

Abstract
The Kaposi's sarcoma-associated herpesvirus (KSHV) delayed-early K-bZIP promoter contains an ORF50/Rta binding site whose sequence is conserved with the ORF57 promoter. Mutation of the site in the full-length K-bZIP promoter reduced Rta-mediated transactivation by greater than 80%. The K-bZIP element contains an octamer (Oct) binding site that overlaps the Rta site and is well conserved with Oct elements found in the immediate-early promoters of herpes simplex virus type 1(HSV-1). The cellular protein Oct-1, but not Oct-2, binds to the K-bZIP element in a sequence-specific fashion in vitro and in vivo and stimulates Rta binding to the promoter DNA. The coexpression of Oct-1 enhances Rta-mediated transactivation of the wild type but not the mutant K-bZIP promoter, and Oct-1 and Rta proteins bind to each other directly in vitro. Mutations of Rta within an amino acid sequence conserved with HSV-1 virion protein 16 eliminate Rta's interactions with Oct-1 and K-bZIP promoter DNA but not RBP-Jk. The binding of Rta to both Oct-1 and DNA contributes to the transactivation of the K-bZIP promoter and viral reactivation, and Rta mutants deficient for both interactions are completely debilitated. Our data suggest that the Rta/Oct-1 interaction is essential for optimal KSHV reactivation. Transfections of mouse embryo fibroblasts and an endothelial cell line suggest cell-specific differences in the requirement for Oct-1 or RBP-Jk in Rta-mediated transactivation of the K-bZIP promoter. We propose a model in which Rta transactivation of the promoter is specified by the combination of DNA binding and interactions with several cellular DNA binding proteins including Oct-1.
Kaposi's sarcoma (KS)-associated herpesvirus (KSHV), or human herpesvirus 8, is the etiologic agent responsible for the endothelial cell neoplasm KS, the B-cell lymphoproliferative disorder primary effusion lymphoma (PEL), and some forms of multicentric Castleman's disease (14, 17, 27, 34, 61, 76). Like other gammaherpesviruses, KSHV is lymphotropic (1, 5, 6, 20, 30, 35, 98) and replicates both latently and lytically (59, 70, 71, 105). Following primary infection, KSHV establishes latency in CD19+ B cells (and possibly other cells types) (7). Reactivation of the virus from latency is critical for the progression to KS, allowing extralymphoid dissemination to endothelial cells and the expression of lytic cycle viral oncogenes (9, 15, 18, 19, 21-23, 33, 38, 48, 66, 75, 87). Treatment of patients with ganciclovir, an antiviral drug that blocks lytic viral replication, reduces the risk of KS development (58), highlighting the importance of reactivation in progression to KS. In clinical KS samples, the virus is predominantly latent in lymphatic endothelial cells (8, 81, 83).
We and others have previously shown that the KSHV protein called “replication and transcriptional activator” (Rta) (encoded by open reading frame 50 [ORF50]) is both necessary and sufficient to induce the entire viral lytic cycle in tissue culture models of latency (29, 53, 54, 84, 102). Mechanistic studies have demonstrated that Rta transactivates transcription in a promoter-specific fashion. ORF50/Rta selects promoters for transactivation by binding promoter DNA independently or in combination with cellular transcription factors. Tetramers of Rta are sufficient to transactivate KSHV promoters and stimulate complete viral reactivation from latency (11).
Among Rta's DNA binding sites, the PAN and kaposin promoters share a 16-bp core sequence that is also found in ori-Lyt (L) (16, 37, 79, 96). That DNA element differs significantly from an Rta-responsive element (RRE) conserved in the ORF57/Mta and K-bZIP (also known as K8 and RAP) promoters (52) and a third element in the viral interleukin-6 promoter (19). The relative strengths of binding of Rta to these promoter elements have been estimated as PAN > kaposin > ORF57 > viral interleukin-6 (77), with a dissociation constant for binding to the PAN element in the nanomolar range (79).
Cellular transcription factors that contribute to promoter-specific Rta transactivation include recombination signal binding protein (RBP)-Jk, octamer 1 (Oct-1), CAAT/enhancer binding protein alpha (C/EBPα), c-Jun, and Sp1 (13, 73, 91-93, 97, 104). Genetic and biochemical experiments demonstrate that RBP-Jk is required for Rta-mediated transactivation of the promoters from the KSHV genes ORF57/Mta, ORF6/single-stranded DNA binding protein, viral G-protein-coupled receptor, K-bZIP/RAP, and Rta itself (autoactivation) (46-48, 52, 97). A central regulatory role of the RBP-Jk/Rta interaction was demonstrated by the inhibition of KSHV reactivation in murine embryo fibroblasts null for RBP-Jk (47).
Rta promotes DNA binding of RBP-Jk, a mechanism that is fundamentally different from that established for other RBP-Jk-activating proteins, including Notch intracellular domain and Epstein-Barr virus EBNA-2 (13). Stimulation of RBP-Jk DNA binding by Rta requires intact DNA binding sites for both proteins in the RRE of the ORF57/Mta promoter and ternary complex formation between the three molecules (Rta/RBP-Jk/DNA) in vitro. In infected PEL cells, chromatin immunoprecipitations (ChIPs) showed that RBP-Jk is virtually undetectable on the ORF57 promoter during latency. However, during viral reactivation, RBP-Jk is significantly enriched on the ORF57 promoter in an Rta-dependent fashion (13).
In vivo, the stimulation of RBP-Jk DNA binding by Rta extended to a series of additional viral and cellular promoters (13), suggesting a general mechanism by which Rta controls KSHV reactivation. One such promoter was that of the KSHV K-bZIP gene, which shares many other molecular features with the ORF57 promoter. The ORF57/Mta and K-bZIP promoters are transactivated to similar magnitudes by ORF50/Rta in transient reporter assays and conserve critical cis-acting transcriptional control elements (52). The TATA-proximal 100 bp of both promoters share three identical sequences: an identical 12-bp partial palindrome (AACAATAATGTT), a 6-bp binding site for the Aml-1a protein, and an 11-bp TATA-box-containing sequence (52). In the ORF57/Mta promoter, the 12-bp palindrome is part of Rta's direct binding site (13, 52); in both promoters, the palindrome contains an A/T trinucleotide (52) that is one unit of a phased repeat of A/T trinucleotides ([A/T]3-N17-[A/T]3) that extends over 93 bp in K-bZIP and 62 bp in ORF57/Mta (49). Phased A/T trinucleotide repeats have been proposed to facilitate the binding of Rta multimers over long stretches of KSHV DNA (49).
Despite these similarities, other observations suggest that the details of Rta-mediated transactivation differ for the ORF57/Mta and K-bZIP promoters. For example, while mutation of the 12-bp palindrome abolishes Rta-mediated transactivation of the ORF57/Mta promoter, the identical mutation severely reduces, but does not eliminate, Rta-mediated transactivation of K-bZIP (52). In the ORF57/Mta promoter, the palindrome overlaps the essential RBP-Jk binding site (13, 46), but an RBP-Jk site is not conserved at a similar position of the K-bZIP promoter (52). The observation that partially pure Rta expressed in insect cells forms a different spectrum of complexes with DNAs containing each of the palindromes suggested that Rta bound to the elements in conjunction with different sets of cellular proteins (52). Thus, many different trans-acting factors and cis elements have been proposed to be crucial for Rta-mediated transactivation of the K-bZIP promoter. We are specifically interested in determining the role of the conserved partial palindrome in this process.
Here, we present evidence that the K-bZIP palindrome contains a binding site for the cellular Oct protein family. Rta transactivation of the K-bZIP promoter is potentiated by coexpressing Oct-1 and requires the Oct site in the palindrome for this effect. The Rta protein contains a domain with homology to two proteins that bind to homeodomain proteins, herpes simplex virus type 1 (HSV-1) virion protein 16 (VP16), and Saccharomyces cerevisiae MATα; point mutations of conserved amino acids eliminate direct interactions of Rta with Oct-1 and DNA and reduce or eliminate Rta-mediated transactivation of the K-bZIP promoter and viral reactivation. We also present evidence that the transactivation of the K-bZIP promoter by Rta is regulated by additional cellular factors, including RBP-Jk, in a cell-specific fashion.
MATERIALS AND METHODS
Plasmids.
All plasmids were propagated as described previously (13). Plasmids not described below have been described previously (13, 46, 52-54).
pcDNA3-50ΔOIDa, expressing Rta with a deletion of amino acids (aa) 134 to 150, was cloned using PCR with the following primers to amplify the Rta C terminus with an SphI site introduced at aa 151: GCGGCATGCCACGAAATATACACTGAAATG (forward) and GATCTCTAGCATTTAGGTGACACTATAGAATAGA (reverse). The product was TOPO-TA cloned into pCR2.1TOPO according to the manufacturer's directions (Invitrogen). The SphI/AatII fragment of that plasmid was swapped with the same fragment of pMalc2X-FLc50 (13) to generate plasmid pMalc2X-50ΔOIDa. The EagI/NcoI fragment of that plasmid was then swapped with the same fragment of pGEM3-FLc50. The full-length insert of that plasmid was subcloned into pcDNA3 between the EcoRI and EcoRV sites.
pcDNA3-50ΔOIDb, expressing Rta with a deletion of aa 584 through 600, was cloned using PCR with the following primers to amplify the N terminus of Rta with a PspOMI site introduced at aa 583: GCAGTTGCCAACAGTAGTCCCT (forward) and GAAGGGCCCGGCGGGGGTAACGACAGTAG (reverse). The product was TOPO-TA cloned into pcR2.1 TOPO according to the manufacturer's directions (Invitrogen). The PspOMI/SacI fragment of that plasmid was then swapped with the same fragment of pRSET0.8 (54) to generate pRSET0.8-ΔOIDb. The BstZI7I/SexAI fragment of the plasmid was then swapped with the same fragment of pGEM3FLc50 to generate pGEM3-50ΔOIDb. The full-length insert of that plasmid was subcloned into pcDNA3 between the EcoRI and EcoRV sites.
Point mutations, or insertions, of Rta were made in vectors pcDNA3 and pMalc2x by site-directed mutagenesis according to the manufacturer's directions (Stratagene) using the following primers (changes are underlined): 5′-GCAGCGGGGTGAGCGCGCCTCCAGCCATATG (forward) and 5′-CATATGGCTGGAGGCGCGCTCACCCCGCTGC (reverse) for ORF50-L136A, 5′-GGGGTGAGCCTGCCTCCAGCCGCATGTAAGCTACTACACGAAATATAC (forward) and 5′-GTATATTTCGTGTAGTAGCTTACATGCGGCTGGAGGCAGGCTCACCCC (reverse) for ORF50-I140A, 5′-GGGGTGAGCCTGCCTCCAGCCATATGTAAGGCACTACACGAAATATAC (forward) and 5′-GTATATTTCGTGTAGTGCCTTACATATGGCTGGAGGCAGGCTCACCCC (reverse) for ORF50-L143A, 5′-GGGGTGAGCCTGCCTCCAGCCATATGTAAGCTAGCACACGAAATATAC (forward) and 5′-GTATATTTCGTGTGCTAGCTTACATATGGCTGGAGGCAGGCTCACCCC (reverse) for ORF50-L144A, 5′-GCCATATGTAAGGCGGACCCTAGGCTACTACACGAAATATAC (forward) and 5′-GTATATTTCGTGTAGTAGCCTAGGGTCCGCCTTACATATGGC (reverse) for ORF50-ADPRins, 5′-GGGGTGAGCCTGCCTCCAGCCGCATGTAAGGCAGCACACGAAATATAC (forward) and 5′-GTATATTTCGTGTGCTGCCTTACATGCGGCTGGAGGCAGGCTCACCCC (reverse) for ORF50-ILL140AAA, and 5′-AGCCTGCCTCCAGCCATATCTAAGCTACTACACGAAATA (forward) and 5′-TATTTCGTGTAGTAGCTTAGATATGGCTGGAGGCAGGCT (reverse) for ORF50-C141S.
pV5-ORF50-ILL140AAA was cloned by inserting the BbvCI/PmeI fragment from pV5-FLc50 (51) into BbvCI/EcoRV-digested pcDNA3-ORF50 ILL140AAA. pMal-50-ILL140AAA was constructed by swapping the EagI/NcoI fragment from pcDNA3-ORF50-ILL140AAA with pMal-50ΔSTAD.
pK-bZIPm34-GL3b was cloned using PCR with plasmid pK-bZIP-GL3basic (53) as a template, a vector-specific reverse primer, and a forward primer that introduced a SacI site replacing positions −235 to −243 (“mutant 34”) (5′-GCGGAGCTCCCATATGCCGAGACTGAAGT-3′). The product was cloned into pGL3basic between the SacI and NcoI sites to generate pK-bZIPm34-3′-GL3b. The 5′ end of the promoter was cloned using PCR with a vector-specific forward primer and a reverse primer that introduced a SacI site replacing positions −235 to −243 (5′-GCGGAGCTCATTGGTGCAGCT-3′). The product was cloned into pGL3basic at the SacI site to generate pK-bZIPm34-5′-GL3b. The insert from pK-bZIPm34-5′-GL3b was then subcloned into pK-bZIPm34-3′-GL3b at the SacI site.
pK-bZIPm1234-GL3b was cloned exactly as described above for pK-bZIPm34-GL3b with the exception that plasmid pK-bZIPm12-GL3b (53) was used as the template in PCRs.
pCG-Oct-1 expresses human Oct-1 from the human cytomegalovirus promoter and was a gift of W. Herr (85). pBS KS II-Oct-1 expresses Oct-1 from the bacteriophage T7 promoter and was cloned by ligating the BamHI/XbaI fragment from pCG-Oct-1 into pBluescript II KS digested with the same enzymes. pGST-Oct-1 was cloned by PCR using primers that introduced BamHI sites into the PCR product and pCG-Oct-1 as a template. The resulting product was digested with BamHI and cloned at the BamHI site of pGex5x-1.
pcDNA3-C/EBPα was a gift of Daniel Tenen (65).
Cell culture, transfections, and reporter assays.
Akata-31 B cells (a gift of P. J. Farrell, Ludwig Institute for Cancer Research) (32), PEL cells (HHB-2 and BCBL-1), and human endothelial SLK cells were maintained in RPMI 1640 containing 10% fetal bovine serum, 2 mM l-glutamine and penicillin-streptomycin, and 0.055 mM β-mercaptoethanol. PEL and Akata-31 cells were transfected by electroporation, as previously described (54), at 150 V and 200 V, respectively.
OT11 cells were maintained as described previously (13).
Oct-1−/− Babe cells, Oct-1−/− Babe-hOct cells (gifts of Dean Tantin and Phillip Sharp) (62, 94), and HeLa cells were maintained in Dulbecco's modified Eagle media supplemented with 10% fetal bovine serum and 2 mM L-glutamine and penicillin-streptomycin.
HeLa, SLK, OT11, Oct-1−/− Babe, and Oct-1−/− Babe-hOct cells were transfected using Transit-LT1 according to the manufacturer's directions (Mirus).
In all transfections, pcDNA3 was used as filler plasmid to normalize the total amount of transfected DNA. pcDNA3.1LacZ was cotransfected to normalize for transfection efficiency.
Luciferase and β-galactosidase assays were performed as described previously for BJAB cells (53), except that cells were lysed in 0.125 ml of reporter lysis buffer.
Immunofluorescence.
For HeLa cells, immunofluorescence was performed exactly as described previously for CV-1 cells (36), with anti-Rta serum (52) diluted 1:300 and a fluorescein isothiocyanate-conjugated anti-rabbit secondary antibody (ICN).
Protein expression and purification.
Glutathione S-transferase (GST)-Oct-1 and GST-RBP-Jk proteins were expressed and purified exactly as described previously (13), with the exception that GST-Oct-1 bacteria were treated with 0.5 U/10 ml DNase for 10 min at 37°C prior to sonication and purification.
His6-N50 (aa 1 to 272) was expressed and purified exactly as described previously (13).
pMal-ORF50ΔSTAD was expressed and purified exactly as described previously for pMal-FLc50 (12).
Nuclear extracts were prepared exactly as described previously (13).
EMSAs.
Electrophoretic mobility shift assays (EMSAs) were performed exactly as described previously (13) by using the following annealed oligonucleotides labeled with 32P: GATCTATTTGTGAAACAATAATGATTAAAGGGGA (forward) and GATCTCCCCTTTAATCATTATTGTTTCACAAATA (reverse) (K-bZIP WT), GATCTATTTGTGACTGCAGTCCGGATAAAGGGGA (forward) and GATCTCCCCTTTATCCGGACTGCAGTCACAAATA (reverse) (K-bZIPm12), GATCTCACCAATGGGCTCCCGTATGCCGAGACTA (forward) and GATCTAGTCTCGGCATACGGGAGCCCATTGGTG (reverse) (K-bZIPm34), GATCTAAACTGGATTGCGCAATAGGAA (forward) and GATCTTCCTATTGCGCAATCCAGTTTA (reverse) (CAAT box), and ORF57/Mta RRE, as described previously (12).
ChIPs.
BCBL-1 cells were induced with 20 ng/ml tetradecanoyl phorbol acetate (TPA) for 40 h or left untreated and then analyzed by ChIP exactly as described previously (13). Chromatin from KSHV-positive BCBL-1 (PEL) cells was harvested from cells induced into the lytic cycle (with TPA) or left uninduced (without TPA). After cross-linking, complexes were immunoprecipitated with 2 μg of Oct-1 (Santa Cruz) antibody, 5 μl of Rta-specific antibody (52), or 2 μg of control rabbit immunoglobulin G (IgG).
ChIP products were analyzed by real-time PCR exactly as previously described (13), with the following primer pairs: GGTGGAGAGTATACGCAACTGCAAC (forward) and GGTTATAGTCGACAACGGAGGAAATAC (reverse) (K-bZIP) GCGTGAGATGTGACCAATAGGGTG (forward) and CGGCATATGGACTAGTTCTAGAGTG (reverse) (Rta), and CGCCTAATAGCTGCTGCTACGG (forward) and TGCATCAGCTGCCTAACCCAG (reverse) (K6).
Molecular beacon sequences used for amplicon detection were as follows (target sequences are underlined): CGCTCGTCTGTGCCTGCGTTAACTTCCGAGCG (pK-bZIP), CGCTCGGTGGGAAGACGATGGGGGAAATGTGCGAGCG (pRta), and CCCCTCCCACCCACCGCCCGTCCAAATTCGGAGGGG (K6).
The ΔΔCT method was used to calculate the enrichment (n-fold) of transcription factor binding in the presence of TPA (see reference 12).
Quantitative RT-PCR.
Total RNA was harvested from untreated or TPA-treated BCBL-1 cells 18 h postinduction exactly as described previously (37). Quantitative reverse transcription-PCR (RT-PCR) was performed with an iScript one-step RT-PCR kit with SYBR green (Bio-Rad) using 5 to 500 ng of RNA according to the manufacturer's recommendations. PCR primers were designed using Primer 3 software (http://frodo.wi.mit.edu) and have the following sequences: 5′-CCAGTGACCAGACGGCAA (forward) and 5′-GAGCTCTAGGCACGTTAAATT (reverse) (Nut-1), 5′-CCAGCAAGAGCACAAGAGGA (forward) and 5′-GGAGATTCAGTGTGGTGGG (reverse) (GAPDH [glyceraldehyde-3-phosphate dehydrogenase]), and 5′-GCCCAGCTCTAATGAGCAAC (forward) and 5′-AAGAGAGCCACCAGAAGCAA (reverse) (Oct-1). Products were quantitated with the ΔΔCT method using GAPDH to correct for input RNA.
GST pull-down assays.
One milliliter of crude Escherichia coli lysate was incubated with 30 μl of preswollen glutathione-Sepharose beads (1:1 [wt/vol] in 1× NETN+ [20 mM Tris {pH 7.5}, 100 mM NaCl, 1 mM EDTA, 0.5% Nonidet P-40]) supplemented with protease inhibitors (Sigma) at 4°C for 2 h. Beads were washed three times in 1× NETN+ to remove unbound proteins. Wild-type (WT) and mutant Rta were expressed in rabbit reticulocyte lysates (RRLs) (TnT-coupled transcription/translation system) in the presence of l-[35S]methionine to label proteins according to the manufacturer's directions (Promega). A total of 0.5 μl was put aside for visualization as 5% input. Ten microliters of each programmed RRL was mixed with the bead-bound GST fusion protein in 250 μl 1× NETN+ and then incubated for 2 h at 4°C with rotation on a nutator (nutation). Alternatively, nuclear extract (100 μg) from BCBL-1 cells treated with TPA or left untreated was incubated with equivalent amounts of bead-bound GST or GST-Oct-1. Complexes were washed extensively, beads were boiled in 2× Laemmli buffer, and bound proteins were displayed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). 35S-labeled proteins were visualized by autoradiography after amplifying signals in 0.5 M salicylic acid.
Viral reactivation assays.
Viral reactivation assays were done as described previously (54), with the following modifications. BCBL-1 cells were electroporated in duplicate with the indicated plasmids and harvested 72 h postelectroporation. Approximately 2.5 × 105 cells were adhered to poly-l-lysine-coated glass slides within 1-in.-diameter circles drawn with a Pap pen (Beckman Coulter). Proteins were detected with mouse anti-V5 serum (diluted 1:200; Bethyl), rabbit anti-Rta serum, rabbit anti-K-bZIP serum (1:300 dilution; a gift of Don Ganem), mouse anti-K8.1 (52) (1:300), tetramethyl rhodamine isocyanate-conjugated anti-rabbit, and fluorescein isothiocyanate-conjugated anti-mouse (1:300 dilution) antibodies. At least 500 cells were scored by visual inspection for the percentage of Rta-positive cells that were also K8.1 positive (Rta positive/K8.1 positive divided by Rta positive) or the percentage of V5-positive cells that were also K-bZIP positive. When anti-Rta serum was used, results from the empty vector transfections (spontaneous reactivation) were subtracted from those of each WT or mutant Rta transfection.
RESULTS
A K-bZIP promoter mutation that severely affects transcription eliminates DNA binding of at least three nuclear proteins from PEL cells.
We previously showed (13) that recombinant ORF50/Rta protein binds to the A/T-rich palindrome from the ORF57/Mta promoter (Fig. 1A, top). The binding of Rta to the palindrome independently of cellular proteins is insufficient for the transactivation of the ORF57 promoter but requires RBP-Jk binding to its overlapping site (13, 46). The palindrome and juxtaposed sequences of the Mta promoter are nearly identical in the K-bZIP promoter (Fig. 1A) (52). Substitution mutations of the palindrome (“K-bZIPm12”) (Fig. 1A) in the full-length K-bZIP promoter (position −722 from the delayed-early start site) reduced Rta-mediated transactivation by 70% in CV-1 cells (52). However, the K-bZIP palindrome lacks an RBP-Jk site that overlaps the Rta binding site.
FIG. 1.

A transcriptional mutation of the K-bZIP promoter eliminates DNA binding of the cellular protein Oct-1. (A) The RRE of the KSHV ORF57/Mta promoter is homologous to the K-bZIP promoter. Alignment of indicated DNA sequences of the top strands of the ORF57/Mta and K-bZIP promoters is shown. cis recognition elements for DNA binding proteins are indicated by open boxes; the Oct site was predicted using the TransFac database (99). A previously described substitution mutation of the K-bZIP promoter (“pK-bZIPm12”) is indicated by gray boxes (52). (B) A transcriptional mutation of the K-bZIP promoter eliminates DNA binding of at least three nuclear proteins from PEL cells. The indicated K-bZIP DNAs (sequences are shown in A) were end labeled with 32P and incubated with increasing amounts of nuclear extracts (N.E.) from TPA-treated BCBL-1 cells. The migration of the free DNA alone is shown in the lanes marked “0.” Complexes labeled with 1, 2, and * form only on the WT but not on mutant DNAs. (C) The K-bZIP promoter. Shown is a schematic of the K-bZIP promoter fragment used in all reporter assays. Established or predicted cis-regulatory elements are indicated on the center line by the letters O (octamer), R (RBP-Jk/CSL) (97), A (AP-1) (91), and C (C/EBP) (92, 93). In the expanded view, open boxes indicate cis elements, and gray boxes indicate substitution mutations. Numbers refer to the 5′-most base pairs of particular indicated features. The triangles represent the positions of each A/T trinucleotide in the phased repeat conforming to the consensus [A/T]3-N17-[A/T]3 or [A/T]3-N7-[A/T]3-N7-[A/T]3, in which N is any nucleotide (49). The arrows indicate positions of IE or delayed-early (DE) transcriptional start sites, with genomic coordinates listed below the arrows (95). (D) Alignment of established or predicted Oct sites. The predicted Oct elements in the K-bZIP promoter are compared to those previously described for the Rta (73), K1 (10, 88), and HSV IE (26, 55-57) promoters and the consensus Oct site (101). (E) Oct-1 from PEL cells binds to the K-bZIP WT12 promoter element. The indicated K-bZIP DNAs (sequences are shown in A) were end labeled with 32P and incubated with increasing amounts of nuclear extracts from HH-B2 cells treated with (+) or without (−) TPA. The migration of the free DNA alone is shown in the lanes marked “0.” The Oct-1 or Oct-2 antibody was added to the indicated reaction mixtures. A shorter exposure corresponding to the portion of the gel indicated by the vertical bar is shown in the bottom panel. The Oct-1/DNA complex is indicated.
The 25-bp ORF57/Mta DNA (Fig. 1A) forms six complexes with nuclear extract proteins from PEL cells in EMSAs (13). To determine whether the K-bZIP promoter DNA (Fig. 1A) also bound to multiple proteins, we performed EMSAs using nuclear extracts from TPA-treated BCBL-1 (PEL) cells. As shown in Fig. 1B, at least seven protein complexes bound to WT K-bZIP DNA in a dose-dependent fashion. The 10-bp substitution mutation (m12) (Fig. 1A) of the DNA eliminates three of the major complexes (Fig. 1B). The complex labeled * in Fig. 1B is the lower band of a doublet; the top band was unaffected by the mutation. The identity of the lower band was investigated further.
Oct-1 and KSHV Rta bind to the delayed-early K-bZIP promoter in vivo and in vitro.
Sequence analysis of the K-bZIP DNA by searching the TransFac database with high stringency (99) revealed a putative binding site for the cellular Oct protein family overlapping the Rta and C/EBPα binding sites at positions −72 to −60 of the K-bZIP promoter (called site “12”) (Fig. 1C). A second Oct site was predicted at positions −241 to −228 (called site “34”).
Both Oct sites are shown in the context of the full-length K-bZIP promoter in Fig. 1C. Site 12 also overlaps a phased repeat of A/T trinucleotides previously identified as being an Rta binding site by Liao et al. (49) (Fig. 1C, triangles). Site 12 is also located in close apposition to AP-1 and RBP-Jk binding sites that regulate activity of the promoter (13, 91, 97). Site 34 is distal to all of these elements. Oct sites 12 and 34 are similar, but not identical, to other Oct binding sites previously described in the Rta and K1 promoters (Fig. 1D) (9, 10, 73, 88). K-bZIP Oct site 12 is more homologous to the Oct-1 elements found in HSV-1 immediate-early (IE) promoters (Fig. 1D).
To determine whether Oct proteins from infected cells could bind to the K-bZIP Oct elements, we performed EMSAs using nuclear extracts from HH-B2 PEL cells and monospecific antibodies for Oct-1 or Oct-2. Without the addition of the antibodies, extracts from latent (without TPA) and reactivating (with TPA) HH-B2 cells formed multiple complexes with the WT K-bZIP promoter element (Fig. 1E, lanes 5 and 12), similar to data shown in Fig. 1B. The addition of the Oct-1-specific antibody to the EMSAs prior to the addition of the labeled DNA caused a dose-dependent elimination of one of the protein-DNA complexes (“Oct-1/DNA”) (Fig. 1E, lanes 6 to 8 and 13 to 15). This complex did not form on the K-bZIP mutant 12 probe (Fig. 1B, asterisk, and E, lanes 2 and 3). Furthermore, the addition of the Oct-2-specific antibody did not eliminate any of the complexes (Fig. 1E, lanes 9 to 11 and 16 to 18). Interestingly, the Oct-1/DNA complex was enriched following the addition of TPA to the cells (Fig. 1E, compare lanes 13 to 15 with 6 to 9), suggesting that Oct-1 is induced during KSHV reactivation. Indeed, real-time RT-PCR analyses demonstrated that the Oct-1 transcript was induced 8.2-fold, relative to GAPDH, following the addition of TPA to BCBL-1 cells (the Nut-1/PAN transcript was induced 31.8-fold) (not shown). Therefore, Oct-1 binds to the K-bZIP promoter in a sequence-specific fashion, corresponding to strong transcriptional activation by Rta. We have been unable to detect a similar Oct-1 interaction with the putative WT34 Oct element from the K-bZIP promoter using PEL extracts and EMSAs. To confirm that Oct-1 binds to the K-bZIP promoter in infected cells, we performed ChIPs using the Oct-1-specific antibody and chromatin purified from untreated or TPA-induced BCBL-1 cells. Chromatin-immunoprecipitated DNA was detected by quantitative real-time PCR using primers that amplified K-bZIP Oct site WT12 or the Oct site from the ORF50/Rta promoter (71). As shown in Table 1, Oct-1 is enriched on both the K-bZIP and Rta promoters during KSHV reactivation about 7- and 17-fold, respectively. As a positive control, the Rta-specific antisera showed that Rta is also enriched on both promoters. As negative controls, the K6 ORF was not chromatin immunoprecipitated using either antiserum, and the IgG antiserum failed to immunoprecipitate any DNA. This proved that both Oct-1 and Rta bind to the K-bZIP promoter during KSHV reactivation in vivo.
TABLE 1.
Oct-1 and Rta are enriched on viral promoters during reactivation
| Target DNA | Fold enrichment for immunoprecipitating antibodya
|
||
|---|---|---|---|
| Anti-Oct-1 | Anti-Rta | Anti-IgG | |
| K-bZIP promoter | 7.4 | 6.7 | 1.2 |
| Rta promoter | 17.4 | 26.7 | 0.6 |
| K6 ORF | 0.8 | 1.3 | 0.8 |
ChIPs, as described in Materials and Methods, are represented as enrichment (n-fold) of Oct-1 or Rta DNA binding in the presence of TPA.
To confirm that Oct-1 binds with sequence specificity to the K-bZIP promoter, we repeated the EMSAs using recombinant Oct-1 expressed as a GST fusion protein in E. coli. As shown in Fig. 2A, GST-Oct-1 bound to the K-bZIP WT12 promoter element in a dose-dependent fashion but did not bind to the m12 element. Importantly, the GST moiety alone was unable to bind to either element (data not shown).
FIG. 2.

Purified Rta and Oct-1 bind to the K-bZIP promoter. (A) Recombinant Oct-1-GST binds to the WT, but not mutant, K-bZIP promoter element 12. Increasing amounts of purified GST-Oct-1 were tested in EMSAs as described in the legend to Fig. 1B. (B) Recombinant ORF50/Rta binds to the WT, but not mutant, K-bZIP promoter element 12. His6-N50 (Rta aa 1 to 272) was preincubated with the indicated unlabeled competitor DNAs, and 32P-end-labeled ORF57 RRE DNA was the added to the mix (sequences of all DNAs are shown in Fig. 1A). Complexes were analyzed by EMSA. (C) Oct-1 enhances DNA binding of ORF50/Rta to K-bZIP element 12. Individual proteins, as indicated (lanes 2, 3, and 4), were tested at suboptimal concentrations by EMSA as described above (A). Alternatively, a constant amount of MBP-ORF50ΔSTAD was incubated with increasing amounts of GST-Oct-1 or corresponding amounts of GST moiety alone, as indicated. (D) Oct-1, but not C/EBPα, binds to the K-bZIP WT12 DNA. RRLs were programmed with the indicated plasmids and then tested for binding to the indicated DNAs by EMSA.
The A/T-rich palindrome in the ORF57 RRE is sufficient for sequence-specific binding of the Rta DNA binding domain expressed recombinantly (13). To determine whether the palindrome of the K-bZIP promoter was also sufficient for sequence-specific Rta binding, we tested the ability of unlabeled K-bZIP WT12 or m12 DNAs to compete for Rta binding to the ORF57 RRE. As shown in Fig. 2B, only the WT12, but not the m12, K-bZIP DNA robustly competed for binding to Rta aa 1 to 272 when preincubated with the protein. The K-bZIP WT12 DNA competed for Rta protein as robustly as the unlabeled ORF57 RRE, confirming that the intact palindrome is an Rta binding site in both promoters.
We previously demonstrated that the A/T-rich palindrome of the ORF57 promoter is necessary for the formation of a ternary Rta/RBP-Jk/DNA complex by EMSA. We were unable to detect a similar ternary Rta/Oct-1/DNA complex formed on the K-bZIP Oct-1 wt12 site. Instead, Oct-1 enhanced Rta binding to the WT12 element. We compared the DNA binding of suboptimal concentrations of GST-Oct-1 and maltose binding protein (MBP) ORF50ΔSTAD proteins individually or together. As shown in Fig. 2C, under these conditions, neither individual protein bound detectably to the K-bZIP WT12 Oct-1 element (lanes 3 and 4). However, when the equivalent amount of ORF50ΔSTAD protein was incubated with increasing amounts of GST-Oct-1, the Rta/DNA complex was detected and increased in a dose-dependent fashion (Fig. 2C, lanes 5 to 8). Repeating this strategy by substituting GST-Oct-1 with the GST moiety alone showed only a modest enhancement of Rta/DNA binding (Fig. 2C, lanes 9 to 12). This modest effect was probably attributable to the effect of nonspecific protein crowding in the solution. We conclude that Oct-1 enhances DNA binding of Rta to the K-bZIP promoter.
The Oct-1 binding site (WT12) overlaps a binding site for C/EBPα (92, 93). To test whether C/EBPα binds to the WT element, we programmed RRLs with an expression vector for C/EBPα. As shown in Fig. 2D (lanes 2 and 3), C/EBPα binds very robustly to a consensus CAAT box DNA. However, when tested for binding to an equivalent amount of K-bZIP WT12 DNA, the equivalent amount of C/EBPα RRL formed only very faint complexes (Fig. 2D, lanes 7 and 8). These data suggest that the binding of C/EBPα to our K-bZIP WT12 DNA is highly inefficient and suboptimal. Indeed, when tested in transient transfection studies in Akata-31 cells, C/EBPα only modestly enhanced Rta-mediated transactivation of the K-bZIP promoter (about 1.5- to 2.5-fold) (data not shown). This effect of C/EBPα was similar when the WT12 promoter was compared to the m12 K-bZIP promoter, demonstrating that the m12 mutation did not disrupt the C/EBPα binding site (data not shown).
Two putative Oct elements are critical for Rta-mediated transactivation of the K-bZIP promoter in B cells.
To test the relative contributions of the two Oct elements in Rta-mediated transactivation of the K-bZIP promoter, we mutated both of the elements, alone or together, in the context of the full-length promoter (Fig. 1C) and cloned them upstream of firefly luciferase as a reporter. We introduced substitution mutations in both elements, rather than deletion mutations, so as not to disrupt the relative spacing of the other cis elements in the promoter. We compared each mutant to the WT promoter for transactivation in cotransfected Akata-31 cells (a subclone of the Akata Burkitt's lymphoma cell line cured of Epstein-Barr virus) (32).
As shown in Fig. 3A, Rta transactivated the K-bZIP WT promoter to a magnitude of about 200-fold. Conversely, Rta transactivation of the K-bZIP promoter was nearly eliminated by the m12 substitution mutation. The m34 mutation has minimal effects on Rta-mediated transactivation: in the context of either the WT or m12 promoter, the m34 mutation reduces Rta-mediated transactivation by about 10%. Taken together, the Oct site at position −72 (site 12) is a greater contributor to Rta transactivation of the K-bZIP promoter than the Oct site at position −241 (site 34). These data agree with the relative amount of Oct-1 binding to the two elements in vitro.
FIG. 3.

Oct-1 enhances Rta-mediated transactivation of the K-bZIP promoter. (A) K-bZIP site 12 is a greater contributor to Rta transactivation of the K-bZIP promoter than site 34. Akata-31 B cells were transfected with increasing amounts of the Rta expression vector (pcDNA3FLg50) and the indicated pK-bZIP reporter plasmids. Results are shown as transactivation (n-fold) versus empty vector alone and were normalized to a cotransfected β-galactosidase expression vector. Error bars represent standard deviations of at least three experiments performed in triplicate. (B and C) Oct-1 enhances Rta-mediated transactivation of the WT12 but not mutant K-bZIP promoter. Akata-31 B cells were transfected with expression vectors for Rta (pcDNA3FLg50) and Oct-1 (pcGOct-1), alone or together, and a promoter-reporter plasmid corresponding to the WT12 (A) or m12 (B) K-bZIP promoter. Analyses were performed as described above (A).
The Oct-1 protein enhances Rta-dependent transactivation of the K-bZIP promoter.
The data described above show that the cellular protein Oct-1 binds to the K-bZIP promoter in vivo and in vitro and stimulates DNA binding of Rta to the promoter. Furthermore, mutations that impair Rta-mediated transactivation of the promoter eliminate Oct-1 DNA binding. We reasoned that if Oct-1 DNA binding contributes to Rta-mediated transactivation of the K-bZIP promoter, then ectopic expression of Oct-1 should stimulate transactivation by Rta.
To test this hypothesis, we transfected the Akata-31 B cells with the Rta expression vector, with and without an Oct-1 expression vector, and the WT K-bZIP promoter-reporter. Transfection of a suboptimal amount of Rta vector alone led to the transactivation of the promoter, while transfection of the Oct-1 vector alone in three different amounts failed to transactivate the promoter (Fig. 3B). When we cotransfected the same amount of Rta vector with increasing amounts of Oct-1 vector, the promoter was transactivated in a dose-responsive fashion up to a maximum of nearly 160-fold (Fig. 3B). This effect was proportional to the amount of Oct-1 expressed in each transfection (data not shown). We saw a similar enhancement of Rta-mediated transactivation by Oct-1 in transfected BL-41 cells (data not shown). Oct-1 had no effect on the expression from the cytomegalovirus promoters in the transfected vectors (data not shown). Thus, Oct-1 enhances Rta-dependent transactivation of the K-bZIP promoter.
When we repeated the experiment but substituted the K-bZIP WT promoter with the K-bZIP m12 promoter, neither Rta nor Oct-1, alone or together, activated the promoter (Fig. 3C). This confirms that the stimulation of Rta-mediated transactivation of the K-bZIP promoter by Oct-1 requires sequence-specific binding to the WT12 Oct element.
A conserved Oct-interacting motif is required for Rta to interact functionally with Oct-1.
To determine whether the Rta and Oct-1 proteins require a direct interaction for transcriptional cooperation, we first asked whether the proteins interacted by performing GST affinity interaction (pull-down) assays. Nuclear extracts were prepared from BCBL-1 cells that were untreated or treated with 20 ng/ml TPA and incubated with GST-Oct-1 immobilized on glutathione agarose. Following washing, proteins that were retained on the GST-Oct-1 matrix were probed by SDS-PAGE/Western blotting. As shown in Fig. 4A, the Rta protein expressed during KSHV reactivation binds to GST-Oct-1. As a control, little Rta from reactivating cells binds to an excess of GST moiety alone (Fig. 4A, right lanes).
FIG. 4.

The Rta N-terminal basic region contains its OID. (A) GST-Oct-1 binds to the ORF50/Rta protein in extracts from infected cells. GST-Oct-1 or GST alone was immobilized on glutathione-agarose beads and incubated with nuclear extracts from untreated or TPA-treated BCBL-1 cells. Bound proteins were analyzed by SDS-PAGE/Western blotting using anti-Rta primary antiserum. (B) Rta interacts with Oct-1 in vitro. GST pull-downs were performed as described above (A), but 35S-labeled Rta (from programmed RRL) was substituted for nuclear extracts. Following washes, beads were boiled in 2× Laemmli buffer, and bound proteins were separated by SDS-PAGE. Fixed, amplified, and dried gels were analyzed by autoradiography. (C) Two regions of Rta are homologous to homeodomain-interacting regions of VP16 and MATα2. Shown is a schematic of the ORF50/Rta primary amino acid sequence. The region of ORF50 contained within the ORF50ΔSTAD mutant (53) is indicated by the bar below the ORF. Predicted OIDs are indicated with arrows. +++, basic amino acid rich; ST, serine/threonine rich; hyd/DE/hyd, hydrophobic/acidic amino acid repeats. (D) Alignment of OIDs. The sequence of each OID aligned with HSV-1 VP16 and S. cerevisiae MATα2 proteins (45, 82) is shown. The box shows the established or predicted α-helix in the four proteins. The amino acids in OIDa in boldface type indicate positions of site-directed mutations and homology in the aligned domains. The arrowhead indicates the site of the ADPR insertion. Numbers represent amino acid positions. (E) OIDa is required for Rta to bind to Oct-1. GST pull-downs were performed as described above (C). WT and mutant ORF50 constructs were used to program RRLs to make the indicated proteins. Input proteins are shown as a reference. (F) OIDa is required for ORF50/Rta to transactivate the K-bZIP promoter. Expression vectors encoding the indicated proteins were transfected into Akata-31 cells and analyzed for transactivation of the K-bZIP WT reporter plasmid, as described in the legend to Fig. 3A. Amounts of each expression vector were 0, 0.1, 0.5, 2.5, and 12.5 μg. (G) ORF50ΔOIDa and ΔOIDb are expressed in cell nuclei. HeLa cells were transfected with the indicated expression vectors and analyzed by indirect immunofluorescence with Rta-specific primary antiserum and fluorophore-conjugated secondary antibodies. Nuclear DNA was stained with DAPI (4′,6′-diamido-2-phenylindole). Images were converted to grayscale for publication.
To determine whether the Rta/Oct-1 interaction could be reproduced with recombinant ORF50/Rta protein, immobilized GST-Oct-1 was tested for binding to full-length WT Rta that was generated in RRLs as a [35S]methionine-labeled protein. As shown in Fig. 4B, Rta was retained on the GST-Oct-1 beads but not on the immobilized GST moiety alone, proving that Rta binds to Oct-1.
HSV-1 virion protein VP16 and S. cerevisiae MATα2 contain short, homologous α-helices that interact directly with heterologous homeodomain proteins like Oct-1 (45, 82). Alignment of these helices with Rta revealed two putative octamer-interacting domains (OIDs) contained within predicted α-helices of Rta: OIDa (within aa 134 to 150) and OIDb (aa 584 to 600) (Fig. 4C and D). We deleted both OIDs independently in the ORF50/Rta cDNA (Fig. 4C) and repeated the GST-Oct-1 pull-down experiments. As shown in Fig. 4E, deletion of OIDa eliminated the interaction of ORF50/Rta with Oct-1. Conversely, deletion of OIDb in either the mutant protein ORF50ΔOIDb or ORF50ΔSTAD (diagrammed in Fig. 4C) had no effect on the interaction of Rta with Oct-1 (Fig. 4E). A deletion of the proline-rich leucine heptapeptide repeat (LR) of ORF50 also eliminated the interaction with Oct-1. The ORF50ΔLR mutant is defective in transcriptional activation due to improper multimerization (12). Taken together, this suggests that OIDa, but not OIDb, is required for Rta to interact directly with Oct-1.
To test the functional consequences of the ORF50 OID mutants, we compared them to WT ORF50 for transactivation of the K-bZIP promoter. As shown in Fig. 4F, deletion of the Oct-1 interaction domain in the mutant ORF50ΔOIDa rendered the protein nonfunctional. This suggested that the Rta/Oct-1 interaction is required for the transactivation of the K-bZIP promoter. However, ORF50ΔOIDb, which retained the ability to interact with Oct-1 (Fig. 4E), was reduced in the transactivation of the promoter by about 60%. We hypothesize that this phenotype is due to a deletion of a portion of the Rta transcriptional activation domain (51) in this mutant (diagrammed in Fig. 4C).
Indirect immunofluorescence verified that the OIDa and OIDb mutants were correctly localized to the nuclei of transfected cells (Fig. 4G). ORF50ΔOIDa also showed weak, diffuse cytoplasmic expression (Fig. 4G, center).
Rta OIDa aa 136, 140, 143, and 144 are essential for functional interaction with Oct-1.
OIDa overlaps the E3 ubiquitin ligase domain required for Rta autoproteolysis (103). Ubiquitin-dependent proteolysis is tightly linked to transcriptional activation by multiple transcription factors (25, 36, 50, 67, 74). To attempt to genetically separate the Rta/Oct-1 interaction from the E3 ubiquitin ligase activity, we introduced alanine substitution mutations of three hydrophobic amino acids in OIDa that are conserved with VP16 and MATα2 (aa 140, 143, and 144 of Rta) (Fig. 4D). This mutant (ORF50-ILL140AAA) was compared to a previously characterized (103) E3 ubiquitin ligase mutant of Rta (ORF50-C141S). As shown in Fig. 5A, ORF50-ILL140AAA failed to bind to GST-Oct-1, while ORF50-C141S retained easily detectable Oct-1 binding activity.
FIG. 5.

Site-specific mutants define the VP16-like amino acid sequence that is essential for the interaction of Oct-1. (A) ORF50-ILL140AAA does not interact with Oct-1 GST. GST pull-downs were performed as described in the legend to Fig. 4E, with RRLs programmed to express the indicated proteins. 50-ILL140AAA contains three alanine substitutions at amino acids 140, 143, and 144; 50-C141S contains a substitution at amino acid 141, changing the Cys to Ser. (B) Additional mutations in OIDa inhibit interactions of ORF50 with Oct-1. GST pull-downs were performed as described above (A) using RRLs programmed to express the indicated ORF50 proteins. (C) ORF50-ILL140AAA retains the ability to interact with RBP-Jk/CSL. GST pull-downs were performed as described above (A) using GST-RBP-Jk immobilized on glutathione beads and the indicated proteins expressed in RRLs. (D) Additional mutations in OIDa have little effect on interactions of ORF50 with RBP-Jk. GST pull-downs were performed as described above (C) using RRLs programmed to express the indicated ORF50 proteins. (E) ORF50-I140A retains a reduced ability to interact with Oct-1. GST pull-downs were performed as described above (B).
We also tested a series of alanine substitution mutants of individual hydrophobic amino acids within Rta OIDa as well as a mutant containing an ADPR insertion within OIDa (ORF50-ADPRins) identical in position to an inactivating mutation of VP16 (79) (Fig. 4D). As shown in Fig. 5B, ORF50-L136A and ORF50-ADPRins lost the ability to bind Oct-1, ORF50-L144A was reduced in Oct-1 interactions, and ORF50-L143A had the WT ability to bind Oct-1. The data are summarized in Table 2. To confirm that the Oct-1-interacting mutants of Rta specifically eliminated Oct-1 binding and were not grossly misfolded, we tested the binding of each mutant to GST-RBP-Jk, a well-established binding partner of Rta (13, 46). As shown in Fig. 5C and D, all of the OIDa mutants maintained the WT ability to interact with RBP-Jk in GST pull-down experiments. An additional Rta OIDa mutant, ORF50-I140A, also maintained full RBP-Jk binding (Fig. 5D) but was reduced in Oct-1 binding (Fig. 5E). These experiments demonstrate that single amino acid mutations within OIDa selectively eliminated the binding of Rta to Oct-1, and the RBP-Jk and Oct-1 interaction domains of Rta are separable.
TABLE 2.
Transactivation and stimulation of reactivation by Rta OIDa mutants that are deficient in DNA and/or Oct-1 binding
| ORF50/Rta | Oct-1 binding resulta | DNA binding resultb | % Transactivationc | % Reactivationd |
|---|---|---|---|---|
| WT | +++ | +++ | 100 | 100 |
| L136A | − | + | 35 | 34 |
| I140A | + | +++ | 104 | 35 |
| L143A | +++ | ++++ | 104 | 117 |
| L144A | + | − | 55 | 51 |
| ILL140AAA | − | − | 1 | 1 |
| ADPRins | − | − | 1 | ND |
GST pull-down (Fig. 5A, B, and E). WT is set at “+++.”
EMSA (Fig. 6). WT is set at “+++.”
Maximal value in transient transactivation of K-bZIP promoter in Akata-31 cells (Fig. 7A), expressed as a percentage compared to the WT (set at 100%).
Induction of K8.1 expression in BCBL-1 cells (Fig. 10), expressed as a percentage compared to the WT (set at 100%). ND, not done.
Site-specific OIDa mutations selectively eliminate binding of Rta to the K-bZIP WT12 promoter element.
HSV-1 VP16 interacts with Oct-1 using a small protein region. Consequently, individual point mutations and small peptide insertions in this region of VP16 reduce or disrupt both Oct-1 and Oct-1/DNA binding of VP16 (44). To determine the consequences of the OIDa mutations on Rta DNA binding, we expressed each of the OIDa mutants as fusions to MBP and compared them to WT Rta for binding to the K-bZIP WT12 promoter element. As shown in Fig. 6, three of the OIDa mutants, ILL140AAA, L144A, and ADPRins, failed to bind to the K-bZIP DNA in EMSA (the ILL140AAA mutation eliminated binding in the context of either full-length ORF50/Rta or truncated ORF50ΔSTAD). Rta mutant L136A was severely reduced in K-bZIP DNA binding, while the L143A mutation enhanced K-bZIP DNA binding by Rta. One mutant, ORF50-I140A, retained K-bZIP DNA binding activity similar to that of WT Rta. Taken together, the OIDa mutations selectively eliminated K-bZIP DNA binding by Rta, and these effects were independent of Oct-1 binding by Rta. The DNA binding results are summarized in Table 2.
FIG. 6.
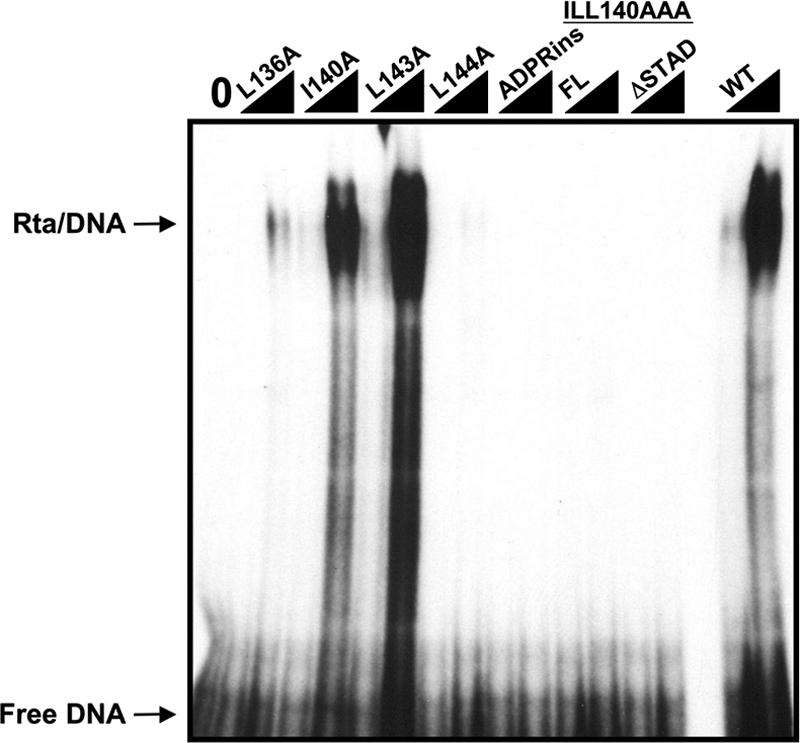
Mutations in ORF50 OIDa have variable effects on DNA binding to the K-bZIP WT12 promoter element. The indicated WT or mutant ORF50 proteins were expressed as N-terminal MBP fusion proteins, and two different amounts were tested for binding to the K-bZIP WT12 element by EMSA as described in the legend to Fig. 2A.
Oct-1 interactions and DNA binding of Rta are required for transactivation of the K-bZIP promoter.
The series of Rta OIDa mutants that we generated demonstrated all of the potential combinations of phenotypes in DNA and Oct-1 binding (summarized in Table 2). Furthermore, all of these mutants maintained the ability to bind to RBP-Jk (Fig. 5). To test the functional consequences of the OIDa mutations, we performed transient transactivation analyses of the K-bZIP promoter and compared each mutant to WT ORF50/Rta. As shown in Fig. 7A, mutant ORF50-L143A, which retained WT levels of DNA and Oct-1 binding, also retained WT levels of transactivation. Curiously, ORF50-I140A also retained WT levels of transactivation of the K-bZIP promoter (Fig. 7A) despite our observation that the binding of the mutant to Oct-1 was reduced (Fig. 5E). ORF50-L144A lost about half of its ability to transactivate (Fig. 7A); that mutant bound Oct-1 quantitatively similarly to I140A but did not bind K-bZIP promoter DNA (Fig. 6). Those observations suggest that the reduction in binding of mutant I140A to Oct-1 is tolerated for transactivation, provided that DNA binding is maintained at WT levels.
FIG. 7.

DNA binding and Oct-1 interactions both contribute to specifying ORF50-mediated transactivation of the K-bZIP promoter. Akata-31 cells were transfected with the indicated plasmids, together with reporter plasmids for the K-bZIP (A), ORF57 (B), or Nut-1/PAN (C) promoters, and analyzed as described in the legend to Fig. 4F. (D) All ORF50 mutants are expressed in cell nuclei. HeLa cells were transfected with the indicated expression vectors and analyzed by indirect immunofluorescence as described in the legend to Fig. 4G.
ORF50 mutant L136A was unable to bind Oct-1, retained some ability to bind K-bZIP DNA, and retained 35% of WT transactivation activity (Fig. 7A). Complete elimination of K-bZIP transactivation required mutations of ORF50/Rta that completely eliminated both K-bZIP DNA and Oct-1 binding, as in mutants ILL140AAA and ADPRins (Fig. 7A). These data are summarized in Table 2. Interestingly, even though the E3 ubiquitin ligase mutant ORF50-C141S could interact with Oct-1, it was reduced in transactivation by about 75% (Fig. 7A). The data suggest that K-bZIP DNA binding, Oct-1 interaction, and E3 ubiquitin ligase activity are critical for optimal transactivation of the K-bZIP promoter by Rta.
To determine whether the OIDa mutations altered the transactivation of other Rta target promoters, we challenged the Nut-1/PAN and ORF57 promoters similarly in Akata-31 cells. As shown in Fig. 7B, mutations of ILL140AAA, L136A, I140A, L143A, and L144A affected Rta's transactivation of the ORF57 promoter in a fashion similar to that of the K-bZIP promoter. As shown in Fig. 7C, mutations of ILL140AAA and C141S affected Rta's transactivation of the Nut-1/Pan promoter similarly to the K-bZIP promoter. Interestingly, Rta mutant I140A, which showed WT activation of the K-bZIP and ORF57 promoters, superactivated the Nut-1/PAN promoter.
Figure 7D shows that all OIDa mutant proteins were correctly localized to the nuclei of transfected cells.
Cell-specific requirement for Oct-1 in Rta-mediated transactivation.
In the experiments described above, we demonstrated the requirement for Oct-1 in Rta-mediated transactivation of the K-bZIP promoter using cis- or trans-acting mutations that eliminated (i) Oct-1 binding to the promoter and (ii) Rta binding to Oct-1. We employed one additional strategy by performing transient transactivation studies using matched mouse embryonic fibroblast (MEF) cells that were either WT or null for Oct-1 (62, 94). Increasing amounts of the WT ORF50/Rta expression vector were cotransfected with the K-bZIP promoter-reporter. Figure 8A shows that Rta transactivated the WT K-bZIP promoter to a maximum of about 20-fold in Oct-1−/− MEFs. The magnitude of transactivation of the WT K-bZIP promoter was nearly identical in the matched MEFs that are WT for Oct-1 (Fig. 8B). This suggested that Oct-1 is not required for Rta-mediated transactivation in MEFs. However, transfection of the Rta vector into an MEF cell line that was null for Oct-1 but that stably expressed human Oct-1 resulted in an increase (about twofold) of K-bZIP promoter activity (Fig. 8A).
FIG. 8.
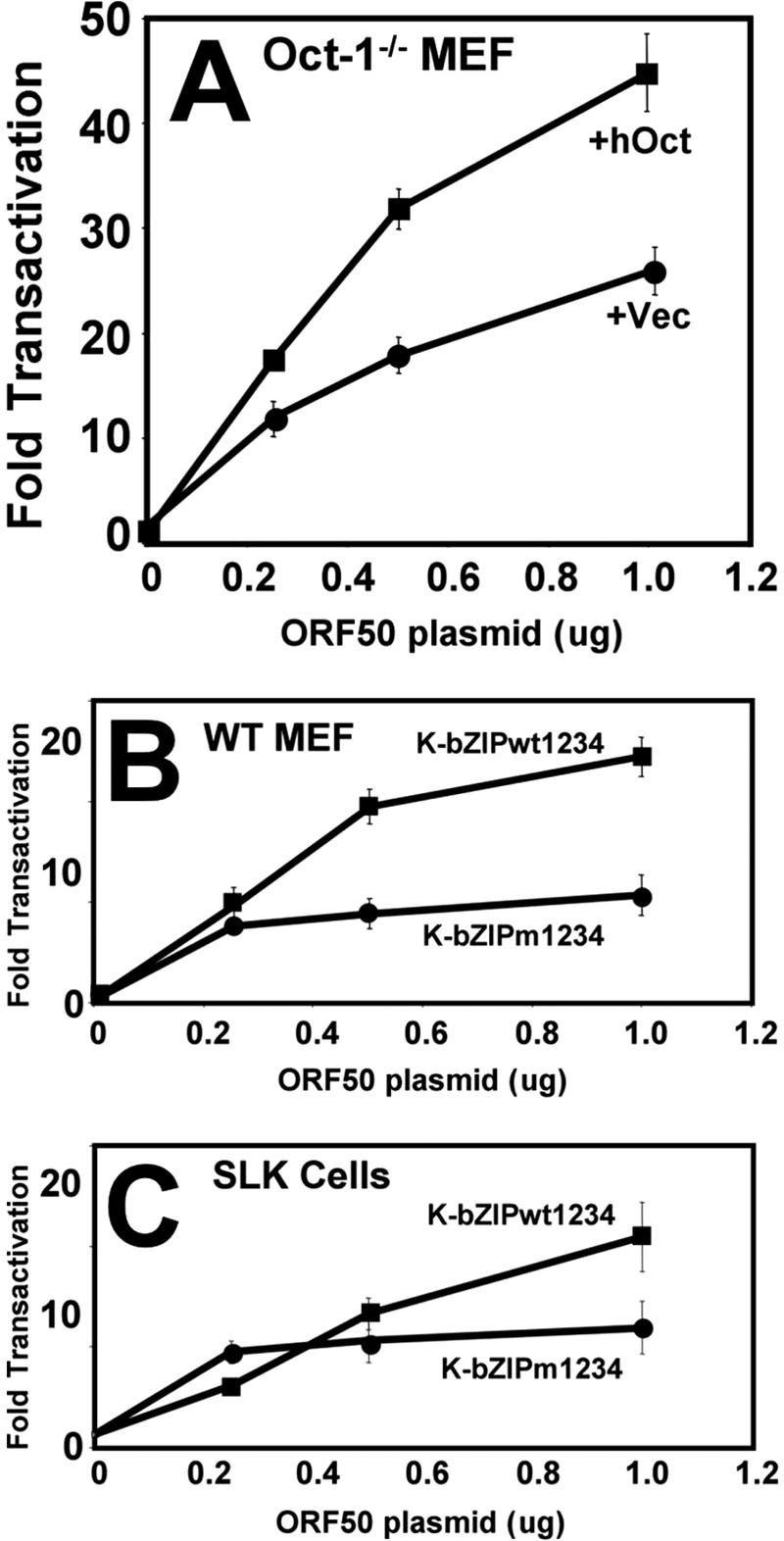
Oct-1 enhances, but is not essential for, Rta-mediated transactivation of the K-bZIP promoter. (A) MEFs null for Oct-1 (+Vec) or null for Oct-1 and stably expressing human Oct-1 (+hOct) were transfected with an Rta expression vector (pcDNA3FLc50) and the WT K-bZIP reporter plasmid. Cells were analyzed for transactivation as described in the legend to Fig. 3A. (B and C) WT MEF cells (B) or SLK cells (C) were tested for the ability of Rta to transactivate the indicated reporter plasmids. Results are shown as described above (A).
We showed in Fig. 3A that mutation of both putative Oct binding sites of the K-bZIP promoter (K-bZIP m1234) virtually eliminated Rta-mediated transactivation in Akata-31 Burkitt's lymphoma cells. In contrast, Rta transactivation of the K-bZIP m1234 reporter was reduced by only about 50% in WT MEF cells (Fig. 8B). These data suggested a cell-specific requirement for Oct-1 in Rta-mediated transactivation, which was not as strict in non-B cells. Results of transient transfections of SLK cells supported this hypothesis. As shown in Fig. 8C, the double Oct site mutation (m1234) reduced Rta-mediated transactivation of the K-bZIP promoter by only 50%.
RBP-Jk, but not Oct-1, is essential for Rta-mediated transactivation of the K-bZIP promoter in MEFs.
We previously showed that Rta stimulates the binding of RBP-Jk to the K-bZIP promoter in vivo (13). Since the requirement for Oct-1 in Rta-mediated transactivation was reduced in MEFs, we asked whether RBP-Jk had a more significant role in those cells. We transfected RBP-Jk null cell lines (OT11) (64) with expression vectors corresponding to Rta and RBP-Jk either alone or together. Similar to previous reports (97), we conclude that Rta transactivation of the K-bZIP promoter in MEF cells requires the expression of RBP-Jk (Fig. 9).
FIG. 9.
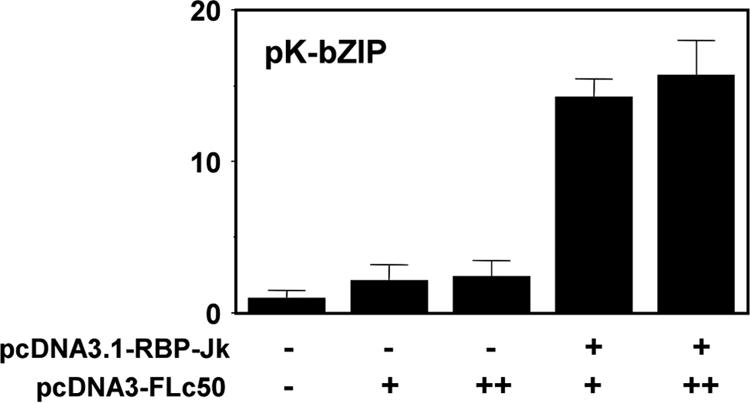
RBP-Jk is essential for Rta-mediated transactivation of the K-bZIP promoter. MEFs null for RBP-Jk (OT11) were transfected with expression vectors corresponding to Rta (pcDNA3FLc50) and RBP-Jk (pcDNA3.1-RBP-Jk). Transfections were analyzed as described in the legend to Fig. 3A.
Interaction of Rta with Oct-1 is required for optimal reactivation of KSHV from latency.
To determine whether binding of Oct-1 and DNA was required for Rta to transactivate the K-bZIP promoter in the context of the authentic viral genome in infected cells, we transfected BCBL-1 cells with WT and mutant Rta vectors and quantitated the induction of the K-bZIP protein. Ectopic Rta proteins were expressed as fusions to the V5 epitope tag to distinguish them from endogenous Rta. As shown in Fig. 10A, approximately 92% of 500 ORF50-transfected (V5-positive) cells also expressed K-bZIP. Conversely, as expected, K-bZIP was expressed in only about 2% of cells transfected with the ORF50-ILL140AAA mutant, which failed to bind both the K-bZIP promoter and Oct-1. This low value was similar to the level of spontaneous reactivation in untransfected cells. Figure 10B shows indirect immunofluorescence of typical fields from these transfections.
FIG. 10.

Interaction of Rta with Oct-1 is required for optimal reactivation from latency. (A and B) Induction of K-bZIP expression. BCBL-1 cells were electroporated in duplicate with plasmids expressing the indicated proteins and analyzed for reactivation by indirect immunofluorescence 72 h later. Cells were scored by the percentage of Rta single-positive cells (detected by anti-V5 antiserum) that also expressed K-bZIP (Rta/K-bZIP double-positive cells). Quantitation is shown in A, and typical fields are shown in B. Arrows point to V5-ORF50-positive cells. The virus is presumably reactivating spontaneously in those cells expressing K-bZIP in the absence of V5-ORF50 (center panels in B). (C) Complete viral reactivation. BCBL-1 cells were electroporated in duplicate with plasmids expressing the indicated proteins and analyzed for reactivation by indirect immunofluo rescence 72 h later. Cells were scored by the percentage of Rta single-positive cells that also expressed K8.1 (Rta/K8.1 double-positive cells; no cells expressed K8.1 in the absence of Rta). To eliminate spontaneously reactivating cells, an identical calculation was performed on cells electroporated with the empty control, the pcDNA3.1 vector; that percentage was subtracted from the values for all of the other transfections.
To test the OIDa mutants for their abilities to reactivate the complete KSHV lytic cycle, we scored reactivation at the single-cell level 72 h after electroporating BCBL-1 cells. Our marker for induction of reactivation was the K8.1 protein, which is expressed in PEL cells with true late kinetics (i.e., K8.1 expression requires viral DNA replication) (51, 54, 100). We calculated the percentage of Rta-expressing cells that also expressed K8.1. Cells that were transfected with the empty expression vector were used to calculate the percentage of spontaneous viral reactivation, which was subtracted from each of the percentages of reactivation induced by WT Rta or mutants. As shown in Fig. 10C, WT ORF50 stimulated K8.1 expression in about 32% of transfected BCBL-1 cells. This value was similar for mutant ORF50-L143A, which had WT K-bZIP promoter transactivation ability (Fig. 7A). Conversely, the ORF50-ILL140AAA mutant (DNA and Oct-1 binding negative, K-bZIP transactivation negative) was completely impaired in its ability to reactivate KSHV from latency. The negative control, ORF50ΔLR (the multimerization mutant of ORF50/Rta) (11), was also impaired in its ability to reactivate KSHV. Viral reactivation stimulated by mutants ORF50-L136A and ORF50-L144A was reduced (34% and 51%, respectively) in a fashion similar to the extent of the reduction of transactivation of the three KSHV promoters by both mutants (approximately 35% and 50%, respectively) (Fig. 7). These mutants affirm the contribution of both Oct-1 and K-bZIP promoter binding of Rta to the optimal stimulation of complete KSHV lytic reactivation. However, even though mutant ORF50-I140A was capable of transcriptional transactivation at WT or greater levels (Fig. 7), it retained only 35% of WT reactivation function.
DISCUSSION
In this article, we have shown that a direct interaction of the KSHV lytic switch protein ORF50/Rta with the cellular protein Oct-1 is one critical component in regulating lytic reactivation of the virus (Fig. 10). Oct-1, but not Oct-2, from KSHV-infected cells binds to the K-bZIP promoter in vitro and in vivo (Fig. 2 and Table 1). Sequence-specific binding of Oct-1 to the promoter corresponds with Rta-mediated transactivation (Fig. 3), and a promoter mutation that eliminates Oct-1 binding is more deleterious for Rta transactivation in B cells than in SLK cells (Fig. 3A and 8C). Ectopic coexpression of Oct-1 augments Rta-mediated transactivation of the WT, but not the mutant, K-bZIP promoter (Fig. 3B and C). A short amino acid signature in the N terminus of Rta that is conserved in HSV-1 VP16 (OIDa) (Fig. 4D) is required for the direct binding of Rta to Oct-1 (Fig. 4E), Rta-mediated transactivation of the K-bZIP promoter (Fig. 4F), and optimal Rta-stimulated reactivation of KSHV from latency (Fig. 10). Selective mutations within OIDa also reduce or eliminate the binding of Rta to K-bZIP promoter DNA (Fig. 6); some of these mutations are independent of Oct-1 binding (Fig. 5A, B, and E), and all have no effect on RBP-Jk binding (Fig. 5C and D). Our data therefore support a model for reactivation that involves sequence-specific interactions of Rta and Oct-1 with DNA and Rta and Oct-1 with each other. Mutations in Rta that eliminate one of the interactions reduce but do not eliminate Rta-mediated reactivation of KSHV from latency (summarized in Table 2). Mutations that eliminate both interactions of Rta also eliminate Rta-mediated reactivation. All of the mutants that we analyzed retained WT levels of interaction with the cellular protein RBP-Jk. Mutations that inhibit the DNA binding of Rta seem to be more deleterious for Rta-mediated transactivation in Akata-31 cells and Rta-mediated reactivation in PEL cells than mutations that inhibit the interaction of Rta with Oct-1 or RBP-Jk. We have not yet detected a ternary Rta/Oct-1/DNA complex with stability sufficient for visualization by EMSA, but Oct-1 stimulates DNA binding of ORF50/Rta in vitro (Fig. 2C).
The relative ability of each Rta protein to reactivate the complete KSHV lytic cycle corresponded directly with its ability to transactivate transcription, except for mutant I140A. That mutant retained WT or greater transactivation of three KSHV promoters but retained only 35% of WT reactivation activity. The I140A mutation therefore appears to define the minimum Oct-1 and/or DNA binding requirements for optimal transactivation of the K-bZIP, ORF57, and Nut-1 promoters but is deficient in optimal stimulation of reactivation. We hypothesize that the isoleucine at aa 140 is required for the transactivation of a promoter that we did not test here, for proper Rta function at a lytic origin of replication (91), and/or for Rta's ubiquitin ligase activity (98).
The Rta/Oct-1 interaction is one of many mechanisms that have been proposed for Rta-mediated transactivation of the K-bZIP promoter. The other previously reported mechanisms involve direct interactions between Rta and cellular proteins (RBP-Jk) (97) (as we see in Fig. 9) and C/EBPα (92, 93) or DNA (a phased, A/T trinucleotide repeat on the K-bZIP promoter DNA) (49). Considered together with our data, we propose that all of these molecular interactions are required for optimal Rta-mediated transactivation of the K-bZIP promoter, but none are individually sufficient.
For example, we uncovered the significance of Oct-1 DNA binding in Rta-mediated transactivation of the K-bZIP promoter by rationally designing the mutation of site 12 based on its high homology to the RRE found in the ORF57/Mta promoter (Fig. 1A) (52). Furthermore, we were careful to introduce the mutation as a sequence-specific substitution in the context of the full-length K-bZIP promoter, without deleting any DNA, to ensure that the phased A/T trinucleotide repeat would not be disrupted (Fig. 1C). Two other studies of the K-bZIP promoter employed a significantly different approach, by analyzing Rta-mediated transactivation of truncation mutants of the K-bZIP promoter (49, 97). Studies of those truncation mutants led those authors to conclude that a mutant promoter containing the Oct-1 site (WT12, identified in our study), the C/EBPα site, and the Rta binding site in the palindrome was insufficient for Rta transactivation in cells other than Akata-31 cells.
The requirement for RBP-Jk in maximal Rta-mediated transactivation of the K-bZIP promoter appears to be cell specific. Nonetheless, RBP-Jk appears to be insufficient for full transactivation. While Rta-mediated transactivation of the K-bZIP promoter is completely abolished in RBP-Jk null cells (Fig. 9) (97), many OIDa mutations that eliminated the Rta/Oct-1 interaction, but not the Rta/RBP-Jk interaction, abolished Rta transactivation of the K-bZIP promoter in B cells (Fig. 5C and D and Table 2). The OIDa mutations that abolished Rta-mediated transactivation of the K-bZIP promoter also inhibited Rta-stimulated reactivation of KSHV (Fig. 10). One potential explanation for the discrepancy is that Oct-1 expression is reduced or eliminated in RBP-Jk null cells.
However, our data from transfections of Oct-1−/− MEF cells suggest that Oct-1 is also not essential for Rta-mediated transactivation of the K-bZIP promoter, because transactivation in those cells was reduced by only 50% (Fig. 8). This result contradicts the apparent requirement for Oct-1 in Akata-31 B cells, since mutation of the Oct-1 site (WT12) virtually eliminated Rta-mediated transactivation of the K-bZIP promoter in those cells (Fig. 3A). This result highlights the insufficiency of RBP-Jk binding to the K-bZIP promoter for Rta transactivation, since the RBP-Jk site is intact in the mutant promoter (Fig. 1C). However, the discrepancy between the MEF and B-cell data might be explained by a cell-specific requirement for C/EBPα (92, 93). Although C/EBPα bound very inefficiently to site 12 in our EMSA DNA probe (Fig. 2D), additional sequences upstream of the palindrome and not included in our EMSA DNA might be required for C/EBPα to bind efficiently to the promoter. However, since some of the Oct-1-interacting mutants of Rta were completely transcriptionally inert in Akata-31 B cells (Fig. 7), and we saw little effect of coexpressing C/EBPα on Rta transactivation of the WT or mutant K-bZIP promoter (not shown), other explanations for the discrepancy between the MEF and B cells cannot be excluded. Such alternative explanations include cell-specific transcriptional activity by Rta, which is supported by our transfections of SLK cells (Fig. 8C), or compensation for the Oct-1 deletion in MEFs by other Oct family members.
To attempt to address the apparently stringent B-cell-specific requirement for Oct-1 in the activation of the K-bZIP promoter by Rta, we attempted to knock down Oct-1 with commercially available small interfering RNAs (not shown) in B cells. We were unsuccessful at eliminating Oct-1 expression. However, with our series of single point mutations in Rta, specifically with mutants I140A and L144A, we were able to determine that even a diminished ability to interact with Oct-1 is sufficient for Rta to strongly transactivate target promoters (summarized in Table 2), provided that DNA binding is maintained at WT levels.
We previously showed that Rta stimulates DNA binding of RBP-Jk to the K-bZIP promoter in vivo, a function of Rta that extends to the promoters of additional KSHV and cellular genes in infected PEL cells (13). In the KSHV ORF57/Mta promoter, stimulation of RBP-Jk DNA binding by Rta requires a juxtaposed A/T-rich binding site for Rta (13). This A/T-rich site is part of the palindrome that is conserved in the ORF57/Mta and K-bZIP promoters (Fig. 1A) (52). In this study, we showed that recombinant Rta binds with sequence specificity to the A/T-rich palindrome in the K-bZIP promoter also (Fig. 2B). In both the ORF57 and K-bZIP promoters, the A/T-rich site comprises one unit of the phased A/T trinucleotide repeat. Liao et al. (49) previously showed that Rta-mediated transactivation increases in direct proportion to the number of such A/T repeats in promoters.
We therefore propose a model in which Rta participates in long-range interactions with the K-bZIP promoter that are specified by a combination of direct binding to the phased A/T trinucleotide repeats and indirect binding by association with Oct-1, RBP-Jk, and C/EBPα. In vitro, Rta's molecular interactions with individual components of this proposed transcriptional complex appear to be relatively weak. In our study, only a small fraction of total Rta generated in RRL was retained on GST-Oct-1 beads (Fig. 4B). Similarly, the binding of Rta to single A/T-rich units of the ORF57 promoter DNA is significantly weaker than the binding to the canonical Rta site in the PAN promoter (77). Furthermore, Rta expressed in RRL fails to detectably bind to the K-bZIP promoter under conditions in which it binds to PAN DNA (92). We propose, therefore, that the sum of these DNA and protein interactions is required for the functional interaction of Rta with the K-bZIP promoter during reactivation. In this regard, the magnitude of activity of this Rta transcription complex could be regulated by the relative kinetics of expression and promoter binding of each of these proteins during KSHV reactivation. Indeed, Oct-1 binding to the K-bZIP promoter is enhanced following the reactivation of KSHV from latency (Fig. 1E), probably as a result of the increase in Oct-1 transcription that we measured (not shown). Our observation that Oct-1 stimulates DNA binding of Rta (Fig. 2C) might result from the ability of Oct-1 to bend target DNA upon binding (89). The cellular architectural DNA bending protein HMG-B1 enhances DNA binding and transcriptional activation by Rta (78). Therefore, an attractive possibility is that Oct-1 bends KSHV promoters to stimulate Rta binding during reactivation. In turn, such a mechanism would facilitate the stimulation of RBP-Jk DNA binding by Rta (12).
Rta's use of Oct-1 for transactivation extends to additional KSHV lytic cycle promoters. Both the K1 and Rta promoters contain functional Oct-1 binding sites that are required for Rta-mediated transactivation (88, 92). Transactivation of the K1 promoter is also specified by direct DNA binding of Rta (10). We characterized the Rta OID mutants by using transactivation of the K-bZIP promoter as a model (Fig. 7A). Nearly all of those transactivation phenotypes of the Rta mutants were identical to their phenotypes in stimulating complete KSHV reactivation (Fig. 10). Taken together, this suggests a broad role for Rta/Oct-1 interactions in KSHV reactivation. Indeed, in Fig. 7B, we demonstrated that Rta mutants deficient for binding Oct-1 and/or DNA were similarly deficient for transactivating the Nut-1/PAN and ORF57 promoters. Assuming that the K-bZIP DNA binding mutants were similarly debilitated in binding the Nut-1/PAN and ORF57 promoters, these data confirmed the requirement for Rta DNA binding in the activation of all three promoters. These data also suggest a previously undescribed requirement for Oct-1 in Rta-mediated transactivation of the ORF57 promoter.
A central role for Oct-1 in replication has been conserved in other members of the Herpesviridae. Oct-1 is essential for VP16-mediated activation of HSV-1 IE promoters (28, 40-42, 56, 63, 69, 86), and Oct-1 specifies transcriptional targets of the varicella-zoster virus ORF10 protein (60). Among other viruses, the mouse mammary tumor virus long terminal repeat is activated by the interaction of Oct-1 with the glucocorticoid receptor (11).
Oct-1 is a member of the POU (for pituitary transcription factor Pit-1, octamer motif binding proteins Oct-1 and Oct-2, and Caenorhabditis elegans UNC-86) class of homeodomain-containing transcription factors. The POU domain provides a bipartite DNA binding domain that contacts DNA flexibly yet sequence specifically via an ATGCAAAT consensus sequence (39, 68). A 20-aa linker region separating each DNA binding domain allows the recognition of sequences that are divergent from the consensus.
The specificity of Oct-1 DNA binding is influenced by interactions with transcriptional coactivator proteins. One well-characterized Oct-1 coactivator is the HSV-1 VP16 protein (101). Following HSV-1 entry of a susceptible host cell, the transactivation of IE promoters requires the formation of the VP16-induced complex. This multiprotein complex consists of Oct-1, VP16, host cell factor (HCF), and DNA (TAATGARAT, where R is purine) (28, 40-42, 56, 63, 69, 86). The cellular protein HCF is required for the nuclear localization of VP16 (43).
Our data suggest that the regulation of KSHV gene expression by Oct-1 partially conserves fundamental molecular mechanisms with HSV-1 but has diverged significantly as well. Similar to VP16 (31, 80), Rta is packaged into the KSHV virion (4), although VP16 is expressed with late, and not IE, kinetics during HSV-1 infection (72). The OIDs of VP16 and Rta are homologous in both primary sequence and putative secondary structure (45, 82) (Fig. 4B), and mutations in both OIDs reduce transactivation by disrupting either Oct-1 binding, DNA binding, or both. Structure-function divergence between the two OIDs, however, is suggested by the reduction of DNA binding in Rta mutant L136A (Fig. 6); the orthologous mutation in VP16 (Y373A) did not disrupt DNA binding by that protein (44).
The Oct elements identified in the KSHV genome demonstrate considerably greater sequence heterogeneity than those in HSV-1 (Fig. 1D). In HSV-1, the TAATGARAT sequence is usually overlapped by an octamer sequence (underlined in ATGCTAATGARAT, where R is purine), but elements that lack the octamer sequence can still be transactivated by the VP16-induced complex albeit with lower efficiency (2, 3). Mutations of the GARAT sequence can eliminate VP16/Oct-1 DNA binding independently of Oct-1 DNA binding (90). The GARAT sequence promotes a VP16-dependent comformational change in Oct-1 that alters the typical Oct-1 contacts on DNA (90). Although the K-bZIP WT12 Oct-1 site contains the overlapping GARAT element (Fig. 1D), it is currently unclear whether the binding of Rta to the promoter requires the GARAT sequence and/or induces conformational changes in Oct-1. However, the lack of a GARAT sequence in the KSHV K1 and Rta Oct-1 elements (Fig. 1D) provides the potential for differential Oct-1-dependent regulation of those promoters by Rta.
Although not formally tested here, the VP16-induced complex protein HCF is not predicted to interact with Rta, since Rta lacks the tetrapeptide sequence ([D/E]HxY) (24, 43, 101) necessary for VP16's interaction with HCF. Nonetheless, since only a small percentage of Rta directly binds to purified GST-Oct-1 (Fig. 4C), heterologous factors might contribute to a more robust Rta/Oct-1 interaction in vivo.
Acknowledgments
This work was supported by the American Cancer Society, the American Heart Association, and the NJ Commission on Cancer Research.
Thanks to P. J. Farrell, Dean Tantin, Winship Herr, Phillip Sharp, and Daniel Tenen for cells and plasmids. Thanks to Don Ganem for the anti-K-bZIP antibody. Thanks to Stephen J. Lynch for technical contributions.
Footnotes
Published ahead of print on 30 May 2007.
REFERENCES
- 1.Ambroziak, J., D. Blackbourn, B. Herndier, R. Glogan, J. Gullet, A. McDonald, E. Lennette, and J. Levy. 1995. Herpesvirus-like sequences in HIV-infected and uninfected Kaposi's sarcoma patients. Science 268:582-583. [DOI] [PubMed] [Google Scholar]
- 2.apRhys, C. M., D. M. Ciufo, E. A. O'Neill, T. J. Kelly, and G. S. Hayward. 1989. Overlapping octamer and TAATGARAT motifs in the VF65-response elements in herpes simplex virus immediate-early promoters represent independent binding sites for cellular nuclear factor III. J. Virol. 63:2798-2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baumruker, T., R. Sturm, and W. Herr. 1988. OBP100 binds remarkably degenerate octamer motifs through specific interactions with flanking sequences. Genes Dev. 2:1400-1413. [DOI] [PubMed] [Google Scholar]
- 4.Bechtel, J. T., R. C. Winant, and D. Ganem. 2005. Host and viral proteins in the virion of Kaposi's sarcoma-associated herpesvirus. J. Virol. 79:4952-4964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bigoni, B., R. Dolcetti, L. de Lellis, A. Carbone, M. Boiocchi, E. Cassai, and D. Di Luca. 1996. Human herpesvirus 8 is present in the lymphoid system of healthy persons and can reactivate in the course of AIDS. J. Infect. Dis. 173:542-549. [DOI] [PubMed] [Google Scholar]
- 6.Blackbourn, D. J., J. Ambroziak, E. Lennette, M. Adams, B. Ramachandran, and J. A. Levy. 1997. Infectious human herpesvirus 8 in a healthy North American blood donor. Lancet 349:609-611. [DOI] [PubMed] [Google Scholar]
- 7.Blasig, C., C. Zietz, B. Haar, F. Neipel, S. Esser, N. H. Brockmeyer, E. Tschachler, S. Colombini, B. Ensoli, and M. Sturzl. 1997. Monocytes in Kaposi's sarcoma lesions are productively infected by human herpesvirus 8. J. Virol. 71:7963-7968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boshoff, C., T. F. Schulz, M. M. Kennedy, A. K. Graham, C. Fisher, A. Thomas, J. O. McGee, R. A. Weiss, and J. J. O'Leary. 1995. Kaposi's sarcoma-associated herpesvirus infects endothelial and spindle cells. Nat. Med. 1:1274-1278. [DOI] [PubMed] [Google Scholar]
- 9.Bowser, B. S., S. M. DeWire, and B. Damania. 2002. Transcriptional regulation of the K1 gene product of Kaposi's sarcoma-associated herpesvirus. J. Virol. 76:12574-12583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bowser, B. S., S. Morris, M. J. Song, R. Sun, and B. Damania. 2006. Characterization of Kaposi's sarcoma-associated herpesvirus (KSHV) K1 promoter activation by Rta. Virology 348:309-327. [DOI] [PubMed] [Google Scholar]
- 11.Bruggemeier, U., M. Kalff, S. Franke, C. Scheidereit, and M. Beato. 1991. Ubiquitous transcription factor OTF-1 mediates induction of the MMTV promoter through synergistic interaction with hormone receptors. Cell 64:565-572. [DOI] [PubMed] [Google Scholar]
- 12.Bu, W., K. Driscoll, D. Palmeri, and D. M. Lukac. 2006. Kaposi's sarcoma-associated herpesvirus ORF50/Rta lytic switch protein functions as a tetramer. J. Virol. 81:5788-5806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carroll, K. D., W. Bu, D. Palmeri, S. Spadavecchia, S. J. Lynch, S. A. Marras, S. Tyagi, and D. M. Lukac. 2006. Kaposi's sarcoma-associated herpesvirus lytic switch protein stimulates DNA binding of RBP-Jk/CSL to activate the Notch pathway. J. Virol. 80:9697-9709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cesarman, E., Y. Chang, P. S. Moore, J. W. Said, and D. M. Knowles. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N. Engl. J. Med. 332:1186-1191. [DOI] [PubMed] [Google Scholar]
- 15.Cesarman, E., and E. Mesri. 2000. Viral G protein-coupled receptor and Kaposi's sarcoma: a model of paracrine neoplasia? J. Exp. Med. 191:417-422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chang, P. J., D. Shedd, L. Gradoville, M.-S. Cho, L.-W. Chen, J. Chang, and G. Miller. 2002. Open reading frame 50 protein of Kaposi's sarcoma-associated herpesvirus directly activates the viral PAN and K12 genes by binding to related response elements. J. Virol. 76:3168-3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chang, Y., E. Cesarman, M. S. Pessin, F. Lee, J. Culpepper, D. M. Knowles, and P. S. Moore. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266:1865-1869. [DOI] [PubMed] [Google Scholar]
- 18.Chen, J., K. Ueka, S. Sakakibara, T. Okuno, and K. Yamanishi. 2000. Transcriptional regulation of the Kaposi's sarcoma-associated herpesvirus viral interferon regulatory factor gene. J. Virol. 74:8623-8634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Deng, H., M. J. Song, J. T. Chu, and R. Sun. 2002. Transcriptional regulation of the interleukin-6 gene of human herpesvirus 8 (Kaposi's sarcoma-associated herpesvirus). J. Virol. 76:8252-8264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dittmer, D., C. Stoddart, R. Renne, V. Linquist-Stepps, M. E. Moreno, C. Bare, J. M. McCune, and D. Ganem. 1999. Experimental transmission of Kaposi's sarcoma-associated herpesvirus (KSHV/HHV-8) to SCID-hu Thy/Liv mice. J. Exp. Med. 190:1857-1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dourmishev, L. A., A. L. Dourmishev, D. Palmeri, R. A. Schwartz, and D. M. Lukac. 2003. Molecular genetics of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) epidemiology and pathogenesis. Microbiol. Mol. Biol. Rev. 67:175-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ensoli, B., M. Sturzl, and P. Monini. 2000. Cytokine-mediated growth promotion of Kaposi's sarcoma and primary effusion lymphoma. Semin. Cancer Biol. 10:367-381. [DOI] [PubMed] [Google Scholar]
- 23.Fakhari, F. D., and D. P. Dittmer. 2002. Charting latency transcripts in Kaposi's sarcoma-associated herpesvirus by whole-genome real-time quantitative PCR. J. Virol. 76:6213-6223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Freiman, R. N., and W. Herr. 1997. Viral mimicry: common mode of association with HCF by VP16 and the cellular protein LZIP. Genes Dev. 11:3122-3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fryer, C. J., J. B. White, and K. A. Jones. 2004. Mastermind recruits CycC:CDK8 to phosphorylate the Notch ICD and coordinate activation with turnover. Mol. Cell 16:509-520. [DOI] [PubMed] [Google Scholar]
- 26.Gaffney, D. F., J. McLauchlan, J. L. Whitton, and J. B. Clements. 1985. A modular system for the assay of transcription regulatory signals: the sequence TAATGARAT is required for herpes simplex virus immediate early gene activation. Nucleic Acids Res. 13:7847-7863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gao, S. J., L. Kingsley, M. Li, W. Zheng, C. Parravicini, J. Ziegler, R. Newton, C. R. Rinaldo, A. Saah, J. Phair, R. Detels, Y. Chang, and P. S. Moore. 1996. KSHV antibodies among Americans, Italians and Ugandans with and without Kaposi's sarcoma. Nat. Med. 2:925-928. [DOI] [PubMed] [Google Scholar]
- 28.Gerster, T., and R. G. Roeder. 1988. A herpesvirus trans-activating protein interacts with transcription factor OTF-1 and other cellular proteins. Proc. Natl. Acad. Sci. USA 85:6347-6351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gradoville, L., J. Gerlach, E. Grogan, D. Shedd, S. Nikiforow, C. Metroka, and G. Miller. 2000. Kaposi's sarcoma-associated herpesvirus open reading frame 50/Rta protein activates the entire lytic cycle in the HH-B2 primary effusion lymphoma cell line. J. Virol. 74:6207-6212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harrington, W. J., Jr., O. Bagasra, C. E. Sosa, L. E. Bobroski, M. Baum, X. L. Wen, L. Cabral, G. E. Byrne, R. J. Pomerantz, and C. Wood. 1996. Human herpesvirus type 8 DNA sequences in cell-free plasma and mononuclear cells of Kaposi's sarcoma patients. J. Infect. Dis. 174:1101-1105. [DOI] [PubMed] [Google Scholar]
- 31.Honess, R. W., and B. Roizman. 1974. Regulation of herpesvirus macromolecular synthesis. I. Cascade regulation of the synthesis of three groups of viral proteins. J. Virol. 14:8-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jenkins, P., U. Binne, and P. Farrell. 2000. Histone acetylation and reactivation of Epstein-Barr virus from latency. J. Virol. 74:710-720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jenner, R., M. Alba, C. Boshoff, and P. Kellam. 2001. Kaposi's sarcoma-associated herpesvirus latent and lytic gene expression as revealed by DNA arrays. J. Virol. 75:891-902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kedes, D., E. Operskalski, M. Busch, R. Kohn, J. Flood, and D. Ganem. 1996. The seroepidemiology of human herpesvirus 8 (Kaposi's sarcoma-associated herpesvirus): distribution of infection in KS risk groups and evidence for sexual transmission. Nat. Med. 2:918-924. [DOI] [PubMed] [Google Scholar]
- 35.Kikuta, H., O. Itakura, K. Taneichi, and M. Kohno. 1997. Tropism of human herpesvirus 8 for peripheral blood lymphocytes in patients with Castleman's disease. Br. J. Haematol. 99:790-793. [DOI] [PubMed] [Google Scholar]
- 36.Kim, S. Y., A. Herbst, K. A. Tworkowski, S. E. Salghetti, and W. P. Tansey. 2003. Skp2 regulates Myc protein stability and activity. Mol. Cell 11:1177-1188. [DOI] [PubMed] [Google Scholar]
- 37.Kirshner, J. R., D. M. Lukac, J. Chang, and D. Ganem. 2000. Kaposi's sarcoma-associated herpesvirus open reading frame 57 encodes a posttranscriptional regulator with multiple distinct activities. J. Virol. 74:3586-3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kirshner, J. R., K. Staskus, A. Haase, M. Lagunoff, and D. Ganem. 1999. Expression of the open reading frame 74 (G-protein-coupled receptor) gene of Kaposi's sarcoma (KS)-associated herpesvirus: implications for KS pathogenesis. J. Virol. 73:6006-6014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Klemm, J. D., M. A. Rould, R. Aurora, W. Herr, and C. O. Pabo. 1994. Crystal structure of the Oct-1 POU domain bound to an octamer site: DNA recognition with tethered DNA-binding modules. Cell 77:21-32. [DOI] [PubMed] [Google Scholar]
- 40.Kristie, T. M., J. H. LeBowitz, and P. A. Sharp. 1989. The octamer-binding proteins form multi-protein-DNA complexes with the HSV alpha TIF regulatory protein. EMBO J. 8:4229-4238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kristie, T. M., and B. Roizman. 1988. Differentiation and DNA contact points of host proteins binding at the cis site for virion-mediated induction of alpha genes of herpes simplex virus 1. J. Virol. 62:1145-1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kristie, T. M., and B. Roizman. 1987. Host cell proteins bind to the cis-acting site required for virion-mediated induction of herpes simplex virus 1 alpha genes. Proc. Natl. Acad. Sci. USA 84:71-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.La Boissiere, S., T. Hughes, and P. O'Hare. 1999. HCF-dependent nuclear import of VP16. EMBO J. 18:480-489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lai, J. S., and W. Herr. 1997. Interdigitated residues within a small region of VP16 interact with Oct-1, HCF, and DNA. Mol. Cell. Biol. 17:3937-3946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li, T., M. Stark, A. Johnson, and C. Wolberger. 1995. Crystal structure of the MATa1/MATa2 homeodomain heterodimer bound to DNA. Science 270:262-269. [DOI] [PubMed] [Google Scholar]
- 46.Liang, Y., J. Chang, S. Lynch, D. M. Lukac, and D. Ganem. 2002. The lytic switch protein of KSHV activates gene expression via functional interaction with RBP-Jk, the target of the Notch signaling pathway. Genes Dev. 16:1977-1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liang, Y., and D. Ganem. 2003. Lytic but not latent infection by Kaposi's sarcoma-associated herpesvirus requires host CSL protein, the mediator of Notch signaling. Proc. Natl. Acad. Sci. USA 100:8490-8495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liang, Y., and D. Ganem. 2004. RBP-J (CSL) is essential for activation of the K14/vGPCR promoter of Kaposi's sarcoma-associated herpesvirus by the lytic switch protein RTA. J. Virol. 78:6818-6826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liao, W., Y. Tang, Y. L. Kuo, B. Y. Liu, C. J. Xu, and C. Z. Giam. 2003. Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8 transcriptional activator Rta is an oligomeric DNA-binding protein that interacts with tandem arrays of phased A/T-trinucleotide motifs. J. Virol. 77:9399-9411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lipford, J. R., G. T. Smith, Y. Chi, and R. J. Deshaies. 2005. A putative stimulatory role for activator turnover in gene expression. Nature 438:113-116. [DOI] [PubMed] [Google Scholar]
- 51.Lu, M., J. Suen, C. Frias, R. Pfeiffer, M. H. Tsai, E. Chuang, and S. L. Zeichner. 2004. Dissection of the Kaposi's sarcoma-associated herpesvirus gene expression program by using the viral DNA replication inhibitor cidofovir. J. Virol. 78:13637-13652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lukac, D., L. Garibyan, J. Kirshner, D. Palmeri, and D. Ganem. 2001. DNA binding by the Kaposi's sarcoma-associated herpesvirus lytic switch protein is necessary for transcriptional activation of two viral delayed early promoters. J. Virol. 75:6786-6799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lukac, D. M., J. R. Kirshner, and D. Ganem. 1999. Transcriptional activation by the product of open reading frame 50 of Kaposi's sarcoma-associated herpesvirus is required for lytic viral reactivation in B cells. J. Virol. 73:9348-9361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lukac, D. M., R. Renne, J. R. Kirshner, and D. Ganem. 1998. Reactivation of Kaposi's sarcoma-associated herpesvirus infection from latency by expression of the ORF 50 transactivator, a homolog of the EBV R protein. Virology 252:304-312. [DOI] [PubMed] [Google Scholar]
- 55.Mackem, S., and B. Roizman. 1982. Differentiation between alpha promoter and regulator regions of herpes simplex virus 1: the functional domains and sequence of a movable alpha regulator. Proc. Natl. Acad. Sci. USA 79:4917-4921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mackem, S., and B. Roizman. 1980. Regulation of herpesvirus macromolecular synthesis: transcription-initiation sites and domains of alpha genes. Proc. Natl. Acad. Sci. USA 77:7122-7126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mackem, S., and B. Roizman. 1982. Structural features of the herpes simplex virus alpha gene 4, 0, and 27 promoter-regulatory sequences which confer alpha regulation on chimeric thymidine kinase genes. J. Virol. 44:939-949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Martin, D. F., B. D. Kuppermann, R. A. Wolitz, A. G. Palestine, H. Li, C. A. Robinson, et al. 1999. Oral ganciclovir for patients with cytomegalovirus retinitis treated with a ganciclovir implant. N. Engl. J. Med. 340:1063-1070. [DOI] [PubMed] [Google Scholar]
- 59.Miller, G., L. Heston, E. Grogan, L. Gradoville, M. Rigsby, R. Sun, D. Shedd, V. M. Kushnaryov, S. Grossberg, and Y. Chang. 1997. Selective switch between latency and lytic replication of Kaposi's sarcoma herpesvirus and Epstein-Barr virus in dually infected body cavity lymphoma cells. J. Virol. 71:314-324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Moriuchi, H., M. Moriuchi, and J. I. Cohen. 1995. Proteins and cis-acting elements associated with transactivation of the varicella-zoster virus (VZV) immediate-early gene 62 promoter by VZV open reading frame 10 protein. J. Virol. 69:4693-4701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nador, R. G., E. Cesarman, A. Chadburn, D. B. Dawson, M. Q. Ansari, J. Sald, and D. M. Knowles. 1996. Primary effusion lymphoma: a distinct clinicopathologic entity associated with the Kaposi's sarcoma-associated herpes virus. Blood 88:645-656. [PubMed] [Google Scholar]
- 62.Nogueira, M. L., V. E. Wang, D. Tantin, P. A. Sharp, and T. M. Kristie. 2004. Herpes simplex virus infections are arrested in Oct-1-deficient cells. Proc. Natl. Acad. Sci. USA 101:1473-1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.O'Hare, P., and C. R. Goding. 1988. Herpes simplex virus regulatory elements and the immunoglobulin octamer domain bind a common factor and are both targets for virion transactivation. Cell 52:435-445. [DOI] [PubMed] [Google Scholar]
- 64.Oka, C., T. Nakano, A. Wakeham, J. L. de la Pompa, C. Mori, T. Sakai, S. Okazaki, M. Kawaichi, K. Shiota, T. W. Mak, and T. Honjo. 1995. Disruption of the mouse RBP-J kappa gene results in early embryonic death. Development 121:3291-3301. [DOI] [PubMed] [Google Scholar]
- 65.Pabst, T., B. U. Mueller, P. Zhang, H. S. Radomska, S. Narravula, S. Schnittger, G. Behre, W. Hiddemann, and D. G. Tenen. 2001. Dominant-negative mutations of CEBPA, encoding CCAAT/enhancer binding protein-alpha (C/EBPalpha), in acute myeloid leukemia. Nat. Genet. 27:263-270. [DOI] [PubMed] [Google Scholar]
- 66.Paulose-Murphy, M., N.-K. Ha, C. Xiang, Y. Chen, L. Gillim, R. Yarchoan, P. Meltzer, M. Bittner, J. Trent, and S. Zeichner. 2001. Transcription program of human herpesvirus 8 (Kaposi's sarcoma-associated herpesvirus). J. Virol. 75:4843-4853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Perissi, V., A. Aggarwal, C. K. Glass, D. W. Rose, and M. G. Rosenfeld. 2004. A corepressor/coactivator exchange complex required for transcriptional activation by nuclear receptors and other regulated transcription factors. Cell 116:511-526. [DOI] [PubMed] [Google Scholar]
- 68.Phillips, K., and B. Luisi. 2000. The virtuoso of versatility: POU proteins that flex to fit. J. Mol. Biol. 302:1023-1039. [DOI] [PubMed] [Google Scholar]
- 69.Preston, C. M., M. C. Frame, and M. E. Campbell. 1988. A complex formed between cell components and an HSV structural polypeptide binds to a viral immediate early gene regulatory DNA sequence. Cell 52:425-434. [DOI] [PubMed] [Google Scholar]
- 70.Renne, R., M. Lagunoff, W. Zhong, and D. Ganem. 1996. The size and conformation of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) DNA in infected cells and virions. J. Virol. 70:8151-8154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Renne, R., W. Zhong, B. Herndier, M. McGrath, N. Abbey, D. Kedes, and D. Ganem. 1996. Lytic growth of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) in culture. Nat. Med. 2:342-346. [DOI] [PubMed] [Google Scholar]
- 72.Roizman, B., and D. M. Knipe. 2001. Herpes simplex viruses and their replication, p. 2399-2459. In D. M. Knipe, P. M. Howley, et al. (ed.), Fields virology, 4th ed., vol. 2. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 73.Sakakibara, S., K. Ueda, J. Chen, T. Okuno, and K. Yamanishi. 2001. Octamer-binding sequence is a key element for the autoregulation of Kaposi's sarcoma-associated herpesvirus ORF50/Lyta gene expression. J. Virol. 75:6894-6900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Salghetti, S. E., M. Muratani, H. Wijnen, B. Futcher, and W. P. Tansey. 2000. Functional overlap of sequences that activate transcription and signal ubiquitin-mediated proteolysis. Proc. Natl. Acad. Sci. USA 97:3118-3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sarid, R., O. Flore, R. A. Bohenzky, Y. Chang, and P. S. Moore. 1998. Transcription mapping of the Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) genome in a body cavity-based lymphoma cell line (BC-1). J. Virol. 72:1005-1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Simpson, G. R., T. F. Schulz, D. Whitby, P. M. Cook, C. Boshoff, L. Rainbow, M. R. Howard, S. J. Gao, R. A. Bohenzky, P. Simmonds, C. Lee, A. de Ruiter, A. Hatzakis, R. S. Tedder, I. V. Weller, R. A. Weiss, and P. S. Moore. 1996. Prevalence of Kaposi's sarcoma associated herpesvirus infection measured by antibodies to recombinant capsid protein and latent immunofluorescence antigen. Lancet 348:1133-1138. [DOI] [PubMed] [Google Scholar]
- 77.Song, M. J., H. Deng, and R. Sun. 2003. Comparative study of regulation of RTA-responsive genes in Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8. J. Virol. 77:9451-9462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Song, M. J., S. Hwang, W. Wong, J. Round, D. Martinez-Guzman, Y. Turpaz, J. Liang, B. Wong, R. C. Johnson, M. Carey, and R. Sun. 2004. The DNA architectural protein HMGB1 facilitates RTA-mediated viral gene expression in gamma-2 herpesviruses. J. Virol. 78:12940-12950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Song, M. J., X. Li, H. J. Brown, and R. Sun. 2002. Characterization of interactions between RTA and the promoter of polyadenylated nuclear RNA in Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8. J. Virol. 76:5000-5013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Spear, P. G., and B. Roizman. 1972. Proteins specified by herpes simplex virus. V. Purification and structural proteins of the herpesvirion. J. Virol. 9:143-159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Staskus, K. A., W. Zhong, K. Gebhard, B. Herndier, H. Wang, R. Renne, J. Beneke, J. Pudney, D. J. Anderson, D. Ganem, and A. T. Haase. 1997. Kaposi's sarcoma-associated herpesvirus gene expression in endothelial (spindle) tumor cells. J. Virol. 71:715-719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Stern, S., and W. Herr. 1991. The herpes simplex virus trans-activator VP16 recognizes the Oct-1 homeodomain: evidence for a homeo domain recognition subdomain. Genes Dev. 5:2555-2566. [DOI] [PubMed] [Google Scholar]
- 83.Sun, R., S.-F. Lin, K. Staskus, L. Gradoville, E. Grogan, A. Haase, and G. Miller. 1999. Kinetics of Kaposi's sarcoma-associated herpesvirus gene expression. J. Virol. 73:2232-2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sun, R., S. F. Lin, L. Gradoville, Y. Yuan, F. Zhu, and G. Miller. 1998. A viral gene that activates lytic cycle expression of Kaposi's sarcoma-associated herpesvirus. Proc. Natl. Acad. Sci. USA 95:10866-10871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tanaka, M., and W. Herr. 1990. Differential transcriptional activation by Oct-1 and Oct-2: interdependent activation domains induce Oct-2 phosphorylation. Cell 60:375-386. [DOI] [PubMed] [Google Scholar]
- 86.Triezenberg, S. J., K. L. LaMarco, and S. L. McKnight. 1988. Evidence of DNA:protein interactions that mediate HSV-1 immediate early gene activation by VP16. Genes Dev. 2:730-742. [DOI] [PubMed] [Google Scholar]
- 87.Ueda, K., K. Ishikawa, K. Nishimura, S. Sakakibara, E. Do, and K. Yamanishi. 2002. Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) replication and transcription factor activates the K9 (vIRF) gene through two distinct cis elements by a non-DNA-binding mechanism. J. Virol. 76:12044-12054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Verma, S. C., K. Lan, T. Choudhuri, and E. S. Robertson. 2006. Kaposi's sarcoma-associated herpesvirus-encoded latency-associated nuclear antigen modulates K1 expression through its cis-acting elements within the terminal repeats. J. Virol. 80:3445-3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Verrijzer, C. P., J. A. van Oosterhout, W. W. van Weperen, and P. C. van der Vliet. 1991. POU proteins bend DNA via the POU-specific domain. EMBO J. 10:3007-3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Walker, S., S. Hayes, and P. O'Hare. 1994. Site-specific conformational alteration of the Oct-1 POU domain-DNA complex as the basis for differential recognition by Vmw65 (VP16). Cell 79:841-852. [DOI] [PubMed] [Google Scholar]
- 91.Wang, S. E., F. Y. Wu, H. Chen, M. Shamay, Q. Zheng, and G. S. Hayward. 2004. Early activation of the Kaposi's sarcoma-associated herpesvirus RTA, RAP, and MTA promoters by the tetradecanoyl phorbol acetate-induced AP1 pathway. J. Virol. 78:4248-4267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wang, S. E., F. Y. Wu, M. Fujimuro, J. Zong, S. D. Hayward, and G. S. Hayward. 2003. Role of CCAAT/enhancer-binding protein alpha (C/EBPα) in activation of the Kaposi's sarcoma-associated herpesvirus (KSHV) lytic-cycle replication-associated protein (RAP) promoter in cooperation with the KSHV replication and transcription activator (RTA) and RAP. J. Virol. 77:600-623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wang, S. E., F. Y. Wu, Y. Yu, and G. S. Hayward. 2003. CCAAT/enhancer-binding protein-α is induced during the early stages of Kaposi's sarcoma-associated herpesvirus (KSHV) lytic cycle reactivation and together with the KSHV replication and transcription activator (RTA) cooperatively stimulates the viral RTA, MTA, and PAN promoters. J. Virol. 77:9590-9612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wang, V. E., T. Schmidt, J. Chen, P. A. Sharp, and D. Tantin. 2004. Embryonic lethality, decreased erythropoiesis, and defective octamer-dependent promoter activation in Oct-1-deficient mice. Mol. Cell. Biol. 24:1022-1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wang, Y., O. T. Chong, and Y. Yuan. 2004. Differential regulation of K8 gene expression in immediate-early and delayed-early stages of Kaposi's sarcoma-associated herpesvirus. Virology 325:149-163. [DOI] [PubMed] [Google Scholar]
- 96.Wang, Y., H. Li, M. Y. Chan, F. X. Zhu, D. M. Lukac, and Y. Yuan. 2004. Kaposi's sarcoma-associated herpesvirus ori-Lyt-dependent DNA replication: cis-acting requirements for replication and ori-Lyt-associated RNA transcription. J. Virol. 78:8615-8629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wang, Y., and Y. Yuan. 2007. Essential role of RBP-Jkappa in activation of the K8 delayed-early promoter of Kaposi's sarcoma-associated herpesvirus by ORF50/RTA. Virology 359:19-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Whitby, D., M. R. Howard, M. Tenant-Flowers, N. S. Brink, A. Copas, C. Boshoff, T. Hatzioannou, F. E. Suggett, D. M. Aldam, A. S. Denton, et al. 1995. Detection of Kaposi sarcoma associated herpesvirus in peripheral blood of HIV-infected individuals and progression to Kaposi's sarcoma. Lancet 346:799-802. [DOI] [PubMed] [Google Scholar]
- 99.Wingender, E., X. Chen, R. Hehl, H. Karas, I. Liebich, V. Matys, T. Meinhardt, M. Prub, I. Reuter, and F. Schacherer. 2000. TRANSFAC: an integrated system for gene expression regulation. Nucleic Acids Res. 28:316-319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wu, L., R. Renne, D. Ganem, and B. Forghani. 2000. Human herpesvirus 8 glycoprotein K8.1: expression, post-translational modification and localization analyzed by monoclonal antibody. J. Clin. Virol. 17:127-136. [DOI] [PubMed] [Google Scholar]
- 101.Wysocka, J., and W. Herr. 2003. The herpes simplex virus VP16-induced complex: the makings of a regulatory switch. Trends Biochem. Sci. 28:294-304. [DOI] [PubMed] [Google Scholar]
- 102.Xu, Y., D. P. AuCoin, A. R. Huete, S. A. Cei, L. J. Hanson, and G. S. Pari. 2005. A Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8 ORF50 deletion mutant is defective for reactivation of latent virus and DNA replication. J. Virol. 79:3479-3487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Yu, Y., S. E. Wang, and G. S. Hayward. 2005. The KSHV immediate-early transcription factor RTA encodes ubiquitin E3 ligase activity that targets IRF7 for proteosome-mediated degradation. Immunity 22:59-70. [DOI] [PubMed] [Google Scholar]
- 104.Zhang, L., J. Chiu, and J. C. Lin. 1998. Activation of human herpesvirus 8 (HHV-8) thymidine kinase (TK) TATAA-less promoter by HHV-8 ORF50 gene product is SP1 dependent. DNA Cell Biol. 17:735-742. [DOI] [PubMed] [Google Scholar]
- 105.Zhong, W., H. Wang, B. Herndier, and D. Ganem. 1996. Restricted expression of Kaposi sarcoma-associated herpesvirus (human herpesvirus 8) genes in Kaposi sarcoma. Proc. Natl. Acad. Sci. USA 93:6641-6646. [DOI] [PMC free article] [PubMed] [Google Scholar]