

Abstract
Retrotransposon L1 is a mobile genetic element of the LINE family that is extremely widespread in the mammalian genome. It encodes a dicistronic mRNA, which is exceptionally rare among eukaryotic cellular mRNAs. The extremely long and GC-rich L1 5′ untranslated region (5′UTR) directs synthesis of numerous copies of RNA-binding protein ORF1p per mRNA. One could suggest that the 5′UTR of L1 mRNA contained a powerful internal ribosome entry site (IRES) element. Using transfection of cultured cells with the polyadenylated monocistronic (L1 5′UTR-Fluc) or bicistronic (Rluc-L1 5′UTR-Fluc) RNA constructs, capped or uncapped, it has been firmly established that the 5′UTR of L1 does not contain an IRES. Uncapping reduces the initiation activity of the L1 5′UTR to that of background. Moreover, the translation is inhibited by upstream AUG codons in the 5′UTR. Nevertheless, this cap-dependent initiation activity of the L1 5′UTR was unexpectedly high and resembles that of the beta-actin 5′UTR (84 nucleotides long). Strikingly, the deletion of up to 80% of the nucleotide sequence of the L1 5′UTR, with most of its stem loops, does not significantly change its translation initiation efficiency. These data can modify current ideas on mechanisms used by 40S ribosomal subunits to cope with complex 5′UTRs and call into question the conception that every long GC-rich 5′UTR working with a high efficiency has to contain an IRES. Our data also demonstrate that the ORF2 translation initiation is not directed by internal initiation, either. It is very inefficient and presumably based on a reinitiation event.
The two major and principally different mechanisms of translation initiation in eukaryotes are cap-dependent scanning and internal ribosome entry. While the first mechanism is believed to be the main way for the majority of cellular mRNAs, the latter is used by some viruses and probably by a specific set of cellular mRNAs that have to be translated under particular conditions such as various environmental stresses, apoptosis, or meiosis.
In the early 1980s, M. Kozak postulated the “scanning model,” which now represents the paradigm and the only existing model for the cap-dependent initiation of translation (18). According to Kozak, the eukaryotic 40S ribosome subunit bearing 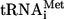 binds at or near the 5′ end of capped mRNA and begins to “scan” through the mRNA 5′ untranslated region (5′UTR) in the 5′-3′ direction, searching for the first AUG codon in a “good” initiation context. During this process, the 40S subunit with the aid of the eIF4 group initiation factors unwinds the secondary structure in the mRNA leader. Once the appropriate codon is found, the 60S subunit joins the complex and translation elongation begins (19). However, in these studies, Kozak used only relatively short and simple leaders such as the reovirus or some artificial 5′UTRs (18, 23). Currently, computer data about the average mammalian 5′UTR offer a different picture: as a rule, it possesses approximately 150 to 200 nucleotides (nt) containing 50 to 60% GC pairs and, in 30 to 45% of cases, one or more upstream AUG (uAUG) codons (14, 43, 62). According to the scanning model, such features should inhibit the initiation of the main open reading frame (ORF) translation or at least make it less efficient.
binds at or near the 5′ end of capped mRNA and begins to “scan” through the mRNA 5′ untranslated region (5′UTR) in the 5′-3′ direction, searching for the first AUG codon in a “good” initiation context. During this process, the 40S subunit with the aid of the eIF4 group initiation factors unwinds the secondary structure in the mRNA leader. Once the appropriate codon is found, the 60S subunit joins the complex and translation elongation begins (19). However, in these studies, Kozak used only relatively short and simple leaders such as the reovirus or some artificial 5′UTRs (18, 23). Currently, computer data about the average mammalian 5′UTR offer a different picture: as a rule, it possesses approximately 150 to 200 nucleotides (nt) containing 50 to 60% GC pairs and, in 30 to 45% of cases, one or more upstream AUG (uAUG) codons (14, 43, 62). According to the scanning model, such features should inhibit the initiation of the main open reading frame (ORF) translation or at least make it less efficient.
Internal ribosome entry is an alternative initiation mechanism which requires specific nonconserved structures known as internal ribosome entry site (IRES) elements. Up to now, the exact mechanism(s) of IRES-dependent translation initiation has been elucidated only for a small set of viral mRNAs (see reference 37 for a review and references 40 and 53 for some novel examples). The conventional approach to identifying new IRES elements is the method of dicistronic constructions. Using this approach, a number of IRES elements have been discovered, not only in uncapped viral mRNAs but also in the 5′UTRs of some cellular mRNAs, especially those which fulfill regulatory roles in eukaryotic cells (reviewed in reference 16). As a rule, the 5′UTRs of such cellular mRNAs are long and highly structured. The existence of cellular IRES elements is now a subject of debate (20, 22, 45). Some researchers claim that the many putative cellular IRES elements identified currently are an artifact of the method of DNA dicistronic constructions (5, 11, 56, 59). On the other hand, it is difficult to understand how the 40S ribosomal subunit is able to traverse long and structured 5′UTRs of some translationally efficient mRNAs if we reject the concept of cellular IRES elements and hold only on the classical scanning mechanism.
Retrotransposon L1, a member of the non-long-terminal repeat (LTR) retrotransposon LINE family, is an extremely widespread mobile element in the mammalian genome. In the course of human evolution, the number of its copies has reached ∼520,000, and in total, they make up about 17% of the human chromosomal DNA. Several dozen copies of the youngest Ta (L1PA1) subgroup still retain their activity, and for this reason, L1 transpositions are thought to cause mutations (in some cases leading to cancer) and hereditary diseases (for a review, see reference 33).
The L1 dicistronic RNA serves as both transposition intermediate and mRNA for the synthesis of two proteins of the retrotransposon. The first cistron encodes an RNA-binding protein of unknown function, and the second one encodes a reverse transcriptase and an endonuclease. In the human L1 mRNA, these two cistrons are separated by just 63 nt. Both proteins are necessary for L1 retrotransposition (32) and act in cis (25, 60). The human L1 mRNA contains a 900-nt-long 5′UTR with high GC (∼60%) content and two short upstream ORFs (uORFs). The first of them starts with AUG in a good context at position +16 and is well conserved between subfamilies. The presence of such a complex leader is accounted for by the fact that the 5′ end of the L1 retrotransposon contains an internal promoter for RNA polymerase II (51), a situation close to that of LTR retrotransposons and retroviruses and also reminiscent of the case of picornavirus and pestivirus mRNAs where the 5′ termini contain the sequences needed for RNA replication. Both picornaviral and pestiviral mRNAs are well known to possess strong IRES elements, a feature which enables them to direct their translation at a high level. More recently, it has been reported that some LTR retrotransposons and many retroviruses also possess IRES elements in their 5′UTR (see references 7, 31, and 44 and references therein).
In the early 1990s, McMillan and Singer (30) demonstrated that a stable hairpin structure inserted at nt 661 of the L1 5′UTR causes a three- to eightfold decrease in ORF1 production. Since this decrease was not as dramatic as expected for such a stable stem-loop, the authors made a cautious conclusion that translation of the ORF1 is most probably 5′-end dependent. Nevertheless, in the rabbit reticulocyte lysate (RRL) cell-free system, the L1 mRNA translation was not stimulated by the 5′ cap (30). Bearing in mind that the introduction of a hairpin into the middle of the 5′UTR might cause the abolition of not only 5′-end-dependent but also IRES-dependent initiation if it distorted any important secondary structure within this region, one may conclude that the mechanism of the L1 ORF1 translation is still poorly understood.
The translation of the ORF1 of L1 mRNA results in the formation of numerous copies of the mRNA-binding protein p40 (3) and leads to assembly of massive cytoplasmic RNP particles (13). At the same time, only few reverse transcriptase molecules are synthesized from ORF2, as the amount of the reverse transcriptase in the cell seems to be very low (15, 34), and probably about one or two ORF2p molecules per RNP particle participate in the L1 reverse transcription reaction (15, 34). The translation initiation at both ORFs of mouse L1 mRNA has recently been studied (28) and is claimed to be directed by two IRES elements positioned upstream of ORF1 and ORF2. As for human L1 mRNA, the mechanism of translation initiation has been elucidated only for the ORF2, using methods of molecular genetics (1).
Here we show that the translation initiation at the ORF1 of human L1 mRNA is directed by a highly efficient cap-dependent initiation. No IRES has been found within this 5′UTR, using both in vitro translation and RNA transfection of living cell approaches. We have found that the capped L1 5′UTR provides a similar level of initiation activity as the 5′UTR of beta-actin mRNA (84-nt length). We also show that either various deletions of 100 to 200 nt from within the L1 5′UTR or the deletion of up to 80% of its nucleotide sequence with most of the stem-loop structures does not significantly change the translation initiation potential of the L1 5′UTR. Finally, we demonstrate that unlike ORF1, the translation of ORF2 proceeds with an extremely low efficiency, thereby providing a physiologically required high ratio of ORF1/ORF2 products. The interpretation of these peculiar features of the translation initiation of the L1 mRNA is presented.
MATERIALS AND METHODS
Cell culture and transfection procedures.
HEK293 and NTera2/D1 cells were cultivated in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum, 2 mM glutamine, and 100 U/ml penicillin and 100 μg streptomycin (PanEco). Cells were kept at 37°C in a humidified atmosphere containing 5% CO2. HEK293 cells were passaged by standard methods; NTera2/D1 cells were maintained in high-density culture by replating at densities of 1:2 to 1:3 with respect to the parent culture. Both DNA and RNA transfectional were performed with Unifectin-56 transfection reagent (RusBioLink) as recommended by the manufacturer. HEK293 cells were transfected at 60 to 70% of confluence and NTera2/D1 at 70 to 80% confluence. For one well of a 24-well plate, a total amount of 0.5 μg of DNA or RNA was used.
Plasmid constructs.
All dicistronic DNA constructs were prepared on the basis of the pGL3R vector (46), hereafter referred to as pRF vector for simplicity. The DNA fragment corresponding to nt 1 to 952 of the human L1 cDNA was obtained from the plasmid p3LZ (27) by PCR with the primers GGCGGAGGAGCCAAGATGG and CGTGGCCAGCTGCGTTTTAGAGTTTCCAG and was blunt-end ligated to pGL3R digested with PvuII and NcoI, resulting in plasmid Rluc-5′UTR-Fluc. Construct Rluc-5′UTRΔ(1-98)-Fluc was obtained from the latter by digestion-religation with ApaI and Acc65I; construct Rluc-L1 5′UTRΔ(133-887)-Fluc was obtained by digestion with PflMI-BstXI and ligation with adapters AAGTAGATAAAACCACAAAG and GTGGTTTTATCTACTTTGT; constructs with 100- to 150-nt step-by-step deletions of the 5′UTR were prepared by digestion-religation by Acc65I-NheI, NheI-StuI, StuI-BglII, and BglII-BstXI (the latter was ligated with adapters AAGTAGATAAAACCACAAAG and GTGGTTTTATCTACTT to sustain the nucleotide context); and constructs L1 5′UTRinsAUG248-Fluc and L1 5′UTRinsAUG473-Fluc were obtained by inverse PCR from the whole plasmid with primers GGACGCACCTGGAAAATCGGGTC and GTCATCCCTTTCTTTGACTCGG (for AUG248) and TGGATAAAAAGCAGCCGGGGAAGCTCG and TGGTTTTAAGCAAGCCTGGGCAATGG (for AUG473). The partial inversion of the L1 5′UTR in L1 5′UTRinv(137-887) was performed by the replacement of the PflMI-BstXI fragment of the vector L1 5′UTR-Fluc with two PCR products obtained with the same plasmid and primers GGTCCCCATCTTTGTGGTTTTATCTACTTATTGCCTCACCTGGGAAGC and AGGCGGGCGCCCCTC and GCGTGCCAGACAGTGGGTCCTTTCTGTTTGTTAGTTTTCC and TTGCCCAGGCTTGCTTAGG, respectively. Such a complicated scheme was necessary to delete a “strong” AUG codon that otherwise would appear in the middle of the reversed strand of the 5′UTR. The plasmid with the beta-actin 5′UTR was prepared by replacement of the SpeI-BstXI fragment of Rluc-L1 5′-UTR-Fluc plasmid with the SpeI-BstXI-digested PCR product obtained from plasmid pAbG (9) with primers CCGGCACTAGTGTAATACGACTCACTATAGG and CCGGGCCATCTTTGTGGCGGCGGGTGTGGAC. To obtain the HCV-Fluc vector, the NcoI-SacI fragment from plasmid pHCV-NS′ (42) was inserted into pGL3R digested with PvuII and NcoI. RhPV-Fluc was prepared by ligation of the SmaI-NarI fragment from the pSRhPV plasmid (53) into the pGL3R vector digested with PvuII and NarI. Rluc-HRV-Fluc plasmid (46) was a gift from A. Willis. All the simian virus 40 (SV40) promoterless derivatives from these plasmids were generated by digestion-religation with SmaI-EcoRV. Constructs pSV40-Fluc and pSV40-Rluc were obtained by digestion-religation of pGL3R vector with EcoRV-PvuII and with XbaI endonucleases, respectively. Plasmid pRLi (56) was a gift from R. E. Lloyd. pRmutF, used for the preparation of normalizing capRluc mRNA with a long 3′UTR, was obtained by digestion-religation of the pGL3R plasmid with SpeI and NarI. The DNA construct for producing dicistronic L1 5′UTR-p40-Fluc mRNA (pLL2) was prepared in the same way as Rluc-5′UTR-Fluc, with the exception that the second PCR primer was CCAGTGTCATTATGATGTTAGCTGG. The pLL2 derivative with ORF1 and the intergenic spacer substitution (L1 5′UTR-lacZ) was obtained as follows: two PCR products were produced, one from the p3LZ plasmid and with primers ATAAAACCACAAAGATGGGGCCGGAGAGCGCCGGGCAA and TTATTTTTGACACCAGACCAACTGG (for the lacZ fragment of exactly the same length as the p40 coding region) and the other from plasmid pGL3R and with primers CTAGCAAAATAGGCTGTCCC and CGTCAGCCATGGCGGCTATGATTCGATAGAGAAATGTTCTGGC (for the nonspecific fragment lacking AUGs). These two products were digested by BstXI-NcoI and ligated into pLL2 digested with the same enzymes. The RR261AA mutation in ORF1 was generated in pLL2 by inverse PCR with primers CAGAGTGGGGGCCAATATTGAAC and CCGCGGCTTGTAGGGTTTC. Substitutions AUG→CCC and AUG→UAG were obtained by ligation of the HindIII-BalI-digested PCR products obtained with primers AACCCAGAATTTCATATCCAGC and CCAGTGTGGGTATGATGTTAGCTGGTGATTTTGC and CCAGTGTCTATATGATGTTAGCTGGTGATTTTGC, respectively, into pLL2, treated in the same way.
mRNA preparation.
To prepare capped polyadenylated mRNAs, PCR products were first obtained from the corresponding plasmids and primers T50AACTTGTTTATTGCAGCTTATAATGG and CCGCCGTAATACGACTCACTATAGGGCGGAGGAGCCAAGATGGCCGAATAGG (in the case of L1 5′UTRΔAUG16-Fluc, the latter primer was substituted for CGCCGTAATACGACTCACTATAGGCGGAGGAGCCAAGTAGGCCGAATAGG). The PCR products were purified by electrophoresis in agarose gel and were used as templates for RNA in vitro transcription by using a T7 RiboMAX large-scale RNA production system kit (Promega). For preparation of m7G- or A-capped transcripts, m7GpppG (Promega) or ApppG (NEB) was added to the transcription mixture in the proportion 10:1 to GTP. The resulting RNAs were purified by LiCl precipitation and checked for integrity by polyacrylamide gel electrophoresis.
Translation in “RRL+HeLa” cell-free system.
For in vitro mRNA translation, we used the cell-free system described in reference 8.
RESULTS
The conventional method of dicistronic DNA constructions does not allow determination of the mode of translation initiation at the ORF1 of L1 mRNA.
The structural organization of the transcript encoded by retrotransposon L1 (L1 mRNA) is shown schematically in Fig. 1A. The translation of the first cistron results in numerous copies of protein p40 per L1 mRNA molecule (3). This agrees poorly with a large size and a GC-rich content of the 5′UTR of the human L1 mRNA (900 nt and 60%, respectively). A computer-assisted folding of this 5′UTR yields the secondary structure with several highly base-paired domains and a ΔG of about −300 kcal/mol (Fig. 1B). Although they were not tested by chemical and enzymatic probing, some of these stem-loop structures are curiously reminiscent of domains characteristic of picornavirus IRES elements. Therefore, one might guess that the translation of ORF1 of L1 mRNA is directed by an efficient IRES. To test this possibility, the conventional method of plasmid-encoded dicistronic mRNAs was applied to the L1 mRNA 5′UTR. Since some IRES elements require the beginning of the corresponding coding region for their activity (42), the first 45 nt of the ORF1 were included in all our constructs. In contrast, as the L1 3′UTR was shown to be dispensable for retrotransposition (32), all our plasmids contained the standard SV40 3′UTR and poly(A) signal instead of those from the L1 mRNA. The corresponding constructs, based on the standard pRF vector (46), are presented in Fig. 2A. In the case of L1 mRNA, the use of this approach was complicated by the presence of an internal promoter in the 5′UTR of retrotransposon L1 which could result in a monocistronic rather than a dicistronic mRNA transcribed from the construct pSV40-Rluc-L1 5′UTR-Fluc. To solve this problem, we used the observation of Swergold and coworkers (51) in which the deletion of the first 100 nt from the 5′end of the retrotransposon completely abolishes the activity of the L1 internal promoter. As shown in Fig. 2B, when such a deletion variant of the 5′UTR of L1 [5′UTRΔ(1-98)] was inserted into the intercistronic position of pRF vector, a very high expression of Fluc (compared to the empty vector) was observed in the human teratocarcinoma NTera2/D1 cell line (the cells for which expression of ORF1p has been documented before [27]). The activity of the L1 5′UTR in this assay was even higher than that of the “classical” picornavirus genus human rhinovirus (HRV) IRES. A further deletion analysis showed the importance of the middle part of the L1 5′UTR [Fig. 2B, constructs Δ(390-526) and Δ(527-662)]. Its removal dramatically decreased but did not completely abolish the Fluc expression from the dicistronic construct. However, a similar if not higher expression was revealed for dicistronic constructs from which the SV40 promoter was excised (Fig. 2C), suggesting the existence of the cryptic promoter activity even in the construct Rluc-L1 5′UTRΔ(1-98)-Fluc. This cryptic promoter activity could not be completely eliminated even after further excising large portions of the 5′-UTRΔ(1-98) (Fig. 2C), indicating that the sequences possessing a promoter activity occupy a large part of the L1 5′UTR. It is also possible that the SV40 enhancer located downstream of the Fluc coding sequence in our pRF vector activates some formerly silent transcription factor binding sites within the L1 5′UTR (5). The small interfering RNA (siRNA) test against Renilla luciferase, as suggested by Van Eden and coworkers (56), showed that the expression of Fluc remained high when an almost complete inhibition of Rluc synthesis was achieved (Fig. 2D). This result confirmed that the Fluc synthesis observed in the initial experiments (Fig. 2B) occurred on a monocistronic mRNA. Therefore, even if an IRES exists within the 5′UTR of L1 mRNA, its sequences may overlap with the L1 promoter, thereby precluding the use of dicistronic DNA constructs for identification of such an IRES. It should be noted that this problem could not be solved by replacing NTera2/D1 cells with HEK293, which are thought to be poor in transcription factors specific for the L1 retrotransposon (52). Qualitatively, similar data were obtained with HEK293 cells, though in this case, the L1 promoter activity was about twofold lower than that with NTera2/D1 cells (data not shown). Thus, the only way to identify an IRES within the L1 5′UTR was the direct transfection of cultured cells with the respective RNA constructs.
FIG. 1.

Structure of the human retrotransposon L1. (A) Schematic representation of the human L1 dicistronic mRNA. (B) Computer-predicted secondary structure of the L1 5′UTR. The overall structure was evaluated using MFold software (64). The potential pseudoknot at the 5′-proximal part was suggested by pknotsRG (41). The last ∼100 nt of the 5′UTR, predicted to form no significant secondary structure, are shown by a dotted line.
FIG. 2.

Method of DNA dicistronic construction produces artifacts with the L1 5′UTR. (A) Schematic presentation of the pRF reporter vector (46). (B) Results of the DNA transfection experiments with constructs based on the pRF vector. NTera2/D1 cells were transfected with corresponding plasmids, harvested 48 h after transfection, and assayed for firefly and Renilla luciferase (Fluc and Rluc, respectively). Fluc/Rluc values were normalized to that of the “empty” pRF vector (taken as 1 unit; the absolute Fluc/Rluc value for this vector was 0.031). (C) Results of NTera2/D1 cell transfection with SV40 promoterless constructs. Corresponding plasmids were cotransfected with pCMV-lacZ vector; firefly luciferase and beta-galactosidase (β-Gal) assays were performed 48 h after transfection. Fluc values were divided by β-Gal values and normalized to values of pRFΔSV40. (D) Effect of siRNA against the Rluc coding region on the pRF-derived plasmid expression levels. Cells were cotransfected with Rluc-L1 5′UTRΔ(1-98)-Fluc, Rluc-HRV-Fluc, or an equimolar mix of pSV40-Rluc plus pSV40-Fluc plasmids and with either pRLi (56) or the corresponding empty vector. pCMV-lacZ was included in the transfection mix as well. Fluc/β-Gal and Rluc/β-Gal values were normalized to that obtained with the empty vector.
Transfection of RNA dicistronic constructs reveals a very low IRES activity of the 5′UTR of L1 mRNA.
The dicistronic RNA constructs for transfection experiments were obtained by in vitro T7 transcription from PCR products prepared on the basis of the DNA dicistronic constructs shown in Fig. 2A, with the exception that in this case, the 5′UTR was complete (i.e., the first 98 nt were included in the L1 construct). 5′ and 3′ PCR primers were supplied with the T7 promoter and the poly(T)50 tail, respectively, to obtain polyadenylated transcripts. Both capped and uncapped dicistronic RNAs were prepared. As follows from the data presented in Fig. 3, the IRES activity of the L1 5′UTR placed in the intercistronic position of uncapped bicistronic RNA was 2- to 2.5-fold higher than the “empty” uncapped dicistronic RNA vector and 2-fold higher than that in the L1 5′UTR with a large deletion [L1 5′UTRΔ(133-887)]. However, the L1 5′UTR worked four to five times less efficiently than even the relatively “weak” IRES from the Rhopalosiphum padi virus (RhPV) RNA (61) or hepatitis C virus (HCV) IRES (the last one in our hands was inefficiently translated with HEK293 cells but translated rather efficiently with NTera2/D1 cells). A similar uncapped construct with the human rhinovirus IRES showed a 50- to 60-fold-higher activity than that of the Rluc-L1 5′UTR-Fluc RNA. When assaying capped dicistronic RNAs, the activity of the HRV IRES was found to be considerably lower (data not shown). This is most probably accounted for by some competition for eIF4F between the cap and the HRV IRES, as may be inferred from a recent report by Svitkin et al. (49). Nevertheless, even in this case, the activity of the HRV IRES was much higher than that of the L1 mRNA 5′UTR. The inevitable conclusion from these data is that even if the 5′UTR of L1 mRNA contains an IRES, its activity is very low and is poorly compatible with the high level of ORF1 translation observed with mammalian cells. In fact, in this case, we face the same problem that other authors studying cellular IRESs have always confronted: on one hand, the IRES activity of the L1 5′UTR is higher than that of the empty RNA vector; on the other hand, it is considerably lower than that of the true IRES elements of RhPV, HCV, and all other HRV RNAs. Therefore, the criteria based on the comparison with an empty vector which are often used to consider whether an RNA sequence may be defined as an IRES suffer from an obvious subjectivity since they aim to elucidate the mechanism by which an RNA might be potentially translated rather than the mode of translation initiation that is actually used in cells. To solve the problem, we compared the activity of the 5′UTR in monocistronic constructs (where this 5′UTR occupied its natural position) with that in the intercistronic position.
FIG. 3.

Results of HEK293 and NTera2/D1 cell transfection with uncapped dicistronic mRNA constructs. Cells were harvested 3 h after transfection. Fluc/Rluc values were normalized to that for RF RNA synthesized in vitro from pRF “empty” vector (its absolute Fluc/Rluc value was 0.003).
Activity of the L1 5′UTR in the internal position of dicistronic RNAs is dramatically lower than in monocistronic RNA constructs.
The experiments were conducted in such a way that the Fluc activity of a dicistronic capped RNA containing the L1 5′UTR in the intercistronic position (construct capRluc-L1 5′UTR-Fluc) was compared to that of an equimolar mixture of capped monocistronic RNA (construct capL1 5′UTR-Fluc) and a reference capRluc mRNA (Fig. 4A). To prevent possible effects of nonequal transfection efficiency of the monocistronic transcripts in the latter case, our reference capRluc mRNA was of a length that was similar to the capL1 5′UTR-Fluc. The Fluc activity for the monocistronic construct was 60-fold higher with HEK293 cells and 45-fold higher with NTera/D1 cells than with cells transfected with the dicistronic mRNA (Fig. 4B). It is important to note that similar ratios for respective pairs of RNAs were obtained when the largest part (∼80%) of the L1 5′UTR sequence was deleted [Fig. 4B, constructs capRluc-L1 5′UTRΔ(133-887)-Fluc and capL1 5′UTRΔ(133-887)-Fluc]. In the same test, however, the HRV IRES showed only a threefold difference, irrespective of whether uncapped (Fig. 4C) or capped (data not shown) RNAs were used. Nevertheless, it is interesting that significant differences exist between the IRES-mediated expression levels from the monocistronic and dicistronic mRNAs for the HRV IRES as well. The possible explanation for this observation may be that the translation of the first cistron in the dicistronic mRNA or the upstream sequences per se have a negative effect on HRV IRES activity.
FIG. 4.

Comparison of activities of various 5′UTRs in monocistronic versus dicistronic contexts. (A) Schematic representation of mono- and dicistronic mRNAs used in the assay. (B and C) HEK293 and NTera2/D1 cells were transfected with either dicistronic mRNA derivatives as indicated or with an equimolar mixture of the corresponding monocistronic reporter (coding for Fluc) and the normalizing mRNA (Rluc). Fluc/Rluc values for monocistronic mRNA were taken as 100 units for each individual construct. (D) Results of mono- and dicistronic mRNA translation in in vitro “RRL+HeLa” cell-free system. Lane 1, Rluc; lane 2, L1 5′UTR-Fluc; lane 3, equimolar mixture of L1 5′UTR-Fluc and Rluc; lane 4, dicistronic Rluc-L1 5′UTR-Fluc; lane 5, mixture of L1 5′UTRΔ(133-887)-Fluc and Rluc; lane 6, dicistronic Rluc-L1 5′UTRΔ(133-887)-Fluc.
Again, these data strongly suggest that the 5′UTR of L1 mRNA does not contain an IRES element, at least an IRES element which could have a functional significance. This conclusion was confirmed by in vitro translation of similar RNA constructs with a combined RRL-HeLa extract (Fig. 4D). This cell-free system was employed as an alternative to the standard RRL translation assay since, as we have recently shown, the RRL system is not an adequate system with which to study the translation of L1 mRNA (8).
Translation initiation activity of the 5′UTR of L1 mRNA is critically dependent on the cap.
The large size of the 5′UTR and its high GC content do not present an impediment for the translation initiation efficiency of L1 mRNA. The data described above allow one to infer that the cap exerts a strong stimulatory effect on the translation of ORF1. This conclusion acquired additional support when we compared the levels of Fluc translation from the capped with those of uncapped monocistronic Fluc mRNAs containing the L1 5′UTR. To perform such a comparison, we first analyzed a time course of capped versus uncapped HRV-Fluc mRNA translation to determine the optimal time for harvesting cells. Figure 5A illustrates the linear increase in the luciferase activity during the first 3 h after transfection. Right after this period, an apparent bend in the curve was observed in the case of uncapped transcripts. These data indicate that little RNA degradation occurs during the first 3 h following transfection, in agreement with results from similar studies (5, 56, 58). The same picture was obtained for other mRNAs tested (data not shown). We therefore established 3-h posttransfection as an appropriate time to harvest the cells for the luciferase assay.
FIG. 5.

Cap dependence of Fluc mRNAs with various 5′UTRs. (A) Time course of luciferase activity in cells transfected with capped versus uncapped monocistronic HRV-Fluc mRNA. Cells from different plates were harvested every hour (1 to 8 h after transfection, as indicated) and assayed for Fluc activity, which was then normalized for each construct to its activity at 3 h. (B and C) Comparison of indicated capped versus uncapped monocistronic transcripts for the RNA transfection assay. Fluc/Rluc values for capped mRNA were taken as 100 units for each individual construct.
In the case of L1 5′UTR, the capped construct was translated 50-fold (HEK293) and 60-fold (NTera2/D1) more efficiently than the uncapped one (Fig. 5B). The same ratios were obtained when the uncapped RNA was replaced by the transcript containing ApppG… at the 5′ end (data not shown). Moreover, the translational level of Fluc for uncapped L1 5′UTR-Fluc mRNA turned out to be only ∼10-fold higher than that for the uncapped dicistronic “empty” pRF vector, representing therefore the level not so far from the background (data not shown). A similar stimulation by the cap was observed for the 5′UTR mutant from which the large part of the GC-rich region was deleted [Fig. 5B, L1 5′UTRΔ(133-887)]. This fact indicates that the translation initiation of the L1 mRNA was critically dependent on the presence of the cap at its 5′ terminus. In contrast, the picture observed in the case of the HRV 5′UTR was that the cap addition turned out to be slightly inhibitory (Fig. 5C). The obvious explanation of this phenomenon is the competition for eIF4F between the eIF4G binding site of the HRV IRES and the 5′ cap structure. Binding of the eIF4F to the latter position seems to lead to a complex which is a dead end for the translation initiation on this 5′UTR. It should be remembered that all these RNA constructs were supplied with the poly(A) tail (50 residues), which is essential for a high efficiency of translation initiation not only on cap-dependent 5′UTRs but also for picornavirus and many other IRES elements (4).
Translation initiation potential of the L1 5′UTR is similar to that of a standard cellular mRNA.
It should be emphasized that not only is the extent of stimulation by the cap high but the absolute level of ORF1 translation in comparison with other mRNAs containing complex 5′UTRs also looks impressive. The capped L1 5′UTR-Fluc mRNA was translated four to five times more effectively than the uncapped HRV-Fluc mRNA, where the translation of Fluc was directed by a rather strong picornavirus IRES (Fig. 6A). Moreover, its translation initiation potential proved to be similar to that of the 5′UTR of human beta-actin mRNA (Fig. 6A). Importantly, in the actin 5′UTR containing mRNA the AUG-proximal 20 nt of the 5′UTR and the first 45 nt of the coding region were the same as those in the L1 5′UTR-Fluc mRNA. This was constructed to avoid any effect of the immediate AUG context on the initiation or any impact of the first 15 codons left from L1 ORF1 in all our L1 5′UTR-derived constructs (see discussion above) on the activity of the firefly luciferase enzyme.
FIG. 6.

Comparison of the Fluc translation directed by various 5′UTRs. (A) Capped L1, uncapped HRV, and capped beta-actin 5′UTRs (the uncapped HRV 5′UTR was used as more “natural” for this mRNA; the capped one also employed in this assay showed lower activity, as shown in Fig. 5C). (B) L1 5′UTR derivatives with various parts deleted or inversed (as indicated). (C) L1 5′UTR derivatives with deletion of uAUG16 or insertion of additional AUG codons at nt 248 or 473. All mRNAs were cotransfected with Rluc mRNA, and Fluc/Rluc values were normalized to that for the wt L1 5′UTR.
According to the classical model of cap-dependent initiation (19), a large and developed secondary structure of 5′UTRs should negatively affect the translational efficiency. Therefore, we expected that shortening the 5′UTR of L1 mRNA or deleting most of its stem-loops would result in a considerable increase of translation efficiency of such mutant constructs compared with that of the capped wild-type (wt) L1 5′UTR-Fluc mRNA. Strikingly, the experiments failed to support this prediction. As can be seen from Fig. 6B [construct L1 5′UTRΔ(133-887)], the deletion of up to 80% of the nucleotide sequence of L1 5′UTR with most of its stem-loops (leaving 133 cap-proximal and 20 AUG-proximal nt unchanged) produced a rather modest effect on the translation efficiency. In addition, stepwise deletions of 100 to 150 nt across the nucleotide sequence of the L1 5′UTR have very little [albeit predominantly negative, with the exception of L1 5′UTRΔ(527-662), see below] effect on the activity, suggesting the absence of specific structural domains within the L1 5′UTR which would facilitate scanning (or shunting) by the 40S ribosomal subunit. Therefore, at least for the 5′UTR of L1 mRNA, neither its large size nor its stem-loop structures (Fig. 1B) appear to be obstacles to the 40S ribosomal subunit crossing over the distance between the cap and the initiation codon of the ORF1.
The start codon of the ORF1 of L1 mRNA is selected by a scanning rather than a shunting mechanism.
The data described above strongly suggest that the 40S ribosome first binds to the cap and then is efficiently transferred to the initiation codon of ORF1 in spite of the length and numerous stem-loops of the L1 5′UTR. The shunting mechanism could be a plausible explanation for this phenomenon. The molecular mechanism of shunting is poorly understood and represented in literature by rare examples. According to Futterer et al. (10), who first suggested this mechanism for the cauliflower mosaic virus gene VII, the 40S ribosomal subunit first initiates the translation of a short uORF in a cap-dependent way and then, after the termination of translation of this uORF, “jumps” over the long stem-loop structure to land near the initiation codon of the main ORF. This jump is promoted by two special sequences termed “starting” and “landing” sequences, which immediately follow the termination codon uORF and precede the main ORF, respectively. The 5′UTR of L1 mRNA contains a conserved uORF with an AUG in a rather “good” nucleotide context (AAGATGG) which could be used in a similar way to transfer the 40S ribosome to the AUG of the first cistron of L1 mRNA. To check whether such a mechanism operates in the case of L1 5′UTR, we mutated this uAUG to UAG (construct L1 5′UTRΔAUG16-Fluc) and examined the translation activity of such a mutant. As seen in Fig. 6C, the mutation of uAUG increased rather than decreased the translation initiation efficiency of L 5′UTR. The effect was modest (less than twofold) but agreed with the scanning rather than the shunting mechanism in the version proposed for cauliflower mRNA (10). It is interesting that among the stepwise deletion mutants (see above), the only construct which had a higher level of translation was L1 5′UTRΔ(527-662) (Fig. 6B), which possesses a deletion of the region containing the second L1 5′UTR uAUG (at position 607). It seems that in spite of a “bad” nucleotide context (TAAAUGT), it is also recognized to some extent by the scanning 40S subunit. The conclusion that a scanning mechanism does exist at the L1 mRNA leader was further supported by the influence of an additional AUG in a good context inserted at position 248 (construct L1 5′UTRinsAUG248-Fluc). This mutant decreased the translation initiation potential of L1 5′UTR, suggesting that the 40S ribosome “inspects” the nucleotide sequence of L1 5′UTR on its way to the AUG of ORF1. The effect of the additional AUG was again not dramatic (Fig. 6C), showing that the initiation at the AUG of ORF1 was highly preferred.
It could be argued that the modest negative effect of uORFs described above is accounted for by the fact that only a portion of ribosomes use the scanning mechanism, whereas other ribosomes employ a highly efficient shunting model. However, it was suspected that the modest effect of natural and of our additional uORFs could be explained simply by a suboptimal structural context of the initiation segments, which included these uAUGs (compared to the authentic start codon AUG908). To check this possibility, we designed one more initiation region in the middle of L1 5′UTR (Fig. 1B, insAUG473). Additional A residues were inserted by mutagenesis both upstream and downstream of uAUG473 to expand the upper loop of the corresponding stem-loop structure. These manipulations aimed to have this AUG restricted as little as possible by base pairing. The uORF thus obtained was rather long (86 nucleotide triplets). This could also enhance its inhibitory effect on the translation initiation at the main ORF. Indeed, as seen from Fig. 6C, the uORF473 construct had a dramatic effect on the translation of the ORF1 (20-fold inhibition).
Another version of shunting was proposed by Yueh and Schneider (63) for the tripartite leader (TPL) of late adenovirus mRNAs. According to this model, the presence of a uORF is not required. The 40S ribosomal subunit first scans the sequence juxtaposed to the cap and is then transferred to the AUG codon, using downstream-specific sequences of the TPL, which were proposed to form Shine-Dalgarno-like duplexes with the respective complementary sequences of 18S rRNA. Although the molecular mechanism of this version of shunting is still very speculative, it is unlikely in the case of L1 5′UTR. This is evidenced by the insignificant effect on the translation efficiency of various large deletions throughout the L1 5′UTR sequence described in the previous section (Fig. 6B). The shunting mechanism is also incompatible with the data from reference 30 indicating that a stable hairpin inserted at position 661 of the 5′UTR strongly inhibits the ORF1 translation. Taken together, these data present compelling evidence that the overwhelming majority of, if not all, 40S ribosomal subunits attain the initiation codon of the ORF1 by a scanning mechanism.
Finally, one could not exclude the possibility that efficient scanning over the entire length of the L1 mRNA 5′UTR was determined by the unique properties of its structure. As shown in Fig. 6B (right columns), this is not the case. The inversion of the sequence from position 137 to position 887 of the L1 5′UTR resulted in the translation activity similar to that of the wt L1 5′UTR.
Efficient translation of the first cistron of L1 mRNA ensures the high ORF1/ORF2 ratio needed for the normal process of retrotransposition.
The mode of translation initiation at the ORF2 was studied by Alisch and coworkers (1), using genetic approaches based on the analysis of efficiency of retrotransposition. They presented strong arguments against the existence of an IRES in their version of L1 mRNA, either within the ORF1 or in the intercistronic spacer, and came to the conclusion that the start of translation of ORF2 was selected by an unconventional mechanism of reinitiation. We reinvestigated the same issue using methods of RNA transfection of cultured cells, followed by analysis of the expression of the Fluc reporter from the construct, shown schematically in Fig. 7A. The translation of Fluc from the capped or uncapped construct L1 5′UTR-p40-Fluc was found to be rather low, comparable to the expression of Fluc from the conventional “empty” pRF vector (Fig. 7B). The Fluc was translated from the capped L1 5′UTR-Fluc RNA in HEK293 cells ∼400 times more efficiently than from the capL1 5′UTR-p40-Fluc, thereby reasonably reflecting the very high ORF1/ORF2 expression ratio reported for LINE transposons (6, 30). For NTera2/D1 cells this difference was somewhat lower (∼130 times) but nevertheless reflected the 2-orders-of-magnitude superiority of ORF1p production (Fig. 7B). The same inefficient ORF2 translation was also observed in vitro with the “RRL+HeLa” (but not in the regular RRL) cell-free system (8).
FIG. 7.

Analysis of the L1 ORF2 translation. (A) Schematic of the dicistronic L1 5′UTR-p40-Fluc mRNA used for these experiments. (B) Comparison of Fluc expression levels directed by the L1 5′UTR and the L1 5′UTR-p40 sequences and the effect of the 5′ cap on the translation of the L1 5′UTR-p40-Fluc transcript. The values of Fluc were normalized to Rluc activity originated from the cotransfected Rluc mRNA and expressed relative to the Fluc/Rluc activity of capped L1 5′UTR-p40-Fluc mRNA (taken as 1 unit). The values are shown on a logarithmic scale. The efficiency of the dicistronic RF mRNA translation is displayed for a background estimation. (C) Effect of point mutations and substitutions on the L1 5′UTR-p40-Fluc translation efficiency. The experiment was performed as described for panel B, with the exception that the Fluc/Rluc value of L1 5′UTR-p40-Fluc was taken as 100 units.
As translation of the L1 bicistronic mRNA results in producing a large amount of ORF1p (p40), which is thought to bind L1 mRNA in cis (3, 13, 60) and may have an effect on the mRNA translation, we tested the impact of ORF1p mutation on ORF2 translation. We used the RR261AA ORF1p mutation, which is known to abolish its RNA-binding capacity (24, 29, 32). This mutation, when introduced into the capL1 5′UTR-p40-Fluc mRNA, does not influence the translation level of the second cistron (Fig. 7C), consistent with the result obtained previously by genetic approaches (1).
As any contribution of ORF1 translation was excluded, we were able to substitute the p40 coding sequence and the intercistronic region for the LacZ-encoding sequence. Such substitution had no significant influence on the expression of ORF2 (Fig. 7C), suggesting the absence of a specific IRES element upstream of the ORF2. On the other hand, omission of the cap at the 5′ end of L1 5′UTR-p40-Fluc did reduce the translation of ORF2 (Fig. 7B), but the effect was much lower than in the case of ORF1 (a difference of 5- to 7-fold versus 50- to 60-fold in the efficiency of translation of ORF1 directed by capped and uncapped L1 5′UTR Fluc constructs, respectively; see discussion above). In agreement with the data of Alisch and coworkers (1), the substitution of the AUG start codon of ORF2 with the CCC triplet resulted in only a twofold reduction of the ORF2 translation. However, a similar reduction, but not abolition, of the translation of ORF2 was observed for the AUG→UGA substitution, the substitution which according to these authors was partially tolerated by mouse L1 mRNA but not by their version of human L1 mRNA. This may be accounted for by differences in the nucleotide context of the start codon of ORF2 in the two studies.
DISCUSSION
The L1 mRNA is an attractive model with which to test the current concepts of mechanisms of translation initiation in eukaryotes. First, it is a dicistronic mRNA, which is exceptionally rare in eukaryotic cellular mRNAs. This allows one to elucidate the translation initiation mechanism of the second cistron by using a natural dicistronic mRNA rather than artificial dicistronic constructs. For all the artificial dicistronic mRNAs studied to date, the translation initiation of the second cistron occurs only if it is directed by an IRES element. Second, the first 800 nt of the sequence of the L1 5′UTR are GC rich and form a series of predicted stem-loop structures. In spite of these features, which are believed to be unfavorable for the 5′-end-dependent translation initiation, the L1 mRNA very actively directs the translation of ORF1. Numerous copies of ORF1p (p40) are formed in the cell per molecule of L1 mRNA, allowing the formation of an L1 mRNP particle competent for subsequent retrotransposition. This suggests that either the 5′UTR of L1 mRNA contains a powerful IRES element whose activity is comparable to that found in some picornavirus RNAs or that the translation initiation of the L1 mRNA ORF1 is directed by an efficient shunting mechanism.
The main focus of this study was the mechanism of translation initiation at the ORF1. As to the mechanism of the human L1 ORF2 translation initiation, it has been recently investigated by analyzing the effect of various mutations at the boundary of ORF1 and ORF2 on the frequency of retrotransposition (1). These authors came to the conclusion that the initiation at the ORF2 is not directed by an IRES and instead employs an unconventional mechanism of translation reinitiation. We have performed analogous investigations but used methods of biochemistry and cell biology and confirmed that there is no IRES element preceding the ORF2, at least for our version of human L1 retrotransposon. The capping of L1 mRNA stimulates the translation of ORF2. Furthermore, the product of the ORF1 (p40) and hence its accumulation in the cell is not required to recruit 40S ribosomes to the ORF2 (Fig. 7B and see reference 1), thereby excluding the possibility that p40 forms an initiation site to promote the 40S ribosome entry onto the start codon of the ORF2. These two facts lend some support to a reinitiation mechanism. However, unlike the data reported in reference 1, our version of human L1 mRNA partially tolerated the substitution of the AUG of ORF2, not only for a missense codon but also for a nonsense one. The possibility that in this case, the translation initiation of ORF2 might occur somewhere downstream, i.e., at an AUG within the coding part of Luc, should be rejected since the firefly luciferase without its N-terminal amino acid residues is totally inactive (57). Our data lend solid support to the unconventional reinitiation mechanism proposed by Alisch and coworkers (1), though more experimentation is still needed to ultimately clarify this issue. Anyway, the extremely inefficient mechanism of translation initiation at the ORF2 and the highly efficient expression of p40 from ORF1 ensure the high ORF1/ORF2 ratio, which is presumably required for the successful retrotransposition of L1 mobile elements of different origins (6).
The classical model of translation initiation in eukaryotes states that the larger the size of the 5′UTR of an mRNA and the more complex its secondary structure, the less efficient is the cap-dependent mechanism of translation initiation on this mRNA (21, 39). This statement is at least one of the reasons why the concept of cellular IRES elements has become so popular among researchers concerned with studies of molecular mechanisms of translation initiation in eukaryotes. When investigators analyze an mRNA with a long and highly structured 5′UTR, the initial interpretation is that an IRES resides within this 5′UTR. During the last decade, the number of putative cellular IRES elements has rapidly accumulated (16, 48). In spite of debates on the existence of cellular IRES elements (20, 22, 45), this concept remains attractive since it allows a mechanism to get around the structural problems mentioned above. In most cases, the only criterion for considering a 5′UTR as harboring an IRES is the comparison between its activity in directing the second cistron translation and the activity of an “empty” vector in the same assay. This criterion is rather subjective, and we propose the comparison of mono- versus bicistronic mRNA translation as a more reliable one. Also, the effect of 5′UTR capping must be taken into account. These two criteria allow one to discriminate between the potential mode of mRNA initiation and its actual mode. When such tests were applied, rare examples indicated that mRNAs possessing long GC-rich leaders are translated much more efficiently in a monocistronic than in a dicistronic context (5).
In the course of this study, we obtained compelling evidence that the 5′UTR of L1 mRNA does not contain an IRES element and its activity is critically dependent on the m7G cap. Indeed, the L1 5′UTR (i) showed a very weak translation initiation potential in RNA transfection assays when placed between two cistrons in the bicistronic mRNA; (ii) directed translation much more efficiently when located in a monocistronic rather than a dicistronic context, both in vivo and in vitro; and (iii) critically required capping of the monocistronic mRNA. In addition, we found that in the modified “RRL+HeLa” cell-free system (8), the L1 mRNA translation is highly sensitive to the addition of the dominant-negative mutant eIF4A R362Q which is a well-known inhibitor of the cap-binding complex eIF4F activity (35, 36) (data not shown). Although this may not be a criterion of cap deficiency per se, since IRES elements show a wide range of sensitivity to inhibition by the mutant eIF4A (50, 54), in some cases reduced sensitivity to the eIF4A mutant points to an internal initiation mechanism (38, 53). Moreover, such high eIF4F dependence for the translation of the L1 mRNA correlates well with the observations of Koromilas and coworkers (17) that overexpression of the cap-binding factor eIF4E preferentially stimulates the translation of cap-dependent mRNAs with complex 5′UTRs in vivo.
Importantly, it should be noted that, although it has not been shown yet directly whether the L1 transcripts are capped or not in living cells, some indirect evidence suggests that they must be capped (2, 26) like other typical mRNAs synthesized (most likely) by RNA polymerase II (33). In fact, our data that uncapped L1 5′UTR directs translation very inefficiently are evidence for the capping of the mRNA in order to achieve high cellular levels of ORF1 protein.
Our conclusion that the L1 mRNA does not contain IRES elements contrasts with a recent report on the existence of IRES elements upstream of both ORF1 and ORF2 in mouse LINE-1 mRNA (28). The organization of the mouse L1 mRNA, especially its 5′UTR, is different from that of human L1, so we cannot exclude the possibility that the translation initiation of human and mouse L1 mRNAs occurs by distinct mechanisms. However, it should be noted that the sequences of mouse L1 mRNA proposed to contain these IRES elements possess a weak promoter activity (28). Moreover, for most of the constructs used in the DNA transfection assay, the level of the 5′UTR IRES activity correlated well with the promoter activity of the same 5′UTR fragment (see Fig. 6 and Fig. 7 in reference 28). In our opinion, it is difficult, if not impossible, to distinguish the Fluc activity which is exclusively accounted for by an IRES element from that originating from the translation of a monocistronic capped mRNA produced from such weak promoters. Dicistronic RNA transfection experiments were also used by those authors. In that case, the Fluc activity directed by the proposed IRES elements was very low. In fact, the values obtained were comparable to those described in the present study for similar constructs (Fig. 3A). Finally, it should be noted that the data of Alisch et al. (1) strongly suggest that the initiation at the ORF2 of mouse L1 mRNA occurs by a reinitiation mechanism rather than by ribosomal internal entry.
One of the most interesting observations from this study is that the efficiency of the translation initiation at the ORF1 does not significantly depend on the length of the 5′UTR and on the number of stem-loop structures it contains. The translation initiation potentials of the beta-actin mRNA 5′UTR (84-nt length) and of the L1 mRNA 5′UTR (900 nt) are similar, and the deletion of up to 80% of the nucleotide sequence of the L1 5′UTR with most of its stem-loops does not significantly change its translation initiation efficiency. This could imply that the selection of the ORF1 start codon is directed by a shunting mechanism. However, our data do not support such an interpretation. By definition, a key feature of shunting mechanisms is that the 40S ribosome skips a part of the untranslated region without examining its nucleotide sequence. This is certainly not the case of the L1 5′UTR since insertion or removal of AUG triplets does not remain “unnoticed” by the ribosome. Thus, it suggests that the 40S ribosomal subunit on its way to the start codon of ORF1 inspects the entire sequence of the L1 5′UTR. This conclusion is reinforced by earlier data obtained by McMillan and Singer (30), who demonstrated that a stable hairpin structure inserted at nt 661 of the L1 5′UTR caused a three- to eightfold decrease in ORF1 production.
The translation initiation properties of the 5′UTR of L1 mRNA discussed above strongly support a scanning mechanism that involves inspection of its entire structure in the 5′-3′ direction. We think that the popular opinion that long and structured 5′UTRs are inefficient in translation is based on a collection of incorrect prerequisites. It is partially based on in vitro experiments with RRL where the inhibitory effect of stem-loop structures is considerably more pronounced than in cells (our own unpublished observations and references 12 and 55). Nevertheless, the conclusions drawn from these experiments have often been extended to the situation in living cells. In other experiments, including those with transfected cells, investigators use, as a rule, extremely stable artificial helixes which rarely occur in natural mRNA. In some reports, the highly stable stem-loop structures are placed either too close to the cap (thereby preventing accommodation of initiation factors of group 4) or too close to the initiation triplet or both. To the best of our knowledge, nobody has ever aimed to quantitatively compare the efficiency of translation of mRNAs with short- versus long-structured 5′UTRs with mammalian cells. However, careful analysis of literature shows that the case of L1 mRNA is not unprecedented, though the authors of the corresponding reports either did not focus on these findings or did not treat it as a general feature (5, 12, 47, 55).
We realize that it is not easy to interpret our data on the basis of the conventional idea of how the scanning process occurs. The scanning mechanism is often imagined as a consecutive unwinding of stem-loops of 5′UTRs which is accompanied by pulling the corresponding regions through the mRNA binding channel of the 40S subunit in the 5′-3′ direction and inspecting their sequences one nucleotide by one until the initiation codon in the optimal context is found. Our working hypothesis is that the final accommodation of the mRNA within the mRNA-binding channel of the 40S ribosome does not occur before the selection of the initiation region is completed. We suggest that the AUG codon and its immediate optimal nucleotide context are not sufficient elements to determine the position of the translation start site on mRNAs with long and highly structured 5′UTRs. Presumably, much larger regions of these 5′UTRs, comparable in size with segments covered by the ribosome, are screened by the 40S ribosomal subunits. Those which are in the optimal conformation and contain the initiation codon in a good immediate nucleotide context are preferentially selected, whereas those which are conformationally restricted have a much lower probability to be placed into the mRNA binding channel of the 40S ribosome. The latter segments of 5′UTRs are rapidly rejected without significant effect on the resulting rate of the 48S complex formation. To recognize all potential initiation codons by the scanning machinery, some consecutive unwinding of stem-loops of a 5′UTR should occur. However, these structural rearrangements do not result in a similar conformation for different regions of the 5′UTR, and hence, only few of these conformations may be readily accepted by the mRNA-binding cleft of the 40S ribosome. To confirm or reject this model, more sophisticated experiments should be performed and the work in this direction is now in progress.
Acknowledgments
We thank G. Swergold for kindly providing plasmids p1LZ and p3LZ, L. Kisselev and E. Alkalaeva for permission to use their laboratory luminometer, R. Lloyd for providing plasmid RLi, S. Akopov and E. Stukacheva for offering us NTera2/D1 cells, and D. Ivanov for assistance at the initial stage of the work.
This study was supported by a Fogarty grant (FIRCA PA-02-057) to W.C.M. and I.N.S. and in part by Russian Foundation of Basic Researches (RFBR) grant 04-04-49507 and President Grant MK- 3155.2005.4 to S.E.D and by RFBR grant 05-04-49366 to V.S.P.
Footnotes
Published ahead of print on 30 April 2007.
REFERENCES
- 1.Alisch, R. S., J. L. Garcia-Perez, A. R. Muotri, F. H. Gage, and J. V. Moran. 2006. Unconventional translation of mammalian LINE-1 retrotransposons. Genes Dev. 20:210-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Athanikar, J. N., R. M. Badge, and J. V. Moran. 2004. A YY1-binding site is required for accurate human LINE-1 transcription initiation. Nucleic Acids Res. 32:3846-3855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Basame, S., L. P. Wai-lun, G. Howard, D. Branciforte, D. Keller, and S. L. Martin. 2006. Spatial assembly and RNA binding stoichiometry of a LINE-1 protein essential for retrotransposition. J. Mol. Biol. 357:351-357. [DOI] [PubMed] [Google Scholar]
- 4.Bergamini, G., T. Preiss, and M. W. Hentze. 2000. Picornavirus IRESes and the poly(A) tail jointly promote cap-independent translation in a mammalian cell-free system. RNA 6:1781-1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bert, A. G., R. Grepin, M. A. Vadas, and G. J. Goodall. 2006. Assessing IRES activity in the HIF-1alpha and other cellular 5′UTRs. RNA 12:1074-1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bouhidel, K., C. Terzian, and H. Pinon. 1994. The full-length transcript of the I factor, a LINE element of Drosophila melanogaster, is a potential bicistronic RNA messenger. Nucleic Acids Res. 22:2370-2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brasey, A., M. Lopez-Lastra, T. Ohlmann, N. Beerens, B. Berkhout, J. L. Darlix, and N. Sonenberg. 2003. The leader of human immunodeficiency virus type 1 genomic RNA harbors an internal ribosome entry segment that is active during the G2/M phase of the cell cycle. J. Virol. 77:3939-3949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dmitriev, S. E., N. V. Bykova, D. E. Andreev, and I. M. Terenin. 2006. Adequate system for investigation of translation initiation of the human retrotransposon L1 mRNA in vitro. Mol. Biol. (Moskva) 40:25-30. (In Russian.) [DOI] [PubMed] [Google Scholar]
- 9.Dmitriev, S. E., I. M. Terenin, Y. E. Dunaevsky, W. C. Merrick, and I. N. Shatsky. 2003. Assembly of 48S translation initiation complexes from purified components with mRNAs that have some base pairing within their 5′ untranslated regions. Mol. Cell. Biol. 23:8925-8933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Futterer, J., Z. Kiss-Laszlo, and T. Hohn. 1993. Nonlinear ribosome migration on cauliflower mosaic virus 35S RNA. Cell 73:789-802. [DOI] [PubMed] [Google Scholar]
- 11.Han, B., and J. T. Zhang. 2002. Regulation of gene expression by internal ribosome entry sites or cryptic promoters: the eIF4G story. Mol. Cell. Biol. 22:7372-7384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hensold, J. O., C. A. Stratton, and D. Barth. 1997. The conserved 5′-untranslated leader of Spi-1 (PU. 1) mRNA is highly structured and potently inhibits translation in vitro but not in vivo. Nucleic Acids Res. 25:2869-2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hohjoh, H., and M. F. Singer. 1996. Cytoplasmic ribonucleoprotein complexes containing human LINE-1 protein and RNA. EMBO J. 15:630-639. [PMC free article] [PubMed] [Google Scholar]
- 14.Iacono, M., F. Mignone, and G. Pesole. 2005. uAUG and uORFs in human and rodent 5′untranslated mRNAs. Gene 349:97-105. [DOI] [PubMed] [Google Scholar]
- 15.Kazazian, Jr., H. H., and J. V. Moran. 1998. The impact of L1 retrotransposons on the human genome. Nat. Genet. 19:19-24. [DOI] [PubMed] [Google Scholar]
- 16.Komar, A. A., and M. Hatzoglou. 2005. Internal ribosome entry sites in cellular mRNAs: mystery of their existence. J. Biol. Chem. 280:23425-23428. [DOI] [PubMed] [Google Scholar]
- 17.Koromilas, A. E., A. Lazaris-Karatzas, and N. Sonenberg. 1992. mRNAs containing extensive secondary structure in their 5′ non-coding region translate efficiently in cells overexpressing initiation factor eIF-4E. EMBO J. 11:4153-4158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kozak, M. 1980. Evaluation of the “scanning model” for initiation of protein synthesis in eucaryotes. Cell 22:7-8. [DOI] [PubMed] [Google Scholar]
- 19.Kozak, M. 1989. The scanning model for translation: an update. J. Cell Biol. 108:229-241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kozak, M. 2001. New ways of initiating translation in eukaryotes? Mol. Cell. Biol. 21:1899-1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kozak, M. 1991. Structural features in eukaryotic mRNAs that modulate the initiation of translation. J. Biol. Chem. 266:19867-19870. [PubMed] [Google Scholar]
- 22.Kozak, M. 2005. A second look at cellular mRNA sequences said to function as internal ribosome entry sites. Nucleic Acids Res. 33:6593-6602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kozak, M., and A. J. Shatkin. 1978. Migration of 40 S ribosomal subunits on messenger RNA in the presence of edeine. J. Biol. Chem. 253:6568-6577. [PubMed] [Google Scholar]
- 24.Kulpa, D. A., and J. V. Moran. 2005. Ribonucleoprotein particle formation is necessary but not sufficient for LINE-1 retrotransposition. Hum. Mol. Genet. 14:3237-3248. [DOI] [PubMed] [Google Scholar]
- 25.Kulpa, D. A., and J. V. Moran. 2006. Cis-preferential LINE-1 reverse transcriptase activity in ribonucleoprotein particles. Nat. Struct. Mol. Biol. 13:655-660. [DOI] [PubMed] [Google Scholar]
- 26.Lavie, L., E. Maldener, B. Brouha, E. U. Meese, and J. Mayer. 2004. The human L1 promoter: variable transcription initiation sites and a major impact of upstream flanking sequence on promoter activity. Genome Res. 14:2253-2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leibold, D. M., G. D. Swergold, M. F. Singer, R. E. Thayer, B. A. Dombroski, and T. G. Fanning. 1990. Translation of LINE-1 DNA elements in vitro and in human cells. Proc. Natl. Acad. Sci. USA 87:6990-6994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li, P. W., J. Li, S. L. Timmerman, L. A. Krushel, and S. L. Martin. 2006. The dicistronic RNA from the mouse LINE-1 retrotransposon contains an internal ribosome entry site upstream of each ORF: implications for retrotransposition. Nucleic Acids Res. 34:853-864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martin, S. L., M. Cruceanu, D. Branciforte, L. P. Wai-lun, S. C. Kwok, R. S. Hodges, and M. C. Williams. 2005. LINE-1 retrotransposition requires the nucleic acid chaperone activity of the ORF1 protein. J. Mol. Biol. 348:549-561. [DOI] [PubMed] [Google Scholar]
- 30.McMillan, J. P., and M. F. Singer. 1993. Translation of the human LINE-1 element, L1Hs. Proc. Natl. Acad. Sci. USA 90:11533-11537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meignin, C., J.-L. Bailly, F. Arnaud, B. Dastugue, and C. Vaury. 2003. The 5′ untranslated region and Gag product of Idefix, a long terminal repeat-retrotransposon from Drosophila melanogaster, act together to initiate a switch between translated and untranslated states of the genomic mRNA. Mol. Cell. Biol. 23:8246-8254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moran, J. V., S. E. Holmes, T. P. Naas, R. J. DeBerardinis, J. D. Boeke, and H. H. Kazazian, Jr. 1996. High frequency retrotransposition in cultured mammalian cells. Cell 87:917-927. [DOI] [PubMed] [Google Scholar]
- 33.Ostertag, E. M., and H. H. Kazazian, Jr. 2001. Biology of mammalian L1 retrotransposons. Annu. Rev. Genet. 35:501-538. [DOI] [PubMed] [Google Scholar]
- 34.Ostertag, E. M., and H. H. Kazazian, Jr. 2001. Twin priming: a proposed mechanism for the creation of inversions in L1 retrotransposition. Genome Res. 11:2059-2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pause, A., N. Méthot, and N. Sonenberg. 1993. The HRIGRXXR region of the DEAD box RNA helicase eukaryotic translation initiation factor 4A is required for RNA binding and ATP hydrolysis. Mol. Cell. Biol. 13:6789-6798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pause, A., N. Methot, Y. Svitkin, W. C. Merrick, and N. Sonenberg. 1994. Dominant negative mutants of mammalian translation initiation factor eIF-4A define a critical role for eIF-4F in cap-dependent and cap-independent initiation of translation. EMBO J. 13:1205-1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pestova, T. V., V. G. Kolupaeva, I. B. Lomakin, E. V. Pilipenko, I. N. Shatsky, V. I. Agol, and C. U. Hellen. 2001. Molecular mechanisms of translation initiation in eukaryotes. Proc. Natl. Acad. Sci. USA 98:7029-7036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pestova, T. V., I. N. Shatsky, S. P. Fletcher, R. J. Jackson, and C. U. Hellen. 1998. A prokaryotic-like mode of cytoplasmic eukaryotic ribosome binding to the initiation codon during internal translation initiation of hepatitis C and classical swine fever virus RNAs. Genes Dev. 12:67-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pickering, B. M., and A. E. Willis. 2005. The implications of structured 5′ untranslated regions on translation and disease. Semin. Cell Dev. Biol. 16:39-47. [DOI] [PubMed] [Google Scholar]
- 40.Pisarev, A. V., L. S. Chard, Y. Kaku, H. L. Johns, I. N. Shatsky, and G. J. Belsham. 2004. Functional and structural similarities between the internal ribosome entry sites of hepatitis C virus and porcine teschovirus, a picornavirus. J. Virol. 78:4487-4497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reeder, J., and R. Giegerich. 2004. Design, implementation and evaluation of a practical pseudoknot folding algorithm based on thermodynamics. BMC Bioinformatics 5:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reynolds, J. E., A. Kaminski, H. J. Kettinen, K. Grace, B. E. Clarke, A. R. Carroll, D. J. Rowlands, and R. J. Jackson. 1995. Unique features of internal initiation of hepatitis C virus RNA translation. EMBO J. 14:6010-6020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rogozin, I. B., A. V. Kochetov, F. A. Kondrashov, E. V. Koonin, and L. Milanesi. 2001. Presence of ATG triplets in 5′ untranslated regions of eukaryotic cDNAs correlates with a “weak” context of the start codon. Bioinformatics 17:890-900. [DOI] [PubMed] [Google Scholar]
- 44.Ronfort, C., B. S. De, V. Sandrin, J. L. Darlix, and T. Ohlmann. 2004. Characterization of two distinct RNA domains that regulate translation of the Drosophila gypsy retroelement. RNA 10:504-515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schneider, R., and M. Kozak. 2001. New ways of initiating translation in eukaryotes. Mol. Cell. Biol. 21:8238-8246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stoneley, M., F. E. Paulin, J. P. Le Quesne, S. A. Chappell, and A. E. Willis. 1998. C-Myc 5′ untranslated region contains an internal ribosome entry segment. Oncogene 16:423-428. [DOI] [PubMed] [Google Scholar]
- 47.Stoneley, M., T. Subkhankulova, J. P. C. Le Quesne, M. J. Coldwell, C. L. Jopling, G. J. Belsham, and A. E. Willis. 2000. Analysis of the c-myc IRES: a potential role for cell-type specific trans-acting factors and the nuclear compartment. Nucleic Acids Res. 28:687-694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stoneley, M., and A. E. Willis. 2004. Cellular internal ribosome entry segments: structures, trans-acting factors and regulation of gene expression. Oncogene 23:3200-3207. [DOI] [PubMed] [Google Scholar]
- 49.Svitkin, Y. V., B. Herdy, M. Costa-Mattioli, A. C. Gingras, B. Raught, and N. Sonenberg. 2005. Eukaryotic translation initiation factor 4E availability controls the switch between cap-dependent and internal ribosomal entry site-mediated translation. Mol. Cell. Biol. 25:10556-10565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Svitkin, Y. V., A. Pause, A. Haghighat, S. Pyronnet, G. Witherell, G. J. Belsham, and N. Sonenberg. 2001. The requirement for eukaryotic initiation factor 4A (elF4A) in translation is in direct proportion to the degree of mRNA 5′ secondary structure. RNA 7:382-394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Swergold, G. D. 1990. Identification, characterization, and cell specificity of a human LINE-1 promoter. Mol. Cell. Biol. 10:6718-6729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tchenio, T., J. F. Casella, and T. Heidmann. 2000. Members of the SRY family regulate the human LINE retrotransposons. Nucleic Acids Res. 28:411-415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Terenin, I. M., S. E. Dmitriev, D. E. Andreev, E. Royall, G. J. Belsham, L. O. Roberts, and I. N. Shatsky. 2005. A cross-kingdom internal ribosome entry site reveals a simplified mode of internal ribosome entry. Mol. Cell. Biol. 25:7879-7888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Thoma, C., G. Bergamini, B. Galy, P. Hundsdoerfer, and M. W. Hentze. 2004. Enhancement of IRES-mediated translation of the c-myc and BiP mRNAs by the poly(A) tail is independent of intact eIF4G and PABP. Mol. Cell 15:925-935. [DOI] [PubMed] [Google Scholar]
- 55.van der Velden, A. W., K. van Nierop, H. O. Voorma, and A. A. Thomas. 2002. Ribosomal scanning on the highly structured insulin-like growth factor II-leader 1. Int. J. Biochem. Cell. Biol. 34:286-297. [DOI] [PubMed] [Google Scholar]
- 56.Van Eden, M. E., M. P. Byrd, K. W. Sherrill, and R. E. Lloyd. 2004. Demonstrating internal ribosome entry sites in eukaryotic mRNAs using stringent RNA test procedures. RNA 10:720-730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang, X. C., J. Yang, W. Huang, L. He, J. T. Yu, Q. S. Lin, W. Li, and H. M. Zhou. 2002. Effects of removal of the N-terminal amino acid residues on the activity and conformation of firefly luciferase. Int. J. Biochem. Cell. Biol. 34:983-991. [DOI] [PubMed] [Google Scholar]
- 58.Wang, Z., and M. Kiledjian. 2001. Functional link between the mammalian exosome and mRNA decapping. Cell 107:751-762. [DOI] [PubMed] [Google Scholar]
- 59.Wang, Z., M. Weaver, and N. S. Magnuson. 2005. Cryptic promoter activity in the DNA sequence corresponding to the pim-1 5′-UTR. Nucleic Acids Res. 33:2248-2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wei, W., N. Gilbert, S. L. Ooi, J. F. Lawler, E. M. Ostertag, H. H. Kazazian, J. D. Boeke, and J. V. Moran. 2001. Human L1 retrotransposition: cis preference versus trans complementation. Mol. Cell. Biol. 21:1429-1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Woolaway, K. E., K. Lazaridis, G. J. Belsham, M. J. Carter, and L. O. Roberts. 2001. The 5′ untranslated region of Rhopalosiphum padi virus contains an internal ribosome entry site which functions efficiently in mammalian, plant, and insect translation systems. J. Virol. 75:10244-10249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yamashita, R., Y. Suzuki, K. Nakai, and S. Sugano. 2003. Small open reading frames in 5′ untranslated regions of mRnas. C. R. Biol. 326:987-991. [DOI] [PubMed] [Google Scholar]
- 63.Yueh, A., and R. J. Schneider. 2000. Translation by ribosome shunting on adenovirus and hsp70 mRNAs facilitated by complementarity to 18S rRNA. Genes Dev. 14:414-421. [PMC free article] [PubMed] [Google Scholar]
- 64.Zuker, M. 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 31:3406-3415. [DOI] [PMC free article] [PubMed] [Google Scholar]