

Abstract
The human scavenger class B type 1 receptor (SR-B1/Cla1) was identified as a putative receptor for hepatitis C virus (HCV) because it binds to soluble recombinant HCV envelope glycoprotein E2 (sE2). High-density lipoprotein (HDL), a natural SR-B1 ligand, was shown to increase the in vitro infectivity of retroviral pseudoparticles bearing HCV envelope glycoproteins and of cell culture-derived HCV (HCVcc), suggesting that SR-B1 promotes viral entry in an HDL-dependent manner. To determine whether SR-B1 participates directly in HCV infection or facilitates HCV entry through lipoprotein uptake, we generated a panel of monoclonal antibodies (MAbs) against native human SR-B1. Two of them, 3D5 and C167, bound to conformation-dependent SR-B1 determinants and inhibited the interaction of sE2 with SR-B1. These antibodies efficiently blocked HCVcc infection of Huh-7.5 hepatoma cells in a dose-dependent manner. To examine the role of HDL in SR-B1-mediated HCVcc infection, we set up conditions for HCVcc production and infection in serum-free medium. HCVcc efficiently infected Huh-7.5 cells in the absence of serum lipoproteins, and addition of HDL led to a twofold increase in infectivity. However, the HDL-induced enhancement of infection had no impact on the neutralization potency of MAb C167, despite its ability to inhibit both HDL binding to cells and SR-B1-mediated lipid transfer. Of note, MAb C167 also potently blocked Huh-7.5 infection by an HCV strain recovered from HCVcc-infected chimpanzees. These results demonstrate that SR-B1 is essential for infection with HCV produced in vitro and in vivo and suggest the possible use of anti-SR-B1 antibodies as therapeutic agents.
Hepatitis C virus (HCV) is the major etiological agent of both community-acquired and posttransfusion non-A, non-B viral hepatitis. Approximately 80% of infected patients develop chronic hepatitis, among which 20% to 30% progress to liver cirrhosis and end-stage liver disease. Chronic infection correlates with an increased risk of hepatocellular carcinoma. Currently available therapies are limited to administration of pegylated alpha interferon in combination with ribavirin (27). Such treatment is expensive, is often unsuccessful, and carries the risk of significant side effects. Consequently, the development of novel therapeutic approaches against HCV remains a high-priority goal.
HCV is an enveloped virus of the family Flaviviridae whose viral genome is a single-stranded, positive-sense RNA of approximately 9.6 kb that encodes a single polyprotein of 3,010 to 3,033 amino acids that is cleaved into nine mature proteins by a combination of host and viral peptidases (24). The predicted structural components comprise the core (C) (∼21 kDa) and two heavily N-glycosylated envelope glycoproteins, E1 (∼31 kDa) and E2 (∼70 kDa). Both E1 and E2 are believed to be type I transmembrane proteins, with N-terminal ectodomains and C-terminal hydrophobic anchors. HCV entry into target cells occurs after attachment to specific cellular receptors via its surface glycoproteins, and much effort is currently devoted to uncovering the mechanism of viral attachment to target cells and elaborating effective strategies for prevention and therapy.
A number of cellular proteins have been proposed as putative HCV receptors (2, 4, 12, 15, 33, 40), but only CD81 has been shown to play an essential role in HCV cell entry through extensive studies using gene expression knockdown by small interfering RNAs (siRNAs), antibody-mediated blocking, competition with soluble receptor homologs, and gain of entry function after transducing CD81 into CD81− cell lines (5, 12, 20, 23, 25, 47, 48, 49).
SR-B1 is a lipoprotein receptor which interacts with high-density lipoprotein (HDL), very-low-density lipoprotein, native low-density lipoprotein (LDL), chemically modified LDL (oxidized LDL [oxLDL] and acetylated LDL), and anionic phospholipids (1, 8, 11, 38). It is a 509-amino-acid cell surface glycoprotein with cytoplasmic C- and N-terminal domains separated by a large extracellular domain. SR-B1 is expressed in the liver and steroidogenic tissues, where it mediates selective cholesteryl ester uptake from HDL and acts as an endocytic receptor (7, 18, 30, 37, 41). SR-B1 was originally identified as a putative receptor for HCV because it binds soluble E2 (sE2), possibly through interaction with E2 hypervariable region 1 (HVR1; 40). Indeed, recombinant E2 lacking HVR1 did not bind SR-B1 and antibodies specific for HVR1 inhibited sE2 binding to SR-B1 (5, 40). Likewise, in vitro experiments in which human hepatoma cells were infected with retroviral pseudoparticles bearing HCV envelope glycoproteins (HCVpp) showed that deletion of HVR1 or competition with anti-HVR1 antibodies strongly reduced virus infectivity (5). Although the interaction between sE2 and SR-B1 was shown to be specific, attempts to demonstrate such an interaction by using recombinant E1-E2 heterodimers isolated from cell lysates have failed (10). Functional evidence for the role of SR-B1 in HCV infection came from partial inhibition of in vitro infectivity of HCVpp and of cell culture-derived HCV (HCVcc) with polyclonal anti-SR-B1 antibodies (5, 16, 21). However, independent studies with siRNAs against SR-B1 have yielded contradictory results. In one report, downregulation of endogenous SR-B1 levels reduced HCVpp infectivity to 30 to 90% of the control level, depending on the HCV genotype (23). In contrast, in a second study SR-B1-targeting siRNA had only a minimal effect on HCVpp infectivity despite efficient silencing of SR-B1 expression (44).
A more complex scenario recently emerged following the observation that serum lipoproteins can modulate HCV infection. In particular, HDL was shown to improve HCVpp infection via SR-B1 (6, 28, 44). On the other hand, oxLDL, another natural ligand of SR-B1, potently inhibited both HCVpp and HCVcc cell entry (46). Moreover, small-molecule inhibitors of SR-B1-mediated lipid transfer (BLT-3 and BLT-4) abrogated the stimulation of HCVpp infectivity by human serum or HDL, suggesting that the enhancement of viral infection might be dependent on the lipid exchange activity of SR-B1 (6, 44). These results led to the hypothesis that SR-B1, rather than being an essential entry factor like CD81, only modulated the efficiency of HCV entry.
To elucidate the importance of SR-B1 in HCV infection and the role of HDL in the entry process, we produced HCVcc in serum-free medium (SFM) and demonstrated that serum lipoproteins are dispensable for HCV infection of Huh-7.5 human hepatoma cells and that HDL can improve infectivity. We generated MAbs specific for human SR-B1 and identified two, 3D5 and C167, which recognized conformation-dependent epitopes on the receptor with high affinity and were capable of inhibiting binding of sE2 and HDL to SR-B1 and interfering with SR-B1-mediated lipid transfer. MAbs 3D5 and C167 potently inhibited HCVcc infectivity of human hepatoma cells regardless of the presence of HDL. The essential role of SR-B1 in HCV infection was further confirmed by MAb C167 inhibition of chimpanzee-derived HCV (ex vivo HCVcc; 26) infection of Huh-7.5 cells.
MATERIALS AND METHODS
Cell lines and antibodies.
Human hepatoma Huh-7.5 cells (29) were grown in Dulbecco's modified essential medium (DMEM; Invitrogen) supplemented with 10% fetal bovine serum (FBS). Chinese hamster ovary (CHO) cells stably transfected with human SR-B1 (CHO/SR-B1; 40) were grown in MEM alpha medium (Invitrogen) supplemented with 10% FBS. HEK-293 EBNA cells were grown in DMEM with 10% FBS, 1% nonessential amino acids, and G418 at 0.25 mg/ml. Murine MAbs 3D5 and 6B8 were raised by GENOVAC GmbH (Freiburg, Germany) after genetic immunization of BALB/c mice with a proprietary vector containing the human SR-B1 gene, followed by cell fusion and hybridoma screening. For the identification of human antibodies from phage display libraries, two rounds of affinity selection of a single-chain Fv (scFv) library were performed with CHO/SR-B1 cells. Subtraction of nonspecific clones and enrichment of SR-B1-specific binders was performed by panning selected phage on untransfected CHO cells after each round of selection. At the end, a representative number of phage (about 200) was tested in a cell-based enzyme-linked immunosorbent assay for reactivity with CHO/SR-B1 versus control untransfected CHO cells. Human MAbs C11 and C167 were generated by subcloning the variable regions from two different phages specifically binding CHO/SR-B1 into two eukaryotic vectors for the expression of heavy (immunoglobulin G4 [IgG4] isotype) and light chains, as previously described (32). These two plasmids were cotransfected into HEK-293 EBNA cells with Lipofectamine (Invitrogen), and whole human IgG4 was purified from culture medium with Hi-Trap protein A columns (Amersham Biosciences). Anti-CD81 MAb was purchased from Santa Cruz Biotechnology (clone 1.3.3.22). Negative control mouse IgG1 was obtained from Serotec, and isotype-matched control human IgG was purchased from The Binding Site.
Binding of anti-SR-B1 MAbs to Huh-7.5 cells and cross-competition analysis.
Huh-7.5 cells (n = 4 × 105) were incubated for 45 min at room temperature with anti-SR-B1 MAbs in phosphate-buffered saline (PBS)-0.2% bovine serum albumin (BSA)-HEPES at 10 mM (fluorescence-activated cell sorter [FACS] buffer). C11 and C167 binding was revealed by biotinylated anti-human IgG4 mouse MAb (BD Pharmingen), followed by streptavidin-phycoerythrin (PE; Serotec), while 3D5 and 6B8 binding was revealed by PE-conjugated anti-mouse antibody. As a control, isotype-matched human IgG4 or mouse IgG1 was used. Mean fluorescence intensities (MFIs) obtained with isotype-matched controls were subtracted from MFIs at each anti-SR-B1 MAb concentration. FACS acquisitions and analysis were performed with a FACScalibur (Becton Dickinson) and CellQuest software. Cells were stained with Sytox (Molecular Probes), and dead cells were excluded from the analyses. Competition between anti-SR-B1 MAbs was also measured by FACS analysis. Huh-7.5 cells were preincubated with a saturating concentration of anti-SR-B1 MAb or an isotype-matched control for 1 h at room temperature in FACS buffer. The competing MAb was directly labeled with a Zenon IgG labeling kit (Molecular Probes) according to the manufacturer's recommendations. Increasing concentrations of the labeled antibodies were incubated with the cells for 45 min at room temperature. Curves determined by measurement of binding in the presence of an isotype-matched control were compared to those determined in the presence of the competing antibody.
Inhibition of sE2 binding by anti-SR-B1 MAbs.
CHO/SR-B1 cells (n = 4 × 105) were incubated with increasing concentrations of anti-SR-B1 MAbs in FACS buffer for 30 min at room temperature. A fixed amount of supernatant from cells expressing recombinant H77 sE2 (39) was added to cells, which were incubated for 1 h at room temperature, and binding was revealed by FACS after incubation with anti-His mouse MAb (QIAGEN) and anti-mouse IgG1-PE (Serotec). As a control, an isotype-matched MAb was preincubated on cells before sE2 binding.
Inhibition of HDL binding to SR-B1 and of SR-B1-mediated cholesterol efflux by anti-SR-B1 MAbs.
HDLs were isolated from plasma of fasting, healthy, normolipidemic individuals by single-spin isopycnic density gradient ultracentrifugation or by sequential ultracentrifugal flotation. Labeling of HDL with the fluorescence probe 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI) was carried out as previously described (34). Inhibition of HDL binding to SR-B1 by MAbs was evaluated by incubating 5 × 105 CHO or CHO/SR-B1 cells in suspension with control IgG or the anti-SR-B1 MAbs at 5 μg/ml or no IgG for 1 h at 4°C in PBS-1% BSA. The cells were then washed, incubated with DiI-labeled HDL (50 μg protein/ml) for 1 h at 4°C, and fixed with 5% formalin neutral buffered solution (Sigma). Cell-associated DiI fluorescence was measured by flow cytometry (Epics XL; Beckman Coulter). SR-B1-mediated free-cholesterol (FC) efflux assays were carried out as previously described (42). Briefly, parental CHO and CHO/SR-B1 cells seeded in 96-well plates were incubated for 24 h in complete F12 medium supplemented with 10% FCS and 2 μg/ml acyl coenzyme A:cholesterol acyltransferase inhibitor (CP113,818). The medium was then replaced with complete F12 medium supplemented with 3 μCi/ml [3H]cholesterol, 20% FCS, and 2 μg/ml acyl coenzyme A:cholesterol acyltransferase inhibitor, and the plates were incubated for 24 h. After washing, the cells were preincubated with various concentrations of the anti-SR-B1 MAbs or control IgG for 30 min and then cholesterol efflux was stimulated by adding SFM containing purified HDL at a final concentration of 20 μg protein/ml. The medium was removed after 2 h, and the cells were washed before extraction of cell lipids with isopropyl alcohol. [3H]cholesterol content in the medium and in the total cell lipid extract was measured by liquid scintillation counting. The percentage of cholesterol efflux was calculated as the amount of [3H]cholesterol recovered in the medium divided by the total label (cells plus medium) in the wells. SR-B1-mediated cholesterol efflux was calculated by subtraction of CHO efflux from CHO/SR-B1 efflux.
HCVcc production and infection.
In vitro-transcribed genomic J6/JFH RNA was delivered to Huh-7.5 cells by electroporation (25). Infectious HCV particles (HCVcc) were recovered from cell culture medium after passaging for 2 weeks. Two days before collection of HCVcc, DMEM was replaced with hybridoma-SFM (Gibco). The virus-containing supernatant was clarified by centrifugation and stored for a few days at +4°C. Huh-7.5 cells were seeded in 24-well plates (2.5 × 104 cells/well) in DMEM-10% FBS 24 h before infection. One hundred microliters of HCVcc-containing supernatant per well (about 3 × 103 IU/well) was added to cells growing in SFM or in SFM with increasing concentrations of freshly prepared native human HDL (Autogen Bioclear, United Kingdom). Cells were incubated at 37°C for 3 h, and then virus was removed and fresh medium was added, followed by incubation for a total of 72 h. Cells were lysed, and RNA was extracted with an RNA extraction kit (Applied Biosystems).
Virus recovery from HCVcc-infected chimpanzees.
Virus recovered from a chimpanzee that had been inoculated with HCVcc is subsequently referred to as ex vivo HCVcc. Generation of ex vivo HCVcc has previously been reported (26). Briefly, 1 × 106 tissue culture infective doses were administered intravenously to two HCV-negative chimpanzees. The virus-containing serum used for this study was recovered from animal 4x0483 by plasmapheresis at 3 weeks postinoculation (26).
Neutralization of HCVcc and ex vivo HCVcc infection.
For HCVcc neutralization assay, Huh-7.5 cells were preincubated for 1 h with increasing concentrations (from 1 pg/ml to 10 μg/ml) of anti-SR-B1 MAb in 250 μl of SFM or SFM supplemented with 1.6 μg/ml HDL, and then 100 μl per well (about 3 × 103 IU/well) HCVcc-containing supernatant was added and the mixture was incubated for 3 h. Subsequently, the virus was removed, the medium was replaced, and MAbs were added again to cells at the same concentration used in the preincubation. Infection was allowed to proceed for 3 days, and then RNA was extracted and quantified. Neutralization assays with ex vivo HCVcc were performed by the same protocol with the following modifications. Heparin at 1 USP U/ml was added to ex vivo HCVcc to prevent coagulation. One hundred microliters of plasma from infected chimpanzee was used per well, and standard HCVcc stocks were diluted to ensure a multiplicity of infection of <0.01 for both HCVcc and ex vivo HCVcc.
Quantitative detection of HCV RNA.
The number of intracellular HCV genomes was determined by TaqMan assay. Briefly, we performed a 384-well plate-based assay and used 20 ng total RNA per well. To quantify HCV in RNA samples, we used oligonucleotides HCV sense (GCGAAAGGCCTTGTGGTACT) and HCV antisense (CACGGTCTACGAGACCTCCC) and probe CCTGATAGGGTGCTTGCGAGTGCC with 5′ 6-carboxyfluorescein and 3′ 6- carboxytetramethylrhodamine. HCV copies were normalized to rRNA amounts with a commercially available 18S rRNA mixture (Applied Biosystems).
Statistical analysis.
Results are presented as means ± standard errors. The statistical significance of the differences between the means of the experimental groups was tested by the Student t test for unpaired data. A difference was considered statistically significant when P was <0.05.
RESULTS
Generation of anti-SR-B1 MAbs and characterization of binding.
As a tool for investigating the role of SR-B1 in HCV infection, we generated MAbs against human SR-B1 by two different approaches. The first one consisted of genetic immunization of mice with a mammalian expression vector that encodes full-length SR-B1. After the resulting hybridomas were screened for binding to CHO cells expressing SR-B1 (CHO/SR-B1; see Materials and Methods), two MAbs, 3D5 and 6B8, were chosen for further characterization given their ability to bind CHO/SR-B1 cells but not parental CHO cells. The second approach consisted of the selection of an scFV library displayed on filamentous bacteriophage M13 (43) by using CHO/SR-B1 cells as the selector. Clones C11 and C167 showed the highest degree of specificity for SR-B1 and were therefore converted into whole human IgG4. Both strategies generated MAbs which bound to SR-B1 conformational epitopes because they failed to recognize the protein after sodium dodecyl sulfate-polyacrylamide gel electrophoresis and Western blotting (data not shown).
All four MAbs, mouse 3D5 and 6B8 and human C11 and C167, also recognized native SR-B1 displayed on the surface of Huh-7.5 cells. The measured half-saturating concentrations (apparent Kds) of the antibodies for Huh-7.5 binding by cytofluorimetric analysis were as follows: C167, 0.8 nM; 3D5, 1.4 nM; C11, 3.5 nM; 6B8, 22.0 nM (Fig. 1A). With a similar assay, we showed that MAbs C167, 3D5, and C11 inhibited sE2 binding to CHO/SR-B1 in a dose-dependent manner, with 50% inhibitory concentrations (IC50s) of 3.0 nM, 2.1 nM, and 11.4 nM, respectively (Fig. 1B). MAb 6B8 displayed only minimal inhibition (20%), even at the highest concentration tested (10 μg/ml = 67 nM; data not shown).
FIG. 1.
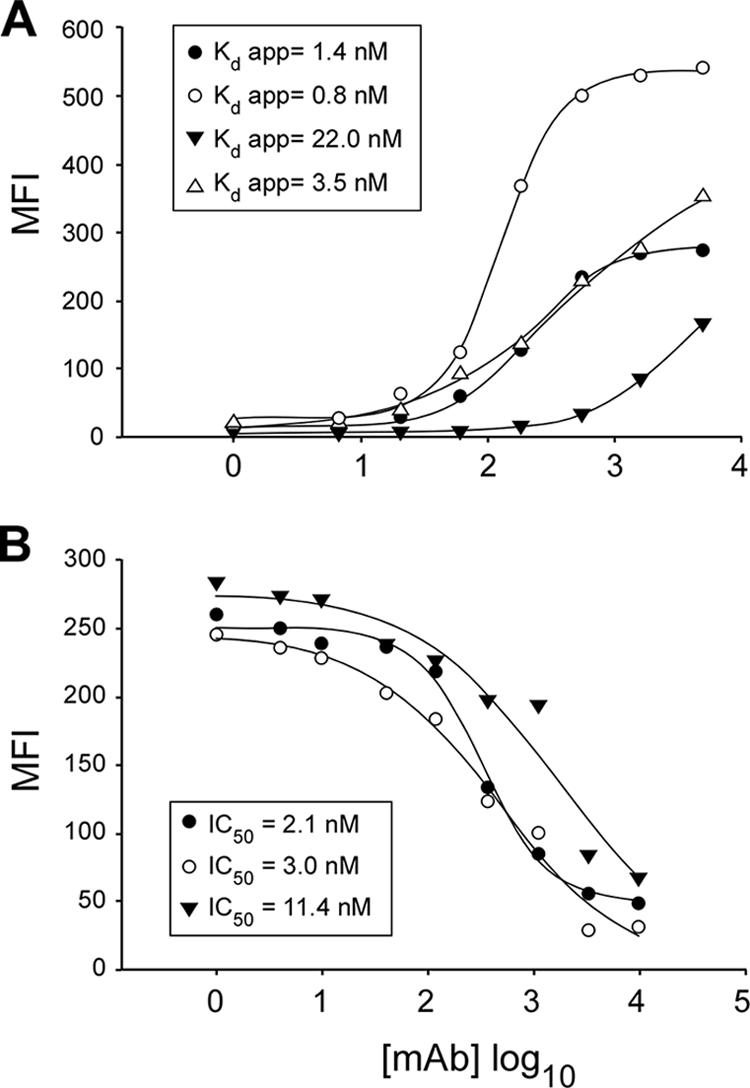
Binding properties of anti-SR-B1 MAbs and neutralization of sE2 binding to stable cell line CHO/SR-B1. (A) Binding of anti-SR-B1 MAbs to Huh-7.5 cells as measured by flow cytometry. Values on the vertical axis are MFIs. Values on the horizontal axis are concentrations (ng/ml) of anti-SR-B1 MAbs on a logarithmic scale. Saturation curves for 3D5 (closed circles), C167 (open circles), 6B8 (closed triangles), and C11 (open triangles) were fitted to experimental data with the Sigma Plot program. MFIs obtained with IgG isotypic controls were subtracted. Apparent Kds are shown in the inset. (B) Inhibition of binding of sE2 to stable cell line CHO/SR-B1 by anti-SR-B1 MAbs as measured by flow cytometry. Concentrations (ng/ml) of 3D5, C167, and C11 are indicated on the horizontal axis on a logarithmic scale. On the vertical axis are reported the MFIs of sE2 binding to the CHO/SR-B1 cell line. Neutralization-of-binding curves for MAbs 3D5 (closed circles), C167 (open circles), and C11 (closed triangles) were fitted to experimental data with the Sigma Plot program, and IC50s of MAbs are shown in the inset.
To investigate whether anti-SR-B1 MAbs recognize different epitopes, we performed cross-competition experiments. Binding of labeled 3D5 or C11 to Huh-7.5 cells was measured after preincubation with saturating concentrations of either a control isotype-matched IgG or MAb C167. Preincubation with MAb C167 reduced the binding of both 3D5 and C11 to cell surface-displayed SR-B1 by almost 80%, while the control IgG did not impair the recognition of Huh-7.5 cells (Fig. 2). These data suggested that all three MAbs recognized overlapping or closely spaced epitopes on the receptor.
FIG. 2.
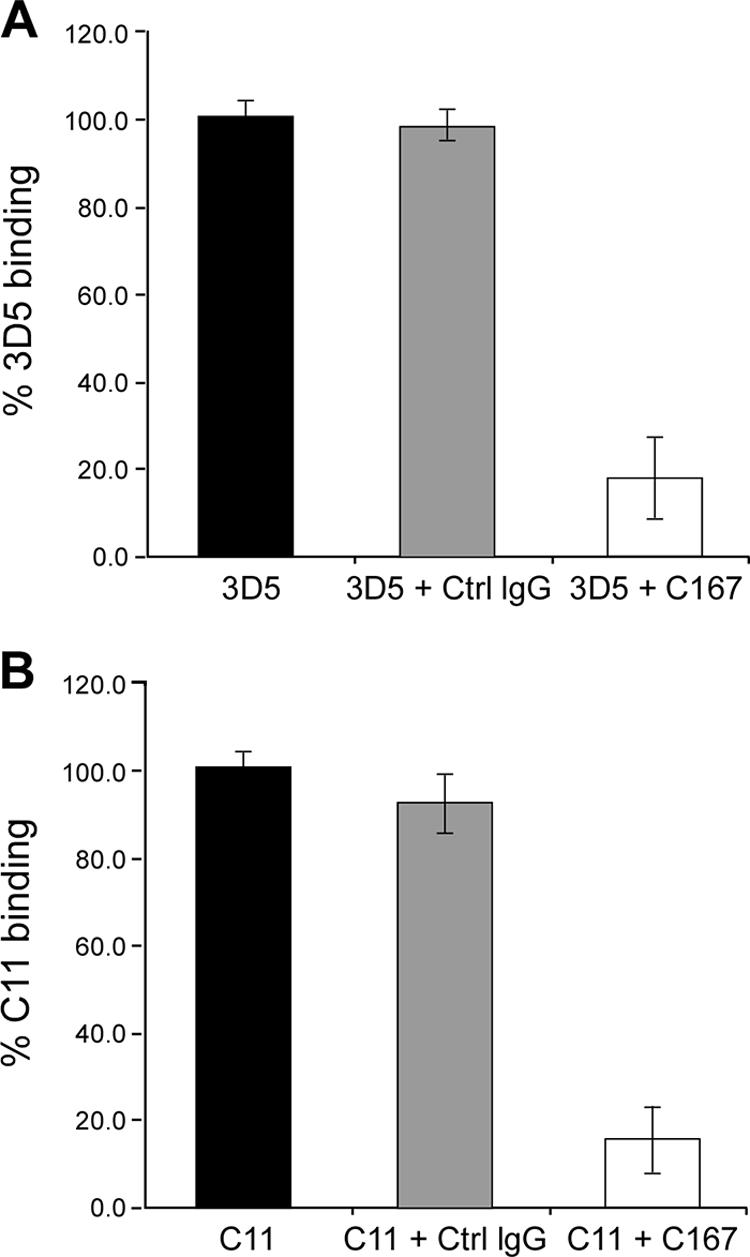
Cross-competition between anti-SR-B1 antibodies. 3D5 (A) and C11 (B) binding to Huh-7.5 cells was measured by flow cytometry in the absence of competing antibodies (black bars) or in the presence of either isotypic control IgG (Ctrl IgG; gray bars) or an anti-SR-B1 antibody (MAb C167; open bars) as a competitor. Values are percentages of MAb binding, relative to the binding of the MAb in the absence of a competing MAb.
MAbs 3D5 and C167 prevented HDL binding to SR-B1 and inhibited SR-B1-mediated cholesterol efflux.
We examined the abilities of anti-SR-B1 MAbs to interfere with HDL binding to SR-B1 and to inhibit cholesterol efflux from cells to lipoprotein acceptors. Expression of human SR-B1 in CHO cells was associated with a marked increase in HDL binding to the cell membrane compared to that in nontransfected CHO cells (Fig. 3A). Preincubation of the cells with anti-SR-B1 MAbs (5 μg/ml) before addition of labeled-HDL resulted in a moderate decrease in HDL binding to CHO/SR-B1 with MAb 6B8, whereas the C11, C167, and 3D5 antibodies significantly reduced cellular HDL binding. The four MAbs did not affect HDL binding to parental CHO cells (Fig. 3A).
FIG. 3.

Influence of anti-SR-B1 MAbs on HDL binding to SR-B1 and on SR-B1-mediated cholesterol transfer. (A) CHO or CHO/hSR-B1 cells were preincubated in PBS-BSA (no IgG; open bars) or in the presence of control IgG (Ctrl IgG; gray bars) or anti-SR-B1 MAbs (black bars, preincubation with 6B8; hatched bars, preincubation with C11; dotted bars, preincubation with C167; checked bars, preincubation with 3D5) at 5 μg/ml prior to incubation with DiI-labeled HDL (50 μg protein/ml) for 1 h at 4°C. Data are the percentages of DiI-HDL-positive cells. Values are the means ± standard deviations of three independent experiments. (B) CHO and CHO/hSR-B1 cells were labeled with [3H]cholesterol, washed, and incubated for 30 min with SFM containing increasing amounts (open bars, 0.3 μg/ml; gray bars, 1 μg/ml; black bars, 3 μg/ml; hatched bars, 9 μg/ml) of control IgG (Ctrl IgG) or anti-SR-B1 MAbs (6B8, C11, C167, and 3D5). After an additional 2 h of incubation in the same medium in the presence HDL (20 μg protein/ml), the amount of [3H]cholesterol present in the medium or remaining in the cells was determined. Data are the percentages of SR-B1-mediated cellular cholesterol efflux and were calculated as the differences between the efflux values determined with CHO/hSR-B1 and those determined with parental CHO cells. Values are the means ± standard deviations of three independent experiments.
The mechanism by which SR-B1 facilitates cholesterol flux between cells and acceptors remains only partially resolved. Nevertheless, some studies have reported that polyclonal antibodies directed against SR-B1 inhibit FC efflux to HDL (17, 42). Moreover, mutations of SR-B1 that block HDL binding are equally associated with a reduced ability of the receptor to mediate FC efflux to HDL (17). These data therefore support the notion that binding of HDL particles to SR-B1 is essential for SR-B1-mediated cellular FC efflux. In agreement with this hypothesis, MAb C11, C167, or 3D5 inhibited cellular FC efflux to HDL via SR-B1 in a dose-dependent manner in our cell-based assay system. In contrast, MAb 6B8 exhibited no blocking activity at the concentrations tested (Fig. 3B). These results suggested that MAb C11, C167, or 3D5 recognizes epitopes on SR-B1 that prevent binding of its natural ligand, HDL, because of steric hindrance or recognition of functional domains and thereby block SR-B1-mediated cholesterol transfer from the cell to HDL acceptor particles.
HCVcc infection occurs in the absence of lipoproteins, but it is enhanced in the presence of HDL.
To further analyze the involvement of SR-B1 and HDL in HCV cell entry, we used the HCV chimeric construct J6/JFH (genotype 2a) to infect Huh-7.5 cells in vitro (25). Our first goal was to evaluate the contribution of human serum or purified HDL to the viral entry process. To this end, we produced infectious HCVcc in SFM. The infectivity of HCVcc produced in the absence of lipoproteins was comparable to that of HCVcc produced in the presence of DMEM-10% FBS (Fig. 4A).
FIG. 4.
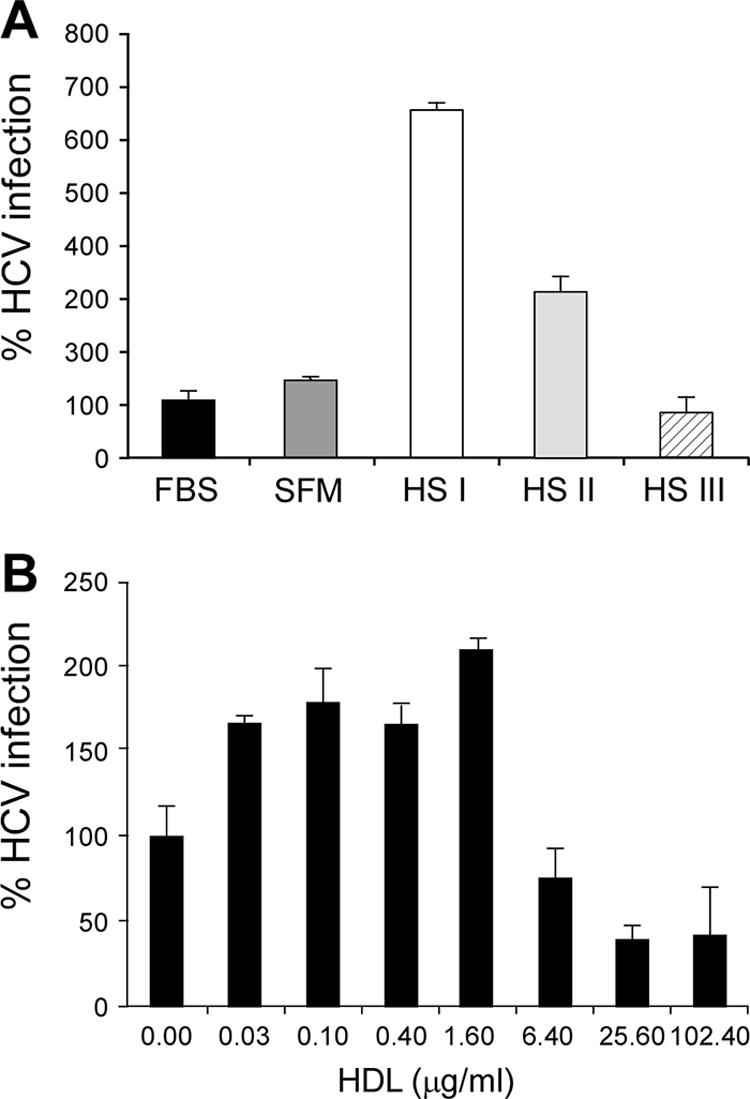
Effects of human serum and HDL on HCVcc infection. (A) J6/JFH HCVcc produced in SFM was used to infect Huh-7.5 cells in SFM (gray bar) or in SFM complemented with 10% FBS (black bar) or human serum (HS) from three different healthy donors (open bar, HS I; light gray bar, HS II; hatched bar, HS III;). Total cellular RNA was analyzed by quantitative RT-PCR for HCV content at day 3 postinfection. On the vertical axis is shown the percentage of HCV copies (% HCV infection) relative to the number of HCV copies measured in cells infected in the presence of 10% FBS. Data are averages and standard deviations of triplicate wells. (B) J6/JFH HCVcc produced in SFM was used to infect Huh-7.5 cells in SFM alone or in the presence of increasing concentrations of HDL. Total cellular RNA was analyzed by quantitative RT-PCR for HCV content at day 3 postinfection. On the vertical axis is shown the percentage of HCV copies (% HCV infection) relative to the number of HCV copies measured in cells infected in SFM. Data are averages and standard deviations of triplicate wells.
We then measured the infection efficiency of HCVcc produced in lipoprotein-free medium by adding 10% human serum from different healthy donors to SFM. The results of this experiment revealed a high degree of variability in the ability to improve infection efficiency by the different human sera, ranging from no effect (or slightly reduced infection) to sevenfold enhancement of HCVcc infection (Fig. 4A). This variability might be due to the contribution of multiple serum components to different steps in viral entry; therefore, we focused our analysis on purified HDL. We performed a dose-response experiment exploring the effects of HDL concentrations ranging from 0.03 μg/ml to 100 μg/ml on HCVcc infection of Huh-7.5 cells. HDL titration showed a maximum twofold enhancement between HDL concentrations of 0.1 μg/ml and 1.6 μg/ml (Fig. 4B). Higher HDL concentrations progressively reduced HCVcc infectivity despite the absence of cellular toxicity (Fig. 4B and data not shown).
SR-B1 mediates HCV infection also in the absence of lipoproteins.
To explore the contribution of SR-B1 in HDL-dependent and -independent HCVcc infections, we evaluated the neutralizing efficiency of anti-SR-B1 MAb C167 in SFM and in HDL-complemented SFM at the highest concentration showing enhanced in vitro infection. HCVcc produced in SFM was used to infect Huh-7.5 cells. Cells were preincubated with increasing concentrations of C167 before virus addition, and the MAb was kept on cells throughout the infection assay to minimize viral spread. We verified that MAb C167 was not toxic for the cells at any of the concentrations tested and did not modify cell growth during the infection assay (data not shown).
Supplementing SFM with 1.6 μg/ml HDL increased J6/JFH HCVcc infection efficiency by twofold. Intracellular HCV RNA levels were reduced by preincubation with MAb C167 in a dose-dependent manner under both culture conditions, with a 70 to 85% reduction in the presence of saturating concentrations of anti-SR-B1 MAb compared to cells infected in the presence of an isotype-matched control antibody (Fig. 5). MAb C167 displayed similar IC50s in SFM alone and in SFM with 1.6 μg/ml HDL (0.07 nM and 0.02 nM, respectively; P > 0.1), indicating that lipoprotein-independent HCVcc infection occurs via SR-B1 and that the ability of C167 to block HCVcc infection is not affected by HDL.
FIG. 5.
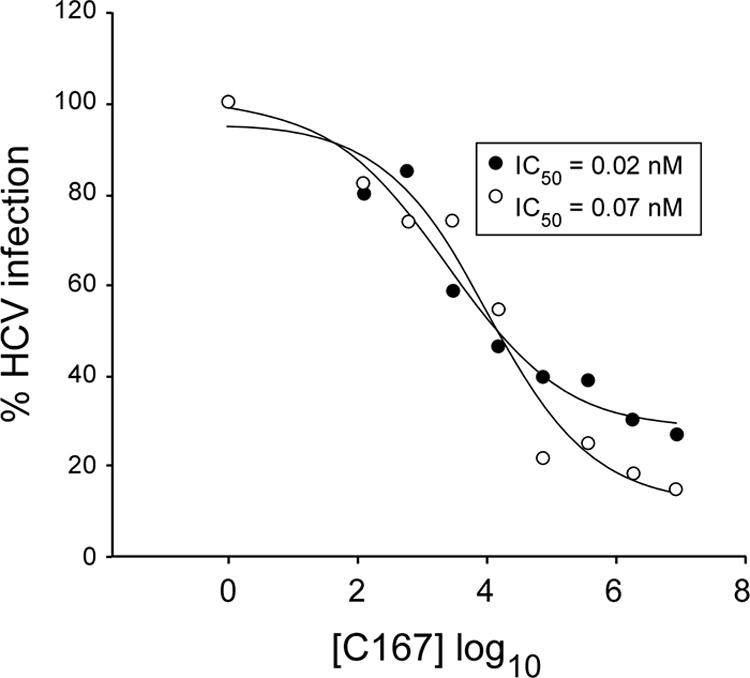
Neutralization of J6/JFH HCVcc infection by anti-SR-B1 MAb in SFM and in the presence of HDL. Huh-7.5 cells were infected with HCVcc produced in SFM upon preincubation with increasing concentrations (pg/ml) of anti-SR-B1 MAb C167 (indicated on the horizontal axis on a logarithmic scale) in SFM or in SFM plus 1.6 μg/ml HDL. Total cellular RNA was analyzed by quantitative RT-PCR for HCV content at day 3 postinfection. On the vertical axis is shown the percentage of HCV copies (% HCV infection) measured in cells infected in the presence of C167, relative to the number of HCV copies measured in control antibody-treated cells either in SFM (open circles) or in the presence of 1.6 μg/ml HDL (closed circles). Neutralization curves were extrapolated by fitting experimental data with the Sigma Plot program, and the IC50s of the MAbs are shown in the inset.
MAb 3D5 was also capable of neutralizing HCVcc infection in SFM with an IC50 lower than that observed with MAb C167 (0.13 nM versus 0.02 nM). This finding is consistent with the lower apparent affinity and lower potency displayed by the former antibody in the sE2-SR-B1 and HDL-SR-B1 binding inhibition assays, as well as in the SR-B1-mediated cholesterol efflux experiments.
To rule out the possibility that steric hindrance imposed by the anti-SR-B1 MAbs could prevent HCV attachment to CD81 because of the association or close proximity of the two receptors, we tested whether the presence of C167 would preclude the binding of a neutralizing anti-CD81 MAb (MAb 1.3.3.22; Santa Cruz Biotechnology; see Materials and Methods) to Huh-7.5 cells. Preincubation of Huh-7.5 cells with saturating amounts of MAb C167 or an unrelated IgG of the same isotype did not affect binding of the anti-CD81 MAb (apparent Kds, 0.97 nM and 1.06 nM, respectively; P > 0.1; data not shown).
MAb C167 inhibits infection of Huh-7.5 with ex vivo HCVcc.
J6/JFH HCVcc was recently shown to be infectious in vivo in chimpanzees and gave rise to in vivo-produced virus that remained infectious in cell culture (ex vivo HCVcc; 26). We wished to determine if blocking of SR-B1 with human MAb C167 could neutralize the infection of bona fide chimpanzee-derived HCV. To this end, we tested acute-phase plasma from a chimpanzee inoculated with J6/JFH HCVcc (26). Infection of Huh-7.5 with ex vivo HCVcc was measured in the presence or absence of increasing concentrations of MAb C167. The anti-SR-B1 MAb reduced ex vivo HCVcc infection at 0.1 μg/ml and nearly abolished ex vivo HCVcc infection at 1 μg/ml (Fig. 6A). As a positive control, an anti-CD81 MAb previously shown to strongly reduce JFH1 infection (47) efficiently blocked CD81 function, leading to 95% inhibition of ex vivo J6/JFH HCVcc infection at 1 μg/ml.
FIG. 6.

Anti-SR-B1 MAbs inhibit infection of Huh-7.5 with chimpanzee-derived HCV. (A) Infection of Huh-7.5 cells with chimpanzee-derived HCV (ex vivo HCVcc) was performed in the presence of anti-SR-B1 antibody (MAb C167; gray bars), anti-human-CD81 (MAb 1.3.3.22; open bars), or control isotypic IgG (Ctrl IgG; black bar) at the concentrations (μg/ml) indicated on the horizontal axis. Total cellular RNA was analyzed for HCV content by quantitative RT-PCR at 3 days postinfection. On the vertical axis is shown the percentage of HCV copies (% HCV infection) relative to the number of HCV copies measured in control antibody-treated cells. Data are averages and standard deviations of triplicate wells. (B) Infection of Huh-7.5 cells in the presence of increasing concentrations (pg/ml) of anti-SR-B1 MAb C167 (indicated on the horizontal axis on a logarithmic scale) was performed either with chimpanzee-derived HCV (ex vivo HCVcc, open circles) or with cell culture-derived HCV (HCVcc, closed circles). RNA was analyzed for HCV content by quantitative RT-PCR at day 3 postinfection. On the vertical axis is shown the percentage of HCV copies (% HCV infection) relative to the number of HCV copies measured in control antibody-treated cells. Neutralization curves were extrapolated by fitting experimental data with the Sigma Plot program, and the IC50s of the MAbs are shown in the inset.
We then repeated the inhibition of ex vivo HCVcc infection with an extended range of anti-SR-B1 MAb C167 concentrations and compared the IC50 of this MAb for the ex vivo HCVcc to that observed on the cell culture-derived virus. The anti-SR-B1 MAb reduced ex vivo HCVcc infection in a dose-dependent manner and nearly abolished ex vivo HCVcc infection at 0.2 μg/ml (Fig. 6B). MAb C167 displayed comparable IC50s on ex vivo HCVcc and cell culture-derived HCVcc (0.04 nM and 0.03 nM, respectively; P > 0.1).
DISCUSSION
It is believed that HCV initiates its life cycle through a multistep process involving the attachment of viral surface proteins to different molecules displayed on target cells, eventually leading to virus entry and release into the cytoplasm. Several cellular molecules have been identified as putative HCV receptors: the tetraspanin CD81, the scavenger class B type 1 receptor (SR-B1), mannose-binding lectins DC-SIGN and L-SIGN, the LDL receptor, heparan sulfate proteoglycans, and the asialoglycoprotein receptor. However, the role, if any, of most of these proteins in the HCV entry process remains to be established, often because of the lack of suitable reagents (i.e., antireceptor MAbs) or inconsistent results obtained with currently available in vitro binding and infection systems. In the present study, we have generated anti-SR-B1 MAbs and have used them to demonstrate that SR-B1 participates in HCV entry by three different approaches, (i) cell binding of sE2, (ii) human hepatoma cell infection with in vitro-produced HCVcc, and (iii) Huh-7.5 cell infection with ex vivo-derived HCVcc.
We previously showed that polyclonal antibodies generated by DNA immunization of mice recognizing the SR-B1 ectodomain were capable of inhibiting sE2 binding to Huh-7 and HepG2 human hepatoma cells (5). In line with this observation, genetic immunization of BALB/c mice with a plasmid expressing human SR-B1 led to the identification of two MAbs with similar inhibitory properties (3D5 and 6B8). By this procedure, we were able to generate only a limited number of different antibodies, possibly because of the high sequence identity between mouse and human SR-B1s (about 80%). Therefore, in an attempt to increase the repertoire of anti-SR-B1 antibodies with biological activity, we screened phage antibody libraries and found two phages that specifically recognized CHO/SR-B1 cells but not parental nontransfected CHO cells (C11 and C167). After conversion into human IgG, these MAbs also efficiently blocked the binding of sE2 to SR-B1 displayed on cells. The hierarchy of the sE2-blocking activities of the four MAbs correlated well with their apparent affinity (C167 > 3D5 > C11 > 6B8), suggesting that their potency might be affinity driven rather than epitope driven. This hypothesis was supported by the findings that all four MAbs recognized conformation-dependent epitopes and that three of them (3D5, C167, and C11) cross-competed for binding to the same or overlapping protein regions. We are currently generating SR-B1 mutants to map the epitopes recognized by these MAbs to help define the region(s) of SR-B1 involved in interacting with the incoming virus. Consistent with their sE2 neutralization of binding activity, both MAbs 3D5 and C167 inhibited infection of Huh-7.5 cells by J6/JFH HCVcc. Also in this assay, MAb C167 demonstrated the highest potency.
It was recently shown that HCVpp entry is facilitated by human serum or HDL in an SR-B1-dependent manner (6, 28, 44). In addition, drugs inhibiting the HDL cholesterol transfer mediated by SR-B1 abolished HDL-mediated enhancement of HCV infection (44). One consequence of SR-B1-mediated lipid uptake is an increase in the cholesterol content of target cell membranes, which is known to facilitate the entry of different viruses such as influenza virus and human immunodeficiency virus (9, 36). On the basis of these observations, it was proposed that SR-B1 is not a classical receptor but rather a molecule that affects HCV entry by modulating plasma membrane lipid composition. To determine if SR-B1 mediates HCV infection by physically interacting with the virus, we examined the ability of anti-SR-BI MAbs to inhibit HCVcc infection in the absence of lipoproteins added to the infection medium. We first established culture conditions allowing the production of J6/JFH HCVcc in SFM and then used the resulting virus for in vitro infection assays of Huh-7.5 in SFM. HCVcc produced in SFM was able to infect Huh-7.5 in the absence of added lipoproteins with an efficiency comparable to or slightly higher than that observed under previously described conditions (10% FBS; 25). Most importantly, infection occurring in the absence of added serum lipoproteins was mediated by SR-B1 because it was efficiently inhibited by anti-SR-B1 MAb C167. This finding, together with the observation that anti-SR-B1 antibodies were capable of interfering with the binding of sE2 to SR-B1 expressed on the cell surface, supports the hypothesis that HCV infection involves a direct molecular interaction between SR-B1 and the viral particle.
By comparing HCVcc infectivity in SFM with that in the presence of 10% human serum from different healthy donors, we observed a high degree of variability in infection efficiency, ranging from no effect (or slightly reduced infection) to a sevenfold increase in infectivity. This variability might be due to competing activities of serum components in HCV entry. Besides positive factors like HDL and apolipoprotein C1 (6, 28, 44), other SR-B1 ligands such as oxLDL or serum amyloid A are potent cell entry inhibitors for a broad range of HCV strains in vitro (22, 46). Thus, it is likely that beyond the basal level of infectivity mediated by CD81 and SR-B1 (as well as other putative coreceptors), the overall efficiency of HCV infection will depend on the relative concentrations of positive and negative modulating factors present in different sera through a complex interplay among serum proteins, the virus particle, and cellular proteins. This hypothesis is consistent with recent evidence indicating the formation of E2-CD81-SR-B1 or HCVpp-CD81-SR-B1 complexes through the lipid membrane layer (19).
The scenario is further complicated by the facts that HCV particles isolated from patients are often associated with plasma lipoproteins such as LDL, very-low-density lipoprotein, and HDL (3) and that via this association HCV might “hitch a ride” into hepatocytes by using receptors recognizing these serum components. While the exact mechanism by which different lipoproteins affect HCV infection is still unknown, it is becoming increasingly clear that HDL can reduce the neutralizing effect of anti-HCV antibodies (13, 45). This phenomenon might be responsible, at least in part, for the limited ability of the humoral immune response to control HCV infection in vivo (14) and raises concerns about the efficacy of anti-HCV antibodies for active or passive immunotherapy.
As an alternative to the development of anti-HCV antibodies, one could consider anti-SR-B1 human MAbs capable of interfering with HCV infection as potential therapeutic leads. The rationale for this approach would be based on antireceptor antibodies not being prone to the problems of viral variability and HDL attenuation of neutralizing activity. Previous observations implicate SR-B1 as important for infection by different HCV subtypes (5) and support the hypothesis that the same SR-B1 protein element is responsible for the recognition of different HCV E2 glycoproteins despite the high level of variability between their amino acid sequences, especially in the HVR1 region previously shown to be involved in interaction with SR-B1 (5, 40). In fact, the chemicophysical properties of HVR1 are highly conserved, suggesting that HCV HVR1, rather than being completely variable, might actually adopt only one conformation or a restricted spectrum of closely related conformations (31, 35).
We found that HDL can improve the efficiency of HCVcc infection in vitro. The strongest enhancement (twofold) of HCVcc infection occurred in the presence of HDL at concentrations ranging from 0.1 μg/ml to 1.6 μg/ml. Similarly, complementation of lipoprotein-depleted serum with 6 and 30 μg/ml HDL resulted in five- and ninefold increases in HCVpp infection, respectively (6, 44). Surprisingly, we observed in our experimental system that higher HDL concentrations (50 to 100 μg/ml), which are closer to physiological levels (0.4 to 0.9 mg/ml), resulted in a reduction of HCV entry efficiency. Whether this dose-response profile actually reflects the true in vivo activity of HDL during HCV infection or is due, rather, to the presence of inhibitory factors in the HDL preparation still remains to be clarified. However, on the basis of these data, we could evaluate the neutralizing activity of anti-SR-B1 antibodies in SFM alone or in SFM with enhancing concentrations of HDL. MAb C167 displayed an IC50 in the presence of 1.6 μg/ml HDL that was comparable to that observed in the absence of HDL. Previous data obtained with HCVpp showed that downregulation of SR-B1 gene expression by siRNA affected only HDL enhancement of infection (6). This apparent discrepancy may be due to the different viral particles and detection methods used, i.e., unmodified HCVcc and reverse transcription (RT)-PCR in the present study and HCVpp and reporter gene expression in the earlier work. Alternatively, the use of MAbs may lead to increased efficiency of blocking of the SR-B1-mediated HCV entry pathway. In support of this conclusion, we have observed that all four of the MAbs used in this study were capable of preventing HDL binding to cells and inhibiting SR-B1-mediated lipid transfer, with MAb C167 displaying the highest potency in these assays, as well as in the sE2-SR-B1 binding inhibition and HCVcc neutralization of infection experiments. Thus, it is conceivable that the efficient blocking activity of MAb C167 in the presence of HDL is due to a dual inhibitory activity of the antibody affecting both HCV particle-SR-B1 and HDL-SR-B1 interactions.
Recently, J6/JFH HCVcc was successfully used to infect chimpanzees (26). The virus grown in vivo was obtained from infected animals and successfully used to reinfect Huh-7.5 cells. Ex vivo HCVcc had higher specific infectivity than virus grown in cell culture, possibly because of increased association with lipoproteins or other biochemical and genetic modifications of the virion (26). Ex vivo HCVcc allowed us to evaluate HCV entry inhibitors in the context of virus produced in vivo. We demonstrated that MAb C167 inhibited infection of Huh-7.5 cells by ex vivo HCVcc with a potency that was comparable to that observed for HCVcc produced in cultured cells. Taken together, these data support the conclusion that SR-B1 plays a key role in HCV infection and suggest that anti-SR-B1 MAbs might prove useful for passive immunotherapy against chronic HCV infection.
Acknowledgments
We thank Manuela Emili for expert graphical work.
This work was supported by funding from the EU (RTD contract QLK2-CT-2001-01120). C.M.R. and T.V.H. were supported by the Greenberg Medical Research Institute, the Ellison Medical Foundation, the Starr Foundation, the Ronald A. Shellow Memorial Fund, the Richard Salomon Family Foundation, and the National Institutes of Health (AI072613). C.M.R. is an Ellison Medical Foundation Senior Scholar in Global Infectious Diseases. T.V.H. was also supported by a postdoctoral fellowship from the Deutsche Forschungsgemeinschaft.
This paper is dedicated to Giovanni Migliaccio, a friend and extraordinary scientist who will be missed by all of us.
Footnotes
Published ahead of print on 16 May 2007.
REFERENCES
- 1.Acton, S., A. Rigotti, K. T. Landschulz, S. Xu, H. H. Hobbs, and M. Krieger. 1996. Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science 271:518-520. [DOI] [PubMed] [Google Scholar]
- 2.Agnello, V., G. Abel, M. Elfahal, G. B. Knight, and Q. X. Zhang. 1999. Hepatitis C virus and other Flaviviridae viruses enter cells via low density lipoprotein receptor. Proc. Natl. Acad. Sci. USA 96:12766-12771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.André, P., F. Komurian-Pradel, S. Deforges, M. Perret, J. L. Berland, M. Sodoyer, S. Pol, C. Brechot, G. Paranhos-Baccala, and V. Lotteau. 2002. Characterization of low- and very-low-density hepatitis C virus RNA-containing particles. J. Virol. 76:6919-6928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barth, H., C. Schafer, M. I. Adah, F. Zhang, R. J. Linhardt, H. Toyoda, A. Kinoshita-Toyoda, T. Toida, T. H. Van Kuppevelt, E. Depla, F. Von Weizsacker, H. E. Blum, and T. F. Baumert. 2003. Cellular binding of hepatitis C virus envelope glycoprotein E2 requires cell surface heparan sulfate. J. Biol. Chem. 278:41003-41012. [DOI] [PubMed] [Google Scholar]
- 5.Bartosch, B., A. Vitelli, C. Granire, C. Goujon, J. Dubuisson, S. Pascale, E. Scarselli, R., Cortese, A. Nicosia, and F. L. Cosset. 2003. Cell entry of hepatitis C virus requires a set of coreceptors that include the CD81 tetraspanin and the SR-B1 scavenger receptor. J. Biol. Chem. 278:41624-41630. [DOI] [PubMed] [Google Scholar]
- 6.Bartosch, B., G. Verney, M. Dreux, P. Donot, Y. Morice, F. Penin, J. M. Pawlotsky, D. Lavillette, and F. L. Cosset. 2005. An interplay between hypervariable region 1 of the hepatitis C virus E2 glycoprotein, the scavenger receptor B1, and high-density lipoprotein promotes both enhancement of infection and protection against neutralizing antibodies. J. Virol. 79:8217-8229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Calvo, D., and M. A. Vega. 1993. Identification, primary structure, and distribution of CLA-1, a novel member of the CD36/LIMPII gene family. J. Biol. Chem. 268:18929-18935. [PubMed] [Google Scholar]
- 8.Calvo, D., D. Gómez-Coronado, M. A. Lasunción, and M. A. Vega. 1997. CLA-1 is a 85 kD plasma membrane glycoprotein which acts as a high affinity receptor for both native (HDL, LDL and VLDL) and modified (OxLDL and AcLDL) lipoproteins. Arterioscler. Thromb. Vasc. Biol. 17:2341-2349. [DOI] [PubMed] [Google Scholar]
- 9.Chazal, N., and D. Gerlier. 2003. Virus entry, assembly, budding, and membrane rafts. Microbiol. Mol. Biol. Rev. 67:226-237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cocquerel, L., C. Voisset, and J. Dubuisson. 2006. Hepatitis C virus entry: potential receptors and their biological functions. J. Gen. Virol. 87:1075-1084. [DOI] [PubMed] [Google Scholar]
- 11.Connelly, M. A., and D. L. Williams. 2004. Scavenger receptor BI: a scavenger receptor with a mission to transport high density lipoprotein lipids. Curr. Opin. Lipidol. 15:287-295. [DOI] [PubMed] [Google Scholar]
- 12.Cormier, E. G., F. Tsamis, F. Kajumo, R. J. Durso, J. P. Gardner, and T. Dragic. 2004. CD81 is an entry coreceptor for hepatitis C virus. Proc. Natl. Acad. Sci. USA 101:7270-7274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dreux, M., T. Pietschmann, C. Granier, C. Voisset, S. Ricard-Blum, P. E. Mangeot, Z. Keck, S. Foung, N. Vu-Dac, J. Dubuisson, R. Bartenschlager, D. Lavillette, and F. L. Cosset. 2006. High density lipoprotein inhibits hepatitis C virus-neutralizing antibodies by stimulating cell entry via activation of the scavenger receptor BI. J. Biol. Chem. 281:18285-18295. [DOI] [PubMed] [Google Scholar]
- 14.Dustin, L. B., and C. M. Rice. 2007. Flying under the radar: the immunobiology of hepatitis C. Annu. Rev. Immunol. 25:71-99. [DOI] [PubMed] [Google Scholar]
- 15.Gardner, J. P., R. J. Durso, R. R. Arrigale, G. P. Donovan, P. J. Maddon, T. Dragic, and W. C. Olson. 2003. L-SIGN (CD 209L) is a liver-specific capture receptor for hepatitis C virus. Proc. Natl. Acad. Sci. USA 100:4498-4503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grove, J., T. Huby, Z. Stamataki, T. Vanwolleghem, P. Meuleman, M. Farquhar, A. Schwarz, M. Moreau, J. S. Owen, G. Leroux-Roels, P. Balfe, and J. A. McKeating. 2007. Scavenger receptor BI and BII expression levels modulate hepatitis C virus infectivity. J. Virol. 81:3162-3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gu, X., K. Kozarsky, and M. Krieger. 2000. Scavenger receptor class B, type I-mediated [3H]cholesterol efflux to high and low density lipoproteins is dependent on lipoprotein binding to the receptor. J. Biol. Chem. 275:29993-30001. [DOI] [PubMed] [Google Scholar]
- 18.Harder, C. J., G. Vassiliou, H. M. McBride, and R. McPherson. 2006. Hepatic SR-BI-mediated cholesteryl ester selective uptake occurs with unaltered efficiency in the absence of cellular energy. J. Lipid Res. 47:492-503. [DOI] [PubMed] [Google Scholar]
- 19.Heo, T. H., S. M. Lee, B. Bartosch, F. L. Cosset, and C. Y. Kang. 2006. Hepatitis C virus E2 links soluble human CD81 and SR-B1 protein. Virus Res. 121:58-64. [DOI] [PubMed] [Google Scholar]
- 20.Hsu, M., J. Zhang, M. Flint, C. Logvinoff, C. Cheng-Mayer, C. M. Rice, and J. A. McKeating. 2003. Hepatitis C virus glycoproteins mediate pH-dependent cell entry of pseudotyped retroviral particles. Proc. Natl. Acad. Sci. USA 100:7271-7276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kapadia, S. B., H. Barth, T. Baumert, J. A. McKeating, and F. V. Chisari. 2007. Initiation of HCV infection is dependent on cholesterol and cooperativity between CD81 and scavenger receptor B type I. J. Virol. 81:374-383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lavie, M., C. Voisset, N. Vu-Dac, V. Zurawski, G. Duverlie, C. Wychowski, and J. Dubuisson. 2006. Serum amyloid A has antiviral activity against hepatitis C virus by inhibiting virus entry in a cell culture system. Hepatology 44:1626-1634. [DOI] [PubMed] [Google Scholar]
- 23.Lavillette, D., A. W. Tarr, C. Voisset, P. Donot, B. Bartosch, C. Bain, A. H. Patel, J. Dubuisson J. K. Ball, and F. L. Cosset. 2005. Characterization of host-range and cell entry properties of the major genotypes and subtypes of hepatitis C virus. Hepatology 41:265-274. [DOI] [PubMed] [Google Scholar]
- 24.Lindenbach, B. D., and C. M. Rice. 2001. Flaviviridae: the viruses and their replication, p. 991-1041. In D. M. Knipe and P. M. Howley (ed.), Fields virology, 4th ed. Lippincott Williams & Wilkins, Philadelphia, PA.
- 25.Lindenbach, B. D., M. J. Evans, A. J. Syder, B. Wolk, T. L. Tellinghuisen, C. C. Liu, T. Maruyama, R. O. Hynes, D. R. Burton, J. A. McKeating, and C. M. Rice. 2005. Complete replication of hepatitis C virus in cell culture. Science 309:623-626. [DOI] [PubMed] [Google Scholar]
- 26.Lindenbach, B. D., P. Meuleman, A. Ploss, T. Vanwolleghem, A. J. Syder, J. A. McKeating, R. E. Lanford, S. M. Feinstone, M. E. Major, G. Leroux-Roels, and C. M. Rice. 2006. Cell culture-grown hepatitis C virus is infectious in vivo and can be recultured in vitro. Proc. Natl. Acad. Sci. USA 103:3805-3809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McHutchison, J. G., S. C. Gordon, E. R. Schiff, M. L. Shiffman, W. M. Lee, V. K. Rustgi, Z. D. Goodman, M. H. Ling, S. Cort, and J. K. Albrecht. 1998. Interferon alfa-2b alone or in combination with ribavirin as initial treatment for chronic hepatitis C. N. Engl. J. Med. 339:1485-1492. [DOI] [PubMed] [Google Scholar]
- 28.Meunier, J. C., R. E. Engle, K. Faulk, M. Zhao, B. Bartosch, H. Alter, S. U. Emerson, F. L. Cosset, R. H. Purcell, and J. Bukh. 2005. Evidence for cross-genotype neutralization of hepatitis C virus pseudo-particles and enhancement of infectivity by apolipoprotein C1. Proc. Natl. Acad. Sci. USA 102:4560-4565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakabayashi, H., K. Taketa, K. Miyano, T. Yamane, and J. Sato. 1982. Growth of human hepatoma cell lines with differentiated functions in chemically defined medium. Cancer Res. 42:3858-3863. [PubMed] [Google Scholar]
- 30.Pagler, T. A., S. Rhode, A. Neuhofer, H. Laggner, W. Strobl, C. Hinterndorfer, I. Volf, M. Pavelka, E. R. Eckhardt, D. R. van der Westhuyzen, G. J. Schutz, and H. Stangl. 2006. SR-BI mediated HDL endocytosis leads to HDL resecretion facilitating cholesterol efflux. J. Biol. Chem. 281:11193-11204. [DOI] [PubMed] [Google Scholar]
- 31.Penin, F., C. Combet, G. Germanidis, P. O. Frainais, G. Deleage, and J. M. Pawlotsky. 2001. Conservation of the conformation and positive charges of hepatitis C virus E2 envelope glycoprotein hypervariable region 1 points to a role in cell attachment. J. Virol. 75:5703-5710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Persic, L., A. Roberts, J. Wilton, A. Cattaneo, A. Bradbury, and H. R. Hoogenboom. 1997. An integrated vector system for the eukaryotic expression of antibodies or their fragments after selection from phage display libraries. Gene 10:9-18. [DOI] [PubMed] [Google Scholar]
- 33.Pileri, P., Y. Uematsu, S. Compagnoli, G. Galli, F. Falugi, R. Petracca, A. J. Weiner, M. Houghton, D. Rosa, G. Grandi, and S. Abrignani. 1998. Binding of hepatitis C virus to CD81. Science 282:938-941. [DOI] [PubMed] [Google Scholar]
- 34.Pitas, R. E., T. L. Innerarity, J. N. Weinstein, and R. W. Mahley. 1981. Acetoacetylated lipoproteins used to distinguish fibroblasts from macrophages in vitro by fluorescence microscopy. Arteriosclerosis 1:177-185. [DOI] [PubMed] [Google Scholar]
- 35.Puntoriero, G., A. Meola, A. Lahm, S. Zucchelli, B. B. Ercole, R. Tafi, M. Pezzanera, M. U. Mondelli, R. Cortese, A. Tramontano, G. Galfre, and A. Nicosia. 1998. Towards a solution for hepatitis C virus hypervariability: mimotopes of the hypervariable region 1 can induce antibodies cross-reacting with a large number of viral variants. EMBO J. 17:3521-3533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rawat S. S, M. Viard, S. A. Gallo, A. Rein, R. Blumenthal, and A. Puri. 2003. Modulation of entry of enveloped viruses by cholesterol and sphingolipids. Mol. Membr. Biol. 20:243-254. [DOI] [PubMed] [Google Scholar]
- 37.Rhainds, D., and L. Brissette. 2004. The role of scavenger receptor class B type I (SR-BI) in lipid trafficking. Defining the rules for lipid traders. Int. J. Biochem. Cell Biol. 36:39-77. [DOI] [PubMed] [Google Scholar]
- 38.Rigotti, A., S. L. Acton, and M. Krieger. 1995. The class B scavenger receptors SR-BI and CD36 are receptors for anionic phospholipids. J. Biol. Chem. 276:16221-16224. [DOI] [PubMed] [Google Scholar]
- 39.Roccasecca, R., H. Ansuini, A. Vitelli, A. Meola, E. Scarselli, S. Acali, M. Pezzanera, B. B. Ercole, J. A. McKeating, A. Yagnik, A. Lahm, A. Tramontano, R. Cortese, and A. Nicosia. 2003. Binding of the hepatitis C virus E2 glycoprotein to CD81 is strain specific and is modulated by a complex interplay between hypervariable regions 1 and 2. J. Virol. 77:1856-1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Scarselli, E., H. Ansuini, R. Cerino, R. M. Roccasecca, S. Acali, G. Filocamo, C. Traboni, A. Nicosia, R. Cortese, and A. Vitelli. 2002. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J. 21:5017-5025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Silver, D. L., N. Wang, X. Xiao, and A. R. Tall. 2001. High density lipoprotein (HDL) particle uptake mediated by scavenger receptor class B type 1 results in selective sorting of HDL cholesterol from protein and polarized cholesterol secretion. J. Biol. Chem. 276:25287-25293. [DOI] [PubMed] [Google Scholar]
- 42.Tréguier, M., M. Moreau, A. Sposito, M. J. Chapman, and T. Huby. 2007. LDL particle subspecies are distinct in their capacity to mediate free cholesterol efflux via the SR-BI/Cla-1 receptor. Biochim. Biophys. Acta 1771:129-138. [DOI] [PubMed] [Google Scholar]
- 43.Vaughan, T. J., A. J. Williams, K. Pritchard, J. K. Osbourn, A. R. Pope, J. C. Earnshaw, J. McCafferty, R. A. Hodits, J. Woilton, and K. S. Johnson. 1996. Human antibodies with sub-nanomolar affinities isolated from a large non-immunized phage display library. Nat. Biotechnol. 14:309-314. [DOI] [PubMed] [Google Scholar]
- 44.Voisset, C., N. Callens, E. Blanchard, A. Op De Beeck, J. Dubuisson, and N. Vu-Dac. 2005. High density lipoproteins facilitate hepatitis C virus entry through the scavenger receptor class B type I. J. Biol. Chem. 280:7793-7799. [DOI] [PubMed] [Google Scholar]
- 45.Voisset, C., A. Op de Beeck, P. Horellou, M. Dreux, T. Gustot, G. Duverlie, F. L. Cosset, N. Vu-Dac, and J. Dubuisson. 2006. High-density lipoproteins reduce the neutralizing effect of hepatitis C virus (HCV)-infected patient antibodies by promoting HCV entry. J. Gen. Virol. 87:2577-2581. [DOI] [PubMed] [Google Scholar]
- 46.von Hahn, T., B. D. Lindenbach, A. Boullier, O. Quehenberger, M. Paulson, C. M. Rice, and J. A. McKeating. 2006. Oxidized low-density lipoprotein inhibits hepatitis C virus cell entry in human hepatoma cells. Hepatology 43:932-942. [DOI] [PubMed] [Google Scholar]
- 47.Wakita, T., T. Pietschmann, T. Kato, T. Date, M. Miyamoto, Z. Zhao, K. Murthy, A. Habermann, H. G. Krausslich, M. Mizokami, R. Bartenschlager, and T. J. Liang. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 11:791-796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang, J., G. Randall, A. Higginbottom, P. Monk, C. M. Rice, and J. A. McKeating. 2004. CD81 is required for hepatitis C virus glycoprotein-mediated viral infection. J. Virol. 78:1448-1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhong, J., P. Gastaminza, G. Cheng, S. Kapadia, T. Kato, D. R. Burton, S. F. Wieland, S. L. Uprichard, T. Wakita, and F. V. Chisari. 2005. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. USA 102:9294-9299. [DOI] [PMC free article] [PubMed] [Google Scholar]