

Abstract
The mechanisms of human papillomavirus (HPV) neutralization by antibodies are incompletely understood. We have used HPV16 pseudovirus infection of HaCaT cells to analyze how several neutralizing monoclonal antibodies (MAbs) generated against HPV16 L1 interfere with the process of keratinocyte infection. HPV16 capsids normally bind to both the cell surface and extracellular matrix (ECM) of HaCaT cells. Surprisingly, two strongly neutralizing MAbs, V5 and E70, did not prevent attachment of capsids to the cell surface. However, they did block association with the ECM and prevented internalization of cell surface-bound capsids. In contrast, MAb U4 prevented binding to the cell surface but not to the ECM. The epitope recognized by U4 was inaccessible when virions were bound to the cell surface but became accessible after endocytosis, presumably coinciding with receptor detachment. Treatment of capsids with heparin, which is known to interfere with binding to cell surface heparan sulfate proteoglycans (HSPGs), also resulted in HPV16 localization to the ECM. These results suggest that the U4 epitope on the intercapsomeric C-terminal arm is likely to encompass the critical HSPG interaction residues for HPV16, while the V5 and E70 epitopes at the apex of the capsomer overlap the ECM-binding sites. We conclude that neutralizing antibodies can inhibit HPV infection by multiple distinct mechanisms, and understanding these mechanisms can add insight to the HPV entry processes.
Human papillomavirus (HPV) infections are extremely common, with estimates suggesting that approximately 75% of women will become infected with one or more of the sexually transmitted HPV types at some point after initiating sexual activity (1). Infection with a subset of sexually transmitted HPVs, especially HPV16, is considered a necessary factor in the development of virtually all cases of cervical cancer (3). A neutralizing antibody response to L1, the major structural viral protein, is known to effectively prevent papillomavirus (PV) infection, as demonstrated by studies in animal models and the successes of the recently developed HPV vaccine (33). However, the mechanisms by which these neutralizing antibodies act to prevent infection are unclear.
L1 can self-assemble to form empty capsids known as virus-like particles that resemble authentic capsids morphologically and immunologically (28) and are the basis for current HPV vaccines. Although L1 is overall a highly conserved PV protein, anti-L1 neutralizing antibodies are type restricted, because the conserved residues are largely confined to the portions of L1 that are poorly exposed on the surface of the capsid, whereas the antibody responses are typically generated against epitopes found on the external loops, where the L1 sequences are highly divergent (11, 38).
These external loops, which form the apex of the L1 capsomer, are relatively unstructured and, based on analogy to the polyomavirus major structural protein, VP1, have been proposed as candidate regions for receptor interaction (10). In fact, despite minimal sequence homology among their major capsid proteins, the PV capsid structure is very similar to that of polyomavirus and simian virus 40 (SV40). However, unlike for polyoma and SV40, no experimental data have supported the involvement of these regions in receptor binding. In contrast, interaction with cell surface heparan sulfate proteoglycans (HSPGs) has been shown to be critical for HPV33 infection in vitro (46), and a conserved, canonical heparin interaction domain has been described at the carboxyl terminus of HPV11 L1 distal to the region that comprises the β sandwich capsomer core (27). These data indicate that a portion of L1 lying distal to the capsid surface may play a role in cell binding. Despite these studies, it remains unknown which portions of L1 are actually responsible for binding to HSPGs or other cell surface receptor molecules.
Virus neutralization is defined as the abrogation of virus infectivity by the association of antibody with the viral particle. In principle, neutralization can inhibit infectivity by several mechanisms (25, 29). Obviously, prevention of virus attachment would preempt the entry process, and many neutralizing antibodies act by abrogating virion attachment to the host cells. Antibodies can also potentially interfere with postattachment interactions of a virus with its receptors or coreceptors or with the cellular endocytic machinery. Neutralizing antibodies have also been described that act after viral endocytosis by negatively affecting trafficking, membrane penetration, uncoating, nuclear import, or viral transcription (9, 41, 49, 50, 55).
In this study, we have examined the neutralization by three anti-HPV16 neutralizing monoclonal antibodies (MAbs). The epitopes to which these antibodies bind on the HPV capsid have been previously described. Two of the antibodies, H16.V5 (V5) and H16.E70 (E70), recognize overlapping epitopes present on the apex of the L1 capsomers (7, 54). The epitope of the third antibody, H16.U4 (U4), has been mapped to a carboxy-terminal portion of L1, which has been proposed to extend between adjacent capsomers (7, 37).
MATERIALS AND METHODS
Cells and antibodies.
HaCaT cells were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum. The polyclonal antiserum raised in rabbits against HPV16 capsids was previously described (43). MAbs against HPV16 were obtained from Neil Christensen (Department of Pathology, College of Medicine, Pennsylvania State University, Hershey, PA) and have been previously described (12).
Pseudovirus production.
Pseudovirus stocks were produced as previously described (5, 39), with the exception of the pseudovirions utilized for the enzyme-linked immunosorbent assay (ELISA). For that assay, pseudovirions were generated by a serial propagation system described on our laboratory's website (http://home.ccr.cancer.gov/lco/synthetichpv.htm) (C. B. Buck, N. Cheng, C. D. Thompson, D. R. Lowy, A. C. Steven, J. T. Schiller, and B. L. Trus, unpublished data). For neutralization assays, the plasmid encoding secreted alkaline phosphatase, pYSEAP, was packaged. For Alexa Fluor 488-coupled pseudovirions, the red fluorescent protein plasmid, ptwB, was packaged. Dye coupling was performed with Alexa Fluor 488 carboxylic acid tetrafluorophenyl ester (A10235; Molecular Probes) according to the manufacturer's suggestions and is detailed on the laboratory's website. For all other assays, pseudovirions were prepared with the green fluorescent protein-expressing plasmid, pfwB, packaged. All plasmids and production methods are fully described on the laboratory's website (http://home.ccr.cancer.gov/lco/default.asp).
Virus neutralization.
The PV neutralization assay was performed as previously described (6, 39, 40). Briefly, diluted pseudovirus stocks were combined with diluted MAb and placed on ice for 1 h. The pseudovirus-antibody mixture was then transferred onto 293TT cells and incubated for 72 h. At the end of the incubation, the secreted alkaline phosphatase (SEAP) content in the culture supernatant was determined using the Great EscAPe SEAP chemiluminescence kit (BD Clontech) as directed by the manufacturer. Samples were read using an MLX microplate luminometer (Dynex Technologies) set at Glow-Endpoint.
ELISA.
MAb binding to pseudovirions was determined by a capture ELISA. Immulon 2HB plates (Thermo Corporation) were coated with 500 ng/well of purified rabbit anti-HPV16 L1 antiserum and incubated overnight at 4°C. Plates were then washed three times with phosphate-buffered saline (PBS) and blocked in PBS containing 0.5% powdered milk and 0.1% fetal bovine serum for 2 h at room temperature. Plates were rewashed, and 150 ng/well of pseudovirions was added in PBS-0.5% powdered milk for 2 h at room temperature. Unbound particles were removed by washing, and MAbs, diluted in PBS-0.5% milk, were added for 2 h at room temperature. After washing, a horseradish peroxidase-conjugated goat anti-mouse kappa chain antiserum (Caltag) was added at a 1/2,000 dilution in PBS-0.5% milk for 1 h at room temperature. The plates were washed again and developed with the peroxidase substrate, ABTS [2,2′-azinobis(3-ethylbenzthiazolinesulfonic acid)] (Roche).
Flow cytometric analysis.
Pseudovirus was bound to 1 × 105 HaCaT cells for 1 h at 4°C in FC buffer (PBS containing 2% fetal bovine serum and 0.01% sodium azide). Following the removal of unbound particles by washing, the MAbs were used at a final concentration of 1 μg/ml and incubated for 1 h at 4°C in FC buffer. Cells were washed again and incubated with a fluorescein isothiocyanate-conjugated donkey anti-mouse immunoglobulin G (IgG) (Jackson Immunoresearch) for 1 h at 4°C. Cells were washed, and cell-associated fluorescence was quantified on a BD FACSCaliber (Becton Dickinson).
Immunofluorescent staining.
Cells were seeded onto glass no. 01 coverslips in a 24-well plate at a density of 1 × 105 cells/well and cultured overnight. For studies to evaluate the capsid binding pattern, 50 ng of pseudovirus was incubated with a given MAb at roughly 2.5 μg/ml, with heparin at 10 μg/ml (Sigma H-4794), or without treatment and then added to the cells for 7 h unless otherwise noted. Cells were fixed in ice-cold ethanol containing 15 mM glycine. For detection of antibody-neutralized particles, the cells were stained with Alexa Fluor 488-conjugated donkey anti-mouse IgG (Molecular Probes). For detection of nonneutralized particles or particles neutralized with heparin, the cells were stained at a 1/1,000 dilution with a rabbit polyclonal antiserum raised against HPV16 particles and subsequently stained with Alexa Fluor 488-conjugated donkey anti-rabbit IgG (Molecular Probes). Rhodamine-conjugated phalloidin (Molecular Probes) was included in the secondary antibody stain. Coverslips were inverted onto DAPI (4′,6′-diamidino-2-phenylindole)-containing mounting solution (Prolong Gold; Molecular Probes). For detection of the U4 epitope during endocytosis, 50 ng of capsids was added, and cells were fixed after 1 h and 7 h. Cells were costained with the anti-HPV16 rabbit polyclonal antiserum and U4 at a 1/500 dilution and subsequently stained with Alexa Fluor 594-conjugated donkey anti-rabbit IgG and Alexa Fluor 488-conjugated donkey anti-mouse IgG (Molecular Probes). All images were acquired with a Zeiss LSM 510 confocal system interfaced with a Zeiss Axiovert 100 M microscope. Images were collated with the Adobe Photoshop software.
Preparation of ECM.
Extracellular matrix (ECM)-coated coverslips were prepared according to a modification of a previously published method (51). Briefly, HaCaT cells were seeded onto glass no. 01 coverslips in a 24-well plate at a density of 3 × 104 cells/well and cultured for 5 days in growth medium supplemented with 50 μg/ml ascorbic acid. Cells were lysed in PBS containing 0.5% Triton X-100 and 20 mM NH4OH. Solubilized material was removed by washing, and coverslips were incubated with DNase I (10 μg/ml; Sigma) for 30 min at 37°C. Coverslips were washed again, and Alexa Fluor 488-coupled pseudovirions were added to extracted matrices in PBS containing 2% bovine serum albumin and bound for 3 h at 37°C. Unbound capsids were removed by washing. Coverslips were fixed with 2% paraformaldehyde for 20 min at room temperature. Laminin 5 was detected with a rabbit polyclonal antiserum (Abcam; ab14509) and Alexa Fluor 594-conjugated donkey anti-rabbit IgG (Molecular Probes). Coverslips were mounted in DAPI-containing mounting solution to ensure detection of any intact cells.
RESULTS
Analysis of relative epitope abundance.
To investigate the relative abundance of the cognate epitopes of MAbs V5, E70, and U4, we performed comparative capture ELISA experiments. Since the MAbs are of different heavy-chain isotypes, a secondary polyclonal antiserum specific for the murine κ light chain (which all three MAbs utilize) was used for detection.
As seen in Fig. 1A, saturating doses of MAbs V5 and E70 displayed a roughly twofold greater signal than saturating doses of MAb U4. This result is consistent with a model in which U4 recognizes a less-abundant epitope than either V5 or E70. It is also important to note that the slope of the U4 binding curve was relatively shallow (Hill coefficient ± the standard error, 0.79 ± 0.08) compared with those of V5 and E70. A simple hypothesis to explain these results would be that U4 binds with various affinities to epitopes in a limited subset of environments, while V5 and E70 bind with higher affinity to a better-exposed set of epitopes.
FIG. 1.

(A) Binding of MAbs to HPV16 pseudovirions. The results represent the ELISA reactivity of V5, E70, and U4. Pseudovirions were captured with purified IgG from an anti-HPV16 polyclonal antiserum, and serial dilutions of MAbs were subsequently applied. The 95% confidence interval is indicated by the dotted lines bracketing the solid line for each antibody. This experiment is representative of multiple experiments. The concentration of MAb is indicated on the x axis. The optical density is indicated on the y axis. (B) Neutralization of HPV16 infection. Inhibition of infection of the HPV16-SEAP pseudovirus was evaluated by SEAP concentration. The assay was performed in triplicate. The concentration of MAb is indicated on the x axis. The percentage of pseudovirus neutralization is indicated on the y axis.
Under some circumstances, the concentration at which 50% of maximum antibody binding occurs in an ELISA will faithfully reflect the overall affinity of the antibody-epitope interaction. For this to be true, the concentration of target antigen must be lower than the affinity constant of the antibody-epitope interaction, such that antibody binding events remove only a negligible fraction of the total antibody pool. If these conditions are satisfied, then varying the amount of antigen in the well does not shift the ELISA binding curve, since the curve is a reflection of the intrinsic affinity of the antibody-epitope interaction. When we reduced the dose of capsids used in our ELISA by threefold, the U4 binding curve did not shift, the E70 binding curve shifted slightly leftward, and the V5 curve shifted leftward by exactly threefold (data not shown). These results show that, particularly for V5, the ELISA does not satisfy the law of mass action (30). This implies that the affinity constant for V5 is lower, perhaps substantially lower, than the 160 pM 50% effective concentration value observed using the standard ELISA conditions.
Quantitative neutralization analysis.
Neutralization assays of an HPV16 pseudovirus expressing a SEAP reporter gene were performed using an established method (Fig. 1B) (39). As expected, each of the three MAbs was able to completely neutralize the infectivity of the pseudovirus. Similar to the results obtained in the ELISA experiments, the neutralization curve for MAb U4 exhibited a slope somewhat more shallow than those for V5 or E70, with Hill coefficients of 1.4 ± 0.1 for U4, 2.6 ± 0.3 for V5, and 3.7 ± 0.1 for E70.
For V5, the neutralization curve was shifted dramatically leftward relative to the binding curve observed in the ELISA experiments. This result reflects the fact that the neutralization assay (unlike the ELISA) can be performed using very low concentrations of capsids (<1 pM), such that antibody is in molar excess over capsids at all points on the curve, thereby satisfying the law of mass action. The use of the highly sensitive chemiluminescent system for detecting SEAP is critical to allow such low amounts of input pseudovirions. When the assay was repeated at a second, lower concentration of viral capsids, no shift in the curves was observed, confirming that antibody concentration was in excess of capsid concentration at all points on the curve (data not shown).
V5 and E70 do not block interaction with the cell surface.
We were interested in determining the mechanism of viral neutralization by V5 and E70, both of which bind to the apex of the capsomers. As both of these antibodies can inhibit mouse red blood cell agglutination (42), it has been assumed that they neutralize infection by preventing virus particles from binding the cell surface. V5 is especially interesting, as it has been previously shown in a competitive ELISA to block binding of most of the HPV16 virus-like particle-specific IgG in the sera of humans who have been infected with HPV16 (53).
For these analyses, we utilized the normal human keratinocyte cell line, HaCaT. It has been recently demonstrated that PV virions can bind to the ECM deposited by HaCaT cells in addition to the cell surface (17). This pattern of binding is evident for particles detected with a rabbit polyclonal serum raised against intact particles, as shown in Fig. 2, panel PV. Surprisingly, when we examined the localization of particles that had been incubated with a neutralizing quantity of either V5 or E70, strong cell surface association was evident (Fig. 2), indicating little or no diminution of cell binding ability by these neutralizing antibodies. Interestingly, treatment with either V5 or E70 reduced the ECM binding of the particles to undetectable levels. These results suggest that V5 and E70 neutralize PV virions via a post-cell attachment mechanism. Given these results, it is likely that the region of the capsid involved in ECM binding is situated on the top surface of the capsomers (10).
FIG. 2.

V5 and E70 do not prevent surface association. The first panel, PV, shows the typical binding pattern of HPV16 capsids to HaCaT cells. Capsids were detected with a rabbit anti-HPV16 antiserum and donkey anti-rabbit Alexa Fluor 488 (green channel). The cell body is delineated by actin staining with rhodamine-phalloidin (red channel). The second panel shows the pattern of virion binding after preincubation of the capsids with V5. The V5-bound capsids were detected with donkey anti-mouse Alexa Fluor 488. The third panel shows the pattern of virion association after E70 binding. The E70-bound capsids were detected with donkey anti-mouse Alexa Fluor 488.
V5- or E70-bound particles are not efficiently internalized.
HPV16 particles are apparently internalized by an atypical clathrin-dependent endocytosis pathway, in that the profile of biochemical inhibitors and subcellular markers supports a clathrin-mediated pathway, but the kinetics of internalization (half-life of 4 h) are unusually slow (4, 19). To determine if virion endocytosis was prevented in the presence of V5 and E70, we compared the viral entry process for untreated capsids with that for capsids that had been neutralized with either antibody. Virions were incubated with cells for 7 h to allow ample time for passage into endosomes. As shown in the first panel of Fig. 3, untreated capsids were clearly evident in perinuclear vesicles, as previously described. However, the capsids that were neutralized with either of the neutralizing antibodies continued to be found only at the cell surface (Fig. 3), strongly suggesting that V5 and E70 prevent viral infection by blocking endocytosis. For comparison, an early time point demonstrating the surface staining of untreated capsids is shown in the first panel of Fig. 8.
FIG. 3.

V5 and E70 prevent internalization of capsids. HPV16 pseudovirions were first either untreated or incubated with either V5 or E70, as indicated, and then incubated on HaCaT cells for 7 h. Untreated capsids were detected with a rabbit anti-HPV16 antiserum and donkey anti-rabbit Alexa Fluor 488 (panel PV). Capsids in the MAb-bound conditions were detected with donkey anti-mouse Alexa Fluor 488.
FIG. 8.

U4 epitope exposure during endocytosis. Capsids were bound to HaCaT cells for 1 h at 37°C. Unbound capsids were removed by washing, and the cells were either fixed (top panels) or incubated for 6 additional hours at 37°C and then fixed (bottom panels). Virions were stained with both a rabbit polyclonal antiserum raised against HPV16 capsids (panels labeled PV) and the U4 MAb (panels labeled U4). The merged channels are shown in the third panels for each time point.
U4 sequesters virions on the ECM.
We then examined the binding pattern of capsids that had been treated with the U4 MAb, which recognizes an epitope within the carboxyl region of the L1 protein that would be present on the intercapsomeric arm as described in the model presented by Modis and colleagues (37; see also reference 7). When pseudovirus was incubated with this antibody and binding to HaCaT cells and ECM was examined, the antibody-bound virus particles were found exclusively on the ECM, rather than on the cell surface, in dramatic contrast to the pattern observed with V5 and E70. This result is seen in Fig. 4, where the swath of virions is extended away from the cell body, as delineated by actin staining, and no binding to the cell surface is evident. Thus, U4 appears to inhibit infection by inhibiting the binding of the capsid to the cell surface, although the capacity to bind ECM is retained.
FIG. 4.

U4 sequesters capsids on the ECM. The first panel shows the pattern of virion binding after preincubation of the capsids with U4. The U4-bound capsids were detected with donkey anti-mouse Alexa Fluor 488. The cell body is delineated by actin staining with rhodamine-phalloidin. The merged image is shown in the third panel.
Binding of V5-, E70-, and U4-bound particles to isolated ECM.
To confirm the disparate effects between U4 and those of V5 and E70 on the ECM-binding capability of the viral particles, we examined the association of antibody-bound capsids to extracted ECM. ECM was prepared from cultures of HaCaT cells that had been plated at low density and grown for 5 to 6 days to allow for a heavy matrix deposition. The cells were subsequently removed by detergent lysis, and matrix was treated with DNase according to standard methodology (51). For direct visualization of virion binding, we used Alexa Fluor 488-conjugated pseudovirions. This conjugation had no effect on the infectivity of the virus, and microscopic analysis of the MAbs with these particles yielded results similar to those observed using indirect immunofluorescence (data not shown). Dye-conjugated particles were incubated with no antibody or with one of the neutralizing MAbs and then incubated with the isolated matrices. As illustrated in Fig. 5, the untreated capsids interacted well with the HaCaT ECM, as anticipated. The capsids that had been incubated with U4 also showed strong binding to the ECM, similar to what we had demonstrated, as described above, with intact cells. However, the particles that had been preincubated with either V5 or E70 showed negligible binding to the matrices, confirming the staining observations with intact HaCaT cultures. Detection of the ECM marker, laminin 5, is shown in the upper panels of Fig. 5 for all conditions to demonstrate equivalent ECM deposition. Polybrene (10 μg/ml) was included in some analyses (not shown) and did not affect the binding of capsids to the ECM, indicating that a heparan sulfate moiety is unlikely to be involved in this interaction.
FIG. 5.

Binding of capsids to HaCaT ECM. ECM was prepared from HaCaT cell cultures. Alexa Fluor 488-conjugated HPV16 pseudovirions were either untreated (UNTX.) or incubated with either V5, E70, or U4, as indicated, prior to addition to the matrix preparation. The upper panels show the extent of ECM deposition as determined by laminin 5 (LAM.5) staining.
Soluble heparin sequesters capsids on the ECM.
The above data indicate that the epitope on the viral capsid that is recognized by the U4 antibody overlaps with, or sterically hinders access to, the site necessary for cell surface binding. As the carboxyl terminus of L1 has been described to contain a canonical heparin sulfate binding motif (27) and as HSPGs have been demonstrated to play a role in PV cell binding and infection (24, 45), we used microscopy to analyze the neutralization of virus infection with soluble heparin, which contains the same polymeric backbone as heparan sulfate but is more highly modified. As shown in Fig. 6, treatment of viral particles with 10 μg/ml of heparin resulted in sequestration of the particles on the ECM of HaCaT cells in a pattern that was indistinguishable from the one observed with the U4 blocking. These data are consistent with the model that binding of U4 prevents the interaction of the capsid with cell surface HSPGs.
FIG. 6.

Soluble heparin sequesters capsids on the ECM. The first panel shows the pattern of virion binding after incubation of the capsids with HaCaT cells in the presence of 10 μg/ml of exogenous heparin. The capsids were detected with a rabbit anti-HPV16 antiserum and donkey anti-rabbit Alexa Fluor 488. The cell body is delineated by actin staining with rhodamine-phalloidin in the second panel. The merged image is shown in the third panel.
U4 epitope is not exposed on cell surface-bound virions.
As further evidence that the cell surface receptor binding site and the U4 epitope are sterically related, we examined the accessibility of the U4 epitope on the cell surface and during virion endocytosis. As heparin binding mimicked the binding of U4, implying that both might bind similar regions on L1, we reasoned that interaction of the capsid with the cell surface, which is HSPG dependent, could prevent binding of the U4 antibody to its epitope, but that U4 binding might be reestablished after virion uptake and receptor dissociation. This model is consistent with the staining data shown in Fig. 7 and 8. First, we assessed the binding of the MAbs to cell surface-bound virions. The antibody binding was detected by flow cytometry. These data, shown in Fig. 7, clearly indicate that while V5 and E70 can efficiently bind to virions bound to the HaCaT cell surface, the U4 MAb does not recognize these particles. Second, we used confocal microscopy to study exposure of the U4 epitope, as shown in Fig. 8. After binding to the cells for 1 h, the particles were readily detected with a polyclonal antiserum, but no binding of the U4 antibody was detected (Fig. 8, upper panels). At this time, the majority of the capsids were still on the cell surface, and few particles had been internalized. However, at a later time point, after the virions have entered the endocytic pathway, the U4 epitope became accessible, and a strong vesicular pattern was observed (Fig. 8, lower panels). Importantly, the U4 antibody staining colocalized with the anti-L1 polyclonal serum staining only in the internalized vesicles and not with the residual particles on the cell surface (see merged pane in lower panels). In contrast, the V5 epitope, which is not obscured on cell surface-bound virions, could be detected throughout the time course of binding and entry, indicating that the surface of at least some capsomers remains exposed after cell surface binding (data not shown).
FIG. 7.

Detection of capsids on the cell surface. Pseudovirions were bound to HaCaT cells for 1 h at 4°C in suspension. Unbound virions were removed by extensive washing, and bound virions were detected with an irrelevant antibody, 5B6, as a negative control, or one of the three MAbs of interest and a fluorescein isothiocyanate-conjugated secondary antibody. The mean fluorescent signal is shown in parentheses below the antibody indicated. The data presented are representative of the results of three separate experiments.
DISCUSSION
Our results, which report at least two unexpected observations, have implications for the mechanisms by which PVs infect cells and may be neutralized. First, we determined that V5 and E70 do not interfere with the virion binding to the cell surface. Instead, they apparently neutralize infection by interfering with the internalization of bound particles. Second, we found that U4 interfered with infection by preventing cell surface binding, but it did not interfere with the ability of particles to bind ECM. These data allow us to hypothesize where some functional regions lie on the PV particle.
PVs share a common structure composed of 72 L1 pentamers located at the vertices of a T = 7 icosahedral lattice. A quasiatomic model of the PV capsid was previously generated by fitting the X-ray crystal structure of an aberrant T = 1 capsid into the high-resolution cryoelectron microscopic image reconstruction generated from native BPV1 virions (10, 37, 48). In this model, the carboxyl-terminal residues were rebuilt to fit in a new conformation constrained by the requirement that cysteine 428 in the C-terminal arm must form a disulfide bond with cysteine 175 in the EF loop of an adjacent L1 (Fig. 9). This model is consistent with the “invading arm” model, as seen in the polyomavirus VP1 subunits, in which the carboxyl terminus forms the principal interpentamer contact by extending from one L1 subunit to an adjacent pentamer (32). In the PV model, residues 430 to 446 would descend into the intercapsomeric cleft and extend partway around the circumference of the adjacent pentamer, with residues 447 to 474 extending into the adjacent pentamer between two L1 subunits (Fig. 9). The outermost surface of the L1 pentamer has five broad pockets, created by the BC, EF, and FG loops with the HI loops intertwined across the top of the pentamer.
FIG. 9.
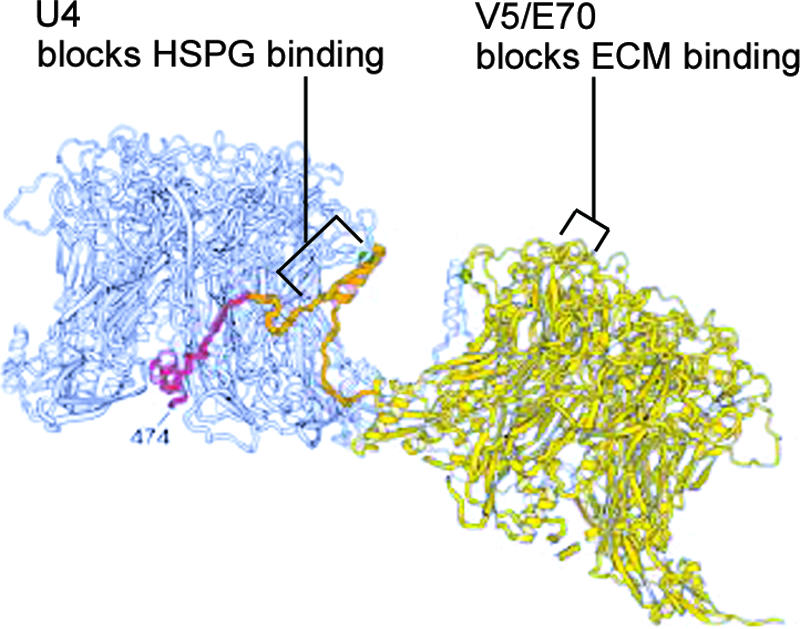
MAb binding epitopes shown on the three-dimensional model of Modis et al. (37). Two adjacent L1 pentamers are shown in gray and yellow. The carboxyl-terminal arm, originating from the yellow pentamer and extending toward the gray pentamer, is indicated in orange and red. MAb binding epitopes are indicated, in addition to our proposed ECM- and HSPG-binding regions. (Reprinted from the EMBO Journal [37] with permission from the publisher.)
The MAbs V5 and E70 bind overlapping, but distinct, epitopes on the top surface of L1 (Fig. 9). The FG loop, with a contribution from the HI loop, was shown to be important for the binding of both antibodies (7, 11). Evidence for their binding distinct epitopes includes biosensor analysis that determined V5 binding could completely obscure the E70 binding epitope (54), while the inverse experiment resulted in incomplete interference of the V5 binding by E70. Additionally, substitution of a number of single amino acids within the FG loop of L1 prevented binding of E70 but did not affect the interaction of V5 (7, 42). The epitope recognized by V5, and to a lesser extent the E70 epitope, overlaps with those recognized by most sera obtained from human subjects with HPV16 antibodies (53). Thus, a polyclonal immune response is largely to the region exposed on the top surface of the capsomers with the FG, HI, and DE loops all contributing to the immunodominant epitopes (8, 52).
Although the carboxyl arm in the Modis model is in the groove between the pentameric protrusions, it was proposed that an antibody could bind the site, particularly the loop at the crown of the pentamer, which includes residues 420 to 429 (37). Cryoelectron microscopic reconstruction image of a neutralizing anti-BPV1 monoclonal, 5B6, complexed to BPV1 virions, clearly shows that it binds the side walls and straddles the interpentameric grooves between the hexavalent capsomers (2). In a later study, it was shown that the epitope of U4 requires a segment of L1 encompassing residues 427 to 445 (Fig. 9) (7). Consistent with these findings, biosensor analysis indicates that the U4 epitope is completely distinct from the V5 epitope (54). Interestingly, that study also found that U4 bound, but did not neutralize, the aberrant HPV16-Rochester variant. A recent report has suggested that U4 neither bound nor neutralized HPV16-114K pseudovirions, the “standard” HPV16 also used in the current study (18). However, that study utilized unquantified hybridoma supernatants. We clearly show that although it has relatively low affinity, U4 can bind and neutralize when sufficient antibody is employed. In the same study by Culp et al., it was shown that anti-HPV6 and HPV11 antibodies previously found to be nonneutralizing when tested as hybridoma supernatants (13, 14) in fact neutralized well when sufficient quantities of antibodies were used.
Inhibition of cell surface interaction, which is an effective mechanism for antibody-mediated neutralization, has been demonstrated for enveloped viruses, including rabies virus (23), dengue virus (26), and influenza virus (31), and for the nonenveloped picornaviruses, of which rhinovirus neutralization is the best understood (15, 47). The “canyon hypothesis” mechanism of rhinovirus neutralization proposes that the receptor-interacting residues are on the floor of a depression on the rhinovirus capsid (15, 16). The small size of this canyon tends to limit its access to antibodies, thereby protecting the receptor site from immune surveillance, while the small receptor molecule, ICAM-1, is not excluded (34, 44). However, the ability of a subset of neutralizing antibodies to restrict entrance of the receptor protein into the opening of this canyon probably accounts for their neutralizing capacity (15, 47). This mechanism may share similarities with U4 neutralization, albeit the rhinovirus canyon is substantially narrower than the PV surface intercapsomeric grooves.
The marked ECM association of U4-bound capsids is probably a simple reflection of the inability of the virions to access the cell surface. It is noteworthy that the same pattern was observed when virus particles were exposed to heparin. The carboxyl terminus of HPV11 L1 was shown to contain a heparin-binding domain that is conserved among PV types (27), implicating this region as playing a critical role in the binding of capsids to glycosaminoglycans on the cell surface. However, utilization of this site for cell interaction is inconsistent with the prevailing model of the PV capsid structure in which this region would be buried within the lumen of the capsid. Given the striking finding that U4 binding prevents cell surface association, we propose that a heparin-binding domain exists within the intercapsomeric cleft, although it may not be the site originally proposed by Joyce et al. (27). It is possible that there is one HSPG-interacting site within the cleft and that cell-induced conformational changes in the virion can expose the extreme carboxyl terminus of L1, resulting in a higher affinity binding. Consistent with this scenario, HPV33 capsids have been shown to transition, while on the cell surface, from a heparin-sensitive interaction to an insensitive one (24). Alternatively, the extreme carboxyl end of L1 may not participate in HSPG binding, but deletion of this region could have indirectly affected the capsid's ability to bind heparin in the previous study. The direct interaction of HPV capsids with purified heparan sulfate derived from the appropriate cells (keratinocytes) versus heparin, or heparan sulfate from other sources, will be important to assess in future experiments. Using the correctly modified HSPG would be critical for these in vitro experiments, as HSPGs contain specific arrangements of variably modified glucosamine and uronic acid residues that occur in a lineage-specific fashion (22).
As the U4 epitope is present near the top of the intercapsomeric cleft, the inhibition of cell surface binding may be attributable to steric hindrance, with the actual HSPG binding motif(s) lying within the cleft. It is also possible that the U4 epitope could physically overlap with the HSPG binding site. This latter interpretation is consistent with the inability of the U4 MAb to recognize surface-bound particles, as the access of U4 to its epitope is presumably blocked by the binding of the particles to HSPG on the cell surface. Following particle internalization, the U4 epitope was again accessible, presumably because the capsids are no longer bound to HSPG. The explanation that U4 directly interferes with HSPG binding could also explain why the neutralizing BPV1 MAb, 5B6, which binds across the cleft, does not prevent binding of the viral particles to the cell surface (2). The observation that the U4 epitope is completely occluded on surface-bound particles was somewhat surprising but reproducibly observed and consistent between the flow cytometric analysis (Fig. 7) and microscopy studies (Fig. 8). The likeliest explanation is that the viral particles are nestled within the thick glycocalyx and, in fact, contacted by HSPG on all faces. Also, consistent with the inaccessibility of the U4 binding epitope, no neutralization of cell-associated pseudovirus was observed (data not shown).
The precise mechanism of neutralization by V5 and E70 remains to be elucidated. It is possible that occupancy of these epitopes prevents a necessary conformational change in capsid morphology, as subtle conformational changes have been reported to occur after HSPG engagement at the cell surface (45), or that antibody binding may itself induce a conformational change within the capsid, as has been shown for some neutralizing anti-picornavirus antibodies (21, 35). Alternatively, the bound antibody might block critical interactions with the endocytic machinery or with a second receptor, following binding to HSPGs. Incubation of the pseudovirions with isolated HaCaT-derived ECM prior to interaction with either V5 or E70 did not affect the ability of these antibodies to neutralize infection of subsequently added cells (data not shown). Therefore, we believe it is unlikely that the neutralizing capacity of V5 and E70 is due solely to their interference with capsid-ECM interactions.
The ability of some PV-neutralizing antibodies, including V5 and E70, to inhibit mouse red blood cell hemagglutination has been interpreted as meaning that neutralization by these antibodies would be via the inhibition of cell surface interaction (42). However, our data indicate that this inference was not correct. V5 and E70 prevent ECM binding but not cell surface binding, while U4, which does not inhibit hemagglutination, has no effect on ECM binding but does block cell surface interactions. Therefore, hemagglutination inhibition, at least for these three monoclonal antibodies, actually correlates with inhibition of ECM binding.
As noted above, the canyon theory of rhinovirus neutralization is that the canyon's surface is largely inaccessible to antibodies, an arrangement that permits conservation of residues that may be required for host cell receptor recognition without the possibility of being recognized by the host's immune system. PVs may similarly protect the viral receptor attachment site, in that antibodies to the immunodominant sites as typified by V5 do not prevent attachment to the cell surface. In contrast to the type-specific nature of these immunodominant neutralizing antibodies, it might be possible, at least in theory, to generate more broadly cross-reactive L1 antibodies, if they were directed to conserved epitopes located on the floor and the walls of the cleft that may become better exposed during viral uncoating. Additionally, as with rhinoviruses (20, 36), small molecule inhibitors might be found that interact with the receptor-binding region of the capsid. Such compounds would have the potential to be broadly cross-protective.
Acknowledgments
This research was supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research.
We thank Neil Christensen (Department of Pathology, College of Medicine, Pennsylvania State University, Hershey, PA) for the antibodies H16.V5, H16.U4, and H16.E70. We are grateful to Ted Pierson (National Institute of Allergy and Infectious Diseases, NIH) for helpful discussions on the design and interpretation of the ELISA and neutralization data.
Footnotes
Published ahead of print on 6 June 2007.
REFERENCES
- 1.Baseman, J. G., and L. A. Koutsky. 2005. The epidemiology of human papillomavirus infections. J. Clin. Virol. 32(Suppl. 1):S16-S24. [DOI] [PubMed] [Google Scholar]
- 2.Booy, F. P., R. B. Roden, H. L. Greenstone, J. T. Schiller, and B. L. Trus. 1998. Two antibodies that neutralize papillomavirus by different mechanisms show distinct binding patterns at 13 A resolution. J. Mol. Biol. 281:95-106. [DOI] [PubMed] [Google Scholar]
- 3.Bosch, F. X., A. Lorincz, N. Munoz, C. J. Meijer, and K. V. Shah. 2002. The causal relation between human papillomavirus and cervical cancer. J. Clin. Pathol. 55:244-265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bousarghin, L., A. Touze, P. Y. Sizaret, and P. Coursaget. 2003. Human papillomavirus types 16, 31, and 58 use different endocytosis pathways to enter cells. J. Virol. 77:3846-3850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buck, C. B., D. V. Pastrana, D. R. Lowy, and J. T. Schiller. 2004. Efficient intracellular assembly of papillomaviral vectors. J. Virol. 78:751-757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Buck, C. B., D. V. Pastrana, D. R. Lowy, and J. T. Schiller. 2005. Generation of HPV pseudovirions using transfection and their use in neutralization assays. Methods Mol. Med. 119:445-462. [DOI] [PubMed] [Google Scholar]
- 7.Carter, J. J., G. C. Wipf, S. F. Benki, N. D. Christensen, and D. A. Galloway. 2003. Identification of a human papillomavirus type 16-specific epitope on the C-terminal arm of the major capsid protein L1. J. Virol. 77:11625-11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carter, J. J., G. C. Wipf, M. M. Madeleine, S. M. Schwartz, L. A. Koutsky, and D. A. Galloway. 2006. Identification of human papillomavirus type 16 L1 surface loops required for neutralization by human sera. J. Virol. 80:4664-4672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Che, Z., N. H. Olson, D. Leippe, W. M. Lee, A. G. Mosser, R. R. Rueckert, T. S. Baker, and T. J. Smith. 1998. Antibody-mediated neutralization of human rhinovirus 14 explored by means of cryoelectron microscopy and X-ray crystallography of virus-Fab complexes. J. Virol. 72:4610-4622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen, X. S., R. L. Garcea, I. Goldberg, G. Casini, and S. C. Harrison. 2000. Structure of small virus-like particles assembled from the L1 protein of human papillomavirus 16. Mol. Cell 5:557-567. [DOI] [PubMed] [Google Scholar]
- 11.Christensen, N. D., N. M. Cladel, C. A. Reed, L. R. Budgeon, M. E. Embers, D. M. Skulsky, W. L. McClements, S. W. Ludmerer, and K. U. Jansen. 2001. Hybrid papillomavirus L1 molecules assemble into virus-like particles that reconstitute conformational epitopes and induce neutralizing antibodies to distinct HPV types. Virology 291:324-334. [DOI] [PubMed] [Google Scholar]
- 12.Christensen, N. D., J. Dillner, C. Eklund, J. J. Carter, G. C. Wipf, C. A. Reed, N. M. Cladel, and D. A. Galloway. 1996. Surface conformational and linear epitopes on HPV-16 and HPV-18 L1 virus-like particles as defined by monoclonal antibodies. Virology 223:174-184. [DOI] [PubMed] [Google Scholar]
- 13.Christensen, N. D., R. Hopfl, S. L. DiAngelo, N. M. Cladel, S. D. Patrick, P. A. Welsh, L. R. Budgeon, C. A. Reed, and J. W. Kreider. 1994. Assembled baculovirus-expressed human papillomavirus type 11 L1 capsid protein virus-like particles are recognized by neutralizing monoclonal antibodies and induce high titres of neutralizing antibodies. J. Gen. Virol. 75:2271-2276. [DOI] [PubMed] [Google Scholar]
- 14.Christensen, N. D., C. A. Reed, N. M. Cladel, K. Hall, and G. S. Leiserowitz. 1996. Monoclonal antibodies to HPV-6 L1 virus-like particles identify conformational and linear neutralizing epitopes on HPV-11 in addition to type-specific epitopes on HPV-6. Virology 224:477-486. [DOI] [PubMed] [Google Scholar]
- 15.Colonno, R. J., P. L. Callahan, and W. J. Long. 1986. Isolation of a monoclonal antibody that blocks attachment of the major group of human rhinoviruses. J. Virol. 57:7-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Colonno, R. J., J. H. Condra, S. Mizutani, P. L. Callahan, M. E. Davies, and M. A. Murcko. 1988. Evidence for the direct involvement of the rhinovirus canyon in receptor binding. Proc. Natl. Acad. Sci. USA 85:5449-5453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Culp, T. D., L. R. Budgeon, and N. D. Christensen. 2006. Human papillomaviruses bind a basal extracellular matrix component secreted by keratinocytes which is distinct from a membrane-associated receptor. Virology 347:147-159. [DOI] [PubMed] [Google Scholar]
- 18.Culp, T. D., C. M. Spatz, C. A. Reed, and N. D. Christensen. 2007. Binding and neutralization efficiencies of monoclonal antibodies, Fab fragments, and scFv specific for L1 epitopes on the capsid of infectious HPV particles. Virology 361:435-446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Day, P. M., D. R. Lowy, and J. T. Schiller. 2003. Papillomaviruses infect cells via a clathrin-dependent pathway. Virology 307:1-11. [DOI] [PubMed] [Google Scholar]
- 20.Diana, G. D., D. C. Pevear, M. J. Otto, M. A. McKinlay, M. G. Rossmann, T. Smith, and J. Badger. 1989. Inhibitors of viral uncoating. Pharmacol. Ther. 42:289-305. [DOI] [PubMed] [Google Scholar]
- 21.Emini, E. A., P. Ostapchuk, and E. Wimmer. 1983. Bivalent attachment of antibody onto poliovirus leads to conformational alteration and neutralization. J. Virol. 48:547-550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Esko, J. D., and U. Lindahl. 2001. Molecular diversity of heparan sulfate. J. Clin. Investig. 108:169-173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Flamand, A., H. Raux, Y. Gaudin, and R. W. Ruigrok. 1993. Mechanisms of rabies virus neutralization. Virology 194:302-313. [DOI] [PubMed] [Google Scholar]
- 24.Giroglou, T., L. Florin, F. Schafer, R. E. Streeck, and M. Sapp. 2001. Human papillomavirus infection requires cell surface heparan sulfate. J. Virol. 75:1565-1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hangartner, L., R. M. Zinkernagel, and H. Hengartner. 2006. Antiviral antibody responses: the two extremes of a wide spectrum. Nat. Rev. Immunol. 6:231-243. [DOI] [PubMed] [Google Scholar]
- 26.He, R. T., B. L. Innis, A. Nisalak, W. Usawattanakul, S. Wang, S. Kalayanarooj, and R. Anderson. 1995. Antibodies that block virus attachment to Vero cells are a major component of the human neutralizing antibody response against dengue virus type 2. J. Med. Virol. 45:451-461. [DOI] [PubMed] [Google Scholar]
- 27.Joyce, J. G., J. S. Tung, C. T. Przysiecki, J. C. Cook, E. D. Lehman, J. A. Sands, K. U. Jansen, and P. M. Keller. 1999. The L1 major capsid protein of human papillomavirus type 11 recombinant virus-like particles interacts with heparin and cell-surface glycosaminoglycans on human keratinocytes. J. Biol. Chem. 274:5810-5822. [DOI] [PubMed] [Google Scholar]
- 28.Kirnbauer, R., F. Booy, N. Cheng, D. R. Lowy, and J. T. Schiller. 1992. Papillomavirus L1 major capsid protein self-assembles into virus-like particles that are highly immunogenic. Proc. Natl. Acad. Sci. USA 89:12180-12184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Klasse, P. J., and Q. J. Sattentau. 2001. Mechanisms of virus neutralization by antibody. Curr. Top. Microbiol. Immunol. 260:87-108. [DOI] [PubMed] [Google Scholar]
- 30.Klasse, P. J., and Q. J. Sattentau. 2002. Occupancy and mechanism in antibody-mediated neutralization of animal viruses. J. Gen. Virol. 83:2091-2108. [DOI] [PubMed] [Google Scholar]
- 31.Knossow, M., M. Gaudier, A. Douglas, B. Barrere, T. Bizebard, C. Barbey, B. Gigant, and J. J. Skehel. 2002. Mechanism of neutralization of influenza virus infectivity by antibodies. Virology 302:294-298. [DOI] [PubMed] [Google Scholar]
- 32.Liddington, R. C., Y. Yan, J. Moulai, R. Sahli, T. L. Benjamin, and S. C. Harrison. 1991. Structure of simian virus 40 at 3.8-A resolution. Nature 354:278-284. [DOI] [PubMed] [Google Scholar]
- 33.Lowy, D. R., and J. T. Schiller. 2006. Prophylactic human papillomavirus vaccines. J. Clin. Investig. 116:1167-1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Luo, M., G. Vriend, G. Kamer, I. Minor, E. Arnold, M. G. Rossmann, U. Boege, D. G. Scraba, G. M. Duke, and A. C. Palmenberg. 1987. The atomic structure of Mengo virus at 3.0 A resolution. Science 235:182-191. [DOI] [PubMed] [Google Scholar]
- 35.Mandel, B. 1976. Neutralization of poliovirus: a hypothesis to explain the mechanism and the one-hit character of the neutralization reaction. Virology 69:500-510. [DOI] [PubMed] [Google Scholar]
- 36.McKinlay, M. A., D. C. Pevear, and M. G. Rossmann. 1992. Treatment of the picornavirus common cold by inhibitors of viral uncoating and attachment. Annu. Rev. Microbiol. 46:635-654. [DOI] [PubMed] [Google Scholar]
- 37.Modis, Y., B. L. Trus, and S. C. Harrison. 2002. Atomic model of the papillomavirus capsid. EMBO J. 21:4754-4762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Orozco, J. J., J. J. Carter, L. A. Koutsky, and D. A. Galloway. 2005. Humoral immune response recognizes a complex set of epitopes on human papillomavirus type 6 L1 capsomers. J. Virol. 79:9503-9514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pastrana, D. V., C. B. Buck, Y. Y. Pang, C. D. Thompson, P. E. Castle, P. C. FitzGerald, S. Kruger Kjaer, D. R. Lowy, and J. T. Schiller. 2004. Reactivity of human sera in a sensitive, high-throughput pseudovirus-based papillomavirus neutralization assay for HPV16 and HPV18. Virology 321:205-216. [DOI] [PubMed] [Google Scholar]
- 40.Pastrana, D. V., R. Gambhira, C. B. Buck, Y. Y. Pang, C. D. Thompson, T. D. Culp, N. D. Christensen, D. R. Lowy, J. T. Schiller, and R. B. Roden. 2005. Cross-neutralization of cutaneous and mucosal papillomavirus types with anti-sera to the amino terminus of L2. Virology 337:365-372. [DOI] [PubMed] [Google Scholar]
- 41.Possee, R. D., G. C. Schild, and N. J. Dimmock. 1982. Studies on the mechanism of neutralization of influenza virus by antibody: evidence that neutralizing antibody (anti-haemagglutinin) inactivates influenza virus in vivo by inhibiting virion transcriptase activity. J. Gen. Virol. 58:373-386. [DOI] [PubMed] [Google Scholar]
- 42.Roden, R. B., A. Armstrong, P. Haderer, N. D. Christensen, N. L. Hubbert, D. R. Lowy, J. T. Schiller, and R. Kirnbauer. 1997. Characterization of a human papillomavirus type 16 variant-dependent neutralizing epitope. J. Virol. 71:6247-6252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Roden, R. B., H. L. Greenstone, R. Kirnbauer, F. P. Booy, J. Jessie, D. R. Lowy, and J. T. Schiller. 1996. In vitro generation and type-specific neutralization of a human papillomavirus type 16 virion pseudotype. J. Virol. 70:5875-5883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rossmann, M. G., E. Arnold, J. W. Erickson, E. A. Frankenberger, J. P. Griffith, H. J. Hecht, J. E. Johnson, G. Kamer, M. Luo, A. G. Mosser, et al. 1985. Structure of a human common cold virus and functional relationship to other picornaviruses. Nature 317:145-153. [DOI] [PubMed] [Google Scholar]
- 45.Selinka, H. C., T. Giroglou, T. Nowak, N. D. Christensen, and M. Sapp. 2003. Further evidence that papillomavirus capsids exist in two distinct conformations. J. Virol. 77:12961-12967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Selinka, H. C., T. Giroglou, and M. Sapp. 2002. Analysis of the infectious entry pathway of human papillomavirus type 33 pseudovirions. Virology 299:279-287. [DOI] [PubMed] [Google Scholar]
- 47.Smith, T. J., N. H. Olson, R. H. Cheng, E. S. Chase, and T. S. Baker. 1993. Structure of a human rhinovirus-bivalently bound antibody complex: implications for viral neutralization and antibody flexibility. Proc. Natl. Acad. Sci. USA 90:7015-7018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Trus, B. L., R. B. Roden, H. L. Greenstone, M. Vrhel, J. T. Schiller, and F. P. Booy. 1997. Novel structural features of bovine papillomavirus capsid revealed by a three-dimensional reconstruction to 9 A resolution. Nat. Struct. Biol. 4:413-420. [DOI] [PubMed] [Google Scholar]
- 49.Varghese, R., Y. Mikyas, P. L. Stewart, and R. Ralston. 2004. Postentry neutralization of adenovirus type 5 by an antihexon antibody. J. Virol. 78:12320-12332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Virgin, H. W., M. A. Mann, and K. L. Tyler. 1994. Protective antibodies inhibit reovirus internalization and uncoating by intracellular proteases. J. Virol. 68:6719-6729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vlodavsky, I. 1999. Preparation of extracellular matrices produced by cultured corneal endothelial and PF-HR9 endodermal cells. In J. S. Bonifacino et al. (ed.), Current protocols in cell biology, suppl. 1, unit 10.4. John Wiley and Sons, Philadelphia, PA. [DOI] [PubMed]
- 52.Wang, X., Z. Wang, N. D. Christensen, and J. Dillner. 2003. Mapping of human serum-reactive epitopes in virus-like particles of human papillomavirus types 16 and 11. Virology 311:213-221. [DOI] [PubMed] [Google Scholar]
- 53.Wang, Z., N. Christensen, J. T. Schiller, and J. Dillner. 1997. A monoclonal antibody against intact human papillomavirus type 16 capsids blocks the serological reactivity of most human sera. J. Gen. Virol. 78:2209-2215. [DOI] [PubMed] [Google Scholar]
- 54.White, W. I., S. D. Wilson, F. J. Palmer-Hill, R. M. Woods, S. J. Ghim, L. A. Hewitt, D. M. Goldman, S. J. Burke, A. B. Jenson, S. Koenig, and J. A. Suzich. 1999. Characterization of a major neutralizing epitope on human papillomavirus type 16 L1. J. Virol. 73:4882-4889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhuge, W., F. Jia, G. Mackay, A. Kumar, and O. Narayan. 2001. Antibodies that neutralize SIV(mac)251 in T lymphocytes cause interruption of the viral life cycle in macrophages by preventing nuclear import of viral DNA. Virology 287:436-445. [DOI] [PubMed] [Google Scholar]