

Abstract
The protein kinase regulated by double-stranded RNA (dsRNA), PKR, is implicated in a range of biologic processes, including apoptotic death and interferon antiviral responses, based in part on studies with mouse cells genetically deficient in Pkr. To test the role of the PKR protein in human cells, an RNA interference silencing strategy was used to generate stable HeLa cell lines with less than 2% of the PKR protein (PKR deficient) compared to either parental or control knockdown HeLa lines. Phosphorylation of the α subunit of eukaryotic initiation factor 2 on serine 51 was not detectably increased in response to dsRNA in PKR-deficient HeLa cells but was elevated severalfold in PKR-sufficient cells. PKR-deficient cells displayed reduced dsRNA-induced apoptosis compared to PKR-sufficient cell lines, whereas tumor necrosis factor alpha (TNF-α)-induced apoptosis was comparable between the HeLa lines. NF-κB was activated to a comparable extent in PKR-deficient and PKR-sufficient HeLa cells upon treatment with either dsRNA or TNF-α. The antiviral response against vesicular stomatitis virus was reduced in interferon-treated PKR-deficient compared to PKR-sufficient HeLa cells. However, the growth of two human viruses, adenovirus and reovirus, was unaffected by the PKR knockdown. Surprisingly, the yield of mutant adenovirus that fails to encode VAI RNA was not enhanced in PKR-deficient cells, indicating the importance of host factors in addition to PKR in conferring the VAI RNA phenotype.
PKR, the protein kinase regulated by double-stranded RNA (dsRNA), is well established as an important component of the host response to viral infection (22, 37). The N-terminal region of PKR includes a repeated domain that confers dsRNA-binding activity, and the C-terminal region possesses subdomains necessary for kinase catalytic activity (23, 32). In addition to the autophosphorylation of PKR that occurs during the dsRNA-mediated autoactivation process, the best-characterized PKR substrate is the α subunit of protein synthesis eukaryotic initiation factor 2 (eIF-2α). PKR catalyzes the phosphorylation of eIF-2α on serine 51, a modification that alters the translation pattern within cells and leads to an inhibition of protein synthesis under stress conditions, including virus infection (36, 37). PKR also modulates nuclear factor κB (NF-κB)-mediated signal transduction processes in response to dsRNA (2, 6, 18).
Studies with both cultured mouse cells and intact mice have provided evidence that PKR plays an important role in a variety of physiologic processes, including cell proliferation and death, in addition to the role that PKR plays in the antiviral actions of interferons (5, 22, 31). Overexpression of PKR in eukaryotic cells, both mammalian and fungal, has a strong growth-suppressive effect, while overexpression of a catalytically inactive PKR mutant protein in mouse NIH 3T3 cells, for example, leads to transformation (11, 21, 35). Two genetic disruptions of the mouse Pkr gene have been described, and in both cases the PKR-deficient mice were viable (1, 38). But the phenotypes of the two Pkr genetic knockouts, and their derivative null murine embryonic fibroblast (MEF) cells, differ substantially from each other, and in some instances experimental findings are contradictory with regard to certain apoptotic and virus-host interaction properties attributed to PKR (1, 4, 38). The different genetic backgrounds of the two mouse Pkr knockout strains, together with possible differences in expression of truncated and partially functional PKR proteins, are hypothesized to be responsible for the phenotypic differences seen between the two mouse Pkr knockouts (4). Contrary to the N-terminal-targeted Pkr disruption mouse, in which null MEFs are resistant to apoptotic death in response to both dsRNA and tumor necrosis factor alpha (TNF-α) (8), TNF-α-induced apoptosis and antiviral apoptosis following influenza virus infection are not impaired in the catalytic domain C-terminal-targeted Pkr null (1). Furthermore, MEFs from the C-terminal disruption of Pkr are not defective in their response to dsRNA (14), and the induction levels of IκBα phosphorylation and NF-κB activation are normal (4). By contrast, dsRNA-dependent NF-κB activation and signaling are diminished in MEFs with the N-terminal truncation of PKR (38). PKR is also reported to be a critical mediator of macrophage apoptosis after activation of Toll-like receptor 4 (13).
The kinase activity of PKR is both activated and antagonized by viral RNAs (22). Among the antagonists is adenovirus VAI RNA (20). Adenovirus grows in human but not mouse cells and is comparatively resistant to the antiviral actions of interferon (IFN) (22). Optimal growth of adenovirus in human cells is dependent upon the viral VAI RNA, which is expressed in high amounts at late times after viral infection (20). In the absence of VAI RNA, late adenovirus protein synthesis is impaired, thereby reducing viral yields (17, 33). VAI RNA antagonizes the activation of the PKR kinase and the interferon-induced antiviral state (17).
To test whether the PKR protein is the primary target of the adenovirus VAI RNA and to assess the extent to which the PKR protein is required for apoptotic responses in a human cell line, we generated HeLa cells stably deficient in PKR protein by using a short hairpin RNA interference approach. We established HeLa cell lines in which >98% of PKR protein expression was stably silenced, both basal PKR and IFN-inducible PKR. By comparing wild-type parental HeLa cells and control knockdown cells that are PKR sufficient with PKR-deficient knockdown cells, the roles of human PKR in mediating apoptosis signaling, NF-κB signaling, and virus multiplication were examined using adenovirus, reovirus, and vesicular stomatitis virus (VSV). dsRNA-induced apoptosis, but not TNF-α-induced apoptosis, was impaired in the PKR-deficient cells as measured by caspase activation and poly(ADP-ribose) polymerase (PARP) cleavage. The dsRNA-mediated phosphorylation of eIF-2α was abolished in the PKR-deficient cells, but little difference was seen in NF-κB activation, which was comparable between PKR-sufficient and -deficient cells following treatment with either dsRNA or TNF-α.
MATERIALS AND METHODS
Cell maintenance and viruses.
HeLa, human amnion U, and mouse fibroblast L cells were maintained in Dulbecco's modified Eagle's medium complemented with 5% or 10% (vol/vol) fetal bovine serum (HyClone), 1% sodium pyruvate (Cambrex BioScience), 100 μg/ml of penicillin, and 100 units/ml streptomycin (Invitrogen) as previously described (7, 26). HeLa stable transfectant clones were maintained in the above medium containing 1 μg/ml puromycin (Sigma). The Indiana serotype of VSV and the Dearing strain of reovirus type 3 were as previously described (26). Adenovirus H5dl309, a phenotypically wild-type virus (15) and adenovirus H5dl331, a mutant that does not express VAI RNA (33), were provided by T. Shenk (Princeton, NJ). Interferon treatment was with 1,000 units/ml of alpha IFN (PBL) for 24 h, using recombinant IFN-α A/D (25).
Construction of shRNA expression vector plasmids.
The pSUPER.retro.puro vector with H1 promoter (Oligo Engine) was used for construction of short hairpin expression constructs to silence human PKR. The successful 21-nucleotide human PKR targeting sequence (GCAGGGAGTAGTACTTAAATA) present in the open reading frame of human PKR (34) was identified using Invitrogen online software. The sense strand oligo (5′-GATCCCCGCAGGGAGTAGTACTTAAATATTCAAGAGATATTTAAGTACTACTCCCTGCTTTTTA-3′) and antisense strand oligo (5′-AGCTTAAAAAGCAGGGAGTAGTACTTAAATATCTCTTGAATATTTAAGTACTACTCCCTGCGGG-3′) were annealed, and the resulting duplex was inserted into the pSUPER.retro.puro vector using the BglII and HindIII restriction sites. A control construct (sense strand oligo, 5′-GATCCCCGCTTGTTCGTTGGTAACTACATTCAAGAGATGTAGTTACCAACGAACAAGCTTTTTA-3′; antisense strand oligo, 5′-AGCTTAAAAAGCTTGTTCGTTGGTAACTACATCTCTTGAATGTAGTTACCAACGAACAAGCGGG-3′) that expresses a nontargeting scramble shRNA sequence was prepared in a similar manner. Recombinant plasmid constructs were verified by DNA sequence analysis.
Cell transfection.
HeLa cells were transfected at 80 to 90% confluence using Lipofectamine 2000 (Invitrogen) according to the manufacturer's recommendations. Briefly, Lipofectamine 2000 and plasmid DNA were diluted and mixed in Opti-MEM (Invitrogen), the DNA-Lipofectamine 2000 complexes were applied to the cultures, and incubation continued for 4 h at 37°C, after which the transfection medium was replaced with fresh maintenance medium containing 5% (vol/vol) fetal bovine serum.
To select for stable transfectants, cells were trypsinized 24 h after transfection with the designated shRNA pSUPER.retro.puro construct, seeded at various dilutions, and then carried in the presence of 1 μg/ml puromycin. Puromycin-resistant clones were isolated and screened by Western immunoblot analysis for PKR protein knockdown. To analyze for the effect of dsRNA, either parental HeLa or puromycin-resistant stable clonal lines were transfected with poly(rI)·poly(rC) (0.6 μg per well; 24-well plates) using Lipofectamine 2000 and analyzed at the indicated times after transfection.
Western blot analysis.
Whole-cell extracts were prepared in the presence of 1 mM phenylmethylsulfonyl fluoride and 1% (vol/vol) protease inhibitor mixture (Sigma) as described previously (7). For analysis of eIF-2α phosphorylation, 50 mM NaF and 2 mM Na2VO3 were included in the extract buffer. Proteins were fractionated by sodium dodecyl sulfate-10% polyacrylamide gel electrophoresis, transferred to nitrocellulose membranes, blocked, and then probed with an appropriate dilution of primary antibody in phosphate-buffered saline (PBS) containing 3% (wt/vol) skim milk. Rabbit polyclonal antibodies were used to detect human PKR (sc707; Santa Cruz Biotechnology, Inc.), eIF-2α (Cell Signaling Technology), and phospho-eIF-2α (Ser51) (Cell Signaling Technology); monoclonal antibodies were used to detect human PARP (551025; BD Pharmingen), β-actin (A5441; Sigma), and α-tubulin (T6199; Sigma). Western blot detection was done with horseradish peroxidase-conjugated anti-rabbit immunoglobulin G or anti-mouse immunoglobulin G secondary antibody using an enhanced chemiluminescence detection reagent kit (Amersham Biosciences) according to the manufacturer's protocol. Immunoreactive bands were visualized using a VersaDoc (Bio-Rad) imaging system, and quantitation was carried out using the Quantity One software program.
Caspase-3/7 activation assay.
As one measure of apoptosis, effector caspase-3/7 activation was measured by using the Caspase-Glo 3/7 assay (Promega) according to the manufacturer's recommendations. HeLa cell lines, either parental or stable puromycin-resistant PKR-deficient knockdown or PKR-sufficient control cell lines, were seeded into 96-well plates at a density of 1.5 × 104 cells per well. After overnight culture, cells were treated with 10 ng/ml TNF-α and 1 μg/ml cycloheximide for 18 h or transfected with 60 ng poly(rI)·poly(rC) for 6 h. At the specified time, caspase-3/7 activation was determined; 100 μl of Caspase-Glo 3/7 reagent was added to each well and mixed gently, and after 1 h of incubation at room temperature in the dark, luciferase reporter activity was measured using the Perkin-Elmer Victor 3V luminometer plate reader model 1420.
NF-κB activation assay.
HeLa and stable cell lines were cotransfected with an NF-κB-dependent firefly luciferase reporter plasmid (generously provided by I. Verma, Salk Institute) and Renilla luciferase reporter plasmid (Promega). At 48 h after transfection, cells were incubated with either 20 ng/ml TNF-α for 4 h or 100 μg/ml poly(rI)·poly(rC) plus 50 μg/ml DEAE-dextran for 4, 6, or 8 h. Cells were harvested and lysed in passive lysis buffer (Promega). Following centrifugation at 13,400 × g for 10 min, luciferase activities were determined using the dual luciferase protocol following the manufacturer's recommendations (Promega).
Cell cycle analysis.
Cells at 70 to 80% confluence in monolayer culture were trypsinized, washed with PBS, and resuspended in 250 μl PBS containing 1% bovine serum albumin. Cells were stained overnight with propidium iodide (30 μg/ml; Sigma) at 4°C, following treatment for 3 h with 50 μl of cell cycle buffer (0.11% sodium citrate plus 0.1% Triton X-100) and 10 μl RNase A (10 mg/ml). The percentages of cells in G0/G1, S, and G2/M were determined using a Guava Technologies Easy Cyte instrument and CytoSoft software.
Protein synthesis assay.
HeLa cells were seeded into 24-well plates at a density of 1.2 × 105 cells per well. After overnight culture, cells were transfected with 0.05 μg pGL2-Control and 0.55 μg pGL2-Basic (Promega) using Lipofectamine 2000. After 3 h of incubation, cells were subjected to a second transfection with 0.6 μg poly(rI)·poly(rC). Cells were harvested 8 h post-second transfection, and luciferase activities were determined using the dual luciferase protocol following the manufacturer's recommendations (Promega).
Single-cycle virus growth assay.
HeLa parental and stable transfectant cells were seeded into six-well plates (6 × 105 cells per well) and either pretreated with recombinant IFN-α A/D (1,000 units/ml) for 24 h or left untreated. Infection was at a multiplicity of infection (MOI) of 0.1 or 10 with VSV, an MOI of 10 with reovirus (26), or an MOI of 3 with adenovirus (17, 33). Virus absorption was carried out for 1 h in 0.2 ml Puck's saline A modified to contain 20 mM MgCl2 and 1% fetal bovine serum. The inoculum was then removed, the monolayers were rinsed twice, and the cultures then incubated in 1.5 ml of maintenance medium. Infected cultures were harvested at 16 h postinfection (p.i.) for VSV or 24 h p.i. for reovirus and adenovirus by scraping the infected cells into the medium. Virus yields were determined by plaque titration, using L for VSV and reovirus and U cells for adenovirus (17, 25, 26).
RESULTS
Establishment of HeLa cell lines with stable knockdown of the PKR protein.
We utilized a short hairpin-based RNA interference vector construct to silence human PKR gene expression in HeLa cells. The successful PKR-targeting region was located in the PKR open reading frame at 59 to 80 nucleotides downstream of the translation initiation ATG codon. In preliminary screening, transient transfection of the shRNA pSUPER.retro.puro expression vector specifically reduced the amount of PKR protein detectable in two human cell lines, HeLa cells and U cells, by 70 to 80% after 3 to 4 days transfection as measured by Western blotting (data not shown). To generate human cells in which the PKR protein was stably knocked down, puromycin-resistant clones were isolated and characterized by Western analysis. The PKR protein levels of the stable knockdown HeLa clones varied. Among the clonal lines isolated was clone 14, in which the PKR protein production was substantially reduced relative to the parental HeLa cells.
As measured by Western analysis (Fig. 1A), the PKR-deficient clone (designated PKRkd) showed <2% of the PKR protein level seen in the parental HeLa cell line (designated PKR+) in the absence of IFN treatment (Fig. 1B). Furthermore, the level of PKR was specifically reduced to a nearly undetectable level in PKRkd even after IFN treatment (Fig. 1). By contrast, the basal and IFN-inducible expression levels of two other IFN-regulated proteins, STAT1 and ADAR1 p150, as well as β-actin and α-tubulin, were unaffected in the PKRkd stable knockdown clone (data not shown). PKRkd and a negative control puromycin-resistant clone shRNA pSUPER.retro.puro designated PKRkd-con were selected for further functional analyses to assess the effects of loss of PKR on apoptotic and antiviral responses in a human cell system.
FIG. 1.

Stable knockdown of PKR protein. (A) Western immunoblot analysis comparing PKR expression in wild-type parental (PKR+), PKR-deficient knockdown (PKRkd), and PKR-sufficient control knockdown (PKRkd-con) HeLa cell clones. Cells were mock treated or treated with 1,000 IU/ml of IFN-α A/D for 24 h. Whole-cell extract protein (10 μg) was analyzed in each lane; the membrane was probed with a polyclonal antibody against human PKR and a monoclonal antibody against β-actin as a loading control. (B) Quantification. Western blots were quantified using a VersaDoc (Bio-Rad) imaging system.
Effect of PKR on cell growth.
As measured by propidium iodide staining to assay DNA content, the cell cycle distribution during subconfluent growth in monolayer culture was comparable for the PKRkd cells and PKR+ parental HeLa cells. Approximately 45% of the cells were in G0/G1, 22% were in S, and 20% were in G2/M for PKR+ parental cells, compared to 46%, 23%, and 21%, respectively, for the PKRkd cells when analyzed in parallel. In three independent analyses, the cycle distribution seen between the PKRkd and PKR+ cells did not differ significantly.
The PKR protein enhances apoptosis in response to double-stranded RNA stimuli.
Conflicting evidence for a role of PKR as a proapoptotic factor came from virus-infected Pkr null mouse cell systems (1, 8). While PKR was required to mediate stress-related apoptosis in Pkr−/− MEF cells derived from knockout mice in which the dsRNA-binding domain was disrupted (8), Pkr−/− MEF cells from kinase catalytic domain-disrupted mice showed a normal apoptotic response to dsRNA-mediated stress (1). To clarify in human cells the possible role of PKR in dsRNA-stimulated apoptosis, the PKR-deficient knockdown HeLa cell clone PKRkd was tested. At 6 h after transfection with dsRNA, both the HeLa parental line and the puromycin-resistant negative control clone PKRkd-con showed characteristic blebbing, and by 24 h most (>90%) of these PKR-sufficient cells were apoptotic. By contrast, the apoptotic blebbing phenotype was greatly reduced (<40%) in the PKR-deficient PKRkd cell line (data not shown).
Caspases-3 and -7 play key effector roles in apoptosis in animal cells. As shown in Fig. 2A, caspase-3/7 activation as an indicator of apoptosis was significantly reduced in the PKRkd HeLa cells in response to dsRNA stimulus compared to either the parental PKR+ or the negative control PKRkd-con lines. Furthermore, as an independent apoptosis indicator, cleavage of PARP was measured by immunoblot analysis. Substantially more intact 116-kDa PARP was seen in the PKRkd cells at 24 h after transfection of dsRNA than in PKR-sufficient cells, either the parental PKR+ HeLa line or the PKRkd-con negative knockdown control (Fig. 2B). Conversely, both of the PKR-sufficient lines, PKR+ parental and PKRkd-con, had a higher amount of the 85-kDa PARP cleavage fragment than did the PKRkd cells deficient in PKR. Finally, in the absence of dsRNA transfection, no significant PARP cleavage was seen and the levels of intact PARP were comparable in the PKR+, PKRkd, and PKRkd-con cells (Fig. 2B).
FIG. 2.
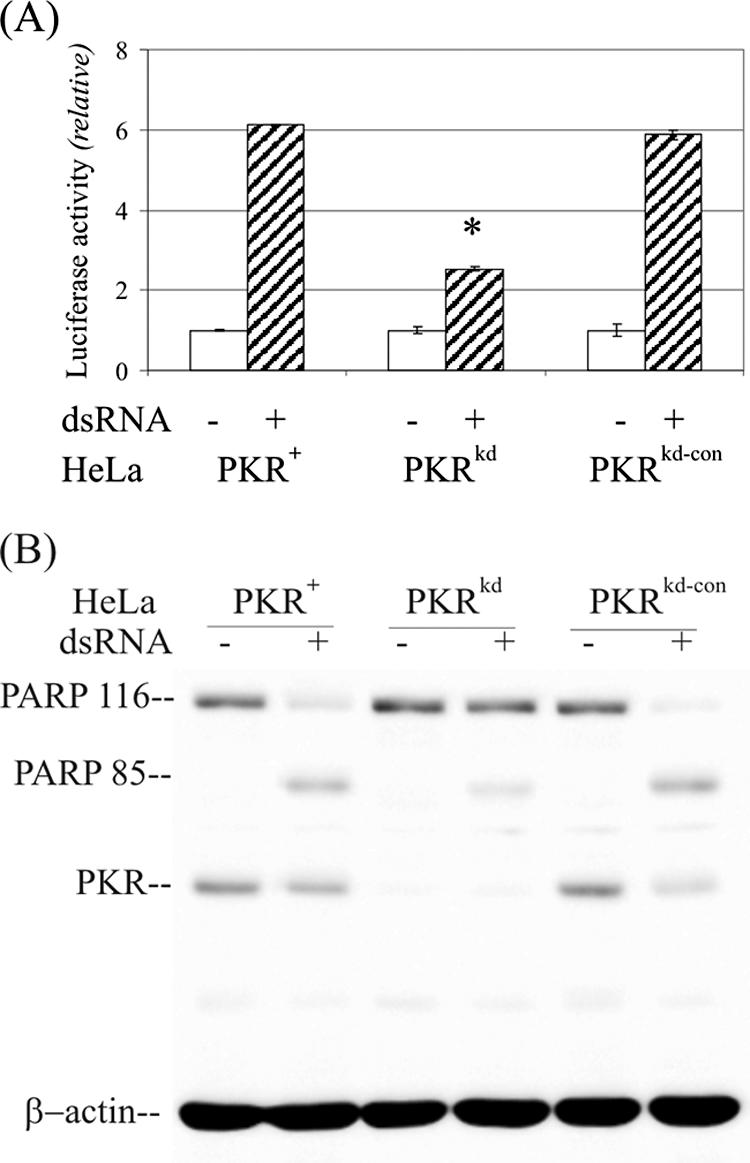
Double-stranded RNA-mediated apoptosis is impaired in PKR-deficient HeLa cells. (A) The effects of dsRNA on apoptosis induction in PKR+, PKRkd, and PKRkd-con HeLa cells were measured in 96-well plates, either mock transfected or transfected with 60 ng dsRNA. Apoptosis was measured using a Caspase-Glo 3/7 kit at 6 h posttransfection. Each experiment was repeated a minimum of three times. *, P < 0.0001, Student's t test, PKRkd compared to the PKR+ parent; P was >0.4 for PKRkd-con compared to the PKR+ parent. (B) Western immunoblot measurement of PARP cleavage at 8 h after dsRNA treatment. Ten μg of whole-cell extract protein was analyzed in each lane.
Analysis of PARP cleavage at various times after treatment with dsRNA showed that PARP protein cleavage was clearly detectable in the parental PKR+ HeLa cells by 4 h after dsRNA transfection and further increased after 6 and 8 h compared to the PKRkd cells (Fig. 3A). Quantitation of the Western blots established that the cleaved 85-kDa PARP fragment in the PKR+ HeLa cells was four- to fivefold more abundant in PKR+ compared to PKRkd cells at the 6- and 8-h time points (Fig. 3B).
FIG. 3.
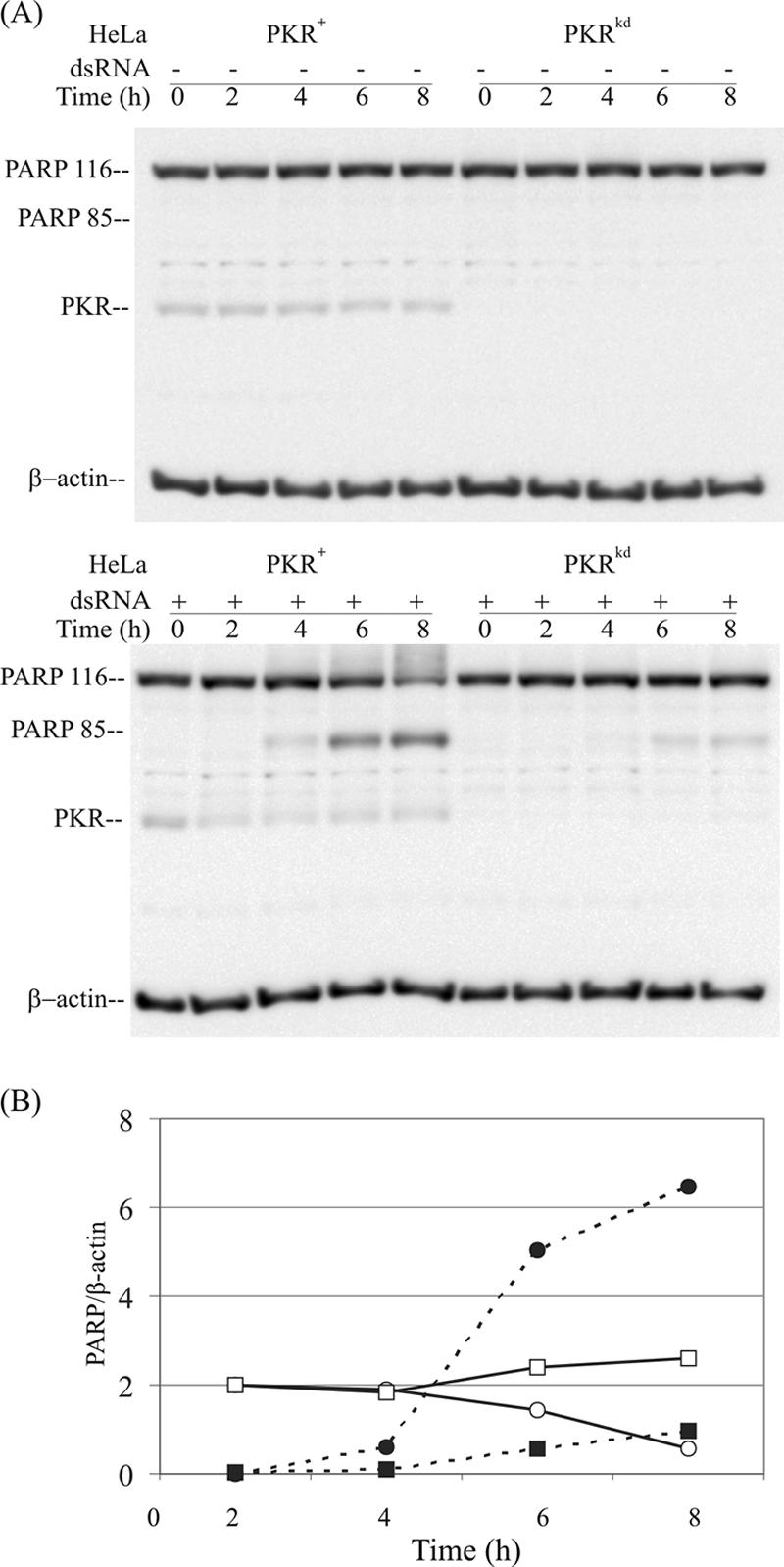
Time course of PARP cleavage following dsRNA treatment in PKR+ and PKRkd HeLa cells. (A) Western blot analysis. Whole-cell extracts were prepared at 0, 2, 4, 6, and 8 h after transfection with dsRNA (+) or mock transfected (-). Ten μg of whole-cell extract protein was loaded in each lane. The membranes were probed with antibodies against PARP and PKR and against β-actin as a loading control. (B) Quantification of PARP. Western blots were quantified using a VersaDoc imager. Intact PARP116 (solid line) and the cleavage product PARP85 (broken line) quantitations from PKR+ (circles) and PKRkd (squares) HeLa cells are relative to β-actin.
Impaired eIF-2α phosphorylation in response to dsRNA in PKR-deficient HeLa cells.
One of the mechanisms for regulation of the translational pattern in eukaryotic cells is through the phosphorylation of eIF-2α at serine 51, which leads to an inhibition of translation (10). While eIF-2α is the best-characterized substrate of PKR, additional eIF-2 kinases also are known (HRI, PERK, and GCN2) that phosphorylate eIF-2α at serine 51 (23). Under conditions of IFN treatment and virus infection, the eIF-2α-P(Ser51) levels in the two Pkr−/− MEF cell lines differed significantly (1, 4), raising the possibilities of either redundancy of kinase function or incomplete genetic disruption of eIF-2α kinase function. Because eIF-2α is hypothesized to be an important downstream factor in the PKR-mediating apoptosis-signaling pathway, a time course was carried out to examine the phosphorylation level of eIF-2α in PKRkd cells upon dsRNA treatment compared to the parental PKR+ HeLa cells.
As measured by immunoblot analysis with a tubulin internal control (Fig. 4A), the amount of eIF-2α protein was comparable in the PKR+ parental and PKRkd cells (Fig. 4B, upper). By contrast, in the presence of dsRNA, the amount of phosphorylated eIF-2α-P(Ser51) was increased about fourfold in the parental PKR+ HeLa cells, whereas there was no significant increase of the low but detectable level of Ser51-phosphorylated eIF-2α in the PKR-deficient PKRkd cells (Fig. 4B, lower). When compared to PKR-sufficient HeLa cells, these findings indicate that the reduced dsRNA-mediated apoptosis seen in the PKRkd HeLa cells measured by either caspase activation (Fig. 2) or PARP cleavage (Fig. 3) correlated with an impaired phosphorylation of eIF-2α in HeLa PKRkd cells in the presence of dsRNA (Fig. 4B). Finally, protein expression from a luciferase reporter was comparably high in the presence and absence of dsRNA in the PKRkd cells but was significantly reduced by dsRNA in the PKR+ parental and PKRkd-con cells (Fig. 4C).
FIG. 4.

eIF-2α phosphorylation and protein expression following dsRNA treatment of PKR+ and PKRkd cells. (A) Western blot analysis. Whole-cell extracts were made at 0, 2, 4, 6, and 8 h after transfection with dsRNA, and 30 μg protein was analyzed in each lane. The membranes were probed with antibodies against eIF-2α or phospho-eIF-2α(Ser 51) and against α-tubulin as a loading control. (B) Quantification. Western blots were quantified by scanning densitometry. The upper panel shows the eIF-2α protein profile, and the lower panel shows the phosphorylated eIF-2α protein profile. Circles, HeLa PKR+; squares, HeLa PKRkd. (C) Protein expression. Luciferase reporter activity was measured in cells transfected in the absence (-) or presence (+) of dsRNA for 8 h, following transfection with pGL2-Control reporter DNA. *, P < 0.0001, Student's t test, PKRkd compared to PKR+ parent or PKRkd-con.
The PKR protein does not play a significant role in TNF-α-mediated apoptosis.
TNF-α, an inflammatory cytokine, is a well-established stimulator of apoptosis via the FADD adaptor to activate initiator caspase 8 (3). Pkr−/− MEF cells genetically deficient in PKR due to the targeted disruption of the N-terminal RNA-binding domain region were reported to be resistant to TNF-α-induced apoptosis (8), whereas Pkr−/− MEF cells derived from the mouse disrupted in the C-terminal catalytic domain region of PKR were found to have a normal apoptotic response to TNF-α (1). To examine the role of PKR on TNF-α-mediated apoptosis in human cells, PKRkd, parental PKR+, and PKRkd-con HeLa cells were treated with TNF-α and cycloheximide. Microscopic examination of the cells revealed characteristics of apoptotic death by 18 h after treatment. However, the percentage of apoptotic cells appeared comparable for all three kinds of HeLa cells, those deficient in PKR (PKRkd) and the two PKR-sufficient HeLa lines (PKRkd-con and PKR+). When quantified by activation of the effector caspase-3/7-dependent luciferase reporter, the extent of apoptosis was low and comparable in the three lines in the absence of TNF-α treatment (Fig. 5A). Treatment with TNF-α elevated caspase-3/7-dependent reporter activity to a comparable extent, about eightfold, in all three of the HeLa lines (Fig. 5A). However, TNF-α treatment did not affect either the amount of eIF-2α or the phosphorylation of eIF-2α (Fig. 5B). The phospho-eIF-2α/actin ratio was comparable in the PKR+ parental, PKRkd, and PKRkd-con cells in the absence and presence of TNF-α treatment (Fig. 5B, right). As a control in the same experiment, dsRNA caused a significant increase in phosphorylation of eIF-2α in the PKR+ parental and PKRkd-con cells but not the PKRkd cells (Fig. 5B, left). These results indicate that the PKR protein does not play a significant role in TNF-α-mediated apoptosis in HeLa cells.
FIG. 5.

TNF-α-mediated apoptosis is normal in PKR-deficient HeLa cells. (A) The effect of TNF-α treatment on apoptosis induction in PKR+, PKRkd, and PKRkd-con HeLa cells was measured using the Caspase-Glo 3/7 kit at 18 h after treatment. Results shown are the means ± standard deviations determined from a minimum of six independent experiments. Based on Student's t test, P was >0.3 for TNF-α treatments between cell pairs. (B) Effects of TNF-α (right) and dsRNA (left) on eIF-2α phosphorylation. Whole-cell extracts were made at 4 h after transfection with dsRNA or treatment with TNF-α (10 ng/ml), and 30 μg protein was analyzed in each lane. The membranes were probed with antibodies against eIF-2α or phospho-eIF-2α(Ser 51) and against β-actin as a loading control.
Effect of PKR protein on activation of NF-κB-dependent signaling.
The role of PKR in the activation of transcription factor NF-κB in response to treatment with either TNF-α or dsRNA was assessed using an NF-κB-dependent luciferase reporter in PKR-sufficient and PKR-deficient HeLa cells. PKR+ and PKRkd cells were transiently transfected with an NF-κB-dependent firefly luciferase reporter plasmid and then treated with either TNF-α or dsRNA. As shown in Fig. 6A, TNF-α treatment activated reporter expression to a similar level in the PKR+ parental HeLa line and the PKR-deficient stable knockdown PKRkd cells. Similarly, no significant difference in NF-κB activation was found between PKRkd and PKR+ HeLa cells when they were treated with dsRNA for 4, 6, or 8 h (Fig. 6B). The above results indicate that the PKR protein is not an obligatory component in the TNF-α/NF-κB and dsRNA/NF-κB signaling pathways in HeLa cells.
FIG. 6.

Role of PKR in the NF-κB activation pathway. PKR+ and PKRkd HeLa cells were transfected with an NF-κB-dependent firefly luciferase reporter plasmid. Transfected cells were mock treated or treated with either 10 ng/ml TNF-α or 100 μg/ml dsRNA and 50 μg/ml DEAE-dextran at 48 h after transfection. (A) TNF-α. Luciferase activity was measured at 4 h after TNF-α treatment (hatched bars) or in cells left untreated (open bars). (B) dsRNA. Luciferase activity was measured in dsRNA mock-treated and treated cells at 4, 6, and 8 h after dsRNA treatment of PKR+ (circles) and PKRkd (squares) HeLa cells. Results shown are the means ± standard deviations determined from a minimum of six independent experiments.
The PKR protein plays a virus type-dependent role in the antiviral action of type I interferon in HeLa cells.
PKR has a well-established role in the antiviral actions of type I interferons (22). While Pkr-null MEFs and mice have proved valuable reagents for the study of the role of PKR in the innate defense against viruses in murine cells, little is known regarding the effect that loss of the PKR protein has on the antiviral action of IFN in human cells infected with a human virus. To address this question, we examined the ability of PKRkd HeLa cells (Fig. 1) to mount an IFN-induced antiviral state first against VSV, a widely utilized virus in IFN studies, and then against two human viruses, adenovirus type 5 and reovirus type 3. As shown in Fig. 7, the VSV single-cycle yields were comparable in PKR+ and PKRkd HeLa cells in the absence of IFN treatment. Treatment of the PKR-sufficient parental cells with recombinant IFN-α reduced the yield of infectious VSV about 100- to 300-fold (∼2 to 2.5 logs), but only about a 10- to 30-fold (∼1 to 1.5 log) reduction was obtained in the PKRkd cells at an MOI of 10 (data not shown) or 0.1 (Fig. 7A). These results indicate that PKR contributes in part to the IFN-induced antiviral state against VSV in HeLa cells but that IFN-induced gene products other than PKR also contribute to the reduction in VSV growth.
FIG. 7.

Single-cycle multiplication of vesicular stomatitis virus in PKR+, PKRkd, and PKRkd-con HeLa cells. Cells were mock treated (-) or treated with 1,000 units/ml IFN-α A/D (+) for 24 h and then infected with VSV. Cells were harvested at 16 h p.i., and virus yields were determined by plaque titration. Based on Student's t test, P was >0.05 for PKRkd compared to PKR+ parent and PKRkd-con without IFN-α A/D pretreatment. *, P < 0.0001, PKRkd compared to PKR+ parent and PKRkd-con with IFN-αA/D pretreatment.
Human reovirus type 3 Dearing, a double-stranded RNA virus, grew comparably well in PKRkd cells and PKR+ HeLa cells and, consistent with earlier findings, with human U cells (26) single-cycle yields were not reduced by IFN-α pretreatment (data not shown).
Human adenovirus type 5 was shown earlier to grow equally well in human 293 cells pretreated with IFN-α and in untreated cells (17, 20). By contrast, mutant adenovirus that does not produce VAI RNA (dl331) grows poorly relative to wild-type virus, and IFN-α further reduces the yield of the VAI RNA deletion mutant virus. VAI RNA was subsequently shown to be an antagonist of PKR (20). Similar to the results reported earlier with 293 cells, we found that wild-type adenovirus growth was not reduced by IFN treatment of HeLa cells, in either PKR+ cells or PKRkd cells deficient in the PKR protein (Fig. 8A). We also observed that the VAI RNA deletion mutant adenovirus (dl331) grew poorly, producing ∼2 logs less infectious virus than wild-type virus in the PKR+ parental HeLa cells. Somewhat surprisingly, the absence of PKR protein in HeLa cells did not eliminate the defective growth phenotype observed for the VAI RNA mutant virus (Fig. 8B). PKRkd cells deficient in PKR protein (Fig. 1) did not produce higher yields of the VAI RNA deletion mutant virus than did the PKR+ parental cells; the yield of the VAI deletion mutant remained low in the PKRkd cells and comparable to the PKR+ cells (Fig. 8B). These results suggest that functionally redundant host components, in addition to the IFN-inducible PKR protein, are present in human cells that are targeted by VAI RNA.
FIG. 8.
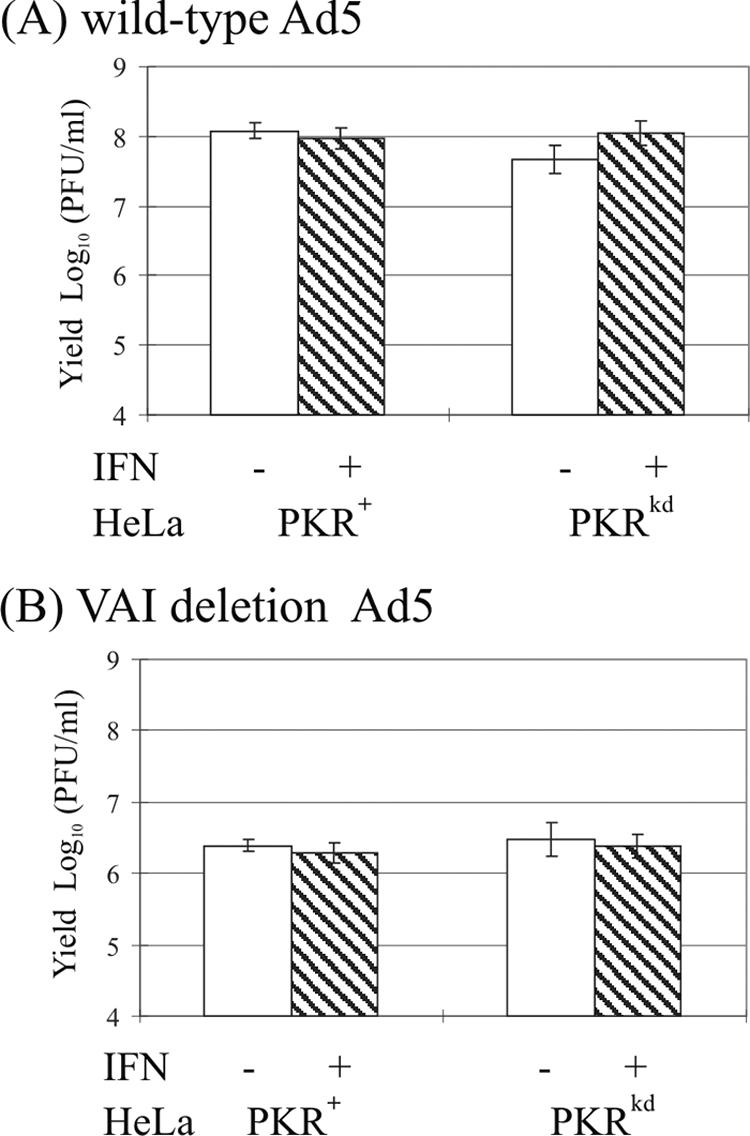
Single-cycle multiplication of adenovirus in PKR+ and PKRkd HeLa cells. Cells were either mock treated (-) or treated with 1,000 units/ml IFN-α A/D (+) for 24 h and then infected with either wild-type adenovirus (A) or VAI deletion mutant adenovirus (B). At 36 h p.i., cells were harvested and virus yields determined by plaque titration. Based on Student's t test, P was >0.18 between infected cell pairs.
DISCUSSION
PKR has been well characterized as an IFN-inducible antiviral protein (22). PKR also is implicated as an important sentinel kinase in signal transduction pathways that mediate cellular responses to intracellular and environmental stresses (16, 23, 37). However, studies carried out with two independently generated PKR knockout mice and their derivative MEF cells gave contradictory results when signaling and biochemical activities attributed to loss of PKR function were analyzed. In this study, we successfully established human cells in which >98% of the PKR protein was depleted by an RNA interference strategy, and we used these knockdown cells and parallel control clones to further investigate the roles of PKR in stress-induced cell death, signaling pathways, and viral growth. Several important points emerged from our findings.
First, the substantial reduction of PKR protein from HeLa cells achieved in the PKRkd cells impaired the apoptotic response elicited by dsRNA as measured both by caspase activation and PARP cleavage. However, the depletion of PKR from HeLa cells did not significantly affect TNF-α-mediated apoptosis or cell growth in culture. These results suggest that PKR is an obligate proapoptotic mediator of dsRNA-mediated but not TNF-α-mediated apoptosis. In the context of prior findings with mouse Pkr knockout cells (1, 8), our observations are most consistent with those obtained using PKR knockout MEF cells derived from mice engineered with an N-terminally disrupted PKR (38).
Protein synthesis initiation factor eIF-2α remains the best-characterized substrate of PKR and, when phosphorylated on serine 51, leads to an inhibition of translation (22, 24). In the PKR-mediated cell death signaling pathway, eIF-2α is presumed to be an essential downstream target of PKR kinase function, leading to the inhibition of protein synthesis based on the phenotypic changes observed for cells overexpressing eIF-2α mutants that cannot be phosphorylated (12, 29). The phosphorylation of eIF-2α was proposed to induce apoptosis by allowing preferential translation of selective mRNAs that encode proteins with proapoptotic functions or by inhibiting the translation of the short-lived antiapoptotic proteins (16). Our findings with HeLa cells are consistent with this notion. The amount of eIF-2α phosphorylated in the PKR-deficient PKRkd cells that displayed less apoptosis in response to dsRNA was not elevated upon dsRNA treatment. By contrast, the eIF-2α phosphorylation level in PKR-sufficient parental cells was increased in response to dsRNA treatment. These results provide additional evidence that, in response to dsRNA, increased eIF-2α phosphorylation correlates with increased apoptosis (16, 39). However, PKR did not play a significant role in TNF-α-mediated apoptosis in HeLa cells, where apoptosis in response to TNF-α treatment was not accompanied by a detectable increase in eIF-2α phosphorylation. Our results with HeLa cells differ from those reported for MEF cells with an N-terminal disruption of Pkr, where TNF-α-induced apoptosis required translational attenuation through PKR-dependent eIF-2α phosphorylation (27). Whether the difference reflects differences between MEF and HeLa cells is unclear, as even between the two different MEF Pkr null mouse cells differences in TNF-α-induced apoptosis have been reported (1, 8).
Our results with HeLa clones are consistent with the notion that PKR is the principal kinase that phosphorylates eIF-2α in response to dsRNA treatment. Three kinases in addition to PKR also phosphorylate eIF-2α on serine 51: HRI, PERK, and GCN2. These multiple eIF-2α kinases constitute a universal mechanism by which translational control occurs in response to different physiological stresses (23). Conceivably one or more of the other known eIF-2α kinases may compensate for the loss of PKR and contribute to the basal level of eIF-2α phosphorylation seen in the absence of dsRNA. However, our PKRkd cell results suggest that these other eIF-2α kinases are unable to substitute for PKR in response to dsRNA treatment. Furthermore, a functional consequence of the reduced phosphorylation of eIF-2α seen in the PKRkd knockdown cells compared to the parental PKR+ and control PKRkd-con cells was illustrated by the increased reporter protein expression seen in the presence of dsRNA in the PKRkd cells.
Although we found that PKR plays an obligatory role in dsRNA-mediated eIF-2α phosphorylation and dsRNA-mediated apoptosis, our results suggest that PKR is not required for either dsRNA- or TNF-α-mediated NF-κB activation. NF-κB-dependent expression of a reporter firefly luciferase activity was comparably enhanced in PKR-sufficient parental and PKR-deficient PKRkd knockdown HeLa cells by dsRNA or TNF-α treatment, suggesting that pathways for NF-κB activation other than PKR are operative. The fact that NF-κB was activated to a comparable extent in response to dsRNA independent of the level of PKR protein present suggests that NF-κB might not modulate the apoptotic response in HeLa cells triggered by dsRNA. This conclusion is independently supported by the observation that cleaved PARP protein was detected in dsRNA-treated cells as soon as 4 h after treatment, while only a 0.2-fold elevation of NF-κB activation was detected at this early time point (Fig. 3).
PKR displayed a role in the host response to infection that was dependent upon the virus examined. Following IFN treatment, a higher single-cycle yield of VSV was produced by the PKRkd cells than the PKR-sufficient parental or PKRkd-con HeLa cells. However, VSV grew comparably well in the PKRkd and PKR+ cells in the absence of IFN treatment. The parental HeLa cells and the control PKRkd-con subline, while sensitive to IFN-α, both showed about a 100- to 300-fold reduction in VSV, which is less sensitive than that seen in some other human cell lines with VSV (26). The relative IFN resistance of the HeLa line makes the PKR effect appear less impressive than it actually may be as part of the antiviral VSV effect of IFN. In prior studies utilizing catalytic domain-disrupted Pkr knockout mice and derivative MEF cells, PKR was found to be a crucial component of IFN-mediated resistance to VSV infection (30), whereas contrary findings were seen with double-stranded RNA-binding-disrupted PKR knockout mice, which survived a high inoculum of VSV as wild-type mice did (38). These results taken together suggest that additional and functionally redundant antiviral pathways independent of PKR act to collectively constitute the antiviral state (22). This is not surprising, as more than 100 genes are known to be significantly upregulated by IFN (9, 22). The precise contributions that the different IFN-inducible proteins play in the inhibition of virus growth no doubt depend on the type of cell, kind of virus, and conditions of infection. In the absence of IFN, the PKR-independent antiviral components could function effectively to compensate for the loss of PKR, depending upon their combined basal level of expression. After treatment with IFN-α, PKR-sufficient HeLa cells acquire an enhanced antiviral state most likely involving both PKR-independent and PKR-dependent pathways, compared to PKRkd cells lacking the PKR-dependent pathways.
An additional important finding comes from our adenovirus growth determinations, where the multiplication levels of wild-type and VAI deletion mutant viruses were compared in parental and PKRkd HeLa cells. The mutant that does not synthesize VAI RNA grew comparably in the PKR-sufficient and PKR-deficient cells, but growth of the mutant was poor in both types of cells relative to wild-type virus. These observations were surprising. Because the VAI viral RNA inhibits PKR activation (17), we anticipated that the knockdown of PKR might complement the loss of VAI RNA, but this was not observed. These results suggest that PKR might not be the centrally important target of VAI RNA as previously believed (20). Alternatively, the residual 2% or less of PKR remaining in the PKRkd cells may still be sufficient to inhibit the mutant virus that does not express VAI RNA. However, a more exciting possibility is that another cellular protein, for example, ADAR1, which also is known to be inhibited by VAI RNA (19), conceivably may be responsible for the VAI mutant virus phenotype normally attributed to PKR. PKR likewise was found in our studies not to play a significant role in affecting the multiplication of reovirus serotype 3, in agreement with early conclusions from studies in human U cells where IFN induced PKR but did not affect reovirus growth (26) and more recent studies where reovirus replication was actually found to be more efficient in the presence of eIF-2α kinases and phosphorylatable eIF-2α (28).
In summary, we have established a stable HeLa cell clone in which the steady-state basal and IFN-inducible levels of human PKR protein are substantially reduced by a hairpin RNA interference strategy to less than 2% of parental or control knockdown protein levels. Our data obtained with the PKRkd (PKR-deficient) and PKRkd-con control and parental (PKR-sufficient) cells clarified some of the apparent contradictions concerning the requirement for PKR function in signaling, apoptosis, and virus growth obtained from studies of the two independent PKR knockout mouse lines and MEF cells. Our loss-of-function analyses in HeLa cells demonstrated that PKR plays a distinct proapoptotic role and an exclusive capacity to mediate phosphorylation of eIF-2α in response to dsRNA but that PKR also plays a minimal, if any, role in TNF-α-induced apoptosis signaling and NF-κB activation in HeLa cells. As a well-established antiviral protein, sole loss of PKR did not broadly affect virus growth in HeLa cells, further illustrating the functional redundancy of antiviral gene products inducible by IFN.
Acknowledgments
This work was supported in part by research grant AI-20611 from the National Institute of Allergy and Infectious Diseases, U.S. Public Health Service.
Footnotes
Published ahead of print on 23 May 2007.
REFERENCES
- 1.Abraham, N., D. F. Stojdl, P. I. Duncan, N. Methot, T. Ishii, M. Dube, B. C. Vanderhyden, H. L. Atkins, D. A. Gray, M. W. McBurney, A. E. Koromilas, E. G. Brown, N. Sonenberg, and J. C. Bell. 1999. Characterization of transgenic mice with targeted disruption of the catalytic domain of the double-stranded RNA-dependent protein kinase, PKR. J. Biol. Chem. 274:5953-5962. [DOI] [PubMed] [Google Scholar]
- 2.Alexopoulou, L., A. C. Holt, R. Medzhitov, and R. A. Flavell. 2001. Recognition of double-stranded RNA and activation of NF-κB by Toll-like receptor 3. Nature 413:732-738. [DOI] [PubMed] [Google Scholar]
- 3.Ashkenazi, A., and V. M. Dixit. 1998. Death receptors: signaling and modulation. Science 281:1305-1308. [DOI] [PubMed] [Google Scholar]
- 4.Baltzis, D., S. Li, and A. E. Koromilas. 2002. Functional characterization of pkr gene products expressed in cells from mice with a targeted deletion of the N terminus or C terminus domain of PKR. J. Biol. Chem. 277:38364-38372. [DOI] [PubMed] [Google Scholar]
- 5.Barber, G. N. 2005. The dsRNA-dependent protein kinase, PKR and cell death. Cell Death Differ. 12:563-570. [DOI] [PubMed] [Google Scholar]
- 6.Bonnet, M. C., C. Daurat, C. Ottone, and E. F. Meurs. 2006. The N-terminus of PKR is responsible for the activation of the NF-kappaB signaling pathway by interacting with the IKK complex. Cell Signal 18:1865-1875. [DOI] [PubMed] [Google Scholar]
- 7.Das, S., S. V. Ward, R. S. Tacke, G. Suske, and C. E. Samuel. 2006. Activation of the RNA-dependent protein kinase PKR promoter in the absence of interferon is dependent upon Sp proteins. J. Biol. Chem. 281:3244-3253. [DOI] [PubMed] [Google Scholar]
- 8.Der, S. D., Y. L. Yang, C. Weissmann, and B. R. Williams. 1997. A double-stranded RNA-activated protein kinase-dependent pathway mediating stress-induced apoptosis. Proc. Natl. Acad. Sci. USA 94:3279-3283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Veer, M. J., M. Holko, M. Frevel, E. Walker, S. Der, J. M. Paranjape, R. H. Silverman, and B. R. Williams. 2001. Functional classification of interferon-stimulated genes identified using microarrays. J. Leukoc. Biol. 69:912-920. [PubMed] [Google Scholar]
- 10.Dever, T. E. 2002. Gene-specific regulation by general translation factors. Cell 108:545-556. [DOI] [PubMed] [Google Scholar]
- 11.Donze, O., R. Jagus, A. E. Koromilas, J. W. Hershey, and N. Sonenberg. 1995. Abrogation of translation initiation factor eIF-2 phosphorylation causes malignant transformation of NIH 3T3 cells. EMBO J. 14:3828-3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gil, J., J. Alcami, and M. Esteban. 1999. Induction of apoptosis by double-stranded-RNA-dependent protein kinase (PKR) involves the alpha subunit of eukaryotic translation initiation factor 2 and NF-κB. Mol. Cell. Biol. 19:4653-4663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hsu, L. C., J. M. Park, K. Zhang, J. L. Luo, S. Maeda, R. J. Kaufman, L. Eckmann, D. G. Guiney, and M. Karin. 2004. The protein kinase PKR is required for macrophage apoptosis after activation of Toll-like receptor 4. Nature 428:341-345. [DOI] [PubMed] [Google Scholar]
- 14.Iordanov, M. S., J. Wong, J. C. Bell, and B. E. Magun. 2001. Activation of NF-κB by double-stranded RNA (dsRNA) in the absence of protein kinase R and RNase L demonstrates the existence of two separate dsRNA-triggered antiviral programs. Mol. Cell. Biol. 21:61-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jones, N., and T. Shenk. 1979. Isolation of adenovirus type 5 host range deletion mutants defective for transformation of rat embryo cells. Cell 17:683-689. [DOI] [PubMed] [Google Scholar]
- 16.Kaufman, R. J. 1999. Double-stranded RNA-activated protein kinase mediates virus-induced apoptosis: a new role for an old actor. Proc. Natl. Acad. Sci. USA 96:11693-11695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kitajewski, J., R. J. Schneider, B. Safer, S. M. Munemitsu, C. E. Samuel, B. Thimmappaya, and T. Shenk. 1986. Adenovirus VAI RNA antagonizes the antiviral action of interferon by preventing activation of the interferon-induced eIF-2 alpha kinase. Cell 45:195-200. [DOI] [PubMed] [Google Scholar]
- 18.Kumar, A., Y. L. Yang, V. Flati, S. Der, S. Kadereit, A. Deb, J. Haque, L. Reis, C. Weissmann, and B. R. Williams. 1997. Deficient cytokine signaling in mouse embryo fibroblasts with a targeted deletion in the PKR gene: role of IRF-1 and NF-κB. EMBO J. 16:406-416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lei, M., Y. Liu, and C. E. Samuel. 1998. Adenovirus VAI RNA antagonizes the RNA-editing activity of the ADAR adenosine deaminase. Virology 245:188-196. [DOI] [PubMed] [Google Scholar]
- 20.Mathews, M. B., and T. Shenk. 1991. Adenovirus virus-associated RNA and translation control. J. Virol. 65:5657-5662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meurs, E. F., J. Galabru, G. N. Barber, M. G. Katze, and A. G. Hovanessian. 1993. Tumor suppressor function of the interferon-induced double-stranded RNA-activated protein kinase. Proc. Natl. Acad. Sci. USA 90:232-236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Samuel, C. E. 2001. Antiviral actions of interferons. Clin. Microbiol. Rev. 14:778-809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Samuel, C. E. 1993. The eIF-2 alpha protein kinases, regulators of translation in eukaryotes from yeasts to humans. J. Biol. Chem. 268:7603-7606. [PubMed] [Google Scholar]
- 24.Samuel, C. E. 1979. Mechanism of interferon action: phosphorylation of protein synthesis initiation factor eIF-2 in interferon-treated human cells by a ribosome-associated kinase processing site specificity similar to hemin-regulated rabbit reticulocyte kinase. Proc. Natl. Acad. Sci. USA 76:600-604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Samuel, C. E., and G. S. Knutson. 1982. Mechanism of interferon action. Kinetics of decay of the antiviral state and protein phosphorylation in mouse fibroblasts treated with natural and cloned interferons. J. Biol. Chem. 257:11796-11801. [PubMed] [Google Scholar]
- 26.Samuel, C. E., and G. S. Knutson. 1981. Mechanism of interferon action: cloned human leukocyte interferons induce protein kinase and inhibit vesicular stomatitis virus but not reovirus replication in human amnion cells. Virology 114:302-306. [DOI] [PubMed] [Google Scholar]
- 27.Scheuner, D., R. Patel, F. Wang, K. Lee, K. Kumar, J. Wu, A. Nilsson, M. Karin, and R. J. Kaufman. 2006. Double-stranded RNA-dependent protein kinase phosphorylation of the alpha-subunit of eukaryotic translation initiation factor 2 mediates apoptosis. J. Biol. Chem. 281:21458-21468. [DOI] [PubMed] [Google Scholar]
- 28.Smith, J. A., S. C. Schmechel, A. Raghavan, M. Abelson, C. Reilly, M. G. Katze, R. J. Kaufman, P. R. Bohjanen, and L. A. Schiff. 2006. Reovirus induces and benefits from an integrated cellular stress response. J. Virol. 80:2019-2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Srivastava, S. P., K. U. Kumar, and R. J. Kaufman. 1998. Phosphorylation of eukaryotic translation initiation factor 2 mediates apoptosis in response to activation of the double-stranded RNA-dependent protein kinase. J. Biol. Chem. 273:2416-2423. [DOI] [PubMed] [Google Scholar]
- 30.Stojdl, D. F., N. Abraham, S. Knowles, R. Marius, A. Brasey, B. D. Lichty, E. G. Brown, N. Sonenberg, and J. C. Bell. 2000. The murine double-stranded RNA-dependent protein kinase PKR is required for resistance to vesicular stomatitis virus. J. Virol. 74:9580-9585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tanaka, N., M. Sato, M. S. Lamphier, H. Nozawa, E. Oda, S. Noguchi, R. D. Schreiber, Y. Tsujimoto, and T. Taniguchi. 1998. Type I interferons are essential mediators of apoptotic death in virally infected cells. Genes Cells 3:29-37. [DOI] [PubMed] [Google Scholar]
- 32.Taylor, S. S., N. M. Haste, and G. Ghosh. 2005. PKR and eIF2α: integration of kinase dimerization, activation, and substrate docking. Cell 122:823-825. [DOI] [PubMed] [Google Scholar]
- 33.Thimmappaya, B., C. Weinberger, R. J. Schneider, and T. Shenk. 1982. Adenovirus VAI RNA is required for efficient translation of viral mRNAs at late times after infection. Cell 31:543-551. [DOI] [PubMed] [Google Scholar]
- 34.Thomis, D. C., J. P. Doohan, and C. E. Samuel. 1992. Mechanism of interferon action: cDNA structure, expression, and regulation of the interferon-induced, RNA-dependent P1/eIF-2 alpha protein kinase from human cells. Virology 188:33-46. [DOI] [PubMed] [Google Scholar]
- 35.Thomis, D. C., and C. E. Samuel. 1992. Mechanism of interferon action: autoregulation of RNA-dependent P1/eIF-2 alpha protein kinase (PKR) expression in transfected mammalian cells. Proc. Natl. Acad. Sci. USA 89:10837-10841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wek, R. C. 1994. eIF-2 kinases: regulators of general and gene-specific translation initiation. Trends Biochem. Sci. 19:491-496. [DOI] [PubMed] [Google Scholar]
- 37.Williams, B. R. 1999. PKR, a sentinel kinase for cellular stress. Oncogene 18:6112-6120. [DOI] [PubMed] [Google Scholar]
- 38.Yang, Y. L., L. F. Reis, J. Pavlovic, A. Aguzzi, R. Schafer, A. Kumar, B. R. Williams, M. Aguet, and C. Weissmann. 1995. Deficient signaling in mice devoid of double-stranded RNA-dependent protein kinase. EMBO J. 14:6095-6106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yeung, M. C., J. Liu, and A. S. Lau. 1996. An essential role for the interferon-inducible, double-stranded RNA-activated protein kinase PKR in the tumor necrosis factor-induced apoptosis in U937 cells. Proc. Natl. Acad. Sci. USA 93:12451-12455. [DOI] [PMC free article] [PubMed] [Google Scholar]