

Abstract
A uracil-to-cytosine mutation at nucleotide position 472 of oral poliovirus vaccine type 3 (OPV3) contributes to the development of vaccine-associated paralytic poliomyelitis (VAPP). To analyze OPV3 shedding patterns, we previously used the multistep method of mutant analysis by PCR and enzyme cleavage (MAPREC). This involves conventional reverse transcription-PCR to detect OPV3, followed by a restriction digest to quantify position 472 reversion. Real-time PCR detects and quantifies nucleic acid as PCR occurs and avoids postreaction processing. The goal of this study was to compare a real-time PCR method to MAPREC. Seventy-three stool samples from Mexican OPV recipients underwent the reverse transcription-PCR step of MAPREC and real-time PCR. Real-time PCR identified 23% more OPV3-positive samples than conventional reverse transcription-PCR. When reversion was compared, the revertant proportion (RP), defined as the percentage of revertants in a sample, differed by ≤10% in 21/25 (84%) samples. The four samples differing by >10% were obtained within 5 days of OPV administration. The real-time PCR assay identified samples with an RP of ≥85% with 94% sensitivity and 86% specificity compared to MAPREC. The mean difference in RP between the two methods was 3.6% (95% confidence interval, −0.3 to 7.5%). Real-time PCR methods reliably detect OPV3, and reversion estimates correlate more consistently with MAPREC when OPV3 reversion rates are high. Detecting VAPP-related mutations by real-time PCR is rapid and efficient and can be useful in monitoring ongoing global polio eradication efforts.
Vaccination using the trivalent, live attenuated oral poliomyelitis vaccine (OPV) led to the elimination of poliomyelitis in the United States and several regions of the world (15, 26). Specifically, OPV is transmitted by the fecal-oral route and by contact with pharyngeal secretions and thereby confers immunity to unvaccinated individuals (10). However, OPV strains also undergo mutations and recombination during replication in humans (13), with the attenuating OPV sequences often being selected against in the human intestinal tract (21). The transmission of these revertant strains leads to vaccine-associated paralytic poliomyelitis (VAPP) (2).
Although many industrialized countries have adopted inactivated polio vaccine (IPV) into their immunization programs due to the risk of VAPP (7), several resource-poor nations cannot afford this policy change. Currently, the global VAPP burden is estimated to be 250 to 500 cases annually (14), with most cases occurring in resource-poor settings, and the risk of paralytic polio due to OPV will continue until as long as the oral vaccine is in use.
Most cases of VAPP in the United States and in Latin America were due to serotype 3 (1, 7). A point mutation at position 472 of Sabin OPV3 within the 5′ noncoding region and its internal ribosomal entry site is known to be an attenuating sequence for this serotype, and reversion of this nucleotide has been associated with VAPP (9). These back-mutations of OPV occur during replication within the human gut (28). A previous study in our laboratory investigating OPV3 shedding and reversion demonstrated a bimodal shedding pattern of attenuated and revertant viruses in accordance with the selective pressure favoring the reversion of OPV3 (20). Among U.S. infants who received OPV followed by IPV, we observed high OPV3 shedding rates, which peaked on day 3 to 4 after vaccination and rose again during the second week after vaccination. The second increase in OPV shedding was associated with high rates of reversion by day 14 after vaccination and coincided with the development of the revertant mutation at position 472.
These studies utilized a modified method of multistep mutant analysis by PCR and enzyme cleavage (MAPREC) to detect OPV3 shedding and estimate reversion. This method was developed at the Federal Drug Administration to evaluate the quality of vaccine lots (8) and was modified by our lab. Stool samples first underwent direct nucleic acid extraction of viral RNA, thus avoiding cell culture techniques. The extracted RNA then underwent reverse transcription-PCR to detect OPV3. During this step, the primers used for OPV3 detection were common to both the attenuated OPV3 virus and the mutated virus. After amplification, samples which contained either type of OPV3 were identified by gel electrophoresis. A final restriction digest using EcoRI distinguished the nonrevertant strains from the revertant strains. The degree of UV fluorescence observed upon imaging of the gels was used to estimate the revertant proportion (RP), defined as the percentage of revertants in a sample (20).
We developed a real-time PCR assay to compare with the MAPREC method. Using the principles of the mismatch amplification mutation assay (18), primers specific to either the revertant or nonrevertant strain, amplifying each strain individually, were designed. This allows the detection of revertants and nonrevertants separately and the quantitation of PCR products as the reaction is occurring and makes it possible to test a large number of samples concurrently. These properties may prove more efficient in detecting revertant OPV strains in the field. In this paper, we compare the real-time PCR method with the multistep MAPREC technique for analyzing OPV3 reversion.
(Some data were presented at the 43rd Annual Meeting of the Infectious Diseases Society of America, San Francisco, CA, October 2005 [L. J. Wong, S. Biswas, D. Gnanashanmugam, M. Fang, and R. Hammon], and the Pediatric Academic Societies' Annual Meeting, San Francisco, CA, May 2006 [D. Gnanashanmugam, M. Fang, M. Falkovitz-Halpern, A. Dodge, M. Esparza, R. Hammon, L. J. Wong, E. E. Rivas-Merelles, J. Santos, and Y. Maldonado].)
MATERIALS AND METHODS
Clinical samples.
We used archived stool samples from a previous study to evaluate the real-time PCR method. Seventy-three stool samples were obtained from OPV vaccinees in Rio Blanco, Mexico, during the summer of 2004 for a study evaluating excretion and reversion of OPV3. The samples were collected in cryovials (Nunc, Nalge, Rochester, NY) and kept refrigerated until the time of transport to Stanford University for analysis. The samples were transported to Stanford University on dry ice and were frozen at −80°C on arrival. They remained at −80°C until the time of RNA extraction.
Control construct preparation.
Stocks of positive control constructs were created separately for the real-time PCR assays and the MAPREC assay. Wild polio type 3 (P3Leon/37, GenBank accession no. X00925-K00043) and OPV3 vaccine strain (GenBank accession no. AY184221) were obtained from the California Health Department courtesy of David Schnurr and were used as templates for the preparation of constructs. Using Primer Express software v2.0 (Applied Biosystems, Foster City, CA), separate primer sets were created for the real-time PCR assays and the MAPREC assays. Using these primers and OPV3 templates, sequences of interest were amplified, and the PCR products created underwent TOPO TA cloning and transformation into Escherichia coli to create stocks of controls for use in subsequent experiments. The procedure used to create these plasmids for the real-time PCR assay and the MAPREC methods differed only in the primers used (Table 1). Additionally, since the 290-bp sequence used in the MAPREC assay does not contain a restriction site, position 468 of the forward primer was altered to include an EcoRI site used in the restriction digest in the MAPREC protocol. If OPV3 was present in amplified products, the 290-bp sequence would be cleaved; if OPV3 reversion had occurred, then the 290-bp sequence was not cleaved.
TABLE 1.
Primers used to create control constructs and those used for MAPREC and real-time PCR assays
| Purpose and name of primer or probe | Sequence (5′ to 3′)a | Location in genome |
|---|---|---|
| Creation of control constructs for MAPREC | ||
| S3+432/472C | GAGCTACATGAGAGTCCTCCGGCCCCTGAATGCGGCGAATC | 432-472 |
| S3+432/472T | GAGCTACATGAGAGTCCTCCGGCCCCTGAATGCGGCGAATT | 432-472 |
| S3-721 | TCAAACAATTTCAATAG | 721-704 |
| Creation of control constructs for real-time assays | ||
| S3+454/471 | CCCCTGAATGCGGCGAAT | 454-472 |
| S3-509 | CTGGCTGCTGGGTTGCA | 509-493 |
| Reverse transcription-PCR step in MAPREC | ||
| S3+432 | GAGCTACATGAGAGTCCTCCGGCCCCTGAATGCGGCGAAT | 432-471 |
| S3-721 | TCAAACAATTTCAATAAG | 721-704 |
| Real-time assay for revertantsb | ||
| S3+454/472C | CCCCTGAATGCGGCGAAGC | 454-472 |
| S3-509 | CTGGCTGCTGGGTTGCA | 509-493 |
| Probe 473/490 | 6FAM TAA CCA TGG AGC AGG CA MGB | 473-490 |
| Real-time assay for nonrevertantsc | ||
| S3+454/472T | CCCCTGAATGCGGCGAACT | 454-472 |
| S3-509 | CTGGCTGCTGGGTTGCA | 509-493 |
| Probe 473/490 | 6FAM TAA CCA TGG AGC AGG CA MGB | 473-490 |
Underlining shows nucleotide 468, which was mismatched in all forward primers to allow an EcoRI digest when the nonrevertant was present. This mismatch was included in the real-time assays. Italics show mismatches of nucleotides 471 and 472 in the forward primers for the real-time assays for nonrevertants and revertants to increase specificity in distinguishing nonrevertant from revertant OPV3 strains. 6FAM, 6-carboxyfluorescein; MGB, minor groove binder.
Detection and quantification of revertants in clinical samples.
Detection and quantification of nonrevertants in clinical samples.
Reverse transcription was performed using P3Leon (wild type) and the OPV3 vaccine strain as revertant and nonrevertant templates, respectively. A 10-μl reaction mixture was required for reverse transcription, consisting of 2.5 μl of either revertant or nonrevertant template, 2.0 μl of 5× reaction buffer, 0.5 μl porcine RNase inhibitor (Fisher Scientific, Pittsburgh, PA), 1.0 μl deoxynucleoside triphosphate (dNTP) mix (10 mM each dNTP), 50 μM lower primer, 0.4 μl avian myeloblastosis virus (AMV) reverse transcriptase (RT) (Promega, Madison, WI), and distilled H2O (dH2O) for the remaining volume. The reaction mixture was incubated for 10 min at 25°C, 60 min at 42°C, and 5 min at 99°C and then cooled to 4°C for at least 5 min. PCR was completed using 5 μl 10× Taq buffer, 0.25 μl NovTaq (Novagen, San Diego, CA), 50 μM upper primer, and 33.75 μl dH2O for a total 50-μl PCR mixture. The reaction was run with 1 cycle for 2 min at 94°C, 35 cycles of denaturing at 94°C for 30 s, and annealing at 50°C for 30 s. After completion of reverse transcription-PCR, the products were visualized on 2% agarose gels to verify that the 290- and 56-bp sequences had been amplified. The PCR products were concentrated using QIAex kits (QIAGEN, Valencia, CA) per the manufacturer's protocol, and the concentrated products were then cloned into TOPO vectors using TOPO TA cloning kits (Invitrogen, Carlsbad, CA) per the manufacturer's protocol. Plasmid DNA was isolated using QIAprep miniprep kits (QIAGEN) and were sequenced (Elim Pharmaceuticals, Hayward, CA) to verify that the insertions of interest were present. Purified plasmid stocks were stored at −20°C, and glycerol stocks of the bacterial colonies were stored at −80°C for long-term use.
RNA extraction.
The RNA extraction procedure was common to both the real-time PCR assay and the MAPREC protocols. An RNeasy minikit (QIAGEN, Valencia, CA) was used per the manufacturer's protocol. Positive and negative control reactions were included in each set of extractions. Extracted RNA preparations were stored at −80°C.
Modified MAPREC method. (i) Conventional RT PCR of the 290-bp region of samples with AMV RT.
After viral extraction of stool samples, reverse transcription-PCR was carried out by previously published methods (23). The primers used are given in Table 1. Briefly, a total reaction volume of 10 μl was required for reverse transcription, consisting of 2.5 μl of extracted samples (or 2.5 μl of OPV3 or dH2O for the positive and negative controls, respectively), 2.0 μl of 5× reaction buffer, 0.5 μl porcine RNase Inhibitor (Fischer Scientific, Pittsburgh, PA), 1.0 μl dNTP mix (10 mM each dNTP), 50 μM primer S3+432, 0.4 μl AMV RT (Promega, Madison, WI), and dH2O for the remaining volume. The mixture was incubated for 10 min at 25°C, 60 min at 42°C, and 5 min at 99°C and then cooled to 4°C for at least 5 min. PCR was completed using 5 μl 10× Taq buffer, 0.25 μl NovTaq (Novagen, San Diego, CA), 50 μM S3-721 primer, and 33.75 μl dH2O for a total of 50 μl and run with 1 cycle for 2 min at 94°C, 35 cycles of denaturing at 94°C for 30 s, and annealing at 50°C for 30 s.
(ii) Visualization of reverse transcription-PCR results.
PCR products were visualized by gel electrophoresis as previously described (20).
(iii) Quantification of revertants by restriction digestion.
After reverse transcription-PCR, revertants in samples were quantified using a restriction digest (20). The samples underwent restriction digestion with 8.6 μl of the sample, 0.3 μl EcoRI (Promega, Madison, WI), 0.1 μl bovine serum albumin, and 1 μl buffer H (Promega). The same quantity of the sample was combined with 1.4 μl dH2O for the undigested control. After incubation at 37°C for 1 h and at 65°C for 5 min, 10 μl of each reaction was evaluated on 3% low-melting-point agarose gels in a Bio-Rad Sub Cell GT gel box at 90 V for 45 min. Constructs of the revertant and nonrevertant sequences were run in parallel (also having undergone incubation with EcoRI and without the enzyme). Ladders of 50 bp and 100 bp were included in wells for scaling. Quantity One software was used to image the gels and calculate the RP. Detailed statistical methods of the derivation of the RP are provided elsewhere (20).
Real-time PCR method.
After viral extraction, reverse transcription was first performed to create cDNA using AMV RT (Promega, Madison, WI). OPV3 and dH2O were included as positive and negative controls with each set of reactions. The reaction mixture consisted of 2.5 μl of extracted samples, 2.0 μl of 5× reaction buffer, 0.5 μl porcine RNase Inhibitor (Fischer Scientific, Pittsburgh, PA), 1.0 μl dNTP mix (10 mM each dNTP), 20 μM primer S3-509 (Table 1), 0.4 μl AMV RT, and dH2O for a total volume of 10 μl. The mixture was incubated for 10 min at 25°C, 60 min at 42°C, and 5 min at 99°C and then cooled to 4°C for 5 min. Fifteen microliters of dH2O was added to each mixture. Each reverse transcription product then underwent real-time PCR analysis with assays for both revertants and nonrevertants. The primers and probes used are presented in Table 1. Five microliters of the reverse transcription reaction mixtures was combined with 10 μl real-time master mix without UNG (uracil-N-glycosylase; Eurogentec, San Diego, CA), 900 nM forward primer (Eurogentec), 900 nM reverse primer (Eurogentec), 200 nM TaqMan probe (Applied Biosystems, Foster City, CA), and dH2O to make 20 μl. This reaction was performed for both revertant and nonrevertant plasmids, and reactions were cycled at 1 cycle of 2 min at 52°C and 10 min at 95°C, 40 cycles of denaturing at 95°C for 15 s, and annealing at 60°C for 60 s using an ABI Prism 7900HT sequence detection system (Applied Biosystems). Nontemplate controls were included in each set of experiments. The cycle number at which the fluorescence crosses the threshold (CT value) was set at ΔRn 0.2 (ΔRn = Rn+ − Rn− where Rn+ represents fluorescence emitted at any given time in a reaction tube and Rn− represents fluorescence measured prior to PCR amplification in the same reaction tube), with a baseline of 3 to 15 cycles. Samples which returned CT values of ≥35 in both real-time assays and on more than one run were considered negative, reflecting low initial copy numbers and leading to inaccuracy in estimating the RP.
The following formula was used to calculate the RP for the real-time PCR assay: 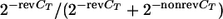 , where revCT and nonrevCT are revertant and nonrevertant CT, respectively.
, where revCT and nonrevCT are revertant and nonrevertant CT, respectively.
Validation of the real-time PCR assays.
In order to validate the plasmids in the real-time assays, dilutions of revertant and nonrevertant plasmids and mixtures with various proportions of revertant and nonrevertant plasmids were tested by both assay methods. Five microliters of the diluted plasmids or mixtures was combined with 10 μl real-time master mix without UNG (Eurogentec, San Diego), 900 nM forward primer (Eurogentec), 900 nM reverse primer (Eurogentec), 200 nM TaqMan probe (Applied Biosystems, Foster City, CA), and dH2O to make 20 μl. Reactions were cycled at 1 cycle of 2 min at 52°C and 10 min at 95°C, 40 cycles of denaturing at 95°C for 15 s, and annealing at 60°C for 60 s using an ABI Prism 7900HT sequence detection system (Applied Biosystems). Nontemplate controls were included in each set of experiments. The formula used for the calculation of RP in clinical samples was used to calculate the RP in these dilutions and mixtures.
Comparison of MAPREC to real-time PCR.
The detection of either revertant or nonrevertant OPV3 by the conventional reverse transcription step of MAPREC was compared to detection by the real-time PCR. OPV3 reversion was compared between the two methods by estimating the RP for OPV3-positive samples. The two methods were also compared based on the difference in RP obtained. The samples which differed by <10% were compared to those with a difference of >10%. The mean of all the differences was estimated.
RESULTS
Validation of the real-time PCR assays.
Experiments were run with the revertant and nonrevertant plasmid construct stocks to validate the real-time assays. First, pure revertant and pure nonrevertant plasmid constructs, which were created as described above, were serially diluted by a factor of 10 from 5 × 107 copies/μl to 5 × 10−7 copies/μl and run against their matching assays to establish the sensitivity and efficiency of the assays. Fecal suspensions known to be negative for OPV3 were spiked with the same serial dilutions of revertant and nonrevertant plasmids and run with the matching assay. The same dilutions of pure revertant and pure nonrevertant plasmids were run against the opposing assay to determine the discrimination of the assays, defined as the ability of the assay for revertants or nonrevertants to distinguish between the revertant and nonrevertant templates. Finally, several mixtures of the plasmids were created with the following proportions: 100% revertant plasmid; 90% revertant plasmid and 10% nonrevertant plasmid; 75% revertant plasmid and 25% nonrevertant plasmid; 50% revertant plasmid and 50% nonrevertant plasmid, 25% revertant plasmid and 75% nonrevertant plasmid; 10% revertant plasmid and 90% nonrevertant plasmid; and 100% nonrevertant plasmid. These mixtures were run against the assays for both nonrevertants and revertants to test discrimination in the presence of the opposing templates.
Sensitivity, efficiency, and discrimination of the real-time PCR assays.
Sensitivity was established by serial dilutions of nonrevertant and revertant construct plasmids in fecal extractions and noted to be 100 copies/μl for both assays (data not shown). To further verify true negatives, three archived stool samples obtained prior to infant vaccination were negative by the real-time PCR assays. The efficiencies of the assays with nonrevertant and revertant templates were determined to be nearly 100%, as determined by slopes of −3.23 and −3.43 for the assays for nonrevertant and revertant strains, respectively, on their standard curves (Fig. 1). Both assays were able to distinguish the matching template from the opposing template, i.e., the assay for revertant virus revealed a lower CT, in the presence of pure revertant plasmid and a higher CT when run against pure nonrevertant plasmid. Thus, discrimination between the two plasmids was seen, with a difference in CT of 3 to 6 for both assays, when the assays were run with their matching templates and against their opposing templates (Fig. 2).
FIG. 1.

Efficiencies of the real-time PCR assays for revertant and nonrevertant strains. Revertant (b) and nonrevertant (a) plasmid control constructs were diluted serially by a factor of 10, and CT values obtained from each dilution were plotted. Both slopes correspond to efficiencies of 90 to 100%.
FIG. 2.

Discrimination of the real-time assays. CT values for revertant and nonrevertant templates differed by 3 to 6 in the assay for nonrevertants (a) and by 5 to 6 in the assay for revertants (b).
The CT values obtained from the mixtures by assays for revertant and nonrevertant strains were used to calculate the proportion of revertants in each mixture. This result was plotted against the known quantity of mixtures (Fig. 3). The mean difference between the RP obtained by real-time PCR and known values was 2.6% (95% confidence interval, 0.4 to 4.8%).
FIG. 3.

Comparison of RPs obtained by real-time assays with known RPs in prepared construct mixtures. Mixtures of revertant and nonrevertant constructs underwent several runs in real-time PCR assays.
Testing of fecal samples from OPV recipients. (i) Comparison of OPV3 detection by the reverse transcription-PCR step in MAPREC with real-time PCR.
A total of 73 samples from 11 OPV vaccinees were analyzed. All vaccinees shed OPV3, as detected by either the reverse transcription-PCR step of MAPREC or real-time PCR. All 73 samples were tested by real-time PCR at least twice, and the mean of the results was used for analysis. Forty-six of 73 (63%) samples tested positive for OPV3 by the reverse transcription-PCR step of MAPREC, and 63 samples (86%) were positive for OPV3 by the real-time PCR assay. All samples positive by reverse transcription-PCR were identified by the real-time assays; no samples were negative by real-time PCR and positive by reverse transcription-PCR. Seven samples were available from children prior to vaccination. Of these, six were negative by reverse transcription-PCR, and the same six samples were negative by real-time PCR. One prevaccination sample was positive by both methods. The four remaining samples negative by real-time PCR were obtained from four vaccinees within 1 week of immunization. Reverse transcription-PCR detected the last positive sample 62 days after OPV administration, whereas real-time PCR detected OPV 77 days after the last OPV dose, which was also the time of the latest postvaccine sample.
(ii) Comparison of OPV3 reversion in fecal samples by MAPREC and real-time PCR.
Twenty-five of the 46 OPV3 positives identified by reverse transcription-PCR underwent the next steps of restriction digest in MAPREC for comparison with real-time PCR (Fig. 4). The RPs obtained by the two methods were compared. MAPREC generally predicted a higher RP than the real-time PCR assay, but as the proportion of revertants in samples increased, the correlation between the two methods became closer. Twenty-one of 25 samples (84%) had a difference in RP of ≤10%. Three of the remaining samples showed a 10 to 20% difference in the RP obtained, and one sample differed by 32%. The mean difference in RP between the two methods was 3.6% (95% confidence interval, −0.3 to 7.5%). The real-time PCR assay detected samples with an RP of ≥85%, with 94% sensitivity and 86% specificity.
FIG. 4.

Comparison of real-time PCR to MAPREC in clinical samples. MAPREC generally predicted a higher RP, but a closer correlation between the two methods was seen when the RP was high.
The median RP of the samples is shown in Table 2. Samples obtained either prevaccination or up to day 13 after OPV administration had a median RP of 92 to 97.3%, depending on the method used to estimate reversion. All later samples had a median RP of nearly 100%. The four samples with a >10% difference in RP were obtained within 5 days after OPV administration. Any sample obtained from vaccine recipients 14 days or later after the last OPV dose had >95% revertants when assessed by either method (13 samples assessed by real-time PCR only and 8 samples assessed by MAPREC and real-time PCR). Ten of eleven vaccinees shed >95% revertants on the last day that OPV3 was detected by either method (range, 6 to 77 days).
TABLE 2.
Median RPs of all samplesa
| Time (days) from OPV vaccination | Median RP (%) by:
|
Difference (%) | |
|---|---|---|---|
| Real-time PCR | MAPREC | ||
| −4 to 13 | 97.3 | 92 | 5.3 |
| 14-77 | 99.2b | 100b | 0.8 |
Median RPs from all samples obtained by MAPREC were compared to those obtained by real-time PCR. Samples with a difference in RP of >10% were obtained within 5 days of OPV administration.
Samples obtained at or after day 14 postvaccination had ≥95% revertants by either method (n = 29).
DISCUSSION
Real-time PCR is more sensitive than conventional PCR (4), and quantitation is recorded during the exponential phase of a PCR when ample reagents and templates are available. We developed a real-time PCR assay to enable identification of OPV3-positive samples and estimation of the RP in one step. This approach avoided the use of postreaction processing and agarose gels, which are required for detection and estimation by the MAPREC method, and made it possible to test the percentage of OPV3 revertants in more samples concurrently. As in our previous studies, direct extraction of nucleic acids from fecal samples was used to avoid the confounding variable of accumulation of mutations during OPV replication in cell culture and to provide a more accurate estimate of in vivo reversions of OPV3 (3). Primers for the real-time PCR assays were designed specifically for OPV3. Since polymerases inefficiently extend beyond a DNA mismatch (11), primers were designed with a mismatch at the terminal and penultimate positions of the sequence of interest in order to distinguish revertant and nonrevertant OPV3 strains, which are divergent by one nucleotide at position 472. Our method employed TaqMan probes, which are more specific than SYBR green dyes (29), and relied on fluorescent resonant energy transfer during amplification to detect OPV3 and estimate the proportion of revertants.
The real-time PCR assays detected as few as 100 copies/μl of construct plasmid in fecal suspensions and detected OPV3 in 17 samples that were negative by the MAPREC assay, demonstrating a higher sensitivity than conventional reverse transcription-PCR. Six of seven prevaccination samples negative by conventional reverse transcription-PCR and three prevaccination samples from a prior study were all negative by real-time PCR, reflecting no false-positive results. The efficiencies of real-time PCR assays for both revertants and nonrevertants approached nearly 100%, and both distinguished revertant from nonrevertant strains in a mixed population of plasmids.
When the efficiencies of the two assays were compared, the dilution curve in the assay for nonrevertants had a slope of −3.23, which showed a lower efficiency than that of the assay for revertants, whose slope was −3.43 (Fig. 1). When mixtures of the plasmids were tested, the assay for nonrevertants also showed less discrimination, with CT differences as low as 3, compared to ∼5 to 6 in the assay for revertants. Previously published studies utilizing TaqMAMA (mismatch amplification mutation assay with Taq) techniques noted higher discrimination between the alleles of interest, with CT values differing by nearly 10 (18). However, as the proportions of revertants were high in given samples, this lack of discrimination was less relevant for purposes of estimating OPV3 reversions.
The rate of OPV3 reversion in clinical samples was estimated by the real-time PCR method and by the MAPREC method. The real-time method showed a high correlation with MAPREC. Only four samples had RP that differed by >10% in the two methods, of which three differed 10 to 20%. This difference was seen in samples obtained within the first week of vaccination, and within the second week, high rates of reversion were seen by both methods (Table 2). This higher concordance between the two methods as time from vaccination increases is clinically relevant, reflecting a high degree of circulating revertant virus regardless of the method employed for its detection.
Previous studies by Maldonado et al. (19) demonstrated peak shedding of OPV3 in 34% of Mexican infants after the first dose of OPV. In addition, a study enrolling 32 children in Belarus (27) demonstrated OPV3 excretion rates of 42% among vaccinees after the first dose of OPV. Both studies investigated only exclusive OPV recipients in resource-poor settings and used tissue cultures for viral isolation. The current study demonstrated that 100% of infants shed OPV3 as estimated by both methods, likely reflecting a higher sensitivity of PCR methods than tissue culture for the detection of OPV3. Data from banked stool samples obtained from first-dose OPV vaccinees in Western countries demonstrated shedding in 95% of vaccinees for all three serotypes (17) and in 65% of vaccinees for OPV3 (22). This study confirms high shedding rates in a country where OPV continues to be used. Whether the use of IPV in developing countries with high rates of OPV shedding will prevent outbreaks and provide adequate immunity against polio outbreaks remains to be seen.
Consistent with data obtained from previous studies (24), our vaccinees demonstrated shedding of OPV3 as late as 10 weeks postvaccination. Detection of OPV3 from vaccinees 11 weeks postvaccination implies its presence in the environment for sustained periods and the possibility of VAPP in poorly immunized communities, a common prerequisite for vaccine-derived poliovirus (VDPV) outbreaks (12). Testing of samples obtained more than 10 to 11 weeks postvaccination may reveal the duration of shedding of OPV and assist in vaccine policy development during the certification era.
These data demonstrate increased reversion of OPV type 3 in stool samples over time, consistent with previous studies (20). This finding may indicate a large amount of revertant virus in communities using OPV and, along with the data indicating prolonged shedding of OPV3, may predict an increased risk of developing VAPP, particularly if immunity to polio declines as eradication efforts move forward. Further evaluation of household members by this sensitive method may delineate patterns of reversion within homes and may show whether revertants are more commonly seen in household members early during the shedding period, possibly implying greater infectivity of revertants than nonrevertants.
Disadvantages of the real-time technique include its greater sensitivity and the ease with which the assay is contaminated. Low initial copy numbers in samples may result in greater differences in RP. Recent studies imply that detection of poliovirus by full-length PCR may have the advantage of better representation of viable virus, which may lead to VAPP (17), although no studies have proven this conclusively. The presence of reversions is believed to increase transmissibility of OPV and may be the first step in the emergence of VDPVs (16), which have been responsible for outbreaks of polio on Hispaniola (12), in Egypt (6), in Madagascar (25), and in the Philippines (5). Whether prerequisite recombination events which lead to VDPV developments in outbreaks require the full virus is unknown.
Real-time PCR offers a rapid and efficient method of detecting OPV and its associated reversion, and this method can be used to assess the risk of VAPP and VDPV outbreaks in communities which continue to use OPV. These data will contribute to informed decisions on the best method of continued immunization against poliomyelitis as global polio eradication continues.
Acknowledgments
This study was supported by a grant from the York Francis Fund.
No author has a commercial or other association that might pose a conflict of interest in this research.
We acknowledge Linda Lew, Marvin Sommer, and Ann Arvin for review of the manuscript.
Informed consent was obtained from parents or guardians; the human experimentation guidelines of the U.S. Department of Health and Human Services and the Committee for the Protection of Human Subjects at Stanford University were followed in the conduct of clinical research.
Footnotes
Published ahead of print on 20 June 2007.
REFERENCES
- 1.Andrus, J. K., P. M. Strebel, C. A. de Quadros, and J. M. Olive. 1995. Risk of vaccine-associated paralytic poliomyelitis in Latin America, 1989-91. Bull. W. H. O. 73:33-40. [PMC free article] [PubMed] [Google Scholar]
- 2.Blume, S., and I. Geesink. 2000. A brief history of polio vaccines. Science 288:1593-1594. [DOI] [PubMed] [Google Scholar]
- 3.Buonagurio, D. A., J. W. Coleman, S. A. Patibandla, B. S. Prabhakar, and J. M. Tatem. 1999. Direct detection of Sabin poliovirus vaccine strains in stool specimens of first-dose vaccinees by a sensitive reverse transcription-PCR method. J. Clin. Microbiol. 37:283-289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bustin, S. A. 2002. Quantification of mRNA using real-time reverse transcription PCR (RT-PCR): trends and problems. J. Mol. Endocrinol. 29:23-39. [DOI] [PubMed] [Google Scholar]
- 5.Centers for Disease Control and Prevention. 2001. Acute flaccid paralysis associated with circulating vaccine-derived poliovirus—Philippines, 2001. Morb. Mortal. Wkly. Rep. 50:874-875. [PubMed] [Google Scholar]
- 6.Centers for Disease Control and Prevention. 2001. Circulation of a type 2 vaccine-derived poliovirus—Egypt, 1982-1993. Morb. Mortal. Wkly. Rep. 50:41-42, 51. [PubMed] [Google Scholar]
- 7.Centers for Disease Control and Prevention. 1997. Poliomyelitis prevention in the United States: introduction of a sequential vaccination schedule of inactivated poliovirus vaccine followed by oral poliovirus vaccine. Recommendations of the Advisory Committee on Immunization Practices (ACIP). Morb. Mortal. Wkly. Rep. Recomm. Rep. 46:1-25. [PubMed] [Google Scholar]
- 8.Chumakov, K. 2000. Mutant analysis by PCR and restriction enzyme cleavage (MAPREC) for oral poliovirus (Sabin) vaccine: standard operating procedure. World Health Organization (WHO) collaborative study on MAPREC, vol. 3.4, p. 1-27. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 9.Evans, D. M., G. Dunn, P. D. Minor, G. C. Schild, A. J. Cann, G. Stanway, J. W. Almond, K. Currey, and J. V. Maizel, Jr. 1985. Increased neurovirulence associated with a single nucleotide change in a noncoding region of the Sabin type 3 poliovaccine genome. Nature 314:548-550. [DOI] [PubMed] [Google Scholar]
- 10.Friedrich, F. 1996. Genomic modifications in Sabin vaccine strains isolated from vaccination-associated cases, healthy contacts and healthy vaccinees. Acta Virol. 40:157-170. [PubMed] [Google Scholar]
- 11.Glaab, W. E., and T. R. Skopek. 1999. A novel assay for allelic discrimination that combines the fluorogenic 5′ nuclease polymerase chain reaction (TaqMan) and mismatch amplification mutation assay. Mutat. Res. 430:1-12. [DOI] [PubMed] [Google Scholar]
- 12.Kew, O., V. Morris-Glasgow, M. Landaverde, C. Burns, J. Shaw, Z. Garib, J. Andre, E. Blackman, C. J. Freeman, J. Jorba, R. Sutter, G. Tambini, L. Venczel, C. Pedreira, F. Laender, H. Shimizu, T. Yoneyama, T. Miyamura, H. van Der Avoort, M. S. Oberste, D. Kilpatrick, S. Cochi, M. Pallansch, and C. de Quadros. 2002. Outbreak of poliomyelitis in Hispaniola associated with circulating type 1 vaccine-derived poliovirus. Science 296:356-359. [DOI] [PubMed] [Google Scholar]
- 13.Kew, O. M., B. K. Nottay, M. H. Hatch, J. H. Nakano, and J. F. Obijeski. 1981. Multiple genetic changes can occur in the oral poliovaccines upon replication in humans. J. Gen. Virol. 56:337-347. [DOI] [PubMed] [Google Scholar]
- 14.Kew, O. M., P. F. Wright, V. I. Agol, F. Delpeyroux, H. Shimizu, N. Nathanson, and M. A. Pallansch. 2004. Circulating vaccine-derived polioviruses: current state of knowledge. Bull. W. H. O. 82:16-23. [PMC free article] [PubMed] [Google Scholar]
- 15.Kim-Farley, R. J., K. J. Bart, L. B. Schonberger, W. A. Orenstein, B. M. Nkowane, A. R. Hinman, O. M. Kew, M. H. Hatch, and J. E. Kaplan. 1984. Poliomyelitis in the USA: virtual elimination of disease caused by wild virus. Lancet 2:1315-1317. [DOI] [PubMed] [Google Scholar]
- 16.Laassri, M., K. Lottenbach, R. Belshe, M. Rennels, S. Plotkin, and K. Chumakov. 2006. Analysis of reversions in the 5′-untranslated region of attenuated poliovirus after sequential administration of inactivated and oral poliovirus vaccines. J. Infect. Dis. 193:1344-1349. [DOI] [PubMed] [Google Scholar]
- 17.Laassri, M., K. Lottenbach, R. Belshe, M. Wolff, M. Rennels, S. Plotkin, and K. Chumakov. 2005. Effect of different vaccination schedules on excretion of oral poliovirus vaccine strains. J. Infect. Dis. 192:2092-2098. [DOI] [PubMed] [Google Scholar]
- 18.Li, B., I. Kadura, D. J. Fu, and D. E. Watson. 2004. Genotyping with TaqMAMA. Genomics 83:311-320. [DOI] [PubMed] [Google Scholar]
- 19.Maldonado, Y. A., V. Pena-Cruz, M. de la Luz Sanchez, L. Logan, S. Blandon, M. F. Cantwell, S. M. Matsui, F. Millan-Velasco, J. L. Valdespino, and J. Sepulveda. 1997. Host and viral factors affecting the decreased immunogenicity of Sabin type 3 vaccine after administration of trivalent oral polio vaccine to rural Mayan children. J. Infect. Dis. 175:545-553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martinez, C. V., M. O. Old, D. K. Kwock, S. S. Khan, J. J. Garcia, C. S. Chan, R. Webster, M. S. Falkovitz-Halpern, and Y. A. Maldonado. 2004. Shedding of Sabin poliovirus type 3 containing the nucleotide 472 uracil-to-cytosine point mutation after administration of oral poliovirus vaccine. J. Infect. Dis. 190:409-416. [DOI] [PubMed] [Google Scholar]
- 21.Minor, P. D., and G. Dunn. 1988. The effect of sequences in the 5′ non-coding region on the replication of polioviruses in the human gut. J. Gen. Virol. 69(Pt. 5):1091-1096. [DOI] [PubMed] [Google Scholar]
- 22.Minor, P. D., G. Dunn, M. E. Ramsay, and D. Brown. 2005. Effect of different immunisation schedules on the excretion and reversion of oral poliovaccine strains. J. Med. Virol. 75:153-160. [DOI] [PubMed] [Google Scholar]
- 23.Old, M. O., C. V. Martinez, D. Kwock, J. Garcia, G. Martin, C. Chan, and Y. A. Maldonado. 2003. Direct extraction of Sabin poliovirus genomes from human fecal samples using a guanidine thiocyanate extraction method. J. Virol. Methods 110:193-200. [DOI] [PubMed] [Google Scholar]
- 24.Parent du Chatelet, I., A. T. Merchant, S. Fisher-Hoch, S. P. Luby, S. A. Plotkin, T. Moatter, M. Agboatwalla, and J. B. McCormick. 2003. Serological response and poliovirus excretion following different combined oral and inactivated poliovirus vaccines immunization schedules. Vaccine 21:1710-1718. [DOI] [PubMed] [Google Scholar]
- 25.Rousset, D., M. Rakoto-Andrianarivelo, R. Razafindratsimandresy, B. Randriamanalina, S. Guillot, J. Balanant, P. Mauclere, and F. Delpeyroux. 2003. Recombinant vaccine-derived poliovirus in Madagascar. Emerg. Infect. Dis. 9:885-887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sabin, A. B., M. Ramos-Alvarez, J. Alvarez-Amezquita, W. Pelon, R. H. Michaels, I. Spigland, M. A. Koch, J. M. Barnes, and J. S. Rhim. 1960. Live, orally given poliovirus vaccine. Effects of rapid mass immunization on population under conditions of massive enteric infection with other viruses. JAMA 173:1521-1526. [DOI] [PubMed] [Google Scholar]
- 27.Samoilovich, E., M. Roivainen, L. P. Titov, and T. Hovi. 2003. Serotype-specific mucosal immune response and subsequent poliovirus replication in vaccinated children. J. Med. Virol. 71:274-280. [DOI] [PubMed] [Google Scholar]
- 28.Svitkin, Y. V., N. Cammack, P. D. Minor, and J. W. Almond. 1990. Translation deficiency of the Sabin type 3 poliovirus genome: association with an attenuating mutation C472—U. Virology 175:103-109. [DOI] [PubMed] [Google Scholar]
- 29.Trujillo, A. A., K. A. McCaustland, D. P. Zheng, L. A. Hadley, G. Vaughn, S. M. Adams, T. Ando, R. I. Glass, and S. S. Monroe. 2006. Use of TaqMan real-time reverse transcription-PCR for rapid detection, quantification, and typing of Norovirus. J. Clin. Microbiol. 44:1405-1412. [DOI] [PMC free article] [PubMed] [Google Scholar]