

Abstract
It has become clear that in Saccharomyces cerevisiae the transcription of ribosomal protein genes, which makes up a major proportion of the total transcription by RNA polymerase II, is controlled by the interaction of three transcription factors, Rap1, Fhl1, and Ifh1. Of these, only Rap1 binds directly to DNA and only Ifh1 is absent when transcription is repressed. We have examined further the nature of this interaction and find that Ifh1 is actually associated with at least two complexes. In addition to its association with Rap1 and Fhl1, Ifh1 forms a complex (CURI) with casein kinase 2 (CK2), Utp22, and Rrp7. Fhl1 is loosely associated with the CURI complex; its absence partially destabilizes the complex. The CK2 within the complex phosphorylates Ifh1 in vitro but no other members of the complex. Two major components of this complex, Utp22 and Rrp7, are essential participants in the processing of pre-rRNA. Depletion of either protein, but not of other proteins in the early processing steps, brings about a substantial increase in ribosomal protein mRNA. We propose a model in which the CURI complex is a key mediator between the two parallel pathways necessary for ribosome synthesis: the transcription and processing of pre-rRNA and the transcription of ribosomal protein genes.
The biosynthesis of ribosomes plays a substantial role in the economy of the yeast cell, being responsible for more than 70% of the total transcription and more than 25% of the total translation of rapidly growing cells (39). Transcription of the 138 genes encoding ribosomal proteins (RP) forms one of the tightest clusters in almost all transcriptome experiments (4, 9), with rapid repression in response to stress and rapid derepression in response to improved conditions. Although much progress has been made in recent years, the molecular basis for the tight coordination of transcription of RP genes and its coupling to rRNA transcription has remained elusive.
It has long been known that the DNA binding protein Rap1 is involved in the transcription of many RP genes (32, 36, 40) and that most but not all RP genes carry a pair of Rap1-binding sites in their promoters (22). However, Rap1 binds to many sites in the yeast genome, acting as an activator at some and as a repressor at others, and indeed is a structural element at the telomeres (25). With the identification of Fhl1 as binding almost exclusively to RP promoters (23), recent work has established that transcription of RP genes is controlled, at least in part, by the interplay of three factors, Rap1, Fhl1, and Ifh1, that are found at most RP promoters when transcription is occurring (27, 33, 35, 38). The association of Ifh1 appears to depend on its interaction with the forkhead-associated (FHA) domain of Fhl1. Repression of transcription leads to the loss of Ifh1, but not of Rap1 and Fhl1, from the RP promoters. Although this has been attributed to competition from Crf1 entering the nucleus as a result of inhibition of TOR, we have found that this is not the case for the W303 strain that is our wild type (42). As this paper was being completed, yet another protein, Hmo1, was identified at the Fhl1/Ifh1/RP-promoter interaction site and was suggested to coordinate rRNA and RP gene transcription (14).
On investigating more thoroughly the basis for the interaction of Rap1, Fhl1, and Ifh1, we have found that most of the cell's Ifh1 is in a complex with three quite different proteins, casein kinase 2 (CK2), Utp22, and Rrp7. Ifh1 can be phosphorylated by CK2 in vitro. Fhl1, but not Rap1, is weakly associated with this complex. An intriguing feature is that both Utp22 and Rrp7 are implicated in the processing of pre-rRNA (2, 3). We present evidence to suggest that this complex is a link between the two parallel pathways leading to ribosome biosynthesis: the transcription and processing of rRNA and the transcription of RP genes leading to the production of ribosomal proteins.
MATERIALS AND METHODS
Strains.
The strains used are listed in Table 1. Strain YZ73 was constructed by introducing the RAP1 open reading frame into plasmid pBS1761 (31) and subsequently replacing the GAL1 promoter with a 300-bp fragment upstream of the RAP1 gene before introducing this into W303. Thus, N-terminally tandem affinity purification (TAP)-tagged RAP1 is under its own promoter. Other tagged strains were constructed as described previously (11, 26). Cultures were grown in YPD (1% yeast extract, 2% Bacto peptone, 2% dextrose) except when, where indicated, dextrose was replaced with galactose (YP-Gal) or raffinose (YP-raffinose).
TABLE 1.
Strains used in this study
| Strain | Genotype | Source or reference |
|---|---|---|
| W303 | ade2-1 leu2-3,112 ura3-1 his3-11,15 trp1-1 can1-100 ssd1-1 | 37 |
| YZ73 | W303 α TAP-RAP1::TRP1 | 33 |
| YZ146 | W303 akanMX4::IFH1-HA3 | This study |
| DR12 | W303 aFHL1-TAP::G418r | 33 |
| DR13 | W303 aFHL1-HA3::G418r | 33 |
| DR14 | W303 α IFH1-TAP::TRP1 | 33 |
| DR23 | W303 α IFH1-TAP::G418rFHL1-HA3::G418r | This study |
| DR34 | W303 α FHL1Δ::HIS3 | 33 |
| DR36 | W303 aIFH1-MYC9::TRP1 FHL1-HA3::G418r | 33 |
| DR47 | W303 FHL1Δ::HIS3 IFH1-MYC9::TRP1 with pRS316 (CEN URA3 FHL1-HA3) | 33 |
| DR48 | As DR47, with pRS316 (CEN URA3 ΔFH-HA3[FHL1Δ440-567]) | 33 |
| DR57 | W303 a/α FHL1Δ::HIS3/FHL1 IFH1-MYC9::TRP1/IFH1 | 33 |
| DR49 | As DR47, with pRS316 (CEN URA3 ΔFHA-HA3[FHL1Δ300-374]) | 33 |
| DR65 | As DR47, with pRS316 (CEN URA3 FHL1-HA3 [S325R]) | 33 |
| DR113 | DR47, UTP22-TAP::G418r | This study |
| DR114 | DR47, RRP7-FLAG::G418r | This study |
| DR115 | DR47, CKB2-FLAG::G418r | This study |
| DR116 | DR34, UTP22-TAP::G418r | This study |
| DR117 | DR34, RRP7-FLAG::G418r | This study |
| DR118 | DR34, CKB2-FLAG::G418r | This study |
| DR127 | W303 a,UTP22-FLAG::G418r | This study |
| JD001 | W303 α/a Fhl1-HA3::G418/Fhl1 Ifh1-Myc13 UTP22-FLAG | This study |
| JD007 | W303 akanMX4::IFH1-HA3GAL-HA-UTP7 | This study |
| JD009 | W303 akanMX4::IFH1-HA3GAL-HA-UTP11 | This study |
| JD010 | W303 akanMX4::IFH1-HA3GAL-HA-UTP14 | This study |
| YSC1178-7500641 | UTP22-TAP | Open Biosystems |
| YSC1178-7499458 | RRP7-TAP | Open Biosystems |
| YSC1178-7502757 | CKB2-TAP | Open Biosystems |
Preparation of yeast cell lysates, immunoprecipitation, and Western blotting.
A 50-ml yeast culture was grown to an optical density at 600 nm (OD600) of∼1.0. Cells were harvested, washed with IP150 buffer (50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 2 mM MgCl2, 0.1% NP40), and lysed by vortexing with glass beads in 300 μl ice-cold IP150 buffer supplemented with Complete Mini protease inhibitor cocktail tablet (Roche) and 1 mM phenylmethylsulfonyl fluoride (PMSF). Lysates were centrifuged at 13,000 × g for 1 min at 4°C to remove debris. For the coimmunoprecipitation (Co-IP) experiments performed in the presence of ethidium bromide, the extract was incubated with 200 μg/ml of ethidium bromide on ice for 30 min followed by centrifugation at 13,000 × g for 1 min. The supernatant was used for immunoprecipitation. The extracts thus prepared were incubated at 4°C with anti-Myc mouse monoclonal antibody (9E10), anti-FLAG polyclonal antibody, anti-Rap1 rabbit polyclonal antibody (a kind gift from D. Shore), preimmune serum, or anti-Nhp2 rabbit polyclonal antibody coupled with protein A-agarose beads (Pierce). To immunoprecipitate TAP-tagged proteins, immunoglobulin G (IgG)-Sepharose beads were used. For the immunoprecipitations done with hemagglutinin (HA) antibody, anti-HA affinity matrix (Roche) was used. Following incubation, beads were washed three times with IP150 buffer. The washed beads containing bound proteins were suspended in 50 μl of 1% sodium dodecyl sulfate (SDS) gel loading buffer and heated at 95°C for 5 min. The released polypeptides in 20 μl of heated sample were resolved in 0.1% SDS-5% polyacrylamide gels. The separated polypeptides were transferred onto a nitrocellulose membrane and analyzed by Western blotting using anti-HA (3F10), anti-c-Myc (9E10), anti-FLAG, or anti-Rap1 polyclonal antibodies wherever applicable.
ChIP.
For chromatin immunoprecipitation (ChIP), a 200-ml culture was grown to early log phase (∼1 × 107 cells/ml) at 30°C. Formaldehyde was added to a final concentration of 1%, and cells were incubated at room temperature for 30 min with occasional swirling. Glycine was added to a final concentration of 360 mM. Cells were washed in 1× Tris-buffered saline and lysed with glass beads in breaking buffer (0.1 M Tris [pH 8.0], 20% glycerol, 1 mM PMSF) using a Mini bead beater (Biospec Products). Cross-linked chromatin was collected by centrifugation at 13,000 rpm for 10 min, and pellets were resuspended in 1 ml FA buffer (50 mM HEPES-KOH [pH 7.5], 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% deoxycholate).
Cells were sonicated until DNA was of an average size of 400 to 600 bp. Soluble chromatin was separated from insoluble material by centrifugation at 13,000 rpm for 10 min and adjusted to 3.5 ml with FA buffer. Chromatin was stored in 800-μl aliquots at −80°C.
Immunoprecipitations of TAP-tagged proteins were performed using IgG-Sepharose beads for 5 h at 4°C. Chromatin was eluted, cross-links were reversed, and DNA was prepared and subjected to quantitative PCR analyses performed in real time using an Applied Biosystems 7700 sequence detector. To calculate the enrichment (fold) of occupancy at an individual promoter, the apparent cross-linking efficiency was determined by dividing the amount of PCR product from the immunoprecipitated sample by the amount of PCR product in the input sample prior to immunoprecipitation and subtracting the apparent cross-linking efficiency of a control promoter of the CYC1 gene that was shown not to be occupied by Fhl1 in a genomewide study (23).
Glycerol gradient analysis and mass spectrometry.
Saccharomyces cerevisiae extracts were loaded onto 10 to 30% glycerol gradients in IP150 buffer and centrifuged at 49,000 rpm for 5 h at 4°C in an SW50.1 rotor. For the experiments in which the first step of TAP purification was followed by glycerol gradient centrifugation, the purification was performed as follows. Two liters of culture from the untagged or TAP-tagged Ifh1 strains (DR13 and DR23, respectively) was grown to an OD600 of ∼1.0. Cells were pelleted, washed first with 500 ml water, and then washed with 50 ml IP150 buffer. Then the cells were resuspended in 10 ml IP150 buffer containing 10% glycerol and 2 mM PMSF and frozen in liquid nitrogen. Extracts were prepared by grinding the frozen cells with a mortar and pestle in the presence of liquid nitrogen. The thawed extracts were centrifuged for 5 min at 7,000 rpm to remove the unbroken cells, and then the supernatant was centrifuged again at 10,000 rpm for 15 min to remove cell debris. Lysates thus prepared were incubated with IgG-Sepharose beads for 2 h at 4°C with tumbling. Beads were washed three times in IP150 and once with TEV cleavage buffer (31) for 5 min each. The beads were resuspended in 400 μl of TEV cleavage buffer and incubated overnight with 100 U of recombinant TEV enzyme (Invitrogen). Three hundred microliters of the TEV eluate was loaded on glycerol gradient for centrifugation described above. Coomassie-stained bands from lanes corresponding to gradient fraction 11 were cut out from the gel and sent for matrix-assisted laser desorption ionization-mass spectrometry peptide mass mapping to the Protein Chemistry core facility, Columbia University, New York.
In vitro phosphorylation studies.
One hundred fifty microliters of fractions 11 and 13 from the glycerol gradients of lysates from the various strains was mixed with an equal volume of 20 mM MgCl2 and incubated with γ-[32P]ATP or γ-[32P]GTP (6,000 Ci/mmol) for 45 min at 30°C and then incubated either with IgG-Sepharose beads or with HA-conjugated affinity matrix for 2 h at 4°C. The beads were centrifuged at 4000 rpm for a minute and washed three times with IP150 buffer. The washed beads with bound proteins were suspended in 50 μl of 1% SDS gel loading buffer and heated at 95°C for 5 min. The released polypeptides were resolved in two 0.1% SDS-5 to 15% gradient polyacrylamide gels. One was dried and subjected to autoradiography. The other was blotted to a nitrocellulose membrane and subjected to Western analysis using anti-HA (3F10) or horseradish peroxidase (HRP)-conjugated anti-protein A antibodies as specified.
RESULTS
Rap1 interacts with Fhl1 and with Ifh1.
Although ChIP experiments clearly demonstrate that Fhl1 is associated with RP gene promoters, we have been unable to detect a direct interaction of Fhl1 with DNA of the RP gene promoter, as measured by gel shift in vitro. Furthermore, deletion of the putative DNA binding domain of Fhl1 does not cause a significant growth defect (33). These results prompted us to ask if Fhl1 is brought to RP genes, not by its FH domain, but by interacting with Rap1. Indeed, Co-IP analysis using anti-Rap1 antibody shows that Rap1 can associate not only with Fhl1 but with Ifh1 as well (Fig. 1A, left). The interaction is unaffected by the presence of ethidium bromide, known to dissociate proteins from DNA (21) (Fig. 1A, right), suggesting that Rap1 can associate with Fhl1 and Ifh1 in a DNA-independent manner. The FHA domain, found in many “forkhead”-type proteins, is known to bind to phosphopeptides (6) and is essential for Fhl1 function and for its interaction with Ifh1. Even a point mutation in the FHA domain (S325R) has a serious impact on its function (33). Interestingly, mutation of neither the FH nor the FHA domains of Fhl1 affects the Co-IP of either Fhl1 or Ifh1 by Rap1 (Fig. 1B, lanes 7, 8, and 9). This observation leads to the unexpected conclusion that Ifh1 can interact with Rap1 independently of Fhl1.
FIG. 1.

Rap1 interacts with both Fhl1 and Ifh1. (A) Co-IP was carried out using rabbit polyclonal anti-Rap1 (αRap1) antibody with extracts prepared from strain DR36 (FHL1-HA3 IFH1-Myc9 double tagged). The immunoprecipitated protein complex was resuspended in SDS loading buffer, boiled, and analyzed by SDS-polyacrylamide gel electrophoresis followed by Western blotting using anti-HA, anti-Myc, or anti-Rap1 antibodies (lane 3). Whole-cell extract was loaded in a separate lane as a loading control (lane 1; Input). Immunoprecipitation with a rabbit polyclonal antibody raised against the yeast protein Nhp2 (αNhp2) was used as a negative control (lane 2). In the right-hand panel, Co-IP was carried out using anti-Rap1 antibody on extracts prepared from DR36 in the presence of 200 μg/ml ethidium bromide (lane 6). In this case, preimmune serum was used as a negative control (lane 5). (B) Co-IP was carried out using rabbit polyclonal anti-Rap1 antibody on extracts prepared from strains harboring HA3-tagged full-length Fhl1 (WT FHL1), with the FH domain deleted (ΔFH), with the FHA domain deleted (ΔFHA), or with the S325R mutant version of Fhl1 (lanes 6, 7, 8, and 9, strains DR47, DR48, DR49, and DR65, respectively). The endogenous copy of FHL1 in these strains is deleted, and Ifh1 is tagged C terminally with Myc9. Rabbit polyclonal antibody against Nhp2 was used as a negative control (lane 5).
Ifh1 is found as a high-MW complex.
To clarify the relationship between Rap1, Fhl1, and Ifh1, we asked whether these three proteins could be found in a stable complex. Glycerol gradient analysis of an extract of wild-type cells is shown in Fig. 2A. Strikingly, Ifh1 sediments as a complex (or complexes) with an apparent molecular mass of about 350 to 400 kDa, significantly larger than expected for a 122-kDa protein. Both Rap1 and Fhl1 sediment primarily as free proteins near the top of the gradient but with a pronounced tail down the gradient. (The significance of the small amount of Rap1 that migrates slightly faster in the gel than most of the Rap1, in fractions 13 and 15, is under investigation.) Since some of the Rap1 and Fhl1 cosediments with Ifh1 in fractions 9 and 11, we asked if they were associated. In the higher-molecular-weight (MW) gradient fractions, not only does Ifh1 coimmunoprecipitate Fhl1, but Rap1 can also coimmunoprecipitate both Fhl1 and Ifh1 (Fig. 2B and C). The efficiency with which Rap1 can coimmunoprecipitate Fhl1 is much greater in the high-MW fractions (fractions 9 and 11) than at the top of the gradient (fractions 3 and 5). Thus, in these fractions Rap1 and Fhl1 are complexed with at least a part of Ifh1.
FIG. 2.
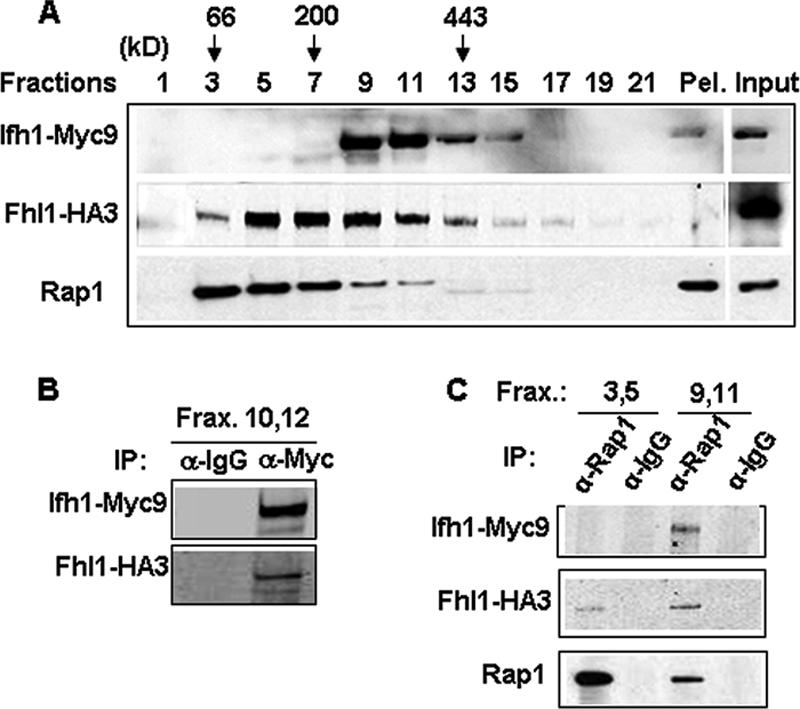
Ifh1 is found as a high-MW complex. (A) Extract prepared from strain DR36 (FHL1-HA3 IFH1-Myc9) was loaded onto a 10 to 30% glycerol gradient and centrifuged for 5 h at 49 krpm using an SW50.1 rotor. Two hundred-microliter fractions were collected. Aliquots (15 μl) from the indicated fractions as well as the whole-cell extract (Input) were analyzed by SDS-polyacrylamide gel electrophoresis followed by Western blotting using anti-HA, anti-Myc, or anti-Rap1 antibodies. The positions of the MW markers bovine serum albumin (66 kDa), β-amylase (200 kDa), and apoferritin (443 kDa), centrifuged in a parallel gradient, are indicated above. Pel, pellet at the bottom of the glycerol gradient. (B) Co-IP was performed on a pool of fractions (Frax) 10 and 12 using anti-Myc (α-Myc) antibody or mouse IgG (α-IgG). This was followed by Western blot analysis of the immunoprecipitated protein complex and probing with anti-HA and anti-Myc antibodies. (C) Co-IP using rabbit polyclonal anti-Rap1 (α-Rap1) antibody or rabbit IgG was performed on a pool of fractions 3 and 5 or 9 and 11 of the glycerol gradient shown in Fig. 2A. Western blotting was performed on the immunoprecipitated protein complex and probed with anti-HA, anti-Myc, or anti-Rap1 antibodies.
Ifh1 is in a complex with rRNA processing factors.
Extracts of cells carrying Fhl1-TAP, Ifh1-TAP, and TAP-Rap1 (the latter tagged at the N terminus [see Materials and Methods] because a C-terminal tag interferes with function) (Fig. 3A) were used to estimate the relative amounts of the three proteins using a slot blot (Fig. 3B) The results suggest that Fhl1 and Ifh1 are approximately equimolar but only 10 to 20% the level of Rap1, in confirmation of the genomewide analysis (12), which is not surprising because Rap1 is found at many sites in the genome. However, since any complex with Fhl1 could account for only a small fraction of the Ifh1, it seemed likely that most of the Ifh1 was associated with something else. To identify other proteins that are complexed with Ifh1, we carried a TAP-tagged Ifh1 through the first step of purification (31), followed by a glycerol gradient, with the hope that most of the proteins that are associated with Ifh1 in fractions 9 to 11 can be detected by silver staining. As is apparent from Fig. 3C, several proteins copurified with Ifh1 on the gradient (fractions 9 and 11). The most prominent of these was identified by mass spectrometry as Utp22, at 140 kDa, consistent with its migration in the denaturing gel. Note that Ifh1, also identified by mass spectrometry, migrates anomalously slowly. In addition, the lower bands contained Rrp7 and the α subunit of CK2. These results are consistent with a recent analysis of TAP-tagged Utp22 in a quite different strain (31) that identified a complex termed UTP-C. This complex contained Rrp7, the four subunits of CK2, and Ifh1 (see supplemental data in reference 19). However, Ifh1 was not visible on the accompanying gel (see Fig. 3C of reference 19), presumably because we find it to be highly labile to proteolytic digestion. Utp22 has also been identified as part of a much larger, 2.2-MDa, complex of proteins associated with U3 snoRNA (the “SSU processome”) and implicated in the early cleavage steps of 35S pre-rRNA (3). Rrp7 has been implicated in a later step in the assembly of 40S ribosomal subunits (2). The absence of either Utp22 or Rrp7 leads to a deficiency in the formation of 40S ribosomal subunits.
FIG. 3.

Ifh1 is in a complex with rRNA processing factors. (A) Western blot analysis performed on whole-cell extracts prepared from the indicated wild-type or TAP-tagged strains, each under its own promoter (see Materials and Methods). HRP-conjugated chicken anti-protein A was used to probe for the TAP-tagged proteins. (B) Slot blot analysis performed on serially diluted amounts of whole-cell extracts prepared from the indicated strains. WT, wild type. (C) The first step of TAP purification (i.e., the eluate derived from an IgG-Sepharose column after cleavage with the TEV protease) was performed using untagged or Ifh1-TAP strains (DR13 and DR23, respectively) as described in Materials and Methods. This was then applied on a glycerol gradient as described in the legend to Fig. 2A, and fractions were analyzed by SDS-polyacrylamide gel electrophoresis and silver staining. Ifh1 and its associated proteins were visualized largely in fractions 9 and 11 as shown. Proteins identified by mass spectrometry are indicated.
To validate the above results and to determine the glycerol gradient pattern of Utp22, Rrp7, and the β subunit of the CK2 protein (Ckb2), we C-terminally FLAG tagged Rrp7 and Ckb2 and TAP tagged Utp22 in a strain carrying Fhl1-HA and Ifh1-Myc (strains DR114, DR115, and DR113, respectively [see Table 1]). A summary of the glycerol gradient patterns from the three strains is shown in Fig. 4A. Both Rrp7-FLAG and Utp22-TAP peak at fractions 9, 11, and 13. A considerable portion of the Ckb2-FLAG protein is also detected in those fractions. It is noteworthy that substantial fractions of Utp22 and Rrp7 are present in the pellet, consistent with the finding that Utp22 is a member of a large nucleolar U3 ribonucleoprotein complex (3) and that both Utp22 and Rrp7 have been identified in a 90S preribosome particle (8). In order to demonstrate that the complex with Ifh1 was distinct from the U3-containing processome, we carried out Co-IP analysis of an extract of strain JD001, carrying Ifh1-Myc9 and Utp22-FLAG. As is apparent from Fig. 4B, Ifh-Myc can coimmunoprecipitate Utp22-FLAG but not U3 RNA (lane 3), while Utp22-FLAG can coimmunoprecipitate both Ifh1 and U3 RNA (lane 4). The results shown in Fig. 4 strongly suggest that much of the Ifh1 in fractions 9 to 13 of the glycerol gradient is present in a complex with CK2, Utp22, and Rrp7. We term this the CURI complex (CK2-Utp22-Rrp7-Ifh1).
FIG. 4.

Portions of Ckb2, Rrp7, and Utp22 cosediment with Ifh1. (A) Glycerol gradient analysis was performed on extracts prepared from cultures of strains DR113, DR114, and DR115. Aliquots (15 μl) from the indicated fractions were analyzed by SDS-polyacrylamide gel electrophoresis followed by Western blotting using anti-HA, anti-Myc, anti-Rap1, or anti-FLAG antibodies for strains DR114 and DR115. For strain DR113, Western blotting was performed using HRP-conjugated chicken IgG to detect Utp22-TAP. A summary of the glycerol gradient pattern from the three strains is shown. Pel, pellet at the bottom of the glycerol gradient. (B) Whole-cell extract was prepared from strains JD001 (Ifh1-Myc9 Utp22-FLAG) (lanes 1, 3, and 4) and W303 (untagged) (lanes 2, 5, and 6) and subjected to immunoprecipitation with anti-myc (α Myc) or anti-FLAG (α-FLAG) antibody, and the bound fraction was subjected to Western (upper two panels) and Northern (lower panel) analyses. α Utp22:FLAG, anti-Utp22-FLAG. Lanes 1 and 2, input (6.5%); lanes 3 and 5, IP with anti-Myc; lanes 4 and 6, IP with anti-FLAG.
Fhl1 is loosely associated with the CURI complex.
Although the purified CURI complex in Fig. 3C showed no silver-stained band corresponding to Fhl1, Western blotting revealed a minute amount of Fhl1 but no Rap1 (data not shown). This suggested that Fhl1 might be loosely associated with the CURI complex. To determine if this is the case, we performed Co-IP experiments on peak fractions containing the CURI complex with the respective FLAG-tagged or TAP-tagged proteins. As expected, Ifh1-Myc9 could be coimmunoprecipitated along with Utp22-TAP by IgG-Sepharose from a strain carrying Utp22-TAP strain (DR113) and not from a strain where Utp22 is untagged (Fig. 5A). Similarly, rabbit anti-FLAG antibody could efficiently coimmunoprecipitate Ifh1-Myc9 from strains DR114 (Rrp7-FLAG) and DR115 (Ckb2-FLAG) (Fig. 5B and C). Furthermore, in all of these cases, a small but reproducible amount of Fhl1 could be coimmunoprecipitated from these fractions with Utp22, Rrp7, and Ckb2 (Fig. 5A to C, middle rows). Note that although the anti-FLAG antibody can immunoprecipitate the small amount of the FLAG-tagged Rrp7 or Ckb2 that is present in the fractions near the top of the gradient, it was unable to coimmunoprecipitate (Fig. 5B and C) any of the relatively large amount of Fhl1 from these fractions (Fig. 4), suggesting that the association of Fhl1 with the CURI complex is authentic. From none of the fractions was Rap1 immunoprecipitated by Utp22, Rrp7, or Ckb2 (data not shown).
FIG. 5.

The CURI complex interacts with Fhl1. (A) Co-IP using IgG-Sepharose was performed on a pool of fractions 9 and 11 of the glycerol gradients from extracts prepared from strains DR113 (UTP22-TAP) and DR47 (as a negative control). Western blotting was performed on the immunoprecipitated proteins using anti-Myc, anti-HA, and HRP-conjugated chicken IgG. (B and C) Co-IP using anti-FLAG rabbit polyclonal antibody was performed on a pool of indicated fractions of the glycerol gradients from extracts prepared from strains DR114 (RRP7-FLAG) and DR115 (CKB2-FLAG). This was followed by Western blotting of the immunoprecipitated protein complexes using the indicated antibodies.
To confirm that Fhl1 indeed associates with the CURI complex within the cell, we performed Co-IP analysis on a whole-cell extract. As shown in Fig. 6A, Fhl1-HA3 can coimmunoprecipitate Rrp7-FLAG (lane 5) or Ckb2-FLAG (lane 6). Conversely, Utp22-TAP or Ckb2-FLAG can coimmunoprecipitate Fhl1-HA3 (Fig. 6B, lane 4, and 6C, lane 3, respectively). It is noteworthy that immunoprecipitation of Utp22 or Ckb2 does not coimmunoprecipitate Rap1 (Fig. 6B and C). Conversely, anti-Rap1 does not detectably coimmunoprecipitate Rrp7 (Fig. 6D, lane 2) or Utp22 (Fig. 6D, lane 5) under conditions where it does coimmunoprecipitate Fhl1 and Ifh1 (lane 2). Taken together, the results shown thus far suggest that (i) a small proportion of Fhl1 and Ifh1 is associated with Rap1, (ii) Ifh1 is mostly associated with the CURI complex, (iii) Fhl1 is weakly associated with the CURI complex, and (iv) Rap1 is not associated with the CURI complex.
FIG. 6.

Fhl1 but not Rap1 interacts with components of the CURI complex in whole-cell extracts. (A) Co-IP was performed using anti-HA (α-HA) antibody or anti-mouse IgG (α-IgG) on strains DR114 (Rrp7-FLAG) and DR115 (Ckb2-FLAG), respectively. This was followed by Western blot analysis and probing with anti-HA and anti-FLAG antibodies to detect the indicated proteins. (B and C) Co-IP followed by Western blot analysis was performed using IgG-Sepharose on strains DR113 (Utp22-TAP) (B) and DR115 (C), respectively. In panel B, immunoprecipitation with IgG-Sepharose performed on strain DR47 was used as a negative control. In panel C, immunoprecipitation with mouse IgG was performed as a negative control. (D) Co-IP was performed using anti-rabbit Rap1 (α-Rap1) antibody or rabbit IgG on whole-cell extracts prepared from DR114 (Rrp7-FLAG) (lanes 1 to 3) or DR127 (Utp22-FLAG) (lanes 4 to 6) strains. The indicated input and immunoprecipitated proteins were detected by Western blot analysis.
Fhl1 influences the stability of the CURI complex.
Cells in which the interaction of Ifh1 with Fhl1 is prevented, either by ablation of Fhl1 (16) or by deletion of the FHA domain of Fhl1 (33), are alive but grow exceedingly slowly. In light of these earlier findings and of the observation that Fhl1 interacts with the CURI complex, we wished to determine the fate of the CURI complex in the ΔFHL1 and ΔFHA strains. As shown in Fig. 7, the deletion of Fhl1 (upper panel) or of the FHA domain (lower panel) has at least three effects on the pattern of migration (compare Fig. 7 with Fig. 4). First, a substantial amount of the Ifh1 now sediments more slowly, although a fraction of the Ifh1 still sediments at its previous location in fractions 9 to 13. Second, most of the Rrp7 sediments towards the top of the gradient. This is in marked contrast to the wild-type cells, where Rrp7 comigrates with Ifh1 in fractions 9 to 13. Third, Rap1 no longer streaks down the gradient. The altered sedimentation patterns of Ifh1 and of Rap1 are remarkably similar in the strain from which the FHA domain has been deleted from Fhl1 (Fig. 7, lower panel). Taken together, these results suggest that Fhl1 not only interacts with the CURI complex but also plays some role in maintaining the stability of the complex. On the other hand, we cannot rule out the possibility that these effects are secondary to the very slow growth and very low ribosome content of ΔFHL1 and ΔFHA cells (33).
FIG. 7.
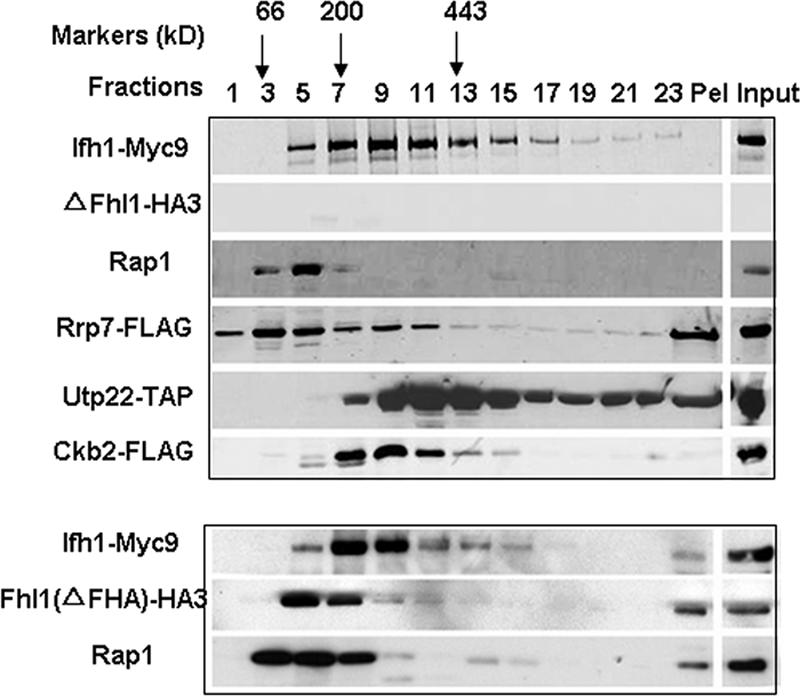
Fhl1 influences the stability of the CURI complex. Glycerol gradient analysis was performed on extracts prepared from DR116, DR117, and DR118 strains. In each, FHL1 is deleted and Utp22, Rrp7, and Ckb2 are tagged as indicated. Aliquots (15 μl) from the indicated fractions as well as the whole-cell extract (Input) were analyzed by SDS-polyacrylamide gel electrophoresis followed by Western blotting using anti-HA, anti-Myc, anti-Rap1, or anti-FLAG antibodies for strains DR117 and DR118. For strain DR116, Western blotting was performed using HRP-conjugated chicken IgG to detect Utp22-TAP. A summary of the glycerol gradient patterns from the three strains is shown. In the lower panel, a glycerol gradient was performed on extracts from strain DR49 (ΔFHA), followed by Western blotting using anti-HA, anti-Myc, or anti-Rap1 antibodies. Pel, pellet at the bottom of the glycerol gradient.
Ifh1 can be phosphorylated by CK2.
The presence of CK2 in the CURI complex is intriguing because CK2 has been implicated in the regulation of transcription by RNA polymerase I (Pol I) (34), RNA Pol II (24), and RNA Pol III (13). To investigate whether CK2 phosphorylates members of the CURI complex, we incubated γ-[32P]ATP with CURI-containing fractions extracted from strains YZ146 and DR23, carrying Ifh1-HA3 and Fhl1-HA3, respectively. The samples were immunoprecipitated with anti-HA antibody, and the result was displayed on two SDS gels (Fig. 8): one subjected to autoradiography (upper) and the other probed with anti-HA antibody after Western blotting (lower). It is evident that Ifh1 is phosphorylated (lane 1), while Fhl1 is not (lane 5). To confirm that CK2 is the kinase responsible, we showed that heparin, a specific inhibitor of CK2 (15), abolishes labeling of Ifh1 (lane 2). Furthermore, the kinase is just as effective using labeled GTP in place of ATP, another diagnostic for CK2 (28) (Fig. 8, lanes 3 and 4). No phosphorylation of other members of CURI was detected in analogous experiments (data not shown).
FIG. 8.

Phosphorylation of Ifh1 by CK2. Fractions of the glycerol gradient containing the CURI complex from strain YZ146 (Ifh1-HA3) were incubated with γ-32P-labeled ATP (lanes 1 and 2) or GTP (lanes 3 and 4), in the absence (lanes 1 and 3) or presence (lanes 2 and 4) of heparin, followed by immunoprecipitation with anti-HA and analysis on two parallel SDS gels. Lanes 5 and 6 are identical to lanes 1 and 2, except that the strain used was DR23 (Fhl1-HA3). One SDS gel (upper panel) was subjected to autoradiography, and the other (lower panel) was Western blotted and probed with anti-HA. MW markers are indicated.
CURI complex and transcription.
To ask if the CURI complex is involved in the activation of transcription of RP genes by Ifh1, we performed ChIP analysis with each of the components of the CURI complex. As seen in Fig. 9, the data show that while Ifh1 is found at the RP genes, the other three components, Ckb2, Rrp7, and Utp22, are not. Thus, Ifh1 seems to appear in two alternative forms: as either a member of the CURI complex or resident at the promoter of an RP gene, presumably in the company of Fhl1 and Rap1.
FIG. 9.

Utp22, Rrp7, and Ckb2 are not present on the promoters of RP genes. ChIP was performed on strains harboring TAP-tagged IFH1 (DR14) and strains from Open Biosystems, Inc., carrying TAP-tagged versions of CKB2, RRP7, and UTP22 (see Materials and Methods.) Following immunoprecipitation, real-time PCR was performed on the samples using primers for the promoters of the indicated genes. Primers specific for the promoters of PGK1 and ACT1 were used as negative controls.
The CURI complex as coordinator of rRNA and RP production.
The presence of both Ifh1 and Utp22 and Rrp7 in the CURI complex led us to hypothesize that CURI might connect Pol I transcription of rRNA genes with Pol II transcription of RP genes. The simplest formulation is shown in Fig. 10. When rRNA transcription is active, much of the Utp22 and Rrp7 is tied up in processing the pre-rRNA, thus freeing Ifh1 to associate with Fhl1 and Rap1 to activate transcription of RP genes. Conversely, when rRNA transcription is reduced, Utp22 and Rrp7 are free to bind more Ifh1, thereby reducing transcription of RP genes.
FIG. 10.

Model of CURI coupling rRNA and RP production. Ifh1 participates in (at least) two interactions. To the left is an RP promoter to which two molecules of Rap1 bind, simultaneously bending the DNA and clearing it of nucleosomes. Fhl1 and Ifh1 bind (not necessarily directly to the DNA) to promote transcription (Tx). When there is active rRNA transcription, Utp22 and Rrp7 are busy processing pre-rRNA. Ifh1 is free to interact with Fhl1 (and Rap1) to facilitate transcription of RP genes. However, when rRNA transcription slows, Utp22 and Rrp7 become available to bind CK2 and Ifh1 in the CURI complex, preventing Ifh1 from associating with RP genes and slowing their transcription. The role of CK2 is thus far unspecified. The arrangement within the CURI complex remains arbitrary pending further experiments. As described more thoroughly in the Discussion section, the association of Fhl1 with CURI and its role in facilitating the exchange of Ifh1 between CURI and the RP genes remain unclear.
As a test of this hypothesis, we generated strains in which either UTP22 or RRP7, and as a control, UTP7, UTP11, or UTP14, were put under GAL control. In galactose medium, these strains grow as wild type. On being shifted to a noninducing, nonrepressive carbon source such as raffinose, growth slows substantially after 12 h and is limited for the protein in question. Analysis of the RNA from such strains under those conditions (Fig. 11A) shows that the level of 20S pre-rRNA is greatly reduced, as described previously for Rrp7 (2) and for many of the Utp proteins (5). For strains limited for Utp22 or Rrp7, the mRNA level of the five RP genes tested was substantially elevated (Fig. 11B), just as we would predict from the hypothesis described above. In contrast, the levels of RP mRNAs in strains limited for Utp7, -11, or -14 showed only a small increase, about 50%. While this result suggests that there may be some feedback from the deprivation of ribosomes, this 50% increase is far less than the three- to fourfold increase in the level of RP mRNAs when the cells are limited for members of the CURI complex, Utp22 or Rrp7. It is noteworthy that limiting either Utp22 or Rrp7 leads to an apparent decrease in the mRNA from ACT1, as well as those from TEF1 or PGK1, whose transcription is also dependent on Rap1.
FIG. 11.

Depletion of Utp22 or Rrp7 leads to overexpression of RP mRNAs. Cells of the indicated genotype (where G-UTP22 means that transcription of UTP22 mRNA was under GAL control, etc.) were grown in YP-Gal, collected by filtration, and then grown in YP-raffinose for 16 h, by which time growth had slowed significantly, limited for Utp7, -11, -14, or -22 or Rrp7. Cultures were harvested, and RNA was prepared and subjected to Northern analysis using 1 μg/lane for the rRNA probes and 7.5 μg/lane for the mRNA probes. (A) 20S and 27S pre-rRNAs. (B) The PhosphorImager data from the mRNAs of the indicated proteins were compared to the level of U3 RNA and then to the wild-type strain (YZ146). (C) Cells of the indicated genotype were grown in YP-raffinose as in panel B. At time zero, a sample was taken and galactose was added to the remainder of the cultures. Samples were taken at the indicated times, and RNA was prepared and subjected to Northern analysis as in panel B. The PhosphorImager data were normalized to the ACT1 signal and then to the wild type.
An additional prediction of the model in Fig. 10 is that restoring Rrp7 or Utp22 would then reduce the level of RP mRNA. As shown in Fig. 11C, this is indeed the case. When galactose was added to restore Rrp7 or Utp22, the level of RP mRNA was reduced in spite of the fact that the growth rate rapidly increased. This experiment is entirely consistent with the model shown in Fig. 10.
The results in Fig. 11B and C are quite remarkable for the following two reasons. (i) The mRNA level of RP genes is usually proportional to growth; in this case, it is the opposite. (ii) The mRNA level of RP genes in normal cells is already very high (17). To increase that level by severalfold means that the RP mRNAs will be crowding out the other mRNAs of the cell. Indeed, the raw data of Fig. 11C suggest that this is the case.
DISCUSSION
Ifh1 has been identified as the major transcription factor responsible for the transcription of RP genes (27, 33, 35, 38, 42). The data presented above demonstrate that Ifh1 is never found as a solo protein but is in a complex with two other sets of proteins, either with Rap1 and Fhl1, presumably at the RP genes, or with CK2, Utp22, and Rrp7 (and perhaps Fhl1) as part of the CURI complex.
CURI complex.
The eclectic set of proteins that make up the CURI complex pose a variety of questions and raise a variety of possibilities. CK2, with hundreds of potential substrates, has been increasingly implicated in numerous gene regulatory functions (reviewed in reference 29). It has been identified in a number of complexes in yeast (10, 18). It has been implicated in the regulation of Pol I transcription (34) and Pol III transcription (13), in the transcription by Pol II of genes employing downstream enhancers (24), and in widespread chromatin remodeling (1). Although potential sites for CK2 phosphorylation exist in several of the proteins in the CURI complex, only Ifh1 appears to be a substrate in vitro (Fig. 8). It will be of interest to determine whether any of the other proteins are substrates in vivo and whether the presence and function of CK2 affect the availability of Ifh1 to drive transcription of RP genes. It is intriguing that the interaction of Ifh1 with Fhl1, necessary for transcription of the RP genes, requires the FHA domain of Fhl1, which usually binds a phosphopeptide (7). Unfortunately, the apparent promiscuity of CK2 makes it difficult experimentally to distinguish primary from secondary effects of manipulating CK2 function.
Utp22 also has been identified in a number of complexes. The predominant one is the very large “SSU processome” containing U3 snoRNA as well as numerous factors explicitly or implicitly implicated in the processing of rRNA and its assembly with RPs (3). In addition, it was found as a complex with Rrp7, CK2, and fragments of Ifh1 in the genomewide analysis of smaller complexes (19). Rrp7 has been found in some but not all of the complexes that contain Utp22. Overall, it seems to have a much more limited repertoire of interactions than Utp22 (http://www.thebiogrid.org). Depletion of either Rrp7 or Utp22 leads to disappearance of the 20S pre-rRNA and to little synthesis of mature 18S rRNA, while synthesis of 25S rRNA appears relatively unperturbed (2, 3). Whether Rrp7 and Utp22 participate in the same reaction is not known, although some evidence suggests that Rrp7 might be a later player in the process. Indeed, we do not even know if the lack of 20S pre-rRNA (Fig. 11A) is due to its lack of synthesis or to its rapid degradation due to improper processing.
Fhl1 and CURI.
As indicated in Fig. 10, the protein whose position remains most ambiguous is Fhl1, a key protein because it seems to be the one whose association with RP genes, by ChIP analysis, is most specific and most comprehensive (23). Glycerol gradient centrifugation shows that most of the Fhl1 remains in monomeric form towards the top of the gradient. However, a substantial amount of it is associated with Ifh1 in a higher-MW complex (Fig. 2A). Subsequent experiments suggest that Fhl1 not only associates with Rap1 and Ifh1 on the RP genes but also appears to be “loosely” associated with CURI (Fig. 5 and 6). Yet the relationship of these experiments is difficult to interpret. The ChIP experiments were carried out with extracts of formaldehyde-fixed cells, while the gradient and Co-IP experiments were carried out on extracts that have suffered from the enormous dilution brought about by opening the cell, as well as from harsh treatments involved in the purification of multiprotein complexes. Thus, although most of the Fhl1 appears not to be associated with anything (Fig. 2A), we have observed it to coimmunoprecipitate with Rap1, Ifh1, Ckb2, Rrp7, and Utp22 (Fig. 6). The interaction between Fhl1 and Ifh1 depends on a functional FHA domain (33) (Fig. 7), which implies that it depends on a specifically phosphorylated residue on Ifh1 (7). CK2 could be the kinase responsible. Since at least a fraction of the Ifh1 in CURI is not phosphorylated, as evident from its availability as a substrate in vitro, perhaps the modulation of its phosphorylation state could affect the interaction with Fhl1. The puzzling observation is that lack of Fhl1 leads to the loss from CURI only of Rrp7 (Fig. 7). This could be due to the severe reduction of rRNA transcription in such a strain, but sirolimus (formerly rapamycin), whose effect on the TOR pathway causes an equally severe reduction of rRNA transcription (30), has no such effect (data not shown).
A second complex, composed of Ifh1, Fhl1, and Rap1, appears to sediment at nearly the same place in a glycerol gradient. We presume that this has been eluted from the RP genes during breakage of the cell since we find little of these proteins in the low-speed pellet that contains nuclei/chromatin. That this complex differs from the CURI complex is apparent from the observation that anti-Rap1 can coimmunoprecipitate Ifh1 and Fhl1 but not Utp22, Rrp7, or Ckb2 (Fig. 2 and 6). Furthermore, the absence of Fhl1 drives the Rap1 molecules to the top of the gradient (compare Fig. 7 with Fig. 4), with an effect on only a small fraction of the Ifh1 molecules and hardly any effect on Utp22 (Fig. 7). Thus, we conclude that Ifh1 participates in two complexes: one with CK2, Utp22, and Rrp7, with Fhl1 loosely associated, and the other with Rap1 and Fhl1, which may have been released from the RP genes during opening of the cell. Unfortunately, the two complexes run together on a glycerol gradient and thus far we have been unsuccessful in separating them by chromatographic means.
The recent comprehensive analyses of interacting factors in yeast (10, 18) has provided a wealth of information, but detailed examination of individual proteins and their complexes shows that the story is often richer. Thus, while the CURI complex was identified in a specific search for relatively small complexes (19), it is not apparent in either of the two global studies. Furthermore, none of the studies identified the interaction of Fhl1 with the CURI complex nor the interaction between Fhl1, Ifh1, and Rap1, which is not sufficiently stable to survive the TAP.
CURI complex coupling rRNA and RP production.
The CURI complex is intriguing not only because it potentially connects RP gene transcription, dependent on Ifh1, with rRNA processing, dependent on Utp22 and Rrp7, but also because CK2 is the potential connector! At least three hypotheses present themselves: CURI could sequester Ifh1 to repress RP gene transcription (Fig. 10). A second model is that Ifh1 must pass through the CURI complex to be activated, perhaps through phosphorylation by CK2. A third is that, through CURI, Ifh1 controls the availability of Utp22 and Rrp7 to carry out pre-rRNA processing. In any case, CK2 could provide either the glue to keep them together or the charge to disperse them. The results in Fig. 11 would argue against the second model. The third remains to be tested.
As shown in Fig. 9, CURI as such is not found at the RP genes. Therefore, its effect as a repressor of transcription occurs elsewhere. We suggest that it acts as a repressor of RP transcription by preventing Ifh1 from interacting with the RP genes. In this view, active rRNA transcription would tie up Utp22 and Rrp7 in processing the new pre-rRNA, freeing Ifh1 to direct more RP mRNA transcription. The recent observation that forced constitutive transcription of rRNA leads to coordinate transcription of RP genes (20) would support this idea. Reduced rRNA transcription would release Utp22 and Rrp7 to sequester Ifh1. In a partial test of the latter, we found that inhibiting rRNA transcription using a ts allele of RRN3 (41) led to substantially lower RP mRNA levels (data not shown). While this is consistent with the model in Fig. 10, it does not implicate Utp22 and Rrp7 directly.
Specific tests of the model of Fig. 10 were carried out by depleting Utp22 or Rrp7. When that is done, the cells show substantially increased levels of RP mRNA, presumably reflecting increased transcription (Fig. 11B and C). Since under normal growing conditions transcripts of RP genes represent 25% of the total mRNA (17), this represents a remarkable amplification of transcription! On the other hand, similar experiments using any of three other members of the processome, Utp7, -11, or -14, do not show such an increase in RP mRNA, suggesting that the effect appears to be specific for these two rRNA processing factors, namely Utp22 and Rrp7. When the synthesis of Utp22 or Rrp7 is restored, the RP mRNA levels decline towards normal. The converse experiment of overproducing Utp22 was unsuccessful because excess Utp22 is rapidly degraded, perhaps due to its need to be complexed with other proteins (data not shown).
A very recent paper, published while the manuscript for this article was being completed, identified Hmo1 as a major player in ribosome synthesis, as it is found at both rRNA and RP genes and its presence is necessary for Fhl1 and Ifh1 to associate with RP genes. It was suggested that Hmo1 is the key player in the coordination of rRNA and RP gene transcription (14). It will be interesting to determine if Hmo1 is associated with any of the complexes we have found and whether its absence has any effect on the CURI complex or on the interaction of Fhl1 with Ifh1.
In summary, the CURI complex, associated with Fhl1 in a yet hazy way, appears to play a role in the coordination of rRNA transcription with RP gene transcription. This is, however, but a first step in our understanding of this critical element in the effective utilization of the cell's resources. What is the role of CK2? Is there two-way communication, as would be suggested by the phenotype of ΔFHL1/ΔIFH1 strains, in which reduced levels of RPs lead to reduced transcription of rRNA (33)? Is Hmo1 involved in some way as a participant in this interaction or as part of a parallel pathway of coordination?
Acknowledgments
We are grateful to David Shore for anti-Rap1 antibody.
This research was supported in part by grants from the NIH: GM-25532 to J.R.W. and CAI-3330 to the Albert Einstein Cancer Center.
Footnotes
Published ahead of print on 23 April 2007.
REFERENCES
- 1.Barz, T., K. Ackermann, G. Dubois, R. Eils, and W. Pyerin. 2003. Genome-wide expression screens indicate a global role for protein kinase CK2 in chromatin remodeling. J. Cell Sci. 116:1563-1577. [DOI] [PubMed] [Google Scholar]
- 2.Baudin-Baillieu, A., D. Tollervey, C. Cullin, and F. Lacroute. 1997. Functional analysis of Rrp7p, an essential yeast protein involved in pre-rRNA processing and ribosome assembly. Mol. Cell. Biol. 17:5023-5032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bernstein, K. A., J. E. G. Gallagher, B. M. Mitchell, S. Granneman, and S. J. Baserga. 2004. The small-subunit processome is a ribosome assembly intermediate. Eukaryot. Cell 3:1619-1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.DeRisi, J. L., V. R. Iyer, and P. O. Brown. 1997. Exploring the metabolic and genetic control of gene expression on a genomic scale. Science 278:680-686. [DOI] [PubMed] [Google Scholar]
- 5.Dragon, F., J. E. Gallagher, P. A. Compagnone-Post, B. M. Mitchell, K. A. Porwancher, K. A. Wehner, S. Wormsley, R. E. Settlage, J. Shabanowitz, Y. Osheim, A. L. Beyer, D. F. Hunt, and S. J. Baserga. 2002. A large nucleolar U3 ribonucleoprotein required for 18S ribosomal RNA biogenesis. Nature 417:967-970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Durocher, D., and S. P. Jackson. 2002. The FHA domain. FEBS Lett. 513:58-66. [DOI] [PubMed] [Google Scholar]
- 7.Durocher, D., I. A. Taylor, D. Sarbassova, L. F. Haire, S. L. Westcott, S. P. Jackson, S. J. Smerdon, and M. B. Yaffe. 2000. The molecular basis of FHA domain:phosphopeptide binding specificity and implications for phospho-dependent signaling mechanisms. Mol. Cell 6:1169-1182. [DOI] [PubMed] [Google Scholar]
- 8.Fatica, A., and D. Tollervey. 2002. Making ribosomes. Curr. Opin. Cell Biol. 14:313-318. [DOI] [PubMed] [Google Scholar]
- 9.Gasch, A. P., P. T. Spellman, C. M. Kao, O. Carmel-Harel, M. B. Eisen, G. Storz, D. Botstein, and P. O. Brown. 2000. Genomic expression programs in the response of yeast cells to environmental changes. Mol. Biol. Cell 11:4241-4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gavin, A. C., P. Aloy, P. Grandi, R. Krause, M. Boesche, M. Marzioch, C. Rau, L. J. Jensen, S. Bastuck, B. Dumpelfeld, A. Edelmann, M. A. Heurtier, V. Hoffman, C. Hoefert, K. Klein, M. Hudak, A. M. Michon, M. Schelder, M. Schirle, M. Remor, T. Rudi, S. Hooper, A. Bauer, T. Bouwmeester, G. Casari, G. Drewes, G. Neubauer, J. M. Rick, B. Kuster, P. Bork, R. B. Russell, and G. Superti-Furga. 2006. Proteome survey reveals modularity of the yeast cell machinery. Nature 440:631-636. [DOI] [PubMed] [Google Scholar]
- 11.Gelbart, M. E., T. Rechsteiner, T. J. Richmond, and T. Tsukiyama. 2001. Interactions of Isw2 chromatin remodeling complex with nucleosomal arrays: analyses using recombinant yeast histones and immobilized templates. Mol. Cell. Biol. 21:2098-2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ghaemmaghami, S., W. K. Huh, K. Bower, R. W. Howson, A. Belle, N. Dephoure, E. K. O'Shea, and J. S. Weissman. 2003. Global analysis of protein expression in yeast. Nature 425:737-741. [DOI] [PubMed] [Google Scholar]
- 13.Ghavidel, A., and M. C. Schultz. 2001. TATA binding protein-associated CK2 transduces DNA damage signals to the RNA polymerase III transcriptional machinery. Cell 106:575-584. [DOI] [PubMed] [Google Scholar]
- 14.Hall, D. B., J. T. Wade, and K. Struhl. 2006. An HMG protein, Hmo1, associates with promoters of many ribosomal protein genes and throughout the rRNA gene locus in Saccharomyces cerevisiae. Mol. Cell. Biol. 26:3672-3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hathaway, G. M., T. H. Lubben, and J. A. Traugh. 1980. Inhibition of casein kinase II by heparin. J. Biol. Chem. 255:8038-8041. [PubMed] [Google Scholar]
- 16.Hermann-Le Denmat, S., M. Werner, A. Sentenac, and P. Thuriaux. 1994. Suppression of yeast RNA polymerase III mutations by FHL1, a gene coding for a fork head protein involved in rRNA processing. Mol. Cell. Biol. 14:2905-2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Holstege, F. C. P., E. G. Jennings, J. J. Wyrick, T. I. Lee, C. J. Hengartner, M. R. Green, T. R. Golub, E. S. Lander, and R. A. Young. 1998. Dissecting the regulatory circuitry of a eukaryotic genome. Cell 95:717-728. [DOI] [PubMed] [Google Scholar]
- 18.Krogan, N. J., G. Cagney, H. Yu, G. Zhong, X. Guo, A. Ignatchenko, J. Li, S. Pu, N. Datta, A. P. Tikuisis, T. Punna, J. M. Peregrin-Alvarez, M. Shales, X. Zhang, M. Davey, M. D. Robinson, A. Paccanaro, J. E. Bray, A. Sheung, B. Beattie, D. P. Richards, V. Canadien, A. Lalev, F. Mena, P. Wong, A. Starostine, M. M. Canete, J. Vlasblom, S. Wu, C. Orsi, S. R. Collins, S. Chandran, R. Haw, J. J. Rilstone, K. Gandi, N. J. Thompson, G. Musso, P. St Onge, S. Ghanny, M. H. Lam, G. Butland, A. M. taf-Ul, S. Kanaya, A. Shilatifard, E. O'Shea, J. S. Weissman, C. J. Ingles, T. R. Hughes, J. Parkinson, M. Gerstein, S. J. Wodak, A. Emili, and J. F. Greenblatt. 2006. Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature 440:637-643. [DOI] [PubMed] [Google Scholar]
- 19.Krogan, N. J., W. T. Peng, G. Cagney, M. D. Robinson, R. Haw, G. Zhong, X. Guo, X. Zhang, V. Canadien, D. P. Richards, B. K. Beattie, A. Lalev, W. Zhang, A. P. Davierwala, S. Mnaimneh, A. Starostine, A. P. Tikuisis, J. Grigull, N. Datta, J. E. Bray, T. R. Hughes, A. Emili, and J. F. Greenblatt. 2004. High-definition macromolecular composition of yeast RNA-processing complexes. Mol. Cell 13:225-239. [DOI] [PubMed] [Google Scholar]
- 20.Laferte, A., E. Favry, A. Sentenac, M. Riva, C. Carles, and S. Chedin. 2006. The transcriptional activity of RNA polymerase I is a key determinant for the level of all ribosome components. Genes Dev. 20:2030-2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lai, J. S., and W. Herr. 1992. Ethidium bromide provides a simple tool for identifying genuine DNA-independent protein associations. Proc. Natl. Acad. Sci. USA 89:6958-6962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lascaris, R. F., W. H. Mager, and R. J. Planta. 1999. DNA-binding requirements of the yeast protein Rap1p as selected in silico from ribosomal gene promoter sequences. Bioinformatics 15:267-277. [DOI] [PubMed] [Google Scholar]
- 23.Lee, T. I., N. J. Rinaldi, F. Robert, D. T. Odom, Z. Bar-Joseph, G. K. Gerber, N. M. Hannett, C. T. Harbison, C. M. Thompson, I. Simon, J. Zeitlinger, E. G. Jennings, H. L. Murray, D. B. Gordon, B. Ren, J. J. Wyrick, J. B. Tagne, T. L. Volkert, E. Fraenkel, D. K. Gifford, and R. A. Young. 2002. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science 298:799-804. [DOI] [PubMed] [Google Scholar]
- 24.Lewis, B. A., R. J. Sims III, W. S. Lane, and D. Reinberg. 2005. Functional characterization of core promoter elements: DPE-specific transcription requires the protein kinase CK2 and the PC4 coactivator. Mol. Cell 18:471-481. [DOI] [PubMed] [Google Scholar]
- 25.Lieb, J. D., X. Liu, D. Botstein, and P. O. Brown. 2001. Promoter-specific binding of Rap1 revealed by genome-wide maps of protein-DNA association. Nat. Genet. 28:327-334. [DOI] [PubMed] [Google Scholar]
- 26.Longtine, M. S., A. McKenzie III, D. J. Demarini, N. G. Shah, A. Wach, A. Brachat, P. Philippsen, and J. R. Pringle. 1998. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14:953-961. [DOI] [PubMed] [Google Scholar]
- 27.Martin, D. E., A. Soulard, and M. N. Hall. 2004. TOR regulates ribosomal protein gene expression via PKA and the Forkhead transcription factor FHL1. Cell 119:969-979. [DOI] [PubMed] [Google Scholar]
- 28.Niefind, K., M. Putter, B. Guerra, O. G. Issinger, and D. Schomburg. 1999. GTP plus water mimic ATP in the active site of protein kinase CK2. Nat. Struct. Biol. 6:1100-1103. [DOI] [PubMed] [Google Scholar]
- 29.Poole, A., T. Poore, S. Bandhakavi, R. O. McCann, D. E. Hanna, and C. V. Glover. 2005. A global view of CK2 function and regulation. Mol. Cell Biochem. 274:163-170. [DOI] [PubMed] [Google Scholar]
- 30.Powers, T., and P. Walter. 1999. Regulation of ribosome biogenesis by the rapamycin-sensitive TOR-signalling pathway in Saccharomyces cerevisiae. Mol. Biol. Cell 10:987-1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Puig, O., F. Caspary, G. Rigaut, B. Rutz, E. Bouveret, E. Bragado-Nilsson, M. Wilm, and B. Seraphin. 2001. The tandem affinity purification (TAP) method: a general procedure of protein complex purification. Methods 24:218-229. [DOI] [PubMed] [Google Scholar]
- 32.Rotenberg, M. O., and J. L. Woolford, Jr. 1986. Tripartite upstream promoter element essential for expression of Saccharomyces cerevisiae ribosomal protein genes. Mol. Cell. Biol. 6:674-687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rudra, D., Y. Zhao, and J. R. Warner. 2005. Central role of Ifh1p-Fhl1p interaction in the synthesis of yeast ribosomal proteins. EMBO J. 24:533-542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Saez-Vasquez, J., M. Meissner, and C. S. Pikaard. 2001. RNA polymerase I holoenzyme-promoter complexes include an associated CK2-like protein kinase. Plant Mol. Biol. 47:449-459. [DOI] [PubMed] [Google Scholar]
- 35.Schawalder, S. B., M. Kabani, I. Howald, U. Choudhury, M. Werner, and D. Shore. 2004. Growth-regulated recruitment of the essential yeast ribosomal protein gene activator Ifh1. Nature 432:1058-1061. [DOI] [PubMed] [Google Scholar]
- 36.Schwindinger, W. F., and J. R. Warner. 1987. Transcriptional elements of the yeast ribosomal protein gene CYH2. J. Biol. Chem. 262:5690-5695. [PubMed] [Google Scholar]
- 37.Thomas, B. J., and R. Rothstein. 1989. Elevated recombination rates in transcriptionally active DNA. Cell 56:619-630. [DOI] [PubMed] [Google Scholar]
- 38.Wade, J. T., D. B. Hall, and K. Struhl. 2004. The transcription factor Ifh1 is a key regulator of yeast ribosomal protein genes. Nature 432:1054-1058. [DOI] [PubMed] [Google Scholar]
- 39.Warner, J. R. 1999. The economics of ribosome biosynthesis in yeast. Trends Biochem. Sci. 24:437-440. [DOI] [PubMed] [Google Scholar]
- 40.Woudt, L. P., A. B. Smit, W. H. Mager, and R. J. Planta. 1986. Conserved sequence elements upstream of the gene encoding yeast ribosomal protein L25 are involved in transcription activation. EMBO J. 5:1037-1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yamamoto, R. T., Y. Nogi, J. A. Dodd, and M. Nomura. 1996. RRN3 gene of Saccharomyces cerevisiae encodes an essential RNA polymerase I transcription factor which interacts with the polymerase independently of DNA template. EMBO J. 15:3964-3973. [PMC free article] [PubMed] [Google Scholar]
- 42.Zhao, Y., K. B. McIntosh, D. Rudra, S. B. Schawalder, D. Shore, and J. R. Warner. 2006. Fine-structure analysis of ribosomal protein gene transcription. Mol. Cell. Biol. 26:4853-4862. [DOI] [PMC free article] [PubMed] [Google Scholar]