). Since ExsE overexpression inhibits T3SS gene transcription in a wild-type background, these analyses were performed with an ΔexsD mutant in which T3SS gene transcription is constitutively derepressed even upon ExsE overexpression (29). To measure ExsE secretion, cells were grown to mid-log phase under high- or low-Ca2+-level conditions, and whole-cell and culture supernatant fractions were immunoblotted using anti-CyaA antibody. In contrast to the observed efficient secretion of the full-length ExsE-CyaA fusion, derivatives lacking the amino-terminal amino acids of ExsE corresponding to codons 2 through 5, 2 through 10, and 3 through 20 were absent from the culture supernatant fractions (Fig. 3B). Comparison of the relative band intensities of ExsE-CyaA proteins in the cell lysate fractions indicated that the ΔN5 and ΔN10 ExsE-CyaA fusion proteins either were expressed at lower levels or were less stable than the full-length ExsE-CyaA fusion. The steady-state expression level of the ΔN20 ExsE-CyaA fusion, however, was similar to that of the full-length fusion.
We previously reported that the putative chaperone activity of ExsC is required for the stability and/or secretion of ExsE (29). To examine this relationship further, the secretion assays were repeated with an exsC exsD double mutant. Consistent with the findings of our previous study, secretion of the ExsE-GSK fusion protein was dependent upon the presence of ExsC (Fig. 3A). The secretion of the ExsE-CyaA fusion protein, however, occurred independently of ExsC. We hypothesize that the addition of the CyaA fusion partner increased the stability of ExsE, and if this hypothesis is true, it would indicate that ExsC functions to stabilize ExsE in the cytoplasm rather than playing a role in targeting ExsE to the secretory apparatus.
We next examined whether deletion of the amino-terminal residues corresponding to codons 3 through 20 affected the ability of ExsE-CyaA to be translocated. Plasmid pUY34ΔN20 was used to transform the ΔexoUT mutant strain carrying pUCPexsC, and the double transformant was tested for translocation of the ExsE fusion protein lacking the specified amino-terminal residues in the cAMP enzyme-linked immunoassay described above. Coculture of CHO cells with the transformant carrying pUY34ΔN20 resulted in a culture lysate cAMP level 10-fold lower than the cAMP level in the lysate from CHO cells cultured with bacteria expressing full-length ExsE-CyaA (Fig. 2C). We conclude that the amino terminus is required for both the secretion and translocation of ExsE.
ExsE secretion is required for the induction of T3SS gene transcription under Ca2+-limiting growth conditions and for full cytotoxicity towards CHO cells.
Our regulatory model predicts that the intracellular depletion of ExsE via secretion into the growth medium or translocation into host cells is required for the induction of T3SS gene transcription. To directly test this prediction, the wild-type chromosomal copy of exsE was replaced with an allele lacking codons 3 through 20 that encodes a nonsecretable ExsE derivative (ExsE*). T3SS transcriptional reporters (PexsD-lacZ, PexoS-lacZ, and PexoU-lacZ) were integrated into the chromosomes of wild-type PA103 and the exsE107ΔN mutant as described previously (17). Strains were grown to mid-log phase under Ca2+-limiting and -replete conditions, and β-galactosidase activity was determined. Consistent with the results of previous studies (17), the expression of T3SS promoters in a wild-type background was induced under Ca2+-limiting growth conditions (Fig. 4A). In contrast, the expression of each of the three reporters in the exsE107ΔN mutant was significantly reduced under Ca2+-limiting growth conditions compared to that in the wild-type parent. Consistent with this result was the failure of the exsE107ΔN mutant to secrete the ExoU effector under Ca2+-limiting growth conditions (immunoblot data not shown). These data suggest that the inability to secrete ExsE* leads to the repression of T3SS gene transcription. It should be noted that under Ca2+-replete conditions, each of the three promoters showed elevated β-galactosidase activity in the exsE107ΔN mutant background compared to that shown in the wild-type background. The increased basal transcription likely results from either a reduced ability of ExsE* to interact with ExsC or a small but significant decrease in the stability of ExsE*. We have not been able to detect ExsE* expressed from the chromosome by using our ExsE antiserum.
FIG. 4.

ExsE secretion is required for the induction of T3SS gene expression and full cytotoxicity towards CHO cells. (A) P. aeruginosa strains carrying the chromosomally integrated T3SS transcriptional reporter PexsD-lacZ, PexoS-lacZ, or PexoU-lacZ were grown under Ca2+-replete (−EGTA) or -limiting (+EGTA) conditions and assayed for β-galactosidase activity (reported as the percentage of activity in the induced [+EGTA] wild-type strain for each reporter). wt, wild type [PA103]; A, exsA mutant; C, exsC mutant; E*, exsE107ΔN mutant. Data presented in panels A and B are from triplicate experiments, and the standard errors are indicated by error bars. Statistical significance was determined using a one-way analysis of variance test. †, P of <0.01 for the exsE107ΔN mutant compared to the wild type (+EGTA); #, P of <0.001. (B) To measure T3SS-dependent cytotoxicity, P. aeruginosa strains were cultured with CHO cells for the indicated times and assayed for the release of LDH. The reported values (percentages of cytotoxicity) are relative to those for Triton X-100-lysed CHO cells. P, <0.05 for each of the mutants compared to the wild type.
Next, we tested the ability of the exsE107ΔN mutant to elicit T3SS-dependent cytotoxicity by using an LDH release assay. We hypothesized that the inability to secrete ExsE* would inhibit virulence. CHO cells were cocultured with P. aeruginosa strains, and the extent of LDH release after 1, 2, and 4 h was determined. As previously reported (4), coculture of CHO cells with wild-type PA103 resulted in significant cytotoxicity at the earliest time point and nearly complete lysis by 4 h (Fig. 4B) whereas an exsA mutant resulted in no T3SS-mediated cell lysis. The exsE107ΔN mutant demonstrated an initial lag in cytotoxicity extending to the 2-h time point but eventually induced significant CHO cell lysis by 4 h. We conclude that ExsE translocation into CHO cells is a triggering event for T3SS induction and is required for the full cytotoxic response of P. aeruginosa to CHO cells.
The lag in the T3SS-dependent cytotoxicity of the exsE107ΔN mutant to CHO cells (Fig. 4B) was similar to that of the previously characterized exsC mutant (5). In bacteriological medium, the exsC mutant displays severely reduced T3SS gene expression that is noninducible (Fig. 4A). We previously hypothesized that the ability of the exsC mutant to exhibit delayed T3SS-dependent cytotoxicity may indicate the existence of second, ExsC-independent induction mechanism specifically activated by host cell contact (4). Using the flow cytometry assay described above, we were able to test whether the observed cytotoxicity of the exsC mutant at 4 h reflects de novo induction of T3SS gene expression. The exsE107ΔN and exsC mutant alleles were transferred into the chromosome of the ΔexoUT strain. The resulting triple mutants were transformed with the PexoS-gfp reporter plasmid and assayed for host contact-dependent gene expression. Surprisingly, the CHO cell-dependent induction of GFP fluorescence in both the exsE107ΔN and exsC mutants was undetectable (Fig. 5). Two explanations may account for the ability of the exsE107ΔN and exsC mutants to exhibit T3SS-dependent cytotoxicity while at the same time demonstrating no induction of GFP fluorescence. First, the transcription of the T3SS may be induced at 2 h by an ExsC-independent mechanism but at levels that are undetectable by the flow cytometry assay. A second possibility is that the basal levels of T3SS gene transcription in the exsE107ΔN and exsC mutants are sufficient to eventually result in T3SS-dependent cytotoxicity.
FIG. 5.
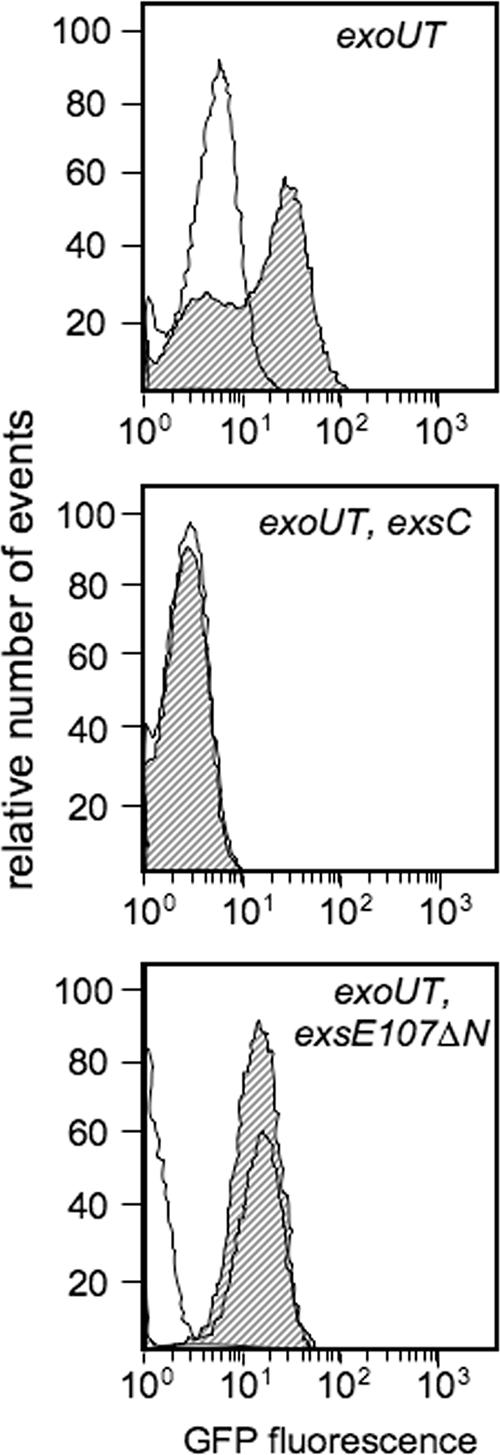
The exsC and exsE107ΔN mutants are defective in the cell contact-dependent induction of T3SS gene transcription. The indicated P. aeruginosa strains carrying pJNE05 were cultured with CHO cells for 4 h and examined by flow cytometry as described in the legend to Fig. 1B. Nonshaded curves, control bacteria incubated in the absence of CHO cells; shaded curves, bacteria incubated in the presence of CHO cells.
To distinguish between these two possibilities, the cytotoxicity assays were repeated using the exsC mutant at the 10-fold-higher MOI of 100. We reasoned that if the low level of cytotoxicity of the exsC mutant at 2 h reflected a requisite lag in transcriptional induction by an ExsC-independent mechanism, then increasing the MOI should not significantly increase the cytotoxicity at 2 h. In contrast, if the low but detectable level of CHO cell lysis observed at 2 h reflected low basal levels of T3SS gene expression, then increasing the MOI 10-fold would increase cytotoxicity. In contrast to the 5% CHO cell lysis obtained with the exsC mutant at an MOI of 10, increasing the MOI to 100 resulted in 44% CHO cell lysis at 2 h. The exsA mutant, however, exhibited no cytotoxicity to CHO cells at the 4-h time point or at an MOI increased to 100. Our data suggest that the basal levels of noninducible T3SS gene expression in the exsC and exsE107ΔN mutants are sufficient to elicit T3SS-dependent cytotoxicity. In support of this conclusion, we previously compared the virulence of wild-type P. aeruginosa to that of exsA and exsC mutants by using a murine pneumonia model (4). Whereas the wild-type strain readily disseminated and replicated to high numbers (>107 CFU/ml) in the blood and the liver, the dissemination of the exsA mutant was undetectable (<103 CFU/ml). The exsC mutant also disseminated into the blood and liver but showed a reduction in cell numbers (>105 CFU/ml) compared to the wild-type parent. It should be noted that the levels of transcription from T3SS promoters in the exsC and exsA mutants were similar (Fig. 1A and 4A). Nevertheless, the slightly higher basal levels of noninducible promoter activity in the exsC mutant were apparently sufficient to support dissemination and replication in vivo. These data suggest that the acquisition of a marginally expressed T3SS gene cluster might have offered a competitive advantage to P. aeruginosa before elaborate T3SS regulatory mechanisms were in place.
The results reported in this work demonstrate that ExsC, ExsD, and ExsE mediate the induction of T3SS gene expression in response to Ca2+ limitation and host cell contact through analogous mechanisms. The secretion and/or translocation of a negative regulatory protein is a common strategy for the coupling of transcription to secretory activity (18). For example, secretion of the FlgM anti-sigma factor following the completion of the flagellar basal body results in the transcriptional induction of the late genes involved in the final stages in flagellar assembly (1). In Yersinia spp., T3SS gene transcription is induced following the translocation of the negative regulators YscM1 and LcrQ into host cells (3, 21).
Effector proteins secreted via the T3SS are modular in structure and function. The first 15 to 20 amino-terminal residues generally contain information required for secretion, the translocation signals reside within the first 80 to 100 amino-terminal residues, and the effector function occupies the carboxy-terminal portion of the protein (9). Given the small size of ExsE (81 amino acids) and the fact that ExsE carries information required for secretion, translocation, and ExsC binding, it is likely that ExsE functions solely in a regulatory capacity. Consistent with this idea, neither the translocation of ExsE into CHO cells by P. aeruginosa nor the transient transfection of CHO cells with an ExsE-expressing plasmid resulted in obvious defects in CHO cell growth or morphology (data not shown). In addition, a mutant lacking exsE showed a high level of cytotoxicity towards CHO cells in the LDH release assay (data not shown). We conclude that ExsE likely has no additional function in mediating cytotoxicity towards host cells once it has been translocated from P. aeruginosa; however, a more subtle role cannot be ruled out at this time. Finally, it is possible the ExsE may have cytotoxic activity towards other types of cells such as macrophages and airway epithelial cells.
Acknowledgments
Support for these studies was provided by the Howard Hughes Medical Institute Biomedical Research Support Faculty Start-up Program and the National Institutes of Health (RO1-AI055042).
We thank the laboratory of Michael Welsh for assistance with the tissue culture studies and Nicholas Carbonetti, Erik Hewlett, Greg Plano, and Matt Wolfgang for their generous gifts of strains, plasmids, and antibodies.
Editor: V. J. DiRita
Footnotes
Published ahead of print on 16 July 2007.
REFERENCES
- 1.Aldridge, P., and K. T. Hughes. 2002. Regulation of flagellar assembly. Curr. Opin. Microbiol. 5:160-165. [DOI] [PubMed] [Google Scholar]
- 2.Barbieri, J. T., and J. Sun. 2004. Pseudomonas aeruginosa ExoS and ExoT. Rev. Physiol. Biochem. Pharmacol. 152:79-92. [DOI] [PubMed] [Google Scholar]
- 3.Cambronne, E. D., L. W. Cheng, and O. Schneewind. 2000. LcrQ/YscM1, regulators of the Yersinia yop virulon, are injected into host cells by a chaperone-dependent mechanism. Mol. Microbiol. 37:263-273. [DOI] [PubMed] [Google Scholar]
- 4.Dasgupta, N., A. Ashare, G. W. Hunninghake, and T. L. Yahr. 2006. Transcriptional induction of the Pseudomonas aeruginosa type III secretion system by low Ca2+ and host cell contact proceeds through two distinct signaling pathways. Infect. Immun. 74:3334-3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dasgupta, N., G. L. Lykken, M. C. Wolfgang, and T. L. Yahr. 2004. A novel anti-anti-activator mechanism regulates expression of the Pseudomonas aeruginosa type III secretion system. Mol. Microbiol. 53:297-308. [DOI] [PubMed] [Google Scholar]
- 6.Frank, D. W. 1997. The exoenzyme S regulon of Pseudomonas aeruginosa. Mol. Microbiol. 26:621-629. [DOI] [PubMed] [Google Scholar]
- 7.Frank, D. W., G. Nair, and H. P. Schweizer. 1994. Construction and characterization of chromosomal insertional mutations of the Pseudomonas aeruginosa exoenzyme S trans-regulatory locus. Infect. Immun. 62:554-563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garcia, J. T., F. Ferracci, M. W. Jackson, S. S. Joseph, I. Pattis, L. R. Plano, W. Fischer, and G. V. Plano. 2006. Measurement of effector protein injection by type III and type IV secretion systems by using a 13-residue phosphorylatable glycogen synthase kinase tag. Infect. Immun. 74:5645-5657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ghosh, P. 2004. Process of protein transport by the type III secretion system. Microbiol. Mol. Biol. Rev. 68:771-795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hoang, T. T., R. R. Karkhoff-Schweizer, A. J. Kutchma, and H. P. Schweizer. 1998. A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212:77-86. [DOI] [PubMed] [Google Scholar]
- 11.Hoang, T. T., A. J. Kutchma, A. Becher, and H. P. Schweizer. 2000. Integration-proficient plasmids for Pseudomonas aeruginosa: site-specific integration and use for engineering of reporter and expression strains. Plasmid 43:59-72. [DOI] [PubMed] [Google Scholar]
- 12.Hornef, M. W., A. Roggenkamp, A. M. Geiger, M. Hogardt, C. A. Jacobi, and J. Heesemann. 2000. Triggering the ExoS regulon of Pseudomonas aeruginosa: a GFP-reporter analysis of exoenzyme (Exo) S, ExoT and ExoU synthesis. Microb. Pathog. 29:329-343. [DOI] [PubMed] [Google Scholar]
- 13.Hovey, A. K., and D. W. Frank. 1995. Analyses of the DNA-binding and transcriptional activation properties of ExsA, the transcriptional activator of the Pseudomonas aeruginosa exoenzyme S regulon. J. Bacteriol. 177:4427-4436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iglewski, B. H., J. Sadoff, M. J. Bjorn, and E. S. Maxwell. 1978. Pseudomonas aeruginosa exoenzyme S: an adenosine diphosphate ribosyltransferase distinct from toxin A. Proc. Natl. Acad. Sci. USA 75:3211-3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee, V. T., D. M. Anderson, and O. Schneewind. 1998. Targeting of Yersinia Yop proteins into the cytosol of HeLa cells: one-step translocation of YopE across bacterial and eukaryotic membranes is dependent on SycE chaperone. Mol. Microbiol. 28:593-601. [DOI] [PubMed] [Google Scholar]
- 16.Lee, V. T., C. Tam, and O. Schneewind. 2000. LcrV, a substrate for Yersinia enterocolitica type III secretion, is required for toxin targeting into the cytosol of HeLa cells. J. Biol. Chem. 275:36869-36875. [DOI] [PubMed] [Google Scholar]
- 17.McCaw, M. L., G. L. Lykken, P. K. Singh, and T. L. Yahr. 2002. ExsD is a negative regulator of the Pseudomonas aeruginosa type III secretion regulon. Mol. Microbiol. 46:1123-1133. [DOI] [PubMed] [Google Scholar]
- 18.Miller, V. L. 2002. Connections between transcriptional regulation and type III secretion? Curr. Opin. Microbiol. 5:211-215. [DOI] [PubMed] [Google Scholar]
- 19.Miller, W. G., J. H. Leveau, and S. E. Lindow. 2000. Improved gfp and inaZ broad-host-range promoter-probe vectors. Mol. Plant-Microbe Interact. 13:1243-1250. [DOI] [PubMed] [Google Scholar]
- 20.Newman, J. R., and C. Fuqua. 1999. Broad-host-range expression vectors that carry the l-arabinose-inducible Escherichia coli araBAD promoter and the araC regulator. Gene 227:197-203. [DOI] [PubMed] [Google Scholar]
- 21.Pettersson, J., R. Nordfelth, E. Dubinina, T. Bergman, M. Gustafsson, K. E. Magnusson, and H. Wolf-Watz. 1996. Modulation of virulence factor expression by pathogen target cell contact. Science 273:1231-1233. [DOI] [PubMed] [Google Scholar]
- 22.Richards, M. J., J. R. Edwards, D. H. Culver, and R. P. Gaynes. 2000. Nosocomial infections in combined medical-surgical intensive care units in the United States. Infect. Control Hosp. Epidemiol. 21:510-515. [DOI] [PubMed] [Google Scholar]
- 23.Richards, M. J., J. R. Edwards, D. H. Culver, R. P. Gaynes, et al. 1999. Nosocomial infections in medical intensive care units in the United States. Crit. Care Med. 27:887-892. [DOI] [PubMed] [Google Scholar]
- 24.Rietsch, A., I. Vallet-Gely, S. L. Dove, and J. J. Mekalanos. 2005. ExsE, a secreted regulator of type III secretion genes in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 102:8006-8011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sato, H., and D. W. Frank. 2004. ExoU is a potent intracellular phospholipase. Mol. Microbiol. 53:1279-1290. [DOI] [PubMed] [Google Scholar]
- 26.Sawa, T., T. L. Yahr, M. Ohara, K. Kurahashi, M. A. Gropper, J. P. Wiener-Kronish, and D. W. Frank. 1999. Active and passive immunization with the Pseudomonas V antigen protects against type III intoxication and lung injury. Nat. Med. 5:392-398. [DOI] [PubMed] [Google Scholar]
- 27.Schesser, K., E. Frithz-Lindsten, and H. Wolf-Watz. 1996. Delineation and mutational analysis of the Yersinia pseudotuberculosis YopE domains which mediate translocation across bacterial and eukaryotic cellular membranes. J. Bacteriol. 178:7227-7233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sory, M. P., A. Boland, I. Lambermont, and G. R. Cornelis. 1995. Identification of the YopE and YopH domains required for secretion and internalization into the cytosol of macrophages, using the cyaA gene fusion approach. Proc. Natl. Acad. Sci. USA 92:11998-12002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Urbanowski, M. L., G. L. Lykken, and T. L. Yahr. 2005. A secreted regulatory protein couples transcription to the secretory activity of the Pseudomonas aeruginosa type III secretion system. Proc. Natl. Acad. Sci. USA 102:9930-9935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vallis, A. J., T. L. Yahr, J. T. Barbieri, and D. W. Frank. 1999. Regulation of ExoS production and secretion by Pseudomonas aeruginosa in response to tissue culture conditions. Infect. Immun. 67:914-920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vogel, H. J., and D. M. Bonner. 1956. Acetylornithinase of Escherichia coli: partial purification and some properties. J. Biol. Chem. 218:97-106. [PubMed] [Google Scholar]
- 32.Wessel, D., and U. I. Flugge. 1984. A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal. Biochem. 138:141-143. [DOI] [PubMed] [Google Scholar]
- 33.Wolfgang, M. C., V. T. Lee, M. E. Gilmore, and S. Lory. 2003. Coordinate regulation of bacterial virulence genes by a novel adenylate cyclase-dependent signaling pathway. Dev. Cell 4:253-263. [DOI] [PubMed] [Google Scholar]
- 34.Yahr, T. L., and D. W. Frank. 1994. Transcriptional organization of the trans-regulatory locus which controls exoenzyme S synthesis in Pseudomonas aeruginosa. J. Bacteriol. 176:3832-3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yahr, T. L., A. K. Hovey, S. M. Kulich, and D. W. Frank. 1995. Transcriptional analysis of the Pseudomonas aeruginosa exoenzyme S structural gene. J. Bacteriol. 177:1169-1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yahr, T. L., A. J. Vallis, M. K. Hancock, J. T. Barbieri, and D. W. Frank. 1998. ExoY, an adenylate cyclase secreted by the Pseudomonas aeruginosa type III system. Proc. Natl. Acad. Sci. USA 95:13899-13904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yahr, T. L., and M. C. Wolfgang. 2006. Transcriptional regulation of the Pseudomonas aeruginosa type III secretion system. Mol. Microbiol. 62:631-640. [DOI] [PubMed] [Google Scholar]
- 38.Zheng, Z., G. Chen, S. Joshi, E. D. Brutinel, T. L. Yahr, and L. Chen. 2007. Biochemical characterization of a regulatory cascade controlling transcription of the Pseudomonas aeruginosa type III secretion system. J. Biol. Chem. 282:6136-6142. [DOI] [PubMed] [Google Scholar]