

Abstract
To explore the pathogenic mechanism of dominant mutations affecting the carboxyl terminus of rhodopsin that cause retinitis pigmentosa, we generated five lines of transgenic mice carrying the proline-347 to serine (P347S) mutation. The severity of photoreceptor degeneration correlated with the levels of transgene expression in these lines. Visual function as measured by the electroretinogram was approximately normal at an early age when there was little histologic evidence of photoreceptor degeneration, but it deteriorated as photoreceptors degenerated. Immunocytochemical staining showed the mutant rhodopsin predominantly in the outer segments prior to histologically evident degeneration, a finding supported by quantitation of signal intensities in different regions of the photoreceptor cells by confocal microscopy. A distinct histopathologic abnormality was the accumulation of submicrometer-sized vesicles extracellularly near the junction between inner and outer segments. The extracellular vesicles were bound by a single membrane that apparently contained rhodopsin as revealed by ultrastructural immunocytochemical staining with anti-rhodopsin antibodies. The outer segments, although shortened, contained well-packed discs. Proliferation of the endoplasmic reticulum as reported in Drosophila expressing dominant rhodopsin mutations was not observed. The accumulation of rhodopsin-laden vesicles likely represents aberrant transport of rhodopsin from the inner segments to the nascent disc membranes of the outer segments. It is possible that photoreceptor degeneration occurs because of a failure to renew outer segments at a normal rate, thereby leading to a progressive shortening of outer segments, or because of the loss of cellular contents to the extracellular space, or because of both.
Mutations in the rhodopsin gene are a cause of dominant and recessive retinitis pigmentosa (RP), accounting for about 10% of all cases. Over 70 dominant alleles have been identified (1), most of which are missense mutations. They do not cause photoreceptor degeneration because of haploinsufficiency, since at least one recessive null allele does not cause disease in heterozygous carriers (2). Dominant rhodopsin mutants are therefore gain-of-function or dominant-negative mutants.
A number of in vitro studies have been carried out to understand the pathogenic mechanism of rhodopsin mutants. On the basis of analyses of rhodopsin mutants expressed in cultured embryonic kidney cell lines, several classes were defined (3, 4, 5). A small number of mutants, designated class I, are like wild type (wt) opsin in their ability to localize to the cytoplasmic membrane and to bind 11-cis-retinaldehyde to form spectrally active rhodopsin. The majority of mutants, designated class II, accumulate abnormally in the rough endoplasmic reticulum, are not transported efficiently to the cytoplasmic membrane as is wt opsin, and fail to bind 11-cis-retinaldehyde to form a spectrally active photopigment. Class II mutants are thought to be defective in protein folding and/or stability. A third class of mutants constitutively activate transducin in vitro (6). These observations form the basis for a number of hypotheses regarding the pathogenic mechanism of dominant rhodopsin mutations (see Discussion).
Among the class I mutants are those affecting proline-347 near the carboxyl terminus. This residue is conserved among all known visual pigments. Six different missense mutations affecting this residue have been identified among patients with RP (1), indicating that proline-347 mediates some vital function of rhodopsin. Proline-347 mutants are of particular interest, since no functional abnormalities of the mutant proteins have been observed in in vitro studies, yet they tend to cause a more severe form of RP than that found in patients with dominant RP due to other mutations (7). This would indicate that expression of the mutant phenotype requires a photoreceptor cell environment and that the pathogenic mechanism of such mutants is best studied in an in vivo setting. Another group has developed a line of transgenic mice expressing a pig rhodopsin with the proline-347 to serine (P347S) mutation. It was found to develop photoreceptor degeneration and to exhibit an increased level of cAMP in the retina, a frequent finding associated with cell death in a variety of tissues (8). Tests of retinal function [i.e., electroretinograms (ERGs)], mutant rhodopsin localization, and ultrastructural features were not described. In the present study, we generated transgenic mice carrying a human P347S allele to investigate the early pathogenic events by which this rhodopsin mutant leads to photoreceptor cell death.
MATERIALS AND METHODS
Generation of Transgenic Mice.
Genomic DNA was purified from the peripheral blood of a patient with dominant RP due to the P347S mutation in the rhodopsin gene. The DNA was partially digested with MboI, and purified fragments between 12 and 20 kb were used to generate a library in the λ bacteriophage vector EMBL3. This library was screened using a human rhodopsin cDNA sequence (9). Phage DNA from positive clones was analyzed by partial DNA sequencing to identify those derived from the P347S allele. One phage clone contained a 13.3-kb insert which encompassed the entire transcriptional unit of the mutant rhodopsin gene with 3.4 kb of upstream and 4.8 kb of downstream flanking sequences. This DNA insert was purified and used for microinjection into fertilized Fvb/n mouse embryos. Founder mice were bred with C57BL/6 (Charles River Laboratories, as were all subsequent generations of transgenic mice. The mutant transcript was verified by sequencing after reverse transcription and PCR amplification. The relative levels of transgene expression were quantified as a ratio of transgenic (human) to endogenous (mouse) rhodopsin mRNA as previously described (10, 11). A previously characterized transgenic mouse line carrying a wt human rhodopsin transgene [NHR, E line (10)] was used as a control in this study.
ERG Testing.
Dark-adapted ERG testing was performed as described (10) except that electrodes were placed in contact with the cornea instead of in the vitreous humor. Full-field ERGs were elicited with 10-μsec flashes of white light [4.6 log footlamberts (1 footlambert = 3.4 cd/m2)] presented from a Ganzfeld dome. In our test system, the a-wave amplitude in adult wt mice is between 150 and 300 μV.
Immunofluorescence and Confocal Microscopy.
Frozen retinal sections were examined under an Olympus IX-70 fluorescence microscope after staining with the monoclonal antibody rho 3A6 and Cy3-conjugated secondary antibody as described (10). Sections were also examined under a Leica TCS 4D confocal laser scanning microscope. Sections were scanned and digitized with a 40× objective in the rhodamine channel. The voltage gains and offset contrast were set so that signal intensities from the outer segments just reached saturation in the NHR-E sections. The average of a series of 16 scans was taken for each image. To plot histograms of signal intensity versus location in the photoreceptors, straight lines 200 μm in length were drawn perpendicular to and originating from the retinal pigment epithelium (RPE) layer. Along these lines, the signal intensities were measured. Measurements along three such lines were superimposed to give each of the plots shown in Fig. 3.
Figure 3.
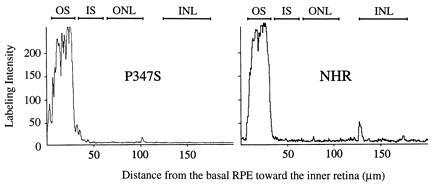
Labeling intensity histograms along the expanse of the retina plotted from digitized images under a confocal microscope. The retinas were from the P347S C1 line and the NHR-E line at 20 days of age. The vertical axis represents signal intensity, saturating at 250 arbitrary units. The horizontal axis extends left to right from the basal RPE toward the inner retina. The approximate locations of the retinal layers are marked at the top. Abbreviations are the same as in Fig. 2.
Light Microscopy and Electron Microscopy (EM).
Light and transmission EM examinations were carried out as described (10). For scanning EM, eye cups were fixed in 2% glutaraldehyde/4% formaldehyde/0.1 M sodium phosphate buffer, pH 7.0, overnight. The retinas were then removed from the eye cup, dehydrated through a graded ethanol series, and critical-point dried. The retinas were then fractured to expose fresh cross sections and sputter-coated with gold/palladium. The specimens were viewed and photographed under an Amray 1400 scanning electron microscope.
For immunocytochemistry under the electron microscope, eyes were fixed in 1% glutaraldehyde/4% formaldehyde in phosphate-buffered saline (PBS). Eye cups were embedded in LR White plastic (Ted Pella, Redding, CA), and thin sections were stained with either one of two monoclonal anti-rhodopsin antibodies, rho 1D4 (specific for the carboxyl terminus) and rho 4D2 (specific for the amino terminus) (12). These two antibodies recognize both human and mouse rhodopsins. This was followed by staining with gold-conjugated goat anti-mouse IgG (Amersham). Sections were then stained with uranyl acetate and lead citrate.
RESULTS
P347S Mutant Rhodopsin Causes Photoreceptor Degeneration.
Five lines of P347S transgenic mice were established. These lines differ in their levels of transgene expression as measured by the ratio of transgene mRNA to endogenous mouse opsin mRNA (Table 1). Light microscopy of retinal sections showed a wide range of photoreceptor degeneration among the lines at a given age, from shortened and disorganized rod outer segments to complete absence of the outer nuclear layer. The severity of retinal degeneration approximately correlated with the level of transgene mRNA (Table 1). In lines A1 and C2 with an intermediate level of expression, approximately four to five rows of photoreceptor nuclei remained at 20–30 days of age. There were many pyknotic nuclei in the outer nuclear layer, indicating ongoing cell death. By 4 months of age, only one to two rows of nuclei remained. In line C1, which had the lowest expression, photoreceptor cell loss was not apparent at 20–30 days, but the outer segments appeared slightly shorter and disorganized. By 4–5 months of age about half of the photoreceptor cells were lost in line C1. Retinal morphology of selected lines and matching ERG recordings are shown in Fig. 1. In all lines, the reduction in ERG amplitudes appeared to correlate with the extent of photoreceptor cell loss. Importantly, at a time when there was little cell loss (e.g., P347S C1 at 30 days), the ERG amplitudes appeared comparable to those of wt (Fig. 1 and Table 1).
Table 1.
Correlation between level of transgene expression and severity of degeneration
| Line | Ratio of transgene to endogenous opsin mRNA | Severity of retinal degeneration measured by ERG a-wave amplitude,* μV |
|---|---|---|
| P347 C1 | 0.25:1 | 191 ± 88.6 (n = 7) |
| P347 A1 | 1:1 | 53 ± 9.0 (n = 9) |
| P347 C2 | 1:1 | 14 ± 12.0 (n = 6) |
| P347 A2 | 5:1 | 8 ± 6.6 (n = 4) |
| P347 E | 5:1 | 3 ± 1.4 (n = 4) |
Measurements were made at 30 days of age. The range for a-wave amplitudes in normal mice is 100–300 μV. Values are mean ± SD.
Figure 1.
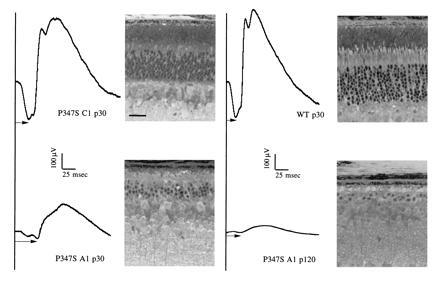
ERG recordings and matching histology of transgenic and control mice. The ages are indicated in postnatal days (e.g., p30). Arrows indicate the measurement of a-wave implicit times. A range of 18–28 msec is typically observed for wt mice. (Bar = 25 μm.)
Over-expression of wt rhodopsin in transgenic mice has been shown to cause retinal degeneration (10, 13). Heterozygous NHR-E transgenic mice with an expression level of 1:1 do not develop degeneration (10) when followed up to 15 months of age. When the mice are bred to homozygosity, transgene expression is increased to 2:1, and the homozygous NHR-E mice exhibit a slow course of photoreceptor degeneration (data not shown). Thus in the C1, C2, and A1 lines with ratios of transgene to endogenous opsin mRNA less than 2:1 (see Table 1), the retinal degeneration can be attributed to a specific pathogenic effect of the mutation, rather than a deleterious effect due to over-expression of rhodopsin. The A2 and E lines likely suffer from a combination of over-expression of rhodopsin and the deleterious effect of the P347S mutation; these lines were not analyzed further.
The P347S Mutant Rhodopsin Localizes Predominantly in the Outer Segments.
To investigate whether the P347S mutant rhodopsin could be transported from the site of synthesis (inner segments) to the outer segments, we examined the distribution of mutant rhodopsin in photoreceptor cells. The monoclonal antibody rho 3A6 (14), which recognizes human but not mouse rhodopsin, was used for immunocytochemical localization of the mutant rhodopsin (which was of human origin) in photoreceptor cells. Fig. 2 shows immunofluorescence images of a retina from line C1 at age 20 days in comparison to NHR-E and wt retinas at the same age. In the P347S C1 retinas, the outer segment layer was brightly stained. Staining of the inner segments and the outer nuclear layer was minimal. Thus, the P347S mutant rhodopsin localized predominantly in photoreceptor outer segments in predegenerate retinas. The normal human rhodopsin in the NHR-E transgenic retinas was found in the outer segments as expected. Retinas from wt mice showed very little staining anywhere, confirming the specificity of the procedures.
Figure 2.
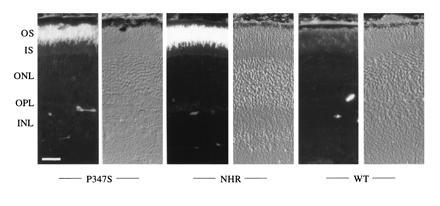
Localization of mutant rhodopsin by immunofluorescence. For each genotype labeled at the bottom of the figure, the left panel is an immunofluorescence image, and the right is the same view photographed under Hoffman interference contrast optics. The P347S retina was from the C1 line, the NHR-E retina was from a heterozygote, and the wt retina was from a C57BL/6 mouse. All mice were 20 days of age. OS, outer segments; IS, inner segments; ONL, outer nuclear layer; OPL, outer plexiform layer; INL, inner nuclear layer. (Bar = 25 μm.)
To evaluate quantitatively the possibility that a small amount of mutant rhodopsin was ectopically distributed but not apparent by regular fluorescence microscopy, rho 3A6-stained retinal sections were examined under a laser confocal microscope. The digitized images of P347S C1 and NHR-E retinas (both at 20 days of age) were plotted as histograms of signal intensity versus distance from the base of the RPE 2. These plots confirmed the presence of mutant rhodopsin predominantly in the outer segments and minimally elsewhere (Fig. 3).
Photoreceptors in Mutant Mice Accumulate Extracellular Vesicles.
Ultrastructurally, P347S retinas were compared with NHR-E and wt retinas. Under transmission EM, there was no visible abnormality in the photoreceptor nuclear layer of P347S C1 retinas at 20 days of age. In the myoid region of the inner segments, the appearance of the rough endoplasmic reticulum and the Golgi apparatus did not notably differ from that in the NHR-E retina (data not shown). The outer segments contained well-packed discs (Fig. 4). The retinal pigment epithelium appeared normal and contained phagosomes encompassing phagocytosed disc material (not shown). A distinct feature in C1 retinas was the presence of vesicular profiles near the base of the outer segments and the apical portion of the inner segments (Fig. 4b). The vesicles were submicrometer-sized and bounded by a single membrane. The space between the vesicles had a much lower electron density than the cytoplasm, and the vesicles appeared outside of the cell membranes. This phenotype was apparently gene dosage-dependent, since more vesicles were seen in P347S C1 mice bred to homozygosity for the transgene (Fig. 4c) and in P347S A1 mice (Fig. 4d). In the latter two retinas, the inner segments were shorter, narrower, and irregular along the borders. The outer segment length was severely reduced.
Figure 4.
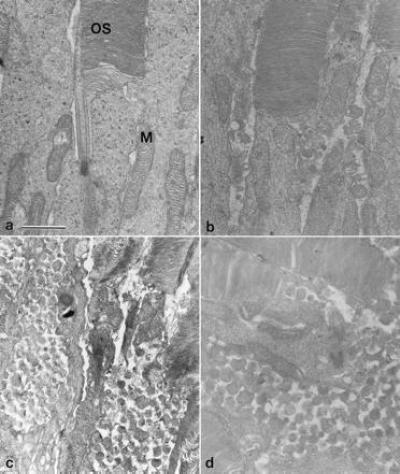
Transmission electron micrographs of the apical inner segments and basal outer segments of photoreceptor cells from 20-day-old retinas. (a) normal morphology in NHR-E retina shown as a control. (b) P347S C1 heterozygote. (c) P347S C1 homozygote. (d) P347S A1. OS, outer segments; M, mitochondria. (Bar = 1 μm.)
To ascertain that these vesicular profiles were spherules and that they were extracellular, the mutant retinas were examined by scanning EM. As shown in Fig. 5a, a wt control retina showed clearly defined, smooth inner and outer segments and connecting cilia. In contrast, clusters of spherules were seen in the P347S A1 retinas, obscuring the inner and outer segments (Fig. 5b). This is consistent with the findings under transmission EM that photoreceptor inner and outer segments were surrounded by a multitude of vesicles. This appearance is also consistent with the vesicles being extracellular.
Figure 5.
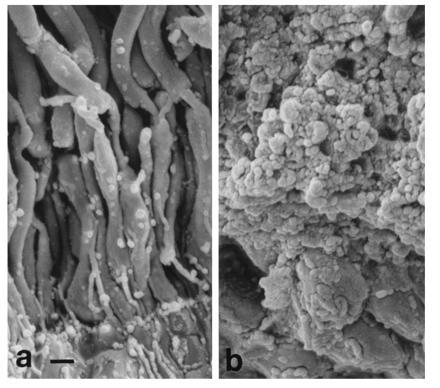
Scanning electron micrographs showing the region of outer retina. (a) Normal morphology in a wt retina shown as a control. Photoreceptor nuclei appear at the very bottom of the photograph. Extending upwards are well-defined inner segments, the thinner connecting cilia, and the outer segments. (b) P347S A1 retina. Note that the inner and outer segments were masked by surrounding vesicles. (Bar = 1 μm.)
Using two monoclonal antibodies against rhodopsin, one specific for the amino terminus (rho 4D2) and the other for the carboxyl terminus (rho 1D4), we performed ultrastructural immunocytochemistry to determine whether the vesicles contained rhodopsin. Both antibodies stained the outer segment and the extracellular vesicles (result with rho 4D2 is shown Fig. 6). The results suggest that the vesicles contain rhodopsin. There was minimal staining over inner segments of the photoreceptors, consistent with the immunocytochemical findings by light microscopy (Fig. 2).
Figure 6.
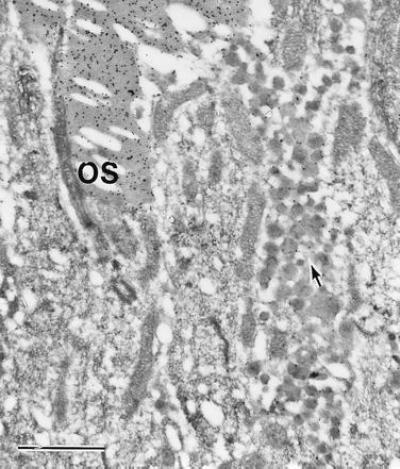
Ultrastructural immunocytochemical examination of a 20-day-old P347S C1 retina. Staining was with rho 4D2 antibody and gold-labeled secondary antibody. A packet of vesicles in the center of view is stained (arrow), but at lower intensity than the outer segments (OS). (Bar = 1 μm.)
DISCUSSION
The objective of this study was to investigate the early pathogenic events in photoreceptor cells expressing the rhodopsin mutant P347S and to assess whether the findings support current hypotheses regarding the pathogenic mechanism of rhodopsin mutants. Three findings are noteworthy. First, there was no gross perturbation of phototransduction, as monitored by ERGs, in the predegenerate or early degenerating retinas. Second, no accumulation of mutant rhodopsin was observed at the sites of synthesis and processing in predegenerate and early degenerating retinas. Third, the mutant retinas exhibited a distinct morphological phenotype—i.e., the extracellular accumulation of submicrometer-sized rhodopsin-containing vesicles in the vicinity of basal outer segments and apical inner segments. This feature has not been reported in mice or humans with any other rhodopsin mutations. In particular, lines of transgenic mice carrying P23H and K296E rhodopsin mutations and exhibiting a similar or faster rate of degeneration clearly did not have this phenotype (refs. 10 and 11, and data not shown).
These findings have relevance to two proposed pathogenic mechanisms for dominant rhodopsin mutants. The equivalent constant light hypothesis postulates that rhodopsin mutants constitutively activate the phototransduction cascade (15, 16), creating a situation that is analogous to constant exposure to environmental light, a known cause of photoreceptor degeneration. So far, studies in vivo have not supported this hypothesis (11, 13, 17, 18, 19). In the current study, the normal ERG a-wave amplitudes seen in the predegenerate P347S mice are inconsistent with constitutive activation of the phototransduction cascade, but they are consistent with the apparently normal ability of P347S rhodopsin to activate transducin, to be phosphorylated by rhodopsin kinase, and to bind arrestin as shown by Weiss et al. (8).
An alternative hypothesis, the cellular congestion hypothesis, suggests that mutant rhodopsins accumulate in and congest the intracellular machinery for protein synthesis, processing, and/or transport. This hypothesis is supported by studies of mutant rhodopsins that cause photoreceptor degeneration in Drosophila (20, 21). Massive proliferation of rough endoplasmic reticulum is observed, presumably due to the accumulation of mutant and wt opsins (20). Such a mechanism has not been established in mammalian photoreceptors. Class II mutants would be expected to display a similar transport defect, given their intracellular accumulation in cultured cells (3, 4, 5). However, in transgenic mice the bulk of P23H (class II) mutant rhodopsin localizes to the outer segments (10). One study of transgenic mice expressing a mutant rhodopsin missing the last five amino acids, Q344ter (class I), found partial retention of the mutant rhodopsin in the inner segments and the perinuclear regions, which led to the suggestion that the carboxyl terminus of rhodopsin might have a role in rhodopsin transport to the outer segments (13). In the current study of another class I mutant involving the carboxyl terminus, P347S, there was no accumulation of mutant rhodopsin in the inner segments. Intracellular congestion is thus unlikely to be a key pathogenic event for this mutation.
The earliest observed pathology in the current study was the accumulation of rhodopsin-containing vesicles in the extracellular space. Three other murine models of photoreceptor degeneration exhibit extracellular vesicles to various degrees. In mice with a defect in the rds gene, whose protein product is required for disc morphogenesis (22, 23), or the rd5 gene, whose product is of unknown function (24), vesicle accumulation is accompanied by absent or poorly formed outer segments (25, 26). In contrast, the P347S retinas contain shortened, but otherwise well-packed, outer segment discs. The retinal pathology in the P347S retina is more similar to that of the pcd mouse (27) retina, due to a defect in an unidentified gene, which accumulates vesicles while maintaining well-packed discs in the outer segments.
Our findings are consistent with the idea that the carboxyl terminus plays a role in the transport of rhodopsin to the outer segments (13). Rhodopsin is normally transported in post-Golgi vesicles (intracellular transport vesicles) to the apical inner segments (28), and it is thought to be transported from there to the nascent disc membranes either through the connecting cilium or through the extracellular space (29). In the intracellular transport vesicles, rhodopsin is embedded in the lipid bilayer with the carboxyl terminus on the cytoplasmic surface (30, 31). Therefore, the carboxyl terminus could conceivably be a necessary motif that interacts with other cellular components to accomplish the complex task of vectorial transport of rhodopsin to the nascent disc membranes. In this context, a plausible explanation for our observations of the P347S mutant retinas is a defect in the vectorial transport of post-Golgi vesicles, causing a portion of the vesicles either to exit inner segments laterally or to fail to reach the nascent discs from apical inner segments. Photoreceptor cell death in the P347S mice could result from either one of two consequences of this primary defect, or their combination. The rate of disc renewal may be reduced due to the loss of material to the extracellular space, hence the shortened outer segments. Maintenance of outer segments at a proper length is likely to be required for photoreceptor survival as exemplified by the photoreceptor degeneration in rds mutant mice. Loss of cellular contents to the extracellular space may additionally contribute to the photoreceptor lethality of the P347S mutation.
Acknowledgments
We thank Dr. Eliot Berson for providing a blood sample of a patient with the P347S mutation, Dr. Robert Molday for providing antibodies, Norman Michaud for assisting with scanning EM, and the Harvard Transgenic Facility for carrying out microinjections. This work was supported by Grant EY10309 from the National Eye Institute.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: ERG, electroretinogram; wt: wild type; EM: electron microscopy; RP, retinitis pigmentosa; RPE, retinal pigment epithelium.
References
- 1.Daiger S, Sullivan L, Rodriguez J. Behav Brain Sci. 1995;18:452–467. [Google Scholar]
- 2.Rosenfeld P J, Cowley G S, McGee T L, Sandberg M A, Berson E L, Dryja T P. Nat Genet. 1992;1:209–213. doi: 10.1038/ng0692-209. [DOI] [PubMed] [Google Scholar]
- 3.Sung C H, Schneider B G, Agarwal N, Papermaster D S, Nathans J. Proc Natl Acad Sci USA. 1991;88:8840–8844. doi: 10.1073/pnas.88.19.8840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sung C H, Davenport C M, Nathans J. J Biol Chem. 1993;268:26645–26649. [PubMed] [Google Scholar]
- 5.Kaushal S, Khorana H G. Biochemistry. 1994;33:6121–6128. doi: 10.1021/bi00186a011. [DOI] [PubMed] [Google Scholar]
- 6.Robinson P R, Cohen G B, Zhukovsky E A, Oprian D D. Neuron. 1992;9:719–725. doi: 10.1016/0896-6273(92)90034-b. [DOI] [PubMed] [Google Scholar]
- 7.Berson E L, Rosner B, Sandberg M A, Weigel-DiFranco C, Dryja T P. Am J Ophthalmol. 1991;111:614–623. doi: 10.1016/s0002-9394(14)73708-0. [DOI] [PubMed] [Google Scholar]
- 8.Weiss E, Hao Y, Dickerson C, Osawa S, Shi W, Zhang L, Wong F. Biochem Biophys Res Commun. 1995;216:755–761. doi: 10.1006/bbrc.1995.2686. [DOI] [PubMed] [Google Scholar]
- 9.Nathans J, Hogness D S. Proc Natl Acad Sci USA. 1984;81:4851–4855. doi: 10.1073/pnas.81.15.4851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Olsson J E, Gordon J W, Pawlyk B S, Roof D, Hayes A, Molday R S, Mukai S, Cowley G S, Berson E L, Dryja T P. Neuron. 1992;9:815–830. doi: 10.1016/0896-6273(92)90236-7. [DOI] [PubMed] [Google Scholar]
- 11.Li T, Franson W K, Gordon J W, Berson E L, Dryja T P. Proc Natl Acad Sci USA. 1995;92:3551–3555. doi: 10.1073/pnas.92.8.3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Molday R. In: Progress in Retinal Research. Osborne N, Chader G, editors. Vol. 8. New York: Pergamon; 1988. pp. 174–209. [Google Scholar]
- 13.Sung C H, Makino C, Baylor D, Nathans J. J Neurosci. 1994;14:5818–5833. doi: 10.1523/JNEUROSCI.14-10-05818.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hodges R, Heaton R, Parker J, Molday L, Molder R. J Biol Chem. 1988;263:11768–11775. [PubMed] [Google Scholar]
- 15.Fain G L, Lisman J E. Exp Eye Res. 1993;57:335–340. doi: 10.1006/exer.1993.1132. [DOI] [PubMed] [Google Scholar]
- 16.Lisman J E, Fain G L. Nat Med. 1995;1:1254–1255. doi: 10.1038/nm1295-1254. [DOI] [PubMed] [Google Scholar]
- 17.Sieving P A, Richards J E, Naarendorp F, Bingham E L, Scott K, Alpern M, Weber B H F, Vogt G, Pruett R C, Stohr H, Felbor U. Proc Natl Acad Sci USA. 1995;92:880–884. doi: 10.1073/pnas.92.3.880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goto Y, Peachey N S, Ripps H, Naash M I. Invest Ophthalmol Visual Sci. 1995;36:62–71. [PubMed] [Google Scholar]
- 19.Goto Y, Peachey N S, Ziroli N E, Seiple W H, Gryczan C, Pepperberg D R, Naash M I. J Opt Soc Am. 1996;13:577–585. doi: 10.1364/josaa.13.000577. [DOI] [PubMed] [Google Scholar]
- 20.Colley N J, Cassill J A, Baker E K, Zuker C S. Proc Natl Acad Sci USA. 1995;92:3070–3074. doi: 10.1073/pnas.92.7.3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kurada P, O’Tousa J. Neuron. 1995;14:571–579. doi: 10.1016/0896-6273(95)90313-5. [DOI] [PubMed] [Google Scholar]
- 22.Travis G H, Brennan M B, Danielson P E, Kozak C A, Sutcliffe J G. Nature (London) 1989;338:70–73. doi: 10.1038/338070a0. [DOI] [PubMed] [Google Scholar]
- 23.Molday R. In: Progress in Retinal and Eye Research. Osborne N, Chader G, editors. Vol. 13. New York: Pergamon; 1994. pp. 272–297. [Google Scholar]
- 24.Heckenlively J R, Chang B, Erway L C, Peng C, Hawes N L, Hageman G S, Roderick T H. Proc Natl Acad Sci USA. 1995;92:11100–11104. doi: 10.1073/pnas.92.24.11100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nir I, Papermaster D S. Invest Ophthalmol Visual Sci. 1986;24:868–878. [PubMed] [Google Scholar]
- 26.Usukura J, Bok D. Exp Eye Res. 1987;45:501–515. doi: 10.1016/s0014-4835(87)80061-1. [DOI] [PubMed] [Google Scholar]
- 27.Blanks J, Mullen R, LaVail M. J Comp Neurol. 1982;212:231–246. doi: 10.1002/cne.902120303. [DOI] [PubMed] [Google Scholar]
- 28.Papermaster D S, Schneider B G, Besharse J C. Invest Ophthalmol Visual Sci. 1985;26:1386–1404. [PubMed] [Google Scholar]
- 29.Besharse J C, Wetzel M G, Janssen J J M, Boveegeurts P H M, Merkx M, Degrip W J. J Neurocytol. 1995;24:371–388. doi: 10.1007/BF01189064. [DOI] [PubMed] [Google Scholar]
- 30.Defoe D, Besharse J. J Neurosci. 1985;5:1023–1034. doi: 10.1523/JNEUROSCI.05-04-01023.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deretic D, Papermaster D S. J Cell Biol. 1991;113:1281–1293. doi: 10.1083/jcb.113.6.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]