

Abstract
We show here that the varicella-zoster virus (VZV) open reading frame 66 (ORF66) protein kinase is one mechanism employed to reduce class I major histocompatibility complex (MHC-I) surface expression in VZV-infected cells. Cells expressing enhanced green fluorescent protein-tagged functional and inactivated ORF66 (GFP-66 and GFP-66kd) from replication-defective adenovirus vectors revealed that ORF66 reduced MHC-I surface levels in a manner dependent on kinase activity. Cells infected with recombinant VZV expressing GFP-66 exhibited a significantly greater reduction in MHC-I surface expression than that observed in cells infected with VZV disrupted in GFP-66 expression. MHC-I maturation was delayed in its transport from the endoplasmic reticulum through the Golgi in both adenovirus-transduced cells expressing only GFP-66 and in VZV-infected cells expressing high levels of GFP-66, and this was predominantly kinase dependent. MHC-I levels were reduced in VZV-infected cells, and analyses of intracellular MHC-I revealed accumulation of folded MHC-I in the Golgi region, irrespective of ORF66 expression. Thus, the ORF66 kinase is important for VZV-mediated MHC-I downregulation, but additional mechanisms also may be involved. Analyses of the VZV ORF9a protein, the ortholog of the bovine herpesvirus 1 transporter associated with antigen processing inhibitor UL49.5 revealed no effects on MHC-I. These results establish a new role for viral protein kinases in immune evasion and suggest that VZV utilizes unique mechanisms to inhibit antigen presentation.
Varicella-zoster virus (VZV) is the human-restricted member of the herpesvirus subfamily Alphaherpesvirinae and causes chicken pox upon primary infection and herpes zoster (shingles) following reactivation from a prolonged period of neuronal latency in the sensory ganglia. Based on the current model of pathogenesis (38), efficient dissemination, disease, and establishment of latency within the infected host requires VZV growth in multiple cell types. Following inhalation, VZV is spread to epidermal sites of replication by a T-lymphocyte-associated viremia. Infection likely occurs in the tonsils, with VZV preferentially infecting memory CD4+ T lymphocytes (39). These T cells can home to and mediate infection of human skin allografts in the severe combined immunodeficient (SCID)-hu model of VZV infection (40). In humans, skin lesions occur 10 to 21 days after the initial inoculation, and VZV DNA is detected in peripheral blood mononuclear cells during primary infection (13, 45). VZV accesses axons of innervating sensory neurons in the skin and establishes a life-long latent infection in neurons of the dorsal root ganglia. In contrast with latency of the closely related herpes simplex virus type 1 (HSV-1), VZV latency is characterized by persistent transcription, and possibly expression, of several viral lytic genes, including open reading frames (ORFs) 4, 21, 29, 62, 63, and 66 (10, 11, 47). This does not appear to stimulate a ganglionic infiltration of T cells (28, 62). VZV may also productively or abortively infect antigen-presenting cells, including immature and mature dendritic cells (2, 27, 50), monocytes, macrophages, and B lymphocytes (4, 20, 30, 35).
The lymphotrophic nature of VZV and its ability to sustain infection in multiple cell types over a prolonged period suggest that VZV pathogenesis benefits from immune evasion mechanisms that limit antigen presentation. Several reports have also indicated a possible chronic, noninflammatory expression of viral lytic antigens during latency (10, 11, 47, 62), which may be suggestive of ongoing virus-directed immune evasion. Accordingly, VZV reduces the surface expression of CD8+ T lymphocyte (CTL)-restricted class I major histocompatibility complexes (MHC-I), both in cultured cells and in thymic T cells in the SCID-hu model (1, 9). Two proteins from closely related alphaherpesviruses, HSV ICP47 and bovine herpesvirus 1 (BHV-1) UL49.5, inhibit the TAP complex using distinct mechanisms to prevent peptide transport into the lumen of the endoplasmic reticulum (ER) (3, 36, 61). VZV does not encode an ICP47 ortholog, and no function in modulation of antigen presentation has been shown for the VZV UL49.5 ortholog, ORF9a (36). Furthermore, in VZV-infected cells, it has been suggested that MHC-I is processed through the cis/medial-Golgi normally but is prevented from accessing the cell surface (1). These observations imply that VZV-encoded mechanisms of MHC-I surface downregulation may be different from those of HSV-1, HSV-2, and BHV-1.
A previous study reported that transient expression of the VZV ORF66 protein kinase resulted in reduced MHC-I surface expression in human foreskin fibroblasts (HFF) (1). The ORF66 protein is a serine/threonine (Ser/Thr)-specific protein kinase with homology to the alphaherpesvirus US3 kinase family. Similar to other US3 kinases, ORF66 recognizes and phosphorylates serines preceded by several basic amino acids (15). While VZV that does not express ORF66 is only moderately impaired for replication in most cell cultures, ORF66 expression is required for efficient replication in cultured T lymphocytes and in thy-liv implants in the SCID-hu model (49, 57, 58). The only known target of ORF66 is the VZV major transcriptional regulatory protein, IE62. ORF66 phosphorylates IE62 adjacent to its nuclear localization signal, leading to IE62 cytoplasmic accumulation in late-stage VZV-infected cells and allowing for IE62 inclusion into the virion tegument (15, 33, 34). However, cellular targets of the ORF66 protein kinase have been suggested from recent studies, in that ORF66 may contribute to antiapoptotic mechanisms and evasion of a host cell response to gamma interferon (γ-IFN) (57).
In this work, the effects of the ORF66 kinase on MHC-I surface expression were characterized both in the context of VZV infection and in the absence of other VZV proteins. Using novel adenovirus vectors and recombinant VZV expressing functional or altered enhanced green fluorescent protein (EGFP)-tagged ORF66 kinase genes, we show that MHC-I surface expression is downregulated by ORF66 in a kinase-dependent manner and that ORF66 contributes to MHC-I downregulation during VZV infection. The ORF66 kinase activity delayed MHC-I transport through the cis/medial-Golgi complex. Inhibition of MHC-I surface expression by intracellular retention in the early secretory compartment is a function that has not been attributed to any other viral protein kinase and represents a novel role for a US3 family kinase in regulating the cellular environment during viral infection. We also show that VZV has ORF66-independent mechanisms that contribute to reduced MHC-I surface expression.
MATERIALS AND METHODS
Cells.
The HEK 293 and HEK 293T cell lines (ATCC, Manassas, VA), MeWo cells (kindly provided by C. Grose, University of Iowa, Iowa City), and MRC-5 human lung fibroblasts (ATCC) were maintained as described previously (15).
Antibodies.
The monoclonal antibodies used for flow cytometric analyses were anti-human transferrin receptor (TfR1 or CD71; clone T56/14; immunoglobulin G1 [IgG1]; Caltag Laboratories, Burlingame, CA), purified, R-phycoerythrin (R-PE), conjugated and used at a 1:100 dilution; an antibody recognizing class I heavy chains in complex with β2-microglobulin (β2-M; clone G46-2.6; IgG1κ; BD Biosciences Pharmingen, San Jose, CA), purified, PE-Cy5 conjugated, and used at a 1:20 dilution; and isotype control antibodies consisting of R-PE-conjugated IgG1 (Caltag Laboratories) and PE-Cy5-conjugated IgG1κ (BD Biosciences Pharmingen), used at the same respective dilutions. A rabbit polyclonal antibody recognizing VZV IE62 and a mouse monoclonal antibody that recognizes the 9-amino-acid epitope (YPYDVPDYA) from influenza virus hemagglutinin (HA) have been previously described (15). The mouse monoclonal antibody W6/32 was obtained from Santa Cruz Biotechnology (Santa Cruz, CA) and used at 1 μg/per immunoprecipitation mixture, or at a dilution of 1:50 for immunofluorescence. Total class I heavy chains were immunoprecipitated with a polyclonal rabbit antibody to class I heavy chain cytoplasmic tail, designated UCSF2, at 1 μl per reaction mixture (a kind gift of R. Salter, University of Pittsburgh). Golgi complexes were identified using rabbit anti-mannosidase II (Mann II) at a 1:50 dilution (Chemicon, Temecula, CA). A mouse monoclonal anti-α-tubulin antibody (Sigma-Aldrich, St. Louis, MO) was used at 1:2,000 for immunoblot assays and at 1 μg per immunoprecipitation mixture. Alexa Fluor-conjugated Fab fragments (goat anti-mouse or goat anti-rabbit; used at 1:400; Invitrogen Corp.) were used for secondary detection in immunofluorescence as previously detailed (15).
Plasmids.
All plasmids were purified using QIAGEN columns (QIAGEN, Inc., Valencia, CA), according to the manufacturer's instructions. All oligonucleotides were obtained from IDT Inc. (Coralville, IA). The proofreading polymerase Expand (Roche Applied Science, Indianapolis, IN) was used to generate all PCR fragments, and accuracy was verified by sequencing. pGK2-HA66 expressing N-terminal HA-tagged ORF66 and a similar plasmid expressing ORF66 kinase inactivated by D206E and K208R mutations (pGK2-HA66kd) have been detailed elsewhere (34). A new derivative, pGK2-HA66s, was generated from pGK2-HA66 by insertion of a double-stranded oligonucleotide that placed stop codons in all three reading frames (5′-CGCGCTAGACTAGTCTAG-3′) into the MluI site at ORF66 amino acid 84. To generate EGFP-tagged ORF66 fusion proteins, the HA-tagged ORF66 genes from pGK2-HA66, pGK2-HA66kd, and pGK2-HA66s were PCR amplified using primers that added a unique BglII site proximal to the initiating ATG and a HindIII site following the stop codon. Digested PCR products were cloned into pEGFP-C1 (Clontech, Mountain View, CA), and the resultant plasmids are referred to as pGFP-66, pGFP-66kd, and pGFP-66s. A plasmid expressing IE62 with an in-frame carboxyl-terminal dsRed2 fusion inserted at amino acid 1309 (pCMV62-dsRed2) was generated in the previously described pK-CMV62 backbone (34), as follows. An MluI-Tth111I fragment representing the C-terminal portion of IE62 was replaced with a PCR-generated product that inserted unique AvrII and BglII sites at the end of the IE62 open reading frame. This allowed the in-frame insertion of an NheI-BglII fragment containing the entire dsRed2 gene from pdsRed2-N1 (Clontech), at the unique AvrII-BglII sites. To express VZV ORF9a and ORF50, each gene was PCR amplified using extended oligonucleotide primers to add EcoRI and NotI overhangs and subsequently cloned into pCDNA3.1 (Invitrogen Corp.) to generate pKORF9a and pKORF50. The transfection reporter plasmid pTK-EGFP, which expresses EGFP under the control of the HSV-1 thymidine kinase (TK) promoter, has been described in detail elsewhere (12). The plasmid expressing the K3 gene from Kaposi's sarcoma herpesvirus (KSHV) in the pCDNA3.1 backbone was a kind gift from F. Jenkins, University of Pittsburgh, Pittsburgh, PA.
Derivation of replication-defective adenoviruses.
Replication-defective adenoviruses expressing GFP-66 or GFP-66kd (referred to as Ad.GFP-66 and Ad.GFP-66kd) were derived using the Adeno-X Tet-Off system (Clontech). Digestion of pGFP-66 and pGFP-66kd with NheI and HindIII released the genes for cloning into the adenovirus intermediate vector pTRE-Shuttle2. Genes were shuttled into the Adeno-X genome as detailed in the manufacturer's instructions, and recombinant adenoviruses were obtained following transfection into HEK 293 cells. All viruses were assayed for the absence of functional replication-competent viruses.
Derivation of recombinant VZV.
Recombinant VZV expressing GFP-66 or GFP-66s was constructed using the VZV parent Oka (pOka) cosmid system (Fig. 1C). To derive pvSpe23 cosmids in which the native ORF66 gene (nucleotides [nt] 113142 to 114323) was replaced by the fluorescent versions, a modified pUC19 vector was made in which the poly-linker between HindIII and EcoRI was replaced by a double-stranded oligonucleotide encoding AvrII and SgrAI sites. This enabled subcloning of the AvrII-SgrAI fragment from the pvSpe23 cosmid (nt 112957 to 117458; pUC19-pvSpe23) (Fig. 1D). Within this construct, the unique AvrII and BamHI (nt 114147) sites were used to remove the native ORF66 promoter and part of the gene. Next, the cytomegalovirus IE promoter in pGFP-66 and pGFP-66s was replaced with a PCR-generated DNA fragment of the ORF66 promoter from the AvrII site to the ORF66 ATG, to give plasmid p66prom-EGFP.C1-HA66 or -66s (Fig. 1D). AvrII and BamHI digestion of these constructs released the ORF66 promoter-GFP/HA-tagged partial ORF66 gene, which was then used to replace the AvrII-BamHI fragment in pUC19-pvSpe23 (Fig. 1D). The entire AvrII-SgrAI fragment was then cloned back into pvSpe23, and recombinant VZV was generated using the three additional pOka cosmids in MeWo cells, as detailed previously (51). VZV plaques exhibiting EGFP fluorescence were grown into stocks and assessed for purity and correct insertion.
FIG. 1.

Schematic for creation of VZV containing GFP-66 or GFP-66s in the native ORF66 locus. (A) Representation of the domains of the 393-amino-acid ORF66 protein, showing the positions of key amino acid residues and a BamHI restriction site, as detailed in the text. (B) Relative position of the ORF66 gene in the structure of the VZV genome. Shown are the unique long (UL) and unique short (US) regions as well as the terminal (TRL and TRS) and internal (IRL and IRS) repeats. (C) Relative positions of the four pOka cosmids used to construct recombinant VZV. Genome coordinates are given with respect to the VZV pOka sequence. (D) Strategy for derivation of recombinant VZV expressing the ORF66 gene as an EGFP-tagged fusion protein. The ORF66 promoter and ORF are fully contained within a unique AvrII-SgrAI fragment of pvSpe23, and this fragment was subcloned into a modified pUC19 vector. The ORF66 promoter-GFP-66 cassette was inserted into this subfragment by using AvrII-BamHI, and then the AvrII-SgrAI subfragment was recloned into pvSpe23 as described in Materials and Methods.
Transfections and infections.
Transfections of MeWo, HEK 293, and HEK 293T cells were performed using the Lipofectamine 2000 reagent (Invitrogen Corp.), according to the manufacturer's instructions and as detailed previously (15). Replication-defective adenovirus-mediated expression of ORF66 was achieved by infecting MRC-5 cells with 5 PFU/cell of trans-gene-expressing virus (Ad.GFP-66 or Ad.GFP-66kd) and 2.5 PFU/cell of Ad-Tet-Off, which expresses the tetracycline-regulated transactivator. Where indicated, 2 μg/ml doxycycline (DOX; Sigma-Aldrich) was added to the medium to fully repress trans-gene expression.
VZV growth curves were performed as detailed in similar studies (57). The exact infection level of the previously frozen input viruses was confirmed by titration on MeWo cells (inoculum) in duplicate. Following fixation in 1% paraformaldehyde, plaques were visualized using EGFP autofluorescence or following immunofluorescent detection of IE62. For VZV flow cytometry analyses, inoculants of VZV.GFP-66 and VZV.GFP-66s were prepared in MRC-5 cells to >75% GFP fluorescence (as gauged by UV microscopy) and mixed with uninfected cells at a ratio of 1 to 10. Cells were gently copelleted by low-speed centrifugation, incubated for 30 min at 25°C, plated, and placed at 37°C. For metabolic labeling studies, trypsinized VZV-infected MRC-5 cells were used to infect confluent fibroblast monolayers at a ratio of 1 infected cell to 3 uninfected cells, to yield 100% EGFP fluorescence by 36 h postinfection (hpi). Studies using 35S labeling components were initiated as detailed elsewhere in the text.
Flow cytometry and immunoblotting.
Cells for flow cytometry were harvested by dislodging following brief exposure to trypsin (MRC-5 cells) or 1× phosphate-buffered saline (PBS)-1 mM EDTA (293T cells). Cells were washed in fluorescence-activated cell sorter (FACS) staining buffer (FSB) (1) and Fc blocked with 15 μg of normal mouse IgG (Caltag Laboratories) for 10 min on ice. Cells were stained in FACS staining buffer containing fluorophore-conjugated antibodies, washed, and fixed in freshly prepared 1% paraformaldehyde in 1× PBS. Isotype control staining was performed for each antibody and each condition. Stained cells were analyzed using a Becton Dickinson FACSAria cell sorter, and data were analyzed using FACSDiva and WinMDI-2.8 software. Surface protein-negative and GFP-negative gates were set using isotype control antibodies and non-GFP-expressing cells. To determine the mean fluorescence intensity (MFI) value for a cell population, the isotype control MFI was subtracted from the test MFI within corresponding GFP-negative or -positive gates. MFI ratios were calculated by dividing the MFI of the test population (adenovirus infected or) by the MFI of the control population (mock infected plasmid transfected) after isotype control background values had been subtracted. Ratios were converted to log values before statistical analysis using a paired Student's t test. For analysis of protein expression in infected or transfected cells, lysates of equivalent numbers of cells from each condition were separated by 7.5% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and analyzed by immunoblotting as previously described (15). Where indicated, membranes were stripped with 0.2 M NaOH for 5 min, rinsed with distilled H2O, and reblotted. Digital images were acquired in the linear range of the film using an Epson Perfection 4990 photo scanner with Silver Fast Ai and Adobe Photoshop CS imaging software.
Immunofluorescence and microscopy.
For immunofluorescence, cells grown on glass coverslips were fixed in 4% paraformaldehyde for 15 min, permeabilized with 0.2% Triton X-100 for 2 min, and stained with the indicated antibodies as described previously (34). Cells for MHC-I localization studies were fixed with 2% paraformaldehyde for 10 min and permeabilized as just described. Where indicated, internal specific MHC-I was detected following a 30-s prefixation treatment with an acid elution buffer (50 mM glycine, 100 mM NaCl, pH 3.3) to remove β2-M from surface class I. Nuclei were stained using Hoechst 33258 (Sigma-Aldrich). Immunofluorescence in fixed cells was observed using a Nikon Eclipse TE2000-E epifluorescence microscope equipped with a xenon lamp and a 40× 1.3 numerical aperture oil objective and recorded with Metamorph 7 software. Where indicated, Z-stacks were acquired at the recommended spacing for each fluorescent channel, and AutoQuant 9.2 was used to deconvolve Z stacks in 10 iterations using a blind deconvolution algorithm. Confocal analyses were performed as previously described (15, 34).
Metabolic labeling and immunoprecipitations.
Pulse-labeling of cells and immunoprecipitations were carried out as previously described (32), with some modifications. Cells were first incubated with complete medium containing 1% fetal bovine serum and lacking cysteine and methionine (Cys/Met) for 20 min at 37°C and then pulsed for 15 min with 0.5 mCi/ml of 35S-labeled Cys/Met in Cys/Met-free medium (Expre35S35S mix; Perkin-Elmer, Waltham, MA). Protein labeling was terminated by incubation in growth medium containing a 10-fold excess of cold methionine and 10% fetal bovine serum and, where indicated, cells were chased in the same medium. Immunoprecipitations were obtained from soluble cell lysates by using an ice-cold NP-40 lysis buffer (32) containing a protease inhibitor cocktail (Complete, EDTA-free; Roche Diagnostics). Soluble fractions were equalized for trichloroacetic acid (TCA)-precipitable radioincorporation and then precleared with an unconjugated mix of proteins A and G and Sepharose (Sigma-Aldrich). Immunoprecipitations were collected using protein A/G-Sepharose and washed in ice-cold radioimmunoprecipitation assay buffer (32) containing protease inhibitors and then once in ice-cold 1× PBS. Precipitates were divided, and one aliquot was subjected to endoglycosidase H (endo H) digestion, according to the manufacturer's suggestions (New England Biolabs). Immunoprecipitates were resolved on 10 to 20% gradient Criterion SDS-PAGE gels (Bio-Rad Laboratories, Hercules, CA), transferred to polyvinylidene difluoride membranes, and analyzed on a Bio-Rad Molecular Imager FX using Quantity One software. In some cases, gels were dried and the signals were enhanced using En3hance solution (Perkin-Elmer, Waltham, MA), captured by autoradiography with Kodak Biomax MP film and Kodak Biomax LE Transcreens, and quantified using ImageJ software.
RESULTS
Expression of functional and point inactivated GFP-tagged ORF66 kinase proteins in VZV-permissive cell types.
An earlier study reported that HFF cells transfected with plasmids expressing ORF66 showed reduced surface MHC-I (1). In that study, it was unclear if the ORF66-induced downregulation of MHC-I surface expression was specific to ORF66-expressing cells, as the protein expression and transfection efficiency were not determined. In particular, HFF cells and lines such as VZV-permissive MRC-5 fibroblasts are considered highly refractory to efficient transfection. To address this issue, we developed EGFP-tagged forms of ORF66 in plasmids, in replication-defective adenoviruses, and in recombinant VZV.
To assess the functionality of an amino-terminally EGFP-tagged ORF66 (GFP-66), we examined its cellular distribution and ability to exclude IE62 from the nucleus in live cells. IE62 nuclear exclusion is dependent on the integrity of ORF66 kinase activity and is induced by a direct ORF66-mediated phosphorylation event adjacent to the IE62 nuclear localization signal (15, 34). We also tested a GFP-66 fusion protein in which two key residues in the ORF66 catalytic domain, which are nearly invariant in all Ser/Thr kinases, were conservatively mutated (D206E, K208R; referred to as GFP-66kd). The HA-tagged version of ORF66-D206E/K208R has been previously shown to lack the abilities to exclude IE62 from the nucleus and phosphorylate IE62 peptides (15, 34). For this, a live cell assay was developed, using a plasmid expressing IE62 with a carboxyl-terminal dsRed2 tag (IE62-dsRed2). In transfected MeWo cells, both GFP-66 and GFP-66kd accumulated in the nucleus but also displayed cytoplasmic forms (Fig. 2A, panels i and ii). When expressed alone, IE62-dsRed2 showed a distribution pattern identical to untagged IE62 and was predominantly nuclear (Fig. 2A, panel iii) (15, 34). When GFP-66 was coexpressed with IE62-dsRed2, most coexpressing cells exhibited strong nuclear exclusion of IE62-dsRed2, which partially overlapped with GFP-66 in the cytoplasm (Fig. 2B). IE62-dsRed2 nuclear exclusion did not occur in cells expressing GFP-66kd, indicating the activity was kinase dependent, as expected from previous studies (Fig. 2C). These results established that amino-terminal EGFP tagging of ORF66 did not interfere with its ability to affect IE62 cellular distribution and that ORF66 kinase activity was intact.
FIG. 2.

Live cell imaging to show that GFP-tagged ORF66 retains the ability to exclude IE62 from the nucleus. (A) Single panels showing cellular distributions of individually transfected GFP-66 (i), GFP-66kd (ii), and IE62-dsRed (iii). (B and C) Images of cells coexpressing IE62-dsRed2 and either GFP-66 (B) or GFP66kd (C). Panel i shows autofluorescence of GFP-66 and GFP-66kd, while panel ii shows IE62-dsRed2 autofluorescence from the same cell. All transfections were performed in MeWo cells, and autofluorescence was visualized and captured at 24 h posttransfection by confocal microscopy. Cells were transfected with expression plasmids at a 1:1 ratio.
The GFP-66 and GFP-66kd genes were subsequently inserted into replication-defective adenovirus vectors under the control of the DOX-repressible promoter (Ad.GFP-66 and Ad.GFP-66kd, respectively) to enable efficient expression in VZV-permissive fibroblasts. The adenovirus vector used lacked large portions of the E1 and E3 genome regions, including the MHC-I-modulating genes E1A and E3/gp19K (7, 18, 54, 63), and does not have any intrinsic effects on MHC-I surface expression. MRC-5 cells required relatively low multiplicities of infection (2 to 5 PFU/cell) to obtain efficient trans-gene expression in over 80% of cells. Expression was dependent on coinfection with adenovirus expressing the tetracycline tTa transactivator and the absence of DOX. Immunoblot analyses of Ad.GFP-66 and Ad.GFP-66kd cell lysates revealed proteins of the expected size (∼75 kDa) which reacted with both anti-HA and anti-GFP antibodies (Fig. 3C; anti-GFP blot not shown). The GFP-66kd protein displayed a slightly faster mobility in SDS-PAGE compared to GFP-66, as reported previously for HA-tagged ORF66kd protein (34, 56). We also verified that adenovirus transduction and GFP-66 or GFP-66kd expression did not result in increased levels of apoptotic cells by using flow cytometric analysis of annexin V surface staining and 7-amino actinomycin D uptake (data not shown). These vectors allow for both efficient expression and quantitative assessment of ORF66 expression in multiple cell types.
FIG. 3.

GFP-66 induces a kinase-dependent and specific downregulation of surface MHC-I. (A) MRC-5 cells were infected with Ad.GFP-66 or Ad.GFP-66kd in conjunction with Ad.Tet-Off, grown in the presence or absence of 2 μg/ml of DOX, and harvested at 36 h postinfection. Cells were stained with fluorophore-conjugated antibodies to MHC-I and TfR1 or isotype controls and analyzed using a Becton Dickinson FACSAria. Histograms of surface fluorescence for MHC-I and TfR1 are shown with the infection condition indicated above each. The shaded gray histograms represent infected cells that were incubated with 2 μg/ml DOX. Cells incubated in the absence of DOX were gated on GFP positivity and are represented by the solid black line histograms. The isotype control antibody levels are shown by dotted lines. (B) Graph depicting the average (± standard error of the mean) MFI ratios of MHC-I and TfR1 surface fluorescence from three independent and identical experiments. (C) Immunoblot analysis of an equal number of cells from the Ad.GFP-66 and Ad.GFP-66kd infections under the four experimental conditions. Blots were probed with antibodies to the HA epitope; the proteins reacting in the size range expected are shown.
GFP-66 induces kinase-dependent downregulation of MHC-I surface expression.
The Ad.GFP-66 and Ad.GFP-66kd viruses were next employed to assess the role of ORF66 kinase activity in downregulation of MHC-I surface expression. We report the studies here in VZV-permissive MRC-5 fibroblasts but have found similar effects in MeWo cells and 293T cells. Surface levels of MHC-I were compared to transferrin receptor (TfR1 or CD71), a control that is widely used in MHC-I surface studies.
MRC-5 fibroblasts infected with Ad.GFP-66 or Ad.GFP-66kd in the presence or absence of 2 μg/ml DOX were assessed for surface MHC-I and TfR1 levels at 36 hpi (Fig. 3A). Representative histograms showing MHC-I fluorescence in GFP-positive cells from the Ad.GFP-66 infection revealed that MHC-I surface expression was reduced when GFP-66 was expressed. Parallel infected cells grown in medium containing DOX showed no MHC-I downregulation, and GFP-66kd expression did not significantly downregulate MHC-I surface expression in the absence or presence of DOX. From three independent and identical experiments, GFP-66 expression induced an average of ∼40% reduction of MHC-I surface levels compared to a minor 5 to 11% reduction in GFP-66kd-expressing cells (Fig. 3B). Analysis of surface TfR1 revealed no significant downregulation under any conditions (Fig. 3A and B). Immunoblot analyses of equal numbers of Ad.GFP-66- and Ad.GFP-66kd-infected cells from the same experiment shown in Fig. 3A indicated that similar levels of GFP-66 and GFP-66kd were expressed (Fig. 3C). These data indicate that functional kinase activity is required for efficient ORF66-mediated MHC-I downregulation and imply that ORF66 phosphorylates a cellular target to induce the reduction in MHC-I surface expression.
Development of recombinant VZV expressing GFP-66 proteins.
In previous studies of VZV and its effects on MHC-I surface expression, the identification of VZV-infected cells required indirect immunofluorescent detection of surface VZV glycoproteins. To more accurately assess infection and correlate the levels of ORF66 expression with levels of surface MHC-I during VZV infection, we generated recombinant VZV expressing GFP-66 (VZV.GFP-66) or one in which protein kinase expression was halted by stop codon insertion at ORF66 amino acid 84 (VZV.GFP-66s). Recombinant viruses were created using wild-type VZV (pOka) cosmids (Fig. 1) and were designed so that GFP-66 or GFP-66s was expressed at the native locus and under the natural ORF66 promoter. Each recombinant was successfully generated twice from cosmid-transfected MeWo cells, yielding green fluorescent plaques with no visible differences in size, morphology, or GFP autofluorescence distribution (data not shown). The GFP tag enabled VZV-infected cells expressing ORF66 to be differentiated in mixed populations of infected and uninfected cells and also indicated the relative amounts of ORF66 expression. Growth curve analysis in MRC-5 fibroblasts of these viruses indicated that VZV.GFP-66 grew with similar kinetics as pOka and achieved equivalent maximal titers (∼1 × 105 infectious foci by 96 hpi) (Fig. 4A). This suggests that the addition of EGFP to ORF66 had only minimal effects, if any, on virus growth. VZV.GFP-66s infections were marginally impaired, similar to observations from a previous study (57), yielding a three- to fourfold reduction in infectious cell titers with respect to pOka and VZV.GFP-66.
FIG. 4.
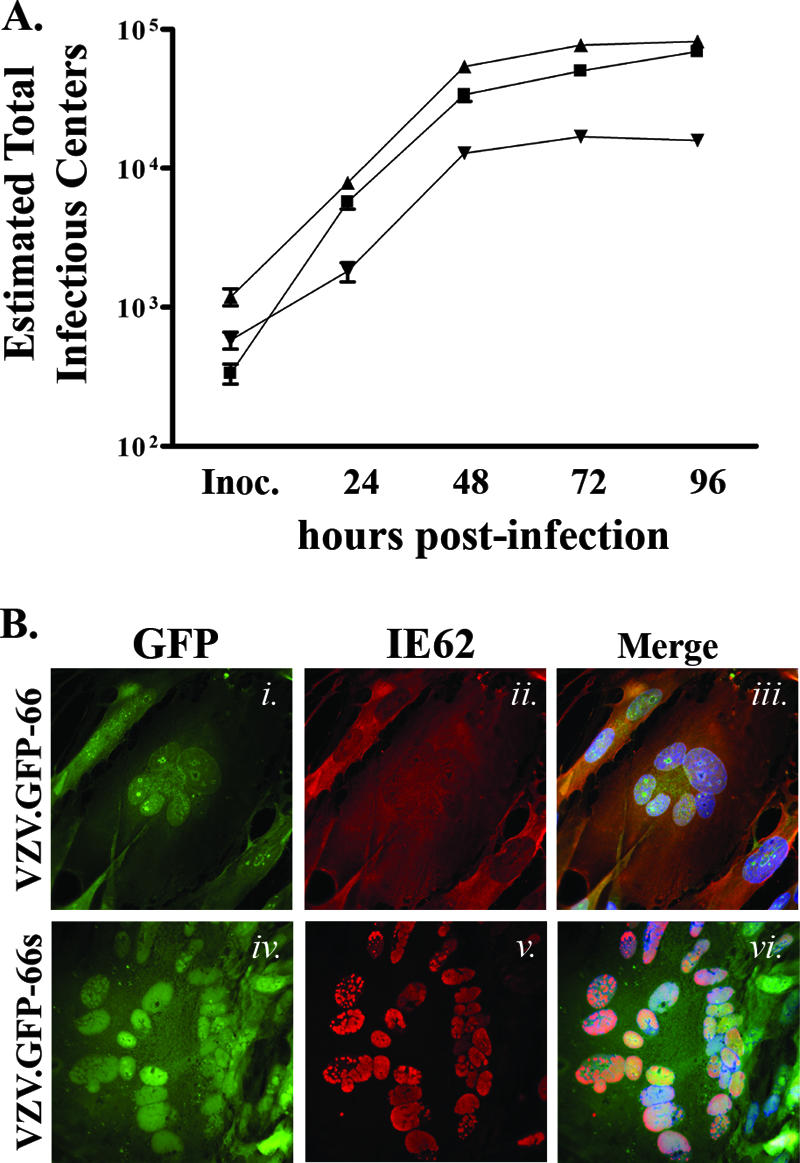
Characterization of recombinant VZV expressing GFP-66 and GFP-66s. (A) Growth curve analyses of VZV.GFP-66 (▴), VZV.GFP-66s (▾), and pOka (▪). Parallel infections were established on confluent monolayers in MRC-5 fibroblasts, and the amount of virus-infected cells in each culture produced by each VZV was titrated on MeWo cells after 24, 48, 72, and 96 hpi. Mean titers and standard errors were deduced from duplicate wells of duplicate titrations. (B) Images showing GFP expression and IE62 cellular localization in fixed VZV.GFP-66-infected (i to iii) and VZV.GFP-66s-infected (iv to vi) MRC-5 cells at 48 hpi. Cells were fixed with 4% paraformaldehyde and stained with rabbit anti-62 and Alexa Fluor 546-conjugated anti-rabbit Fab fragments and Hoechst dye to identify nuclei. The depicted autofluorescence or stained protein is indicated above the panels, and the infection condition is on the left. Images were selected to represent the center of VZV syncytia at approximately the same stage of infection.
The functionality of the GFP-66 kinase in VZV.GFP-66 and the disruption of ORF66 kinase expression in VZV.GFP-66s were assessed by examining IE62 localization in infected MRC-5 cells (Fig. 4B). Consistent with functional ORF66 kinase activity, IE62 was predominantly cytoplasmic in late-stage VZV.GFP-66-infected plaques, and most nuclei showed only low levels of IE62 staining (Fig. 4B, panels ii and iii). Nuclear IE62 was observed at plaque edges where GFP-66 expression was low or absent, as was found for pOka-infected cells (56). GFP-66 exhibited both nuclear and cytoplasmic distributions and was often concentrated in discrete nuclear foci that appeared to surround the nucleolus (Fig. 4B, panel i). In contrast, IE62 in VZV.GFP-66s-infected MRC-5 cells remained strictly nuclear, even in late-stage-infected plaques and syncytia (Fig. 4B, panels iv to vi), consistent with the lack of ORF66 kinase domain expression. Despite including only the first 83 residues of ORF66, we noted a significant overlap between GFP-66s autofluorescence and IE62 immunofluorescence in the nucleus. Together, the growth curve analysis and IE62 nuclear exclusion assay validate the use of VZV.GFP-66 and VZV.GFP-66s to assess the correlation of ORF66 expression with the downregulation of MHC-I surface expression in VZV-infected cells.
VZV has ORF66-dependent and -independent effects on MHC-I and TfR1 surface expression.
The cell-associated nature of VZV precludes large-scale synchronous infection, and so MHC-I and TfR1 surface levels were compared in MRC-5 cells infected with VZV.GFP-66 and VZV.GFP-66s at a moderate multiplicity of infection (uninfected to VZV-infected fibroblasts at a ratio of 10:1) over a 72-hour time course. At 24, 48, and 72 hpi, MHC-I and TfR1 surface expression levels were analyzed by flow cytometry (Fig. 5). Mock-infected cells stained with isotype control antibodies were used to define the GFP-negative quadrant gate at each time point (Fig. 5A and D, panels a to c). Uniform levels of surface MHC-I and TfR1 were consistently displayed in mock-infected cells (Fig. 5A and D, panels d to f). We note that the MHC-I level in mock-infected cells showed a slight increase over time in culture, but we cannot rule out that this was a consequence of experimental variability. At 24 hpi, VZV.GFP-66-infected cells exhibited a range of GFP expression levels (Fig. 5A, panel g). The GFP-negative population had MHC-I surface levels similar to that of uninfected cell controls, whereas most (but not all) cells expressing the higher levels of GFP had reduced MHC-I surface expression. This indicates that ORF66 expression does not have an immediate effect on MHC-I surface levels. At 48 and 72 hpi, most cells expressed high amounts of GFP in VZV.GFP-66 infections and showed more downregulation of MHC-I surface fluorescence (Fig. 5A, panels h to i). The greatest reduction in MHC-I surface expression was consistently observed in high GFP expressers at 72 hpi (Fig. 5A, panel i). Regarding VZV.GFP-66s infections, while fewer cells were infected at 24 hpi, MHC-I was still reduced in a fraction of GFP-positive cells (Fig. 5A, panel j). At later times in very high GFP expressers, reduced MHC-I surface expression was also observed (Fig. 5A, panels k and l). These data show that VZV can downregulate MHC-I in the absence of the expression of the ORF66 kinase domain. We note that the GFP signal in GFP-66s was expressed with similar timing and to greater levels compared to that from GFP-66 during VZV infection (Fig. 5F). The basis of the reduced levels of ORF66 kinase at 72 h is not yet clear, but it is conceivable that the functional kinase has possible autoregulatory effects on its own expression.
FIG. 5.


MHC-I downregulation in VZV infection with and without expression of functional kinase. MRC-5 cells were infected with VZV.GFP-66 or VZV.GFP-66s at a ratio of 10:1, surface stained for MHC-I and TfR1 at 24, 48, and 72 hpi, and analyzed by flow cytometry. (A) Dot plots from a representative experiment depicting MHC-I surface expression as a function of GFP-66 or GFP-66s expression. The analysis time point is shown to the left of each row, and the infection condition is shown above each column. Quadrant gates to identify MHC-I-negative and MHC-I-positive cell populations were established at each time point using mock-infected isotype control-stained cells (a to c). The same quadrant gate was used for mock (d to f), VZV.GFP-66 (g to i), and VZV.GFP-66s (j to l) infections. To quantitatively assess differences between VZV.GFP-66 and VZV.GFP-66s infections, cells were gated on high GFP fluorescence, represented as the right-most vertical line in infected cell dot plots (g to l). The GFP-negative and GFP-high (where relevant) cell populations for each column are indicated below panels c, f, i, and l. The percentages of mock-infected cells exhibiting an MHC-I-negative or MHC-I-positive phenotype are indicated in the upper left and lower left quadrants in panels d to f. Similar MHC-I-negative and MHC-I-positive cell percentages in GFP-high cells are indicated in the upper right and lower right quadrants of panels g to l. (B) The average percentage (± standard error of the mean) of MHC-I-positive (gray) and MHC-I-negative (black) cells in the GFP-high gate under each infection condition was calculated from three independent experiments. (C) MFIs of GFP-high cells in VZV.GFP-66 (black) and VZV.GFP-66s (gray) infections were expressed as a ratio of the MFI of mock-infected cells. Average ratios (± standard error of the mean) from three independent experiments were compared at each time point, and we found that VZV.GFP-66 exhibited a significant reduction in MHC-I surface expression over VZV.GFP-66s at 72 hpi. (D and E) TfR1 surface expression dot plots (D) and MFI ratios (E) are shown exactly as described for MHC-I. (F) Lysates of aliquots of equal numbers of cells from each infection condition at each time point were separated by SDS-PAGE and analyzed by immunoblotting sequentially with mouse anti-HA and then mouse anti-α-tubulin antibodies. *, P ≤ 0.05; **, P ≤ 0.005.
We assume that similar levels of GFP represent similar stages of infection, since the native ORF66 promoter controls both GFP-66 and GFP-66s expression. To determine if there were differences in the abilities of VZV.GFP-66 and VZV.GFP-66s to mediate MHC-I downregulation, we examined cells exhibiting similar relative GFP intensities. The same high GFP gate was used for all infections at all time points and was defined as cells with 10-fold or greater GFP fluorescence compared to mock-infected cells (indicated in each representative infection dot plot, Fig. 5A, panels g to l). Similar approaches were recently used to compare apoptosis and Stat1 phosphorylation in pOka- and pOka66S-infected cells (57). Our data were accumulated in three independent experiments. GFP-high cells in VZV.GFP-66 infections consistently exhibited a more negative MHC-I surface expression phenotype compared to GFP-high cells from VZV.GFP-66s infections (Fig. 5B). On average, ∼75% of GFP-66-high cells fell below the MHC-I-positive gate at both 48 and 72 hpi, while similar GFP-66s-high cells were ∼55 to 60% MHC-I negative. We also calculated the average MHC-I MFI ratio (relative to mock-infected cells) of GFP-high cells from these three experiments and found that GFP-high cells in VZV.GFP-66 infections had a lower average MHC-I MFI ratio at all time points compared to similar cells in VZV.GFP-66s infections (Fig. 5C). This difference was statistically significant at 72 hpi (P ≤ 0.05). These analyses indicate that while ORF66-independent processes negatively influence MHC-I surface expression, more efficient MHC-I surface downregulation in VZV infection requires expression of the functional ORF66 kinase domain.
Surprising results were obtained with TfR1. Both VZV.GFP-66 and VZV.GFP-66s induced an increase in TfR1 surface expression compared to mock-infected cells, with more upregulation occurring when the ORF66 kinase activity was intact (Fig. 5D and E). A 2.5-fold increase in TfR1 expression over mock infection was observed in the VZV.GFP-66 infection at 72 hpi, compared to a 1.8-fold increase in VZV.GFP-66s infection. At all times, the difference was statistically significant. These data indicate that the surface expression of TfR1 is also regulated by both ORF66-dependent and -independent mechanisms during VZV infection but is affected in an opposite manner compared to surface MHC-I. The differential regulation of outcomes for surface MHC-I and TfR1 implies that ORF66 may have multiple effects on other cell surface protein trafficking patterns.
ORF66 kinase delays the biosynthetic maturation of MHC-I.
Molecular mechanisms underlying downregulation of MHC-I by VZV infection and ORF66 have not been well resolved. While one study reported no change in MHC-I synthesis levels or rate of acquisition of endo H resistance in VZV-infected fibroblasts (1), another study observed a modest effect on total class I heavy chain and β2-M synthesis at 48 hpi (9). Our data showed that most MHC-I surface downregulation occurred after 24 hpi. To clearly elucidate the mechanism, we undertook an analysis of MHC-I biogenesis in cells expressing the ORF66 kinase independently of other proteins as well as in the context of VZV infection.
Pulse-chase studies combined with endo H cleavage analysis were used to address MHC-I synthesis and maturation in MRC-5 cells infected with Ad.GFP-66 or Ad.GFP-66kd. Endo H resistance is acquired when cis/medial-Golgi enzymes catalyze replacement of high mannose groups with more complex N-linked glycans on glycoproteins. Mock- or adenovirus-infected MRC-5 cells were subjected to a [35S]Cys/Met 15-min pulse and harvested or chased to allow maturation of labeled protein. Equivalent amounts of labeled protein from each infection condition were subjected to immunoprecipitation with the W6/32 antibody, which only recognizes folded MHC-I molecules that have associated with β2-M. Immunoprecipitations performed with nonspecific isotype control antibodies did not result in significant amounts of precipitated radiolabeled protein (data not shown).
Comparison of the SDS-PAGE resolution of immunoprecipitates obtained at the end of the pulse (0 min) revealed that expression of functional or kinase-inactive ORF66 had no effect on the levels of MHC-I complexes, compared to that obtained from mock-infected cells, and that MHC-I from all conditions was completely sensitive to endo H cleavage (Fig. 6A to C, lanes 1 and 2). These observations indicate that ORF66 does not modulate expression of MHC-I components or its initial assembly into heavy chain (HC)-β2-M hetero-dimers. The amount of MHC-I HC was also found to remain relatively stable under chase conditions up to 120 min, indicating that ORF66 does not induce MHC-I degradation. However, ORF66 expression clearly delayed MHC-I acquisition of endo H resistance. In mock-infected cells, heavy chains were ∼53% resistant to endo H cleavage at 30 min, ∼78% at 60 min, and 98% resistant by 120 min of chase (Fig. 6A and D). In contrast, MHC-I from Ad-GFP-66-infected cells demonstrated only ∼11% and 16% endo H resistance at 30 and 60 min of chase, respectively (Fig. 6B and D). By 120 min of chase, most heavy chains remained sensitive to cleavage, with only 26% exhibiting resistance. In Ad.GFP-66kd-infected cells, MHC-I acquired endo H resistance at a rate more similar to that observed in mock infections, with 35% resistance at 30 min, 52% at 60 min, and 74% at 120 min (Fig. 6C and D). In a similar experiment, Ad.GFP-66 infection induced a comparable reduction in MHC-I maturation relative to mock infection, but no impairment was observed when infections were incubated in the presence of 2 μg/ml DOX (data not shown). These data indicate that MHC-I maturation through the cis/medial-Golgi compartment is impaired predominantly as a result of ORF66 kinase activity.
FIG. 6.
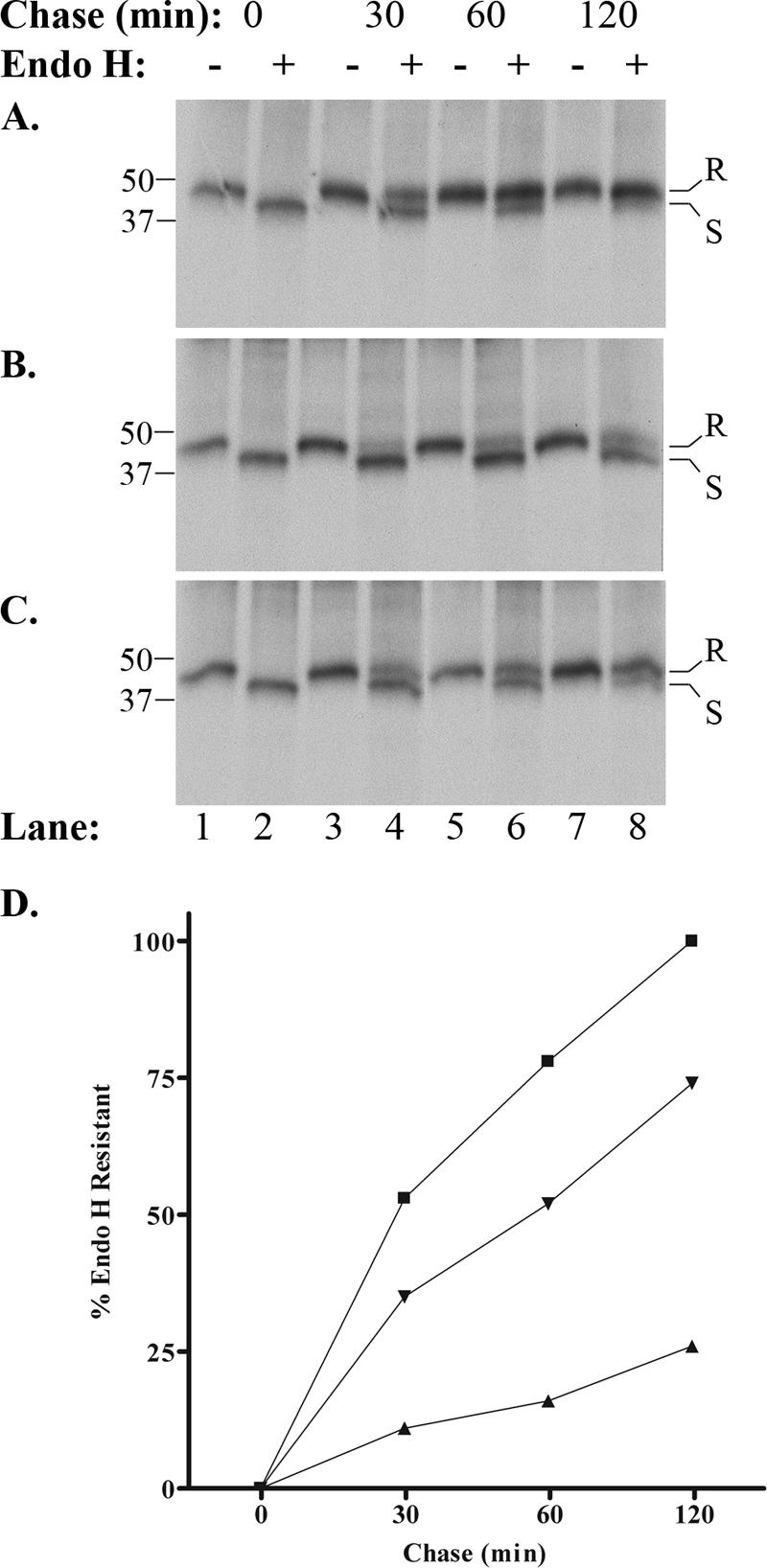
GFP-66 expression induces a delay in MHC-I maturation in MRC-5 fibroblasts. (A) Mock-infected, (B) Ad.GFP-66-infected, or (C) Ad.GFP-66kd-infected cells were pulse-labeled with [35S]Cys/Met and chased in medium containing cold Cys/Met, and folded MHC-I was immunoprecipitated and subjected to endo H digestion. Autoradiographs of proteins separated by SDS-PAGE are shown. The chase time and presence (+) or absence (−) of endo H are indicated above each lane, and lane numbers are shown at the bottom. Class I molecules that are resistant to endo H cleavage migrate more slowly through the gel and are designated by the letter R to the side of each radiograph. Sensitive molecules migrate more quickly and are indicated by the letter S. Protein size standards were run with each gel and are shown (in kDa) to the left. (D) The rate of acquisition of endo H resistance was quantified for mock infections (▪), Ad.GFP-66 infections (▴), and Ad.GFP-66kd infections (▾) using autoradiography. The percentage of the total MHC-I molecules exhibiting resistance was plotted as a function of time.
VZV affects early events in MHC-I biogenesis.
The ORF66-mediated delay of MHC-I maturation prompted a reassessment of MHC-I maturation in VZV-infected cells. Infection conditions for the two VZV were carefully normalized to be equivalent, and VZV-infected MRC-5 fibroblasts were established at a relatively high VZV-infected cell multiplicity (3 uninfected cells to 1 infected cell) to ensure efficient infection. Cells were pulse-labeled at 36 hpi, when all cells were GFP positive. Immunoblot analysis of infection lysate supernatants indicated that GFP-66s was expressed to a greater level than GFP-66 under these conditions (see Fig. 8A, below).
FIG. 8.
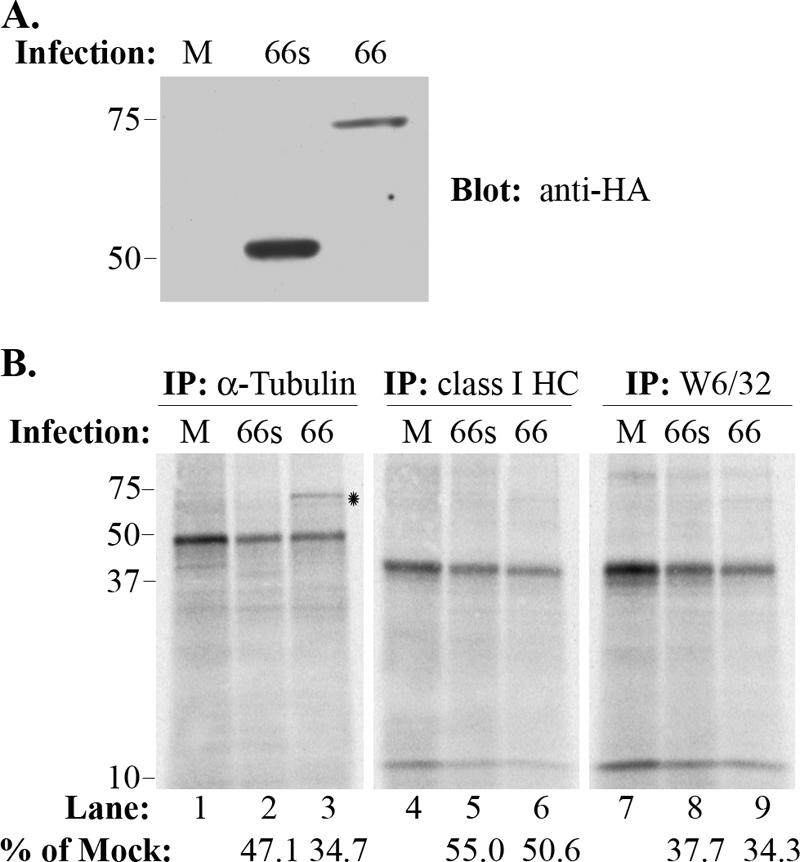
VZV infection reduces class I synthesis and impairs the association of class I heavy chains with β2-M. Total α-tubulin and class I heavy chain levels, as well as folded class I heavy chain levels, were compared by parallel immunoprecipitation from either mock-infected cells (M) or cells infected with VZV.GFP-66 (66) or VZV.GFP-66s (66s), following a 6-hour [35S]Cys/Met label. (A) Immunoblot analysis of cleared infected cell lysates showing the level of GFP-66 and GFP-66s protein expressed under each infection condition. The infection condition is indicated at the top. (B) Autoradiographs of each set of parallel immunoprecipitations. The antibody used and infection condition are indicated above the radiograph, and lane numbers are shown below. Protein size markers (in kDa) are shown to the left. The relative amounts (percentage) of proteins expressed in infected cells compared to uninfected cells for both VZV.GFP-66 and VZV.GFP-66s are shown under lane numbers 2, 3, 5, 6, 8, and 9. The * indicates a reproducible coprecipitating band of ∼75 kDa which was apparent in the α-tubulin immunoprecipitation of VZV.GFP-66-infected cells. This band did not react with antibodies to HA; its identity is currently unknown.
In two independent and identical experiments, SDS-PAGE-separated immunoprecipitates for each infection condition revealed that pulse-labeled class I heavy chains in uninfected cells were completely sensitive to endo H cleavage at 0 min but became almost fully resistant by 180 min of chase (Fig. 7A). Labeled MHC-I levels remained relatively stable for the duration of the experiment, suggesting there was no turnover. In VZV-infected cells, there was a consistent reduction in the rate of class I maturation compared to uninfected cells, with a greater degree of delay in VZV-infected cells expressing the functional ORF66 kinase (Fig. 7B to D). On average, after 30 min of chase only 31% of the total precipitated MHC-I heavy chains from VZV.GFP-66-infected cells exhibited endo H resistance, whereas greater than 60% were resistant in uninfected cells. VZV.GFP-66-infected cells were 51% and 72% resistant at 60 and 180 min of chase, respectively, compared to 78% and 85% resistance in uninfected cells at the same times. MHC-I in VZV.GFP-66s-infected cells was marginally delayed in acquisition of endo H resistance, but not to the level seen in VZV.GFP-66 infection, showing an average 58% resistance at 30 min, 67% at 60 min, and 82% after 180 min of chase. These data indicate that the greatest delay in acquisition of endo H resistance for MHC-I occurs when the ORF66 kinase is expressed and functional. This correlates with the observed delay in cells expressing the ORF66 kinase in the absence of other VZV proteins. Thus, the ORF66 kinase delays MHC-I maturation in a predominantly kinase-dependent manner and contributes to the reduced MHC-I phenotype seen in VZV-infected cells.
FIG. 7.
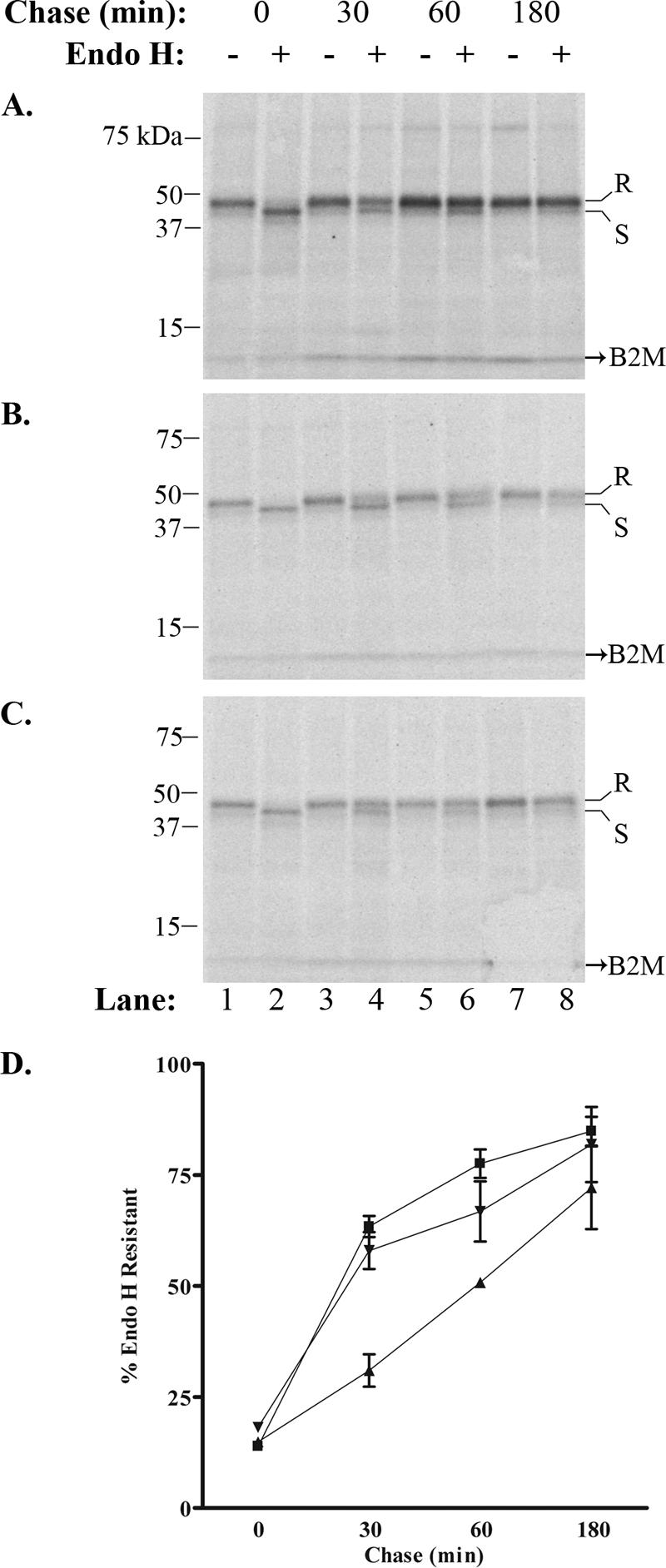
GFP-66 mediates a delay in MHC-I maturation in the context of VZV infection. Endo H resistance analysis was performed for MHC-I molecules from (A) mock-infected, (B) VZV.GFP-66-infected, and (C) VZV.GFP-66s-infected cells. Chase times and treatment with endo H are shown above each radiograph, and lane numbers are shown below. Protein size markers are specified in kDa to the left, and endo H-resistant (R) or -sensitive (S) forms are indicated to the right. (D) The rate of MHC-I maturation was determined by phosphorimager analysis from two independent but identical experiments and is plotted as a function of time for mock infections (▪), VZV.GFP-66 infections (▴), and VZV.GFP-66s infections (▾).
Additional VZV activities contributing to the reduction of MHC-I on the cell surface were also indicated in these studies. Specifically, we noted that the levels of β2-M-associated class I immunoprecipitated by the W6/32 antibody from both VZV.GFP-66 and VZV.GFP-66s infections were greatly reduced at all time points compared to that from uninfected cells, even though all immunoprecipitations were normalized for equal amounts of labeled protein in the cleared lysates. The TCA-precipitable material was substantially reduced in both VZV.GFP-66- and VZV.GFP-66s-infected cells, over that in uninfected cells (data not shown). This likely reflects a VZV-induced global reduction in host protein synthesis that may affect MHC-I component expression. To determine if VZV induces a specific reduction in MHC-I synthesis, we performed parallel immunoprecipitations from longer-term-radiolabeled (6 h) extracts of uninfected cells or cells equally infected with VZV.GFP-66 or VZV.GFP-66s, using antibodies that recognized folded MHC-I (W6/32), total class I heavy chains, or α-tubulin. Equivalent levels of TCA-precipitable counts were used for each immunoprecipitation. The SDS-PAGE-resolved immunoprecipitates revealed that α-tubulin synthesis was reduced under both infection conditions compared to uninfected cells, consistent with a global reduction in host protein expression (Fig. 8B, lanes 1 to 3). Quantification of total MHC-I heavy chain synthesis by phosphorimager analysis revealed a 45 to 50% reduction relative to uninfected cells in both infections (Fig. 8B, lanes 4 to 6). However, folded MHC-I heavy chains precipitated with W6/32 were more reduced (∼65%) than that predicted by heavy chain levels in either VZV-infected cell population (Fig. 8B, lanes 7 to 9). While modest, this difference was consistently detected in replicate experiments, suggesting an additional impairment in the formation of the heavy chain-β2-M hetero-dimer. Altogether, the biochemical analyses of MHC-I from VZV-infected cells strongly indicated that VZV uses multiple processes to impede MHC-I surface levels.
Intracellular retention of MHC-I in VZV-infected cells.
A previous study suggested that MHC-I is processed correctly but retained in the Golgi apparatus in VZV-infected HFF cells (1). Given our new observations that VZV delays MHC-I early maturation, we reevaluated the distribution of MHC-I in VZV infections in both the presence and absence of the ORF66 kinase. Immunolocalization of MHC-I and its association with cis/medial-Golgi in MRC-5 fibroblasts was determined using the conformation-dependent W6/32 antibody and anti-Mann II antibodies. To facilitate specific detection of intracellular MHC-I, cells were treated briefly with a mild acid buffer (59) prior to fixation and permeabilization, to dissociate β2-M from surface class I heavy chains and render surface MHC-I unrecognizable by W6/32. In uninfected, untreated cells, surface MHC-I displayed finely punctate distribution with more concentrated staining at cellular boundaries (Fig. 9A, panel i). Uninfected cells subjected to acid treatment lacked a similar pattern of surface staining and had a characteristic perinuclear distribution of MHC-I that was proximal to, but did not completely overlap, Mann II staining. There were also obvious peripheral vesicular cytoplasmic foci, which may represent MHC-I en route to or recycling from the cell surface (Fig. 9A, panels ii to iv).
FIG. 9.

MHC-I localization in VZV infection with and without ORF66 expression. Cells on coverslips were either surface stained for MHC-I or stripped of surface β2-M, permeabilized, and stained for intracellular folded MHC-I (W6/32) and rabbit anti-Mann II combined with anti-mouse Alexa-Fluor 647 and anti-rabbit Alexa-Fluor 546, respectively. All cells were stained with Hoechst 33258 to identify nuclei. Z-stacks were acquired and deconvolved for each fluorescent channel. MHC-I, Mann II, and Hoechst stains were pseudo-colored to promote simultaneous observance of each channel in the merged images. (A) MHC-I expression profile in uninfected MRC-5 fibroblasts. MHC-I surface fluorescence is shown as an image merged with Hoechst staining (i). A similar staining pattern was not observed in β2-M-stripped cells, which exhibited low levels of perinuclear MHC-I that partially overlapped with mannosidase II staining (ii to iv). Individual stains are indicated above each micrograph panel. (B) Comparison of intracellular MHC-I distribution in late-stage VZV.GFP-66-infected (i to iv) and VZV.GFP-66s-infected (v to viii) syncytia. Each set of panels shows a representative syncytium. Stains are indicated above each micrograph. In both A and B, arrows highlight specific features that are discussed in the text.
For comparison of MHC-I localization in VZV.GFP-66 and VZV.GFP-66s infections, we assessed late-stage multinucleate syncytia, which expressed abundant levels of GFP-66 or GFP-66s and retained clear cellular morphology. Syncytia were randomly selected, and Mann II and MHC-I staining patterns were documented. Representative VZV.GFP-66 and VZV.GFP-66s syncytia are shown in Fig. 9B, with high-level GFP fluorescence indicating similar late infection stages (panels i and v). Surface MHC-I in VZV.GFP-66 and VZV.GFP-66s infections was distributed in a pattern similar to uninfected cells, although the signal was clearly consistently reduced (data not shown). In the VZV.GFP-66 syncytium, nuclei were arranged in central clusters and the Golgi apparatus was significantly rearranged, consistent with previous observations (19, 23, 48). Two populations of Mann II became disjointed: some localized to foci closely apposed to the nucleus, while the remainder was distributed in a ringed structure in the center of the syncytia (Fig. 9B, panel ii). Golgi apparatus rearrangement resulted in a central pool of intracellular MHC-I, which partially overlapped the Mann II staining, but also showed independent accumulation at the center of the Mann II ring (Fig. 9B, panels iii and iv). Little or no MHC-I was observed outside of the Mann II staining region, in contrast to the abundance of punctate foci observed throughout the periphery of uninfected cell cytoplasm. In VZV.GFP-66s syncytia, a similar central Mann II ring formed (Fig. 9B, panel vi). MHC-I showed accumulation in this region but was disorganized and exhibited a remarkable lack of overlap with Mann II staining (Fig. 9B, panels vi to viii). Additional undefined populations of MHC-I were consistently observed throughout the VZV.GFP-66s cytoplasm (Fig. 9B, panel vii). These did not colocalize with Mann II or other cytoplasmic organelle markers for cis-Golgi (GM130), trans-Golgi network (p230), ER (Erp29), early endosomes (EEA-1), or lysosomes (Lamp-1) (data not shown). The localization and organization of MHC-I in infected cells support a role for ORF66 in MHC-I retention in the cis/medial-Golgi and indicate that other factors may influence the distribution of intracellular MHC-I during VZV infection.
Assessment of expression of additional VZV ORFs on MHC-I surface expression.
As our data clearly indicate that ORF66-independent mechanisms regulate MHC-I surface levels, we evaluated select VZV genes for abilities to affect MHC-I. A panel of plasmids expressing VZV ORFs 0, 1, 2, 7, 9, 11, 17, 21, 23, 32, 40, 44, 46, 58, 64, and 65 revealed no ability for any one protein to downregulate MHC-I (data not shown). VZV encodes an ortholog to the BHV-1 UL49.5 TAP inhibitor (VZV ORF9a) (36), and we also addressed the ability of the ORF9a protein to downregulate MHC-I in VZV-permissive cells (Fig. 10). For this study, test plasmids were cotransfected in 293T cells with an equal amount of a plasmid expressing low levels of EGFP under the control of the weakly constitutive HSV-1 thymidine kinase promoter (pTK-EGFP), and MHC-I surface expression was analyzed in EGFP-positive cells by flow cytometry. Cells transfected with plasmid vector lacking any viral ORF were considered the negative control, and we utilized plasmids expressing either ORF66 or the KSHV K3 protein as independent positive controls. EGFP-positive cells from both ORF66 and K3 transfections yielded significantly reduced MHC-I surface levels (P < 0.05) relative to vector control transfections, indicating that the pTK-EGFP cotransfection successfully reported test plasmid uptake. No significant MHC-I downregulation was observed in ORF9a transfections, supporting data from a previous study in which ORF9a failed to mediate MHC-I downregulation in human melanoma (Mel JuSo) cells (36). This indicates that the ability of ORF9a to inhibit TAP either may not be conserved or may require the expression of additional VZV genes. Little is known of the functions of VZV ORF9a during infection, but the pseudorabies virus ORF9a ortholog (gN) is disulfide-linked to pseudorabies virus gM (VZV ortholog: ORF50) in infected cells (31). We considered the potential ORF9a-ORF50 interaction as the most likely to affect ORF9a's functions, and we assessed the effect of combined ORF9a and ORF50 expression on MHC-I surface expression. Neither ORF50 alone nor ORF9a and ORF50 in combination resulted in significant MHC-I downregulation. While we could not confirm the expression of ORF9a due to lack of an ORF9a antibody, similar negative results were observed in cells transfected with an EGFP-tagged version of ORF9a (data not shown). Therefore, VZV genes other than the ORF66 kinase and ORF9a are likely involved in the regulation of MHC-I surface expression.
FIG. 10.
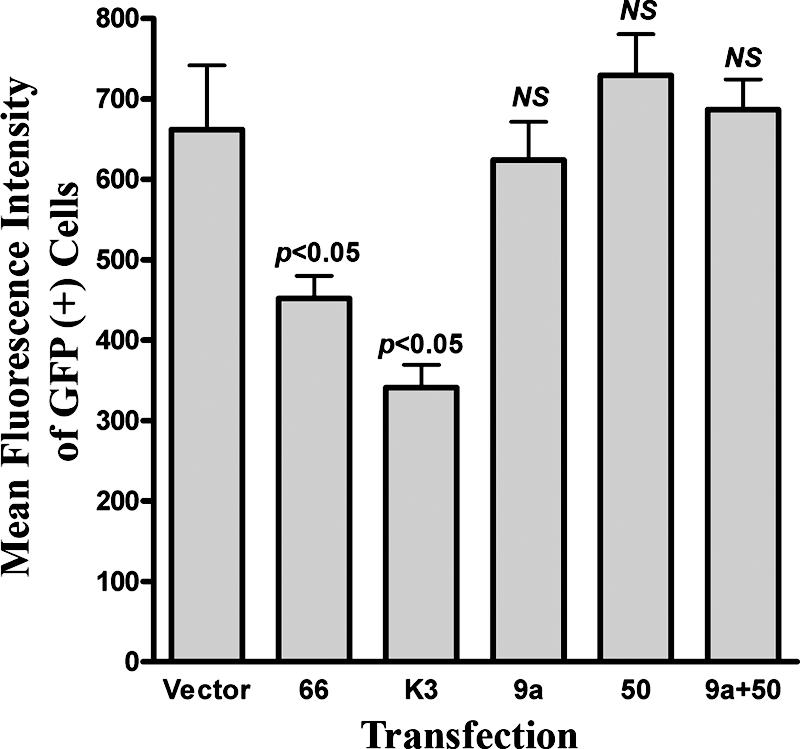
The VZV ortholog to BHV-1 UL49.5, ORF9a, does not mediate downregulation of MHC-I surface expression. HEK 293T cells were cotransfected with an EGFP-expressing marker plasmid (pTK-EGFP) and plasmids expressing untagged versions of ORF9a, ORF50, or ORF9a and ORF50 in combination. Empty vector cotransfection was used as a negative control, and cotransfections with plasmids encoding the KSHV K3 MHC-I downregulator (29) or VZV ORF66 served as the positive controls. At 48 h posttransfection, cells were stained for MHC-I and analyzed by flow cytometry. The average MFIs (± standard error of the mean) of GFP-positive cells from four independent experiments are shown. MFI data from each test transfection condition were compared against those of the negative control using a paired Student's t test, and the results are indicated above the corresponding bars. An equal level of pTK-EGFP was maintained under all transfection conditions. NS, not significant.
DISCUSSION
The data presented here indicate a new role for the VZV ORF66 protein kinase in inhibition of MHC-I surface presentation and also strongly suggest that VZV possesses additional ORF66-independent mechanisms to modulate surface MHC-I in VZV-infected cells. Novel recombinant VZV expressing GFP-66 proteins allowed highly accurate measurement of VZV infection and clearly demonstrated that the most efficient MHC-I downregulation was concurrent with extensive ORF66 expression at later stages of VZV infection. VZV reduced the rate of MHC-I transport from the ER to the cis/medial-Golgi, induced a reduction in MHC-I heavy chain synthesis, and caused a moderate but consistent decrease in levels of folded hetero-dimerized class I heavy chains. ORF66 appeared to act predominantly by inducing a delay in MHC-I transport from the ER to the cis/medial-Golgi. While two previous studies have reported that VZV inhibits MHC-I surface expression, our study provides a more coherent portrait of MHC-I regulation during VZV infection and begins to clarify the VZV genes that are involved.
Abendroth et al. (1) reported no decrease in MHC-I synthesis or transport through the cis/medial-Golgi at 24 hpi but indicated that the major bulk of intracellular MHC-I was retained in the more peripheral stacks of the Golgi complex. It is clear from studies presented here that VZV infection does not immediately induce a surface downregulation of MHC-I, as is seen with ICP47 expression during the immediate-early stage of HSV infection. Rather, the modulation of MHC-I surface expression correlates more closely with later stages of infection and high levels of ORF66 expression. While we did observe intracellular accumulation of MHC-I in the Golgi region, we also detected an ORF66-dependent retention preceding the cis/medial-Golgi complex, which was concomitant with the lowest levels of MHC-I surface expression in VZV-infected cells. Since the former study was performed at 24 hpi, ORF66 expression most likely did not reach significant enough levels to affect MHC-I processing. The data from a second study suggested that MHC-I heavy chain and β2-M synthesis were lower at 48 hpi (9), and our data are in agreement with this, since we observed an ORF66-independent reduction in MHC-I synthesis in cells expressing high levels of GFP. While a nonspecific mechanism of global protein synthesis reduction is almost certainly involved, our data also suggest that VZV may impair MHC-I heavy chain and β2-M association, since levels of folded hetero-dimerized heavy chains were consistently lower than total class I heavy chains. The consistent upregulation of TfR1 surface expression in VZV-infected cells was unexpected and contrasts with data from the two previous studies (1, 9). We suspect that differences in the experimental approaches used may account for this. For example, Abendroth et al. (1) reported no effects on TfR1 surface expression in VZV-infected fibroblasts analyzed at 24 hpi. While we observed some effect on TfR1 at 24 hpi, the greatest upregulation was observed at 48 and 72 hpi, indicating that this is predominantly a late-stage infection phenomenon. This adds to the complexity of the VZV program for controlling surface protein expression in infected cells.
The specific mechanism by which ORF66 affects MHC-I surface levels remains unclear at this time. Expression of functional ORF66 kinase resulted in impaired MHC-I maturation but did not affect MHC-I synthesis, degradation, or association with β2-M. The successful association of class I heavy chains with β2-M suggests that ER resident chaperone machinery (including calnexin, calreticulin, and Erp57) may function normally in the presence of ORF66 expression. Our evidence does not suggest a close association of ORF66 with either folded MHC-I or total class I heavy chains, as ORF66 did not coprecipitate with MHC-I and did not colocalize with MHC-I in VZV-infected cells. Inactivation of the ORF66 kinase domain abrogated most of its ability to mediate MHC-I downregulation, suggesting that phosphorylation of a host cell protein is involved. However, the slight reductions in MHC-I maturation and surface expression observed in the presence of the GFP-66kd protein do imply a possible dominant-negative interaction with a cellular protein involved in MHC-I biosynthesis. Two commonly used viral mechanisms that inhibit MHC-I maturation in the early secretory compartment are inhibition of the TAP complex (16, 24, 25, 36, 42), which transports proteasomally derived peptides into the ER, and inhibition of tapasin (7, 52), which bridges TAP to MHC-I and mediates the transfer of peptides from TAP to the MHC-I binding groove. While neither TAP nor tapasin exhibits motifs similar to those phosphorylated by the ORF66 kinase (15), we hypothesize that ORF66 may activate host kinase cascades with downstream effects that dysregulate TAP or tapasin function. Interestingly, it has been reported that phosphorylated TAP complexes bind peptides and ATP but cannot transport peptides without being dephosphorylated (43). We speculate that ORF66 induces a hyperphosphorylated state of the TAP that may lock the complex in a peptide-bound state. An alternative scenario is that ORF66 causes aberrant phosphorylation of MHC-I cytoplasmic tails or the cellular machinery involved in vesicular transport between the ER and the cis/medial-Golgi. This is prompted by the observations that cellular protein kinase A (PKA) is known to phosphorylate MHC-I tails (22, 53), and the US3 kinase of HSV-1 (orthologous to ORF66) has been shown to induce phosphorylation of some cellular PKA targets (6). Known ORF66 targets are highly similar to the optimized target motif of HSV-1 US3 kinase (41), suggesting that some PKA targets could also be targets of ORF66. MHC-I cytoplasmic tail phosphorylation normally occurs at the cell surface or in late or recycling endosomes (8, 14, 32, 44). Thus, aberrant phosphorylation of pre-Golgi MHC-I could result in deviations from normal trafficking early in its secretory transport. Studies to address ORF66-mediated effects on the peptide loading complex and ORF66-directed phosphorylation of MHC-I are in progress.
The ORF66 kinase modulates several aspects of the cellular environment to facilitate VZV infection, but cellular proteins that are targeted for direct phosphorylation by ORF66 remain unknown. In VZV-infected T lymphocytes, ORF66 inhibits activation of caspase 3 and IFN-γ-mediated Stat1 phosphorylation (57). One consequence of IFN-γ signaling is the upregulation of MHC-I surface expression, through increased transcription from the HLA locus (46). Here, we demonstrate that ORF66 modulates MHC-I surface expression independently of IFN-γ signaling. However, it will be interesting to address the effects of ORF66 on both induced MHC-I transcription and posttranslational membrane trafficking in response to both type I and type II IFN treatment. The identification of host cell proteins phosphorylated in the presence of the ORF66 kinase and the effects of ORF66 expression on the cellular transcriptome are currently under investigation.
VZV lacking ORF66 expression maintains the ability to induce MHC-I downregulation, and therefore VZV must encode one or more additional genes that affect MHC-I surface expression. Several alphaherpesviruses, including HSV-1, negatively regulate MHC-I through the virion host shutoff (vhs) gene, which mediates a global reduction of cellular protein expression (21, 26, 37, 60). VZV also causes a reduction in cellular protein synthesis that may contribute to the reduction in MHC-I surface levels in infected cells. The VZV vhs ortholog (ORF17) is known to induce mRNA degradation, albeit to a lesser extent than its HSV-1 counterpart, but its role in silencing cellular protein expression during VZV infection remains unclear (55). VZV ORF9a, the ortholog of the BHV-1 UL49.5 TAP inhibitor, was previously assessed for effects on MHC-I surface expression in a human melanoma cell line (Mel JuSo) and was found to have no effect (36). Our analyses also indicated no ability to induce MHC-I surface downregulation, even when ORF9a was expressed with its possible chaperone ORF50. However, we note that the HCMV US10 protein is able to bind MHC-I and impede its maturation through the Golgi apparatus but does not affect the surface expression of MHC-I detected by pan-specific MHC-I antibodies (17). The identities of additional VZV genes involved in MHC-I downregulation remain to be elucidated.
We conjecture that ORF66-induced MHC-I downregulation could be important at multiple stages of VZV pathogenesis. Viruses lacking ORF66 expression show only minor reductions in growth in several VZV-permissive cell types in culture but are growth restricted in T lymphocytes in the SCID-hu model of VZV infection (49, 57, 58). This restriction has been attributed to, at least in part, ORF66-mediated inhibition of apoptosis, as a greater percentage of cells showed increased caspase 3 activation when ORF66 expression was lacking (57). Thus, one role for the ORF66 kinase is to promote T-lymphocyte survival and the subsequent transfer of infectious virus to skin cells during the viremic stage of VZV infection. While we do not know whether ORF66 induces MHC-I downregulation in T cells, we suggest that this ORF66 function could serve to further enhance T-cell survival by allowing avoidance of immune surveillance. The ORF66-induced downregulation of MHC-I surface expression may also be critical during VZV growth in skin cell layers. ORF66 is not required for pathogenesis in human skin implants in the SCID-hu mouse (49, 57), but this model lacks a fully competent immune system. Recent evidence suggests that epidermal cells are infected shortly after inoculation and undergo a prolonged infection period before producing infectious lesions at the skin surface (40). Since VZV-specific immunity may become well developed during later parts of the skin infection phase, evasion of adaptive immunity could be important for survival of infected skin cells. Finally, ORF66 may affect MHC-I presentation in VZV-infected sensory neurons during latency. Several VZV lytic antigens, in addition to ORF66, are transcribed and may be expressed during latent infection, and these include ORFs 4, 21, 29, 62, and 63 (11, 47). Memory CTLs that recognize ORFs 4, 10, 29, and 62 can be detected in VZV-immune subjects (5), but human ganglia have been found to lack VZV-specific CTL infiltration (28, 62). The latter study also showed that human ganglia harboring latent HSV-1 contain HSV-1-specific CTL, clearly implying that antigens can be properly presented in neurons. Hence, the expression of ORF66 may be required for two roles during VZV latency: one may be to suppress the nuclear localization and nuclear functions of the major VZV transcriptional transactivator by promoting cytoplasmic location (15), and the other may be to prevent antigen presentation of latency-associated VZV antigens.
In summary, VZV appears to attack MHC-I using a multifaceted approach that includes the ORF66 kinase. Similar functions have not been described for orthologous US3 kinase family proteins, and as no other viral kinase has been shown to impair MHC-I transport through the Golgi complex, this represents a novel role for kinases in viral immune evasion. ORF66's ability to delay MHC-I maturation in VZV-infected cells may contribute to VZV pathogenesis by promoting efficient dissemination, survival during the extended incubation period, and persistence in latently infected ganglia.
Acknowledgments
This work was supported by Public Health Service grant EY09397, a CORE grant for vision research (EY08098), and by funds from The Eye & Ear Foundation of Pittsburgh and Research to Prevent Blindness, Inc. A.J.E. was supported by predoctoral T32 training grant AI49820.
We thank Nancy Zurowski and Kira Lathrop of the University of Pittsburgh Ophthalmology Department CORE modules for their technical assistance with flow cytometry and epifluorescence microscopy, respectively, and Nathan Shively of the University of Pittsburgh Summer Undergraduate Research Program for technical assistance.
Footnotes
Published ahead of print on 13 June 2007.
REFERENCES
- 1.Abendroth, A., I. Lin, B. Slobedman, H. Ploegh, and A. M. Arvin. 2001. Varicella-zoster virus retains major histocompatibility complex class I proteins in the Golgi compartment of infected cells. J. Virol. 75:4878-4888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abendroth, A., G. Morrow, A. L. Cunningham, and B. Slobedman. 2001. Varicella-zoster virus infection of human dendritic cells and transmission to T cells: implications for virus dissemination in the host. J. Virol. 75:6183-6192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ahn, K., T. H. Meyer, S. Uebel, P. Sempe, H. Djaballah, Y. Yang, P. A. Peterson, K. Fruh, and R. Tampe. 1996. Molecular mechanism and species specificity of TAP inhibition by herpes simplex virus ICP47. EMBO J. 15:3247-3255. [PMC free article] [PubMed] [Google Scholar]
- 4.Arbeit, R. D., J. A. Zaia, M. A. Valerio, and M. J. Levin. 1982. Infection of human peripheral blood mononuclear cells by varicella-zoster virus. Intervirology 18:56-65. [DOI] [PubMed] [Google Scholar]
- 5.Arvin, A. M., M. Sharp, M. Moir, P. R. Kinchington, M. Sadeghi-Zadeh, W. T. Ruyechan, and J. Hay. 2002. Memory cytotoxic T cell responses to viral tegument and regulatory proteins encoded by open reading frames 4, 10, 29, and 62 of varicella-zoster virus. Viral Immunol. 15:507-516. [DOI] [PubMed] [Google Scholar]
- 6.Benetti, L., and B. Roizman. 2004. Herpes simplex virus protein kinase US3 activates and functionally overlaps protein kinase A to block apoptosis. Proc. Natl. Acad. Sci. USA 101:9411-9416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bennett, E. M., J. R. Bennink, J. W. Yewdell, and F. M. Brodsky. 1999. Cutting edge: adenovirus E19 has two mechanisms for affecting class I MHC expression. J. Immunol. 162:5049-5052. [PubMed] [Google Scholar]
- 8.Capps, G. G., and M. C. Zuniga. 2000. Phosphorylation of class I MHC molecules in the absence of phorbol esters is an intracellular event and may be characteristic of trafficking molecules. Mol. Immunol. 37:59-71. [DOI] [PubMed] [Google Scholar]
- 9.Cohen, J. I. 1998. Infection of cells with varicella-zoster virus down-regulates surface expression of class I major histocompatibility complex antigens. J. Infect. Dis. 177:1390-1393. [DOI] [PubMed] [Google Scholar]
- 10.Cohrs, R. J., and D. H. Gilden. 2007. Prevalence and abundance of latently transcribed varicella-zoster virus genes in human ganglia. J. Virol. 81:2950-2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cohrs, R. J., D. H. Gilden, P. R. Kinchington, E. Grinfeld, and P. G. Kennedy. 2003. Varicella-zoster virus gene 66 transcription and translation in latently infected human ganglia. J. Virol. 77:6660-6665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Decman, V., P. R. Kinchington, S. A. Harvey, and R. L. Hendricks. 2005. Gamma interferon can block herpes simplex virus type 1 reactivation from latency, even in the presence of late gene expression. J. Virol. 79:10339-10347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Jong, M. D., J. F. Weel, T. Schuurman, P. M. Wertheim-van Dillen, and R. Boom. 2000. Quantitation of varicella-zoster virus DNA in whole blood, plasma, and serum by PCR and electrochemiluminescence. J. Clin. Microbiol. 38:2568-2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eichholtz, T., P. Vossebeld, M. van Overveld, and H. Ploegh. 1992. Activation of protein kinase C accelerates internalization of transferrin receptor but not of major histocompatibility complex class I, independent of their phosphorylation status. J. Biol. Chem. 267:22490-22495. [PubMed] [Google Scholar]
- 15.Eisfeld, A. J., S. E. Turse, S. A. Jackson, E. C. Lerner, and P. R. Kinchington. 2006. Phosphorylation of the varicella-zoster virus (VZV) major transcriptional regulatory protein IE62 by the VZV open reading frame 66 protein kinase. J. Virol. 80:1710-1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fruh, K., K. Ahn, H. Djaballah, P. Sempe, P. M. van Endert, R. Tampe, P. A. Peterson, and Y. Yang. 1995. A viral inhibitor of peptide transporters for antigen presentation. Nature 375:415-418. [DOI] [PubMed] [Google Scholar]
- 17.Furman, M. H., N. Dey, D. Tortorella, and H. L. Ploegh. 2002. The human cytomegalovirus US10 gene product delays trafficking of major histocompatibility complex class I molecules. J. Virol. 76:11753-11756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ge, R., X. Liu, and R. P. Ricciardi. 1994. E1A oncogene of adenovirus-12 mediates trans-repression of MHC class I transcription in Ad5/Ad12 somatic hybrid transformed cells. Virology 203:389-392. [DOI] [PubMed] [Google Scholar]
- 19.Gershon, A. A., D. L. Sherman, Z. Zhu, C. A. Gabel, R. T. Ambron, and M. D. Gershon. 1994. Intracellular transport of newly synthesized varicella-zoster virus: final envelopment in the trans-Golgi network. J. Virol. 68:6372-6390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gilden, D. H., A. R. Hayward, J. Krupp, M. Hunter-Laszlo, J. C. Huff, and A. Vafai. 1987. Varicella-zoster virus infection of human mononuclear cells. Virus Res. 7:117-129. [DOI] [PubMed] [Google Scholar]
- 21.Gopinath, R. S., A. P. Ambagala, S. Hinkley, and S. Srikumaran. 2002. Effects of virion host shut-off activity of bovine herpesvirus 1 on MHC class I expression. Viral Immunol. 15:595-608. [DOI] [PubMed] [Google Scholar]
- 22.Guild, B. C., and J. L. Strominger. 1984. HLA-A2 antigen phosphorylation in vitro by cyclic AMP-dependent protein kinase. Sites of phosphorylation and segmentation in class i major histocompatibility complex gene structure. J. Biol. Chem. 259:13504-13510. [PubMed] [Google Scholar]
- 23.Harson, R., and C. Grose. 1995. Egress of varicella-zoster virus from the melanoma cell: a tropism for the melanocyte. J. Virol. 69:4994-5010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hengel, H., J. O. Koopmann, T. Flohr, W. Muranyi, E. Goulmy, G. J. Hammerling, U. H. Koszinowski, and F. Momburg. 1997. A viral ER-resident glycoprotein inactivates the MHC-encoded peptide transporter. Immunity 6:623-632. [DOI] [PubMed] [Google Scholar]
- 25.Hill, A., P. Jugovic, I. York, G. Russ, J. Bennink, J. Yewdell, H. Ploegh, and D. Johnson. 1995. Herpes simplex virus turns off the TAP to evade host immunity. Nature 375:411-415. [DOI] [PubMed] [Google Scholar]
- 26.Hill, A. B., B. C. Barnett, A. J. McMichael, and D. J. McGeoch. 1994. HLA class I molecules are not transported to the cell surface in cells infected with herpes simplex virus types 1 and 2. J. Immunol. 152:2736-2741. [PubMed] [Google Scholar]
- 27.Hu, H., and J. I. Cohen. 2005. Varicella-zoster virus open reading frame 47 (ORF47) protein is critical for virus replication in dendritic cells and for spread to other cells. Virology 337:304-311. [DOI] [PubMed] [Google Scholar]
- 28.Hufner, K., T. Derfuss, S. Herberger, K. Sunami, S. Russell, I. Sinicina, V. Arbusow, M. Strupp, T. Brandt, and D. Theil. 2006. Latency of alpha-herpes viruses is accompanied by a chronic inflammation in human trigeminal ganglia but not in dorsal root ganglia. J. Neuropathol. Exp. Neurol. 65:1022-1030. [DOI] [PubMed] [Google Scholar]
- 29.Ishido, S., C. Wang, B. S. Lee, G. B. Cohen, and J. U. Jung. 2000. Downregulation of major histocompatibility complex class I molecules by Kaposi's sarcoma-associated herpesvirus K3 and K5 proteins. J. Virol. 74:5300-5309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ito, Y., H. Kimura, S. Hara, S. Kido, T. Ozaki, Y. Nishiyama, and T. Morishima. 2001. Investigation of varicella-zoster virus DNA in lymphocyte subpopulations by quantitative PCR assay. Microbiol. Immunol. 45:267-269. [DOI] [PubMed] [Google Scholar]
- 31.Jons, A., J. M. Dijkstra, and T. C. Mettenleiter. 1998. Glycoproteins M and N of pseudorabies virus form a disulfide-linked complex. J. Virol. 72:550-557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kasper, M. R., J. F. Roeth, M. Williams, T. M. Filzen, R. I. Fleis, and K. L. Collins. 2005. HIV-1 Nef disrupts antigen presentation early in the secretory pathway. J. Biol. Chem. 280:12840-12848. [DOI] [PubMed] [Google Scholar]
- 33.Kinchington, P. R., K. Fite, A. Seman, and S. E. Turse. 2001. Virion association of IE62, the varicella-zoster virus (VZV) major transcriptional regulatory protein, requires expression of the VZV open reading frame 66 protein kinase. J. Virol. 75:9106-9113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kinchington, P. R., K. Fite, and S. E. Turse. 2000. Nuclear accumulation of IE62, the varicella-zoster virus (VZV) major transcriptional regulatory protein, is inhibited by phosphorylation mediated by the VZV open reading frame 66 protein kinase. J. Virol. 74:2265-2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Koenig, A., and M. H. Wolff. 2003. Infectibility of separated peripheral blood mononuclear cell subpopulations by varicella-zoster virus (VZV). J. Med. Virol. 70(Suppl. 1):S59-S63. [DOI] [PubMed] [Google Scholar]
- 36.Koppers-Lalic, D., E. A. Reits, M. E. Ressing, A. D. Lipinska, R. Abele, J. Koch, M. Marcondes Rezende, P. Admiraal, D. van Leeuwen, K. Bienkowska-Szewczyk, T. C. Mettenleiter, F. A. Rijsewijk, R. Tampe, J. Neefjes, and E. J. Wiertz. 2005. Varicelloviruses avoid T cell recognition by UL49.5-mediated inactivation of the transporter associated with antigen processing. Proc. Natl. Acad. Sci. USA 102:5144-5149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Koppers-Lalic, D., F. A. Rijsewijk, S. B. Verschuren, J. A. van Gaans-Van den Brink, A. Neisig, M. E. Ressing, J. Neefjes, and E. J. Wiertz. 2001. The UL41-encoded virion host shutoff (vhs) protein and vhs-independent mechanisms are responsible for down-regulation of MHC class I molecules by bovine herpesvirus 1. J. Gen. Virol. 82:2071-2081. [DOI] [PubMed] [Google Scholar]
- 38.Ku, C. C., J. Besser, A. Abendroth, C. Grose, and A. M. Arvin. 2005. Varicella-zoster virus pathogenesis and immunobiology: new concepts emerging from investigations with the SCIDhu mouse model. J. Virol. 79:2651-2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ku, C. C., J. A. Padilla, C. Grose, E. C. Butcher, and A. M. Arvin. 2002. Tropism of varicella-zoster virus for human tonsillar CD4+ T lymphocytes that express activation, memory, and skin homing markers. J. Virol. 76:11425-11433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ku, C. C., L. Zerboni, H. Ito, B. S. Graham, M. Wallace, and A. M. Arvin. 2004. Varicella-zoster virus transfer to skin by T Cells and modulation of viral replication by epidermal cell interferon-alpha. J. Exp. Med. 200:917-925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Leader, D. P., A. D. Deana, F. Marchiori, F. C. Purves, and L. A. Pinna. 1991. Further definition of the substrate specificity of the alpha-herpesvirus protein kinase and comparison with protein kinases A and C. Biochim. Biophys. Acta 1091:426-431. [DOI] [PubMed] [Google Scholar]
- 42.Lehner, P. J., J. T. Karttunen, G. W. Wilkinson, and P. Cresswell. 1997. The human cytomegalovirus US6 glycoprotein inhibits transporter associated with antigen processing-dependent peptide translocation. Proc. Natl. Acad. Sci. USA 94:6904-6909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li, Y., L. Salter-Cid, A. Vitiello, T. Preckel, J. D. Lee, A. Angulo, Z. Cai, P. A. Peterson, and Y. Yang. 2000. Regulation of transporter associated with antigen processing by phosphorylation. J. Biol. Chem. 275:24130-24135. [DOI] [PubMed] [Google Scholar]
- 44.Lippe, R., E. Luke, Y. T. Kuah, C. Lomas, and W. A. Jefferies. 1991. Adenovirus infection inhibits the phosphorylation of major histocompatibility complex class I proteins. J. Exp. Med. 174:1159-1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mainka, C., B. Fuss, H. Geiger, H. Hofelmayr, and M. H. Wolff. 1998. Characterization of viremia at different stages of varicella-zoster virus infection. J. Med. Virol. 56:91-98. [DOI] [PubMed] [Google Scholar]
- 46.Malmgaard, L. 2004. Induction and regulation of IFNs during viral infections. J. Interferon Cytokine Res. 24:439-454. [DOI] [PubMed] [Google Scholar]
- 47.Mitchell, B. M., D. C. Bloom, R. J. Cohrs, D. H. Gilden, and P. G. Kennedy. 2003. Herpes simplex virus-1 and varicella-zoster virus latency in ganglia. J. Neurovirol. 9:194-204. [DOI] [PubMed] [Google Scholar]
- 48.Moffat, J., C. Mo, J. J. Cheng, M. Sommer, L. Zerboni, S. Stamatis, and A. M. Arvin. 2004. Functions of the C-terminal domain of varicella-zoster virus glycoprotein E in viral replication in vitro and skin and T-cell tropism in vivo. J. Virol. 78:12406-12415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Moffat, J. F., L. Zerboni, M. H. Sommer, T. C. Heineman, J. I. Cohen, H. Kaneshima, and A. M. Arvin. 1998. The ORF47 and ORF66 putative protein kinases of varicella-zoster virus determine tropism for human T cells and skin in the SCID-hu mouse. Proc. Natl. Acad. Sci. USA 95:11969-11974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Morrow, G., B. Slobedman, A. L. Cunningham, and A. Abendroth. 2003. Varicella-zoster virus productively infects mature dendritic cells and alters their immune function. J. Virol. 77:4950-4959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Niizuma, T., L. Zerboni, M. H. Sommer, H. Ito, S. Hinchliffe, and A. M. Arvin. 2003. Construction of varicella-zoster virus recombinants from parent Oka cosmids and demonstration that ORF65 protein is dispensable for infection of human skin and T cells in the SCID-hu mouse model. J. Virol. 77:6062-6065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Park, B., Y. Kim, J. Shin, S. Lee, K. Cho, K. Fruh, S. Lee, and K. Ahn. 2004. Human cytomegalovirus inhibits tapasin-dependent peptide loading and optimization of the MHC class I peptide cargo for immune evasion. Immunity 20:71-85. [DOI] [PubMed] [Google Scholar]
- 53.Pober, J. S., B. C. Guild, and J. L. Strominger. 1978. Phosphorylation in vivo and in vitro of human histocompatibility antigens (HLA-A and HLA-B) in the carboxy-terminal intracellular domain. Proc. Natl. Acad. Sci. USA 75:6002-6006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Proffitt, J. L., E. Sharma, and G. E. Blair. 1994. Adenovirus 12-mediated down-regulation of the major histocompatibility complex (MHC) class I promoter: identification of a negative regulatory element responsive to Ad12 E1A. Nucleic Acids Res. 22:4779-4788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sato, H., L. D. Callanan, L. Pesnicak, T. Krogmann, and J. I. Cohen. 2002. Varicella-zoster virus (VZV) ORF17 protein induces RNA cleavage and is critical for replication of VZV at 37°C but not 33°C. J. Virol. 76:11012-11023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schaap-Nutt, A., M. Sommer, X. Che, L. Zerboni, and A. M. Arvin. 2006. Orf66 protein kinase function is required for T cell tropism of varicella-zoster virus in vivo. J. Virol. 80:11806-11816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schaap, A., J. F. Fortin, M. Sommer, L. Zerboni, S. Stamatis, C. C. Ku, G. P. Nolan, and A. M. Arvin. 2005. T-cell tropism and the role of ORF66 protein in pathogenesis of varicella-zoster virus infection. J. Virol. 79:12921-12933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Soong, W., J. C. Schultz, A. C. Patera, M. H. Sommer, and J. I. Cohen. 2000. Infection of human T lymphocytes with varicella-zoster virus: an analysis with viral mutants and clinical isolates. J. Virol. 74:1864-1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Storkus, W. J., H. J. Zeh III, R. D. Salter, and M. T. Lotze. 1993. Identification of T-cell epitopes: rapid isolation of class I-presented peptides from viable cells by mild acid elution. J. Immunother. 14:94-103. [PubMed] [Google Scholar]
- 60.Tigges, M. A., S. Leng, D. C. Johnson, and R. L. Burke. 1996. Human herpes simplex virus (HSV)-specific CD8+ CTL clones recognize HSV-2-infected fibroblasts after treatment with IFN-gamma or when virion host shutoff functions are disabled. J. Immunol. 156:3901-3910. [PubMed] [Google Scholar]
- 61.Tomazin, R., A. B. Hill, P. Jugovic, I. York, P. van Endert, H. L. Ploegh, D. W. Andrews, and D. C. Johnson. 1996. Stable binding of the herpes simplex virus ICP47 protein to the peptide binding site of TAP. EMBO J. 15:3256-3266. [PMC free article] [PubMed] [Google Scholar]
- 62.Verjans, G. M., R. Q. Hintzen, J. M. van Dun, A. Poot, J. C. Milikan, J. D. Laman, A. W. Langerak, P. R. Kinchington, and A. D. Osterhaus. 2007. Selective retention of herpes simplex virus-specific T cells in latently infected human trigeminal ganglia. Proc. Natl. Acad. Sci. USA 104:3496-3501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhao, B., and R. P. Ricciardi. 2006. E1A is the component of the MHC class I enhancer complex that mediates HDAC chromatin repression in adenovirus-12 tumorigenic cells. Virology 352:338-344. [DOI] [PubMed] [Google Scholar]