

Abstract
BACKGROUND
Timothy syndrome is a multisystem disorder associated with QT interval prolongation and ventricular cardiac arrhythmias. The syndrome has been linked to mutations in CaV1.2 resulting in gain of function of the L-type calcium current (ICa,L). Ranolazine is an antianginal agent shown to exert an antiarrhythmic effect in experimental models of long QT syndrome.
OBJECTIVE
The purpose of this study was to develop and characterize an experimental model of Timothy syndrome by using BayK8644 to mimic the gain of function of ICa,L and to examine the effects of ranolazine.
METHODS
Action potentials from epicardial and M regions and a pseudo-electrocardiogram (ECG) were simultaneously recorded from coronary-perfused left ventricular wedge preparations, before and after addition of BayK8644 (1 μM).
RESULTS
BayK8644 preferentially prolonged action potential duration of the M cell, leading to prolongation of the QT interval and an increase in transmural dispersion of repolarization (from 44.3 ± 7 ms to 86.5 ± 25 ms). Stimulation at cycle lengths of 250 –500 ms led to ST-T wave alternans due to alternation of the plateau voltage of the M cell action potential as well as development of delayed afterdepolarizations in epicardial and M cell action potentials. Ventricular extrasystoles and tachycardia (monomorphic, bidirectional, or torsades de pointes) developed spontaneously or after rapid pacing. Peak and late INa were unaffected by BayK8644. Clinically relevant concentrations of ranolazine (10 μM) suppressed all actions of BayK8644.
CONCLUSION
A left ventricular wedge model of long QT syndrome created by augmentation of ICa,L recapitulates the ECG and arrhythmic manifestations of Timothy syndrome, which can be suppressed by ranolazine.
Keywords: Long QT syndrome, Sudden cardiac death, Arrhythmias, Ion channelopathy, Syndactyly, Left ventricle, Inherited syndrome
Introduction
Timothy syndrome, also referred to as syndactyly-associated long QT syndrome (LQTS) or LQT8, is a multisystem disorder characterized by developmental defects causing dysmorphic facial features including round face, flat nasal bridge, receding upper jaw, thin upper lip, and webbing of the toes and fingers (syndactyly).1 The disorder also is associated with prolongation of the QT interval, development of ventricular arrhythmias, and sudden cardiac death. The syndrome has recently been linked to a missense mutation in CaV1.2, which encodes for the α subunit of the L-type calcium channel, resulting in a gain of function of 1 the L-type calcium current (ICa,L).1
Calcium loading has been shown to contribute to the development of both the trigger [early afterdepolarizations [EADs] and delayed afterdepolarizations (DADs)] and substrate (transmural dispersion of repolarization [TDR]) for torsades de pointes (TdP). Thus, the gain of function in ICa,L is expected to be associated with a high risk for TdP in this form of LQTS.
Ranolazine is a novel antianginal agent capable of producing anti-ischemic effects at plasma concentrations of approximately 1.5–8 μM with minimal or no changes in heart rate or blood pressure.2 In addition to its anti-ischemic effects, ranolazine has been shown to be effective in suppressing arrhythmogenesis in experimental models of LQTS, particularly LQT2 and LQT3.3–5 The antiarrhythmic efficacy of ranolazine has been attributed to its potent block of late sodium channel current (late INa).4 Inhibition of late INa lessens the prolongation of the action potential (AP) of the M cell, the cell type in which late INa is most prominent, thus limiting the increase in TDR and the development of EADs.
In the present study, we used the calcium channel agonist BayK8644 to mimic the gain of function of ICa,L in an attempt to create an experimental model of Timothy syndrome and to elucidate the cellular basis for the ECG features and arrhythmias responsible for sudden cardiac death in this variant of LQTS. We also tested the hypothesis that ranolazine can effectively suppress the arrhythmias observed in the BayK8644 model of Timothy syndrome.
Methods
Dogs weighing 20–35 kg were anticoagulated with heparin (180 IU/kg) and anesthetized with pentobarbital (35 mg/kg IV). The chest was opened via left thoracotomy, and the heart was excised and placed in a cold cardioplegic solution ([K+]o = 8 mmol/L, 4°C). Fourteen animals were used in this study. Nine of 14 wedges were included in the study. Five wedge preparations displayed persistent ST-segment depression indicating the presence of ischemia somewhere in the preparation and were discarded. A total of nine left ventricular (LV) wedge preparations were exposed to BayK8644 to create a model of Timothy syndrome. All protocols were in conformance with guidelines established by the Institutional Animal Care and Use Committee.
Arterially perfused canine LV wedge preparations
Transmural LV wedges with dimensions of approximately 12 mm × 35 mm × 12 mm were dissected from the mid-to-basal anterior region of the LV wall, and a diagonal branch of the left anterior descending coronary artery was cannulated to deliver the perfusate (Tyrode’s solution). The composition of the Tyrode’s solution was as follows (in mM): NaCl 129, KCl 2 or 4, NaH2PO4 0.9, NaHCO3 20, CaCl2 1.8, MgSO4 0.5, and D-glucose 5.5. Intracellular Aps were recorded from epicardial and subendocardial M cell regions using floating microelectrodes. A transmural pseudo-electrocardiogram (ECG) was recorded using two Ag/AgCl half cells (2 mm diameter × 4 mm) placed approximately 1 cm from the epicardial (+) and endocardial (−) surfaces of the preparation and along the same axis as the intracellular recordings. Four intramural unipolar electrograms were recorded using stainless steel wires (120-μm diameter). Ventricular wedges were allowed to equilibrate in the chamber for 2 hours while pacing was performed at basic cycle length (BCL) of 2,000 ms using silver bipolar electrodes contacting the endocardial surface. Perfusion pressure was maintained at 40–50 mmHg and temperature at 37° ± 0.5°C. The preparations were fully immersed in the extracellular solution throughout the course of the experiment.
Epicardial APs were recorded from the epicardial surface of the wedge preparation, and the reported M cell is the one with the longest AP obtained along the same axis as the ECG electrodes. Transmural dispersion of repolarization was defined as the difference between the longest and the shortest repolarization times [activation time plus action potential duration (APD) measured at 90% repolarization (APD90)] of intracellular APs recorded across the wall (typically M cell minus epicardial cell repolarization time). QT interval was defined as the time interval between QRS onset and the point at which the line of maximal downslope of the T wave crossed the isoelectric line.
Canine LV myocyte dissociation
Myocytes were isolated by enzymatic dissociation.6 Adult mongrel dogs of either sex (≥1 year old, weight 20–25 kg) were anesthetized with sodium pentobarbital (35 mg/kg IV), and their hearts were quickly removed and placed in HEPES-buffered Tyrode’s solution containing 2 mM CaCl2. All procedures followed Institutional Animal Care and Use Committee guidelines. The left descending circumflex artery was cannulated and flushed with Ca-free HEPES-buffered Tyrode’s solution supplemented with 0.1% bovine serum albumin (fraction V, Sigma-Aldrich Co, St. Louis MO) and gassed with 100% O2 for 5 minutes at a rate of 12 mL/min. Perfusion was then switched to Tyrode’s solution containing 1 mg/ml bovine serum albumin and 0.1 mg/ml collagenase (CLS 2, Worthington, Lakewood, NJ, USA) for 5–11 minutes at 37°C (100% O2, with recirculation). Following perfusion, thin slices of tissues were dissected from the midmyocardium using a dermatome. Midmyocardial cells were stored in Tyrode’s solution (see following) containing 0.5 mM Ca2+ and 1.5% bovine serum albumin at room temperature for later use. HEPES-buffered Tyrode’s solution contained the following (in mM): NaCl 145, KCl 4, MgCl2 1, HEPES 10, and glucose 10; pH adjusted with NaOH to 7.4.
Whole-cell patch clamp experiments
APs were measured using the perforated patch technique.7 Experiments were performed on cells that had been allowed to adhere to the polylysine-coated floor of a 0.5-mL chamber mounted in a stage heater (model PDMI-2, Harvard Apparatus, Holliston, MA, USA) on a Nikon Eclipse microscope. The chamber was perfused with Tyrode’s solution at 37°C at a rate of 2–3 mL/min before and during gigaseal formation, after which the appropriate experimental solution replaced the Tyrode’s solution. APs were recorded using a Multiclamp 700A amplifier with a CV-7A head stage (Axon Instruments–Molecular Devices Corp., Union City, CA, USA). Stimulations were delivered and data acquired using a DigiData 1322A computer interface and the pClamp 9 program suite (Axon Instruments). Data were acquired at 10 kHz and filtered at 2 kHz.
Cells were stimulated with 3-ms square current pulses of 2.5–2.8 nA. Cells were rested for 15 seconds before evoking a train of 30 APs at 800-ms BCL, followed immediately by five APs at a faster rate, picked to produce stable DADs or alternans of APD in the presence of 1 μM BayK8644. In eight cells in which either DADs >2 mV or alternans of APD were observed, this BCL ranged from 290–450 ms. Once these rates were established for an individual mid-myocardial myocyte, the protocol was repeated in the presence of 1 μM BayK8644 and 10 μM ranolazine.
Voltage clamp
Whole-cell currents were measured using the ruptured membrane patch technique. Experiments were performed on cells that had been allowed to adhere to the polylysine- coated floor of a 0.5-mL chamber mounted in a stage heater (model PDMI-2, Harvard Apparatus) on a Nikon Eclipse microscope. The cells were superfused with Tyrode’s solution at 37°C at a rate of 2–3 mL/min before and during gig seal formation, after which the appropriate experimental solution replaced the Tyrode’s solution. Currents were recorded using a Multiclamp 700A amplifier with a CV-7A head stage (Axon Instruments–Molecular Devices Corp.). Command voltages were delivered and data acquired using a DigiData 1322A computer interface and the pClamp 9 program suite (using Axon Instruments). Data were acquired at 10 kHz and filtered at 2 kHz. Cells were held at −80 mV before evoking 300-ms pulses to voltages between −60 and −20 mV in 10-mV increments at a rate of 0.2 Hz.
BayK8644, in addition to increasing ICa,L,has been shown to augment the inward sodium current in neonatal rat myocytes.8 Because this action of BayK8644 could contribute to APD prolongation and induction of EADs, we examined the effects of BayK8644 on the peak and late INa. Late sodium current (late INa) was quantified as the current blocked by 10 μM tetrodotoxin. Currents during the first 5 ms of the activating pulse were disregarded to reduce contamination from components of the fast sodium current, and the integrated current from 5 ms to the end of the 300-ms test pulse is reported. Decay of the whole-cell current from 5 ms to the end of the 300-ms test pulse was fit to a double exponential. External solution contained the following (mM): CaCl2 2, glucose 10, MgCl2 1, NaCl 140, HEPES 10, pH to 7.4 with NaOH. Internal solution contained (in mM): MgCl2 1, NaCl 10, CsCl 10, Cs-aspartate 130, HEPES 10, EGTA 5, and MgATP 5; pH adjusted to 7.1 with CsOH.
Peak INa was recorded in reduced external sodium concentration. In order to delineate the components of the fast sodium current, the decay of the whole-cell current from 1 to 80 ms was fit to a double exponential. External solution contained the following (in mM): CaCl2 2, glucose 10, MgCl2 1, NaCl 20, N-methyl-D-glucamine 120, and HEPES 10; pH adjusted to 7.4 with HCl. Internal solution contained the following (in mM): MgCl2 1, NaCl 5, Cs-aspartate 145, HEPES 10, EGTA 5, and MgATP 5; pH adjusted to 7.1 with CsOH.
Drugs
BayK8644 was prepared as a 1 mM stock in 100 % Dimethyl Sulfoxide (DMSO) and diluted to 1 μM in external solution. Ranolazine was prepared as a 10 mM stock in water and diluted to 10 μM in external solution.
Statistical analysis
Statistical analysis was performed using one-way repeated measures analysis of variance (ANOVA) followed by Bon-ferroni test. Mean values were considered to be different when P <.05.
Results
The calcium channel agonist BayK8644 (1 μM, 60 minutes of exposure) produced an increase in APD of epicardial (Epi) and M cells in the coronary-perfused LV wedge preparations (Figure 1). The preferential prolongation of the M cell AP induced by BayK8644 led to QT prolongation and an increase in TDR. Also noteworthy is the more prominent plateau and rectangular form of the M cell AP. Ranolazine (10 μM, 30 minutes of exposure) in the continued presence of BayK8644 (1 μM) caused little change in Epi AP but abbreviated the M cell AP, more at 50% repolarization (APD50) than at 90% repolarization (APD90; Figure 1C). This change resulted in a reduction in the prominence of the plateau as well as a reduction in T-wave amplitude and TDR (from 74 to 54 ms at BCL = 2,000 ms). The QT interval was slightly decreased due to the small effect of ranolazine in shortening APD90 of the M cell in this experiment. Figures 1D and E show the rate dependence of the QT interval and TDR in the absence and presence of ranolazine. Ranolazine reduced the BayK8644 –induced increase in TDR but only slightly diminished the increase in QT interval. This effect was observed at slow rates (characteristic of patients affected with Timothy syndrome) as well as more rapid rates (BCL 500 –1,000 ms). Ranolazine induced a small decrease in QT interval (2–13 ms) in four of five experiments and a small increase (9 ms) in one experiment. Composite data of the effects of ranolazine on BayK844-induced QT and TDR prolongation are given in Table 1. In five preparations, ranolazine significantly reduced TDR from 88.4 ms ± 28 to 56.6 ± 12 ms, whereas QT intervals remained relatively unchanged (359.2 ± 18 ms vs 359.8 ± 16 ms).
Figure 1. BayK8644–induced QT prolongation in a left ventricular canine wedge preparation and the effect of ranolazine.
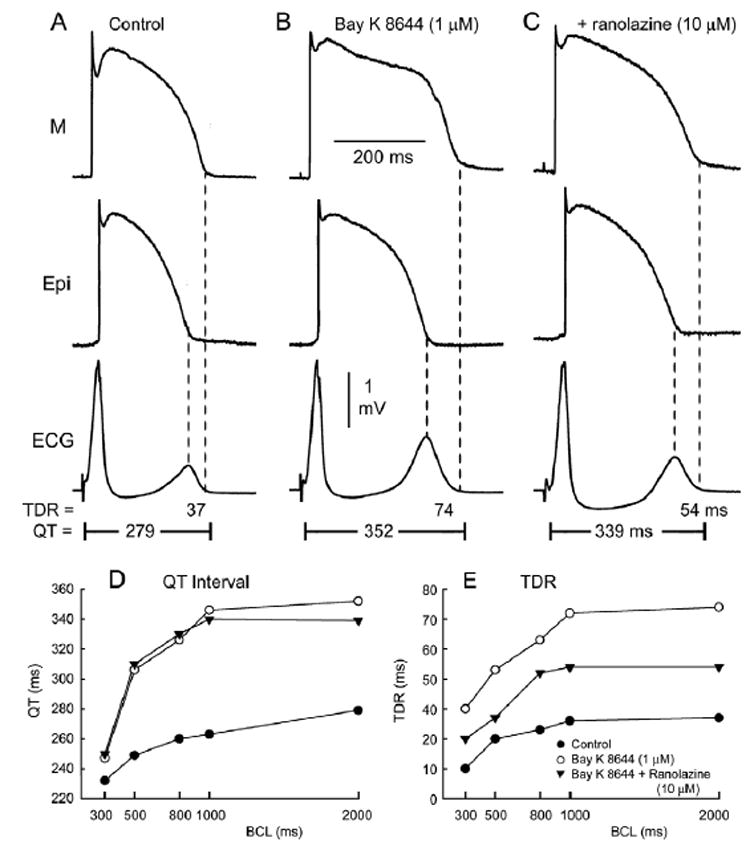
A–C: Action potentials recorded from M and epicardial (Epi) cells together with a transmural ECG. BayK8644 (1 μM, 60 minutes of exposure) produces preferential prolongation of the M cell action potential [M cell: 250 to 325 ms (29.6%); Epi: 200 to 237 ms (18.5%)], leading to prolongation of the QT interval and increase in transmural dispersion of repolarization (TDR). Ranolazine (10 μM, 30 minutes of exposure) in the continued presence of BayK8644 (1 μM) causes little changes in the Epi action potential but abbreviates the duration of the M cell action potential, especially at the plateau level, resulting in a decrease in the amplitude of the T wave and the magnitude of TDR (from 74 to 54 ms). Basic cycle length = 2,000 ms. D, E: Rate dependence of the QT interval and TDR in the absence of presence of BayK8644 (1 μM), with and without ranolazine (10 μM).
Table 1.
Effects of ranolazine (10 μM) on BayK8644 μ M)-induced increase in QT interval and transmural dispersion of repolarization in canine left ventricular wedge prepartions (n = 5)
| Control | BayK8644 | BayK8644 + ranolazine | |
|---|---|---|---|
| QT interval (ms) | 298.8 ± 19 | 359.2 ± 18* | 361.8 ± 1* |
| Transmural dispersion of repolarization (ms) | 41.8 ± 7 | 81.4 ± 26* | 53.4 ± 9*† |
Basic cycle length = 2,000 ms.
p <.05 vs control.
P <.05 vs BayK8644.
BayK8644-induced triggered activity and ventricular tachycardia in LV wedge preparations and its suppression by ranolazine
In LV wedge preparations, BayK8644 (1 μM, 60 minutes of exposure) frequently caused extrasystoles and accelerated idioventricular rhythms (Figure 2A) or monomorphic ventricular tachycardia (VT; Figure 2B). BayK8644–induced triggered activity was observed in five of five experiments after a train of 10 stimuli at BCL = 500, 400, and 300 ms, giving rise to VT lasting 20–80 seconds. This activity was not observed during basic drive at relatively slow rates (BCL ≥800 ms). Ranolazine (10 μM, 30 minutes of exposure) in the continued presence of BayK8644 (1 μM) abolished BayK8644–induced triggered activity and VT (4/4 experiments; Figure 3).
Figure 2.
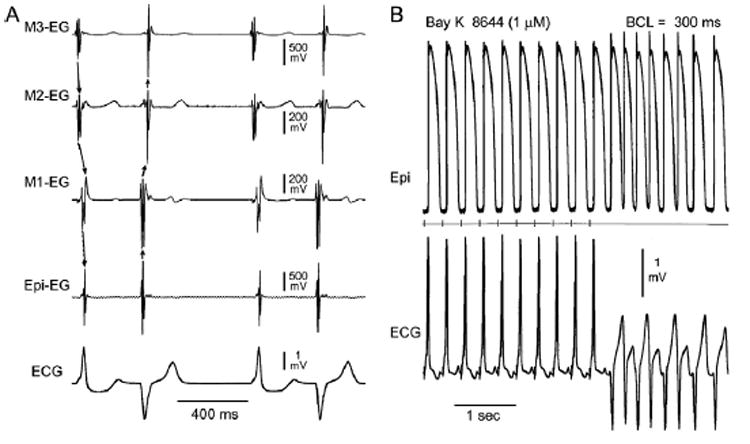
A: BayK8644–induced bidirectional idioventricular rhythm in a left ventricular wedge preparation. Shown are electrograms from epicardial (Epi), subepicardial (M1), midmyocardial (M2), and subendocardial (M3) sites, together with a transmural ECG. BayK8644 (1 μM, 60 minutes of exposure) induces spontaneous bidirectional idioventricular rhythm with ectopic activity alternating between epicardial and endocardial sites of origin. B: BayK8644–induced ventricular tachycardia (VT) of subepicardial origin. Shown are intracellular epicardial (Epi) action potentials and an ECG simultaneously recorded from a left ventricular wedge preparation after 60 minutes of exposure to 1 μM BayK8644. The middle trace is a stimulus marker. VT originating from a subepicardial site developed following a train of 10 stimuli at a basic cycle length (BCL) of 300 ms. T-wave alternans is apparent in stimulated as well as ectopic beats.
Figure 3. BayK8644–induced ventricular tachycardia (VT) of subendocardial origin and suppression by ranolazine.
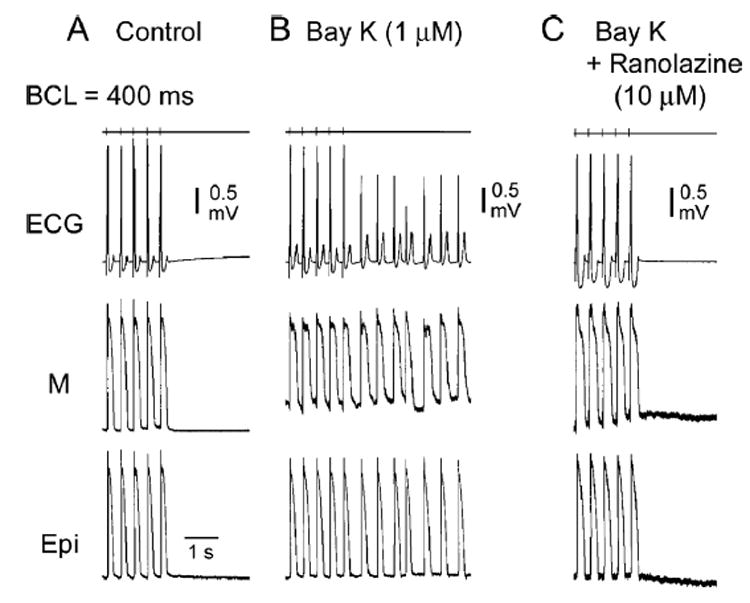
Shown are ECG and intracellular epicardial (Epi) and M cell action potentials simultaneously recorded from a left ventricular wedge preparation before (A) and after 60 minutes of exposure to 1 μM BayK8644 (B) and 20 minutes after the addition of ranolazine (10 μM) in the continued presence of BayK8644. The top trace is a stimulus marker. VT originating from a subendocardial site developed following a train of 10 stimuli at a basic cycle length of 400 ms. T-wave alternans is apparent in the stimulated ectopic beats following exposure to BayK8644, but not in control or after ranolazine.
BayK8644-induced TdP and its suppression by ranolazine
BayK8644 induced polymorphic VT resembling TdP under conditions of markedly augmented TDR. TdP was observed in two of nine preparations after exposure to BayK8644. In the example shown in Figure 4, a bigeminal rhythm developed, giving rise to a short-long-short sequence terminating in a closely coupled spontaneous extrasystole, which precipitates TdP. Tpeak-Tend, an index of TDR, was 126 ms. Ranolazine (10 μM, 30 minutes of exposure) suppressed TdP (Figure 5) in two of two preparations in which TdP was observed.
Figure 4. BayK8644 (1 μM, 60 minutes of exposure)-induced torsades de pointes in a left ventricular wedge preparation.
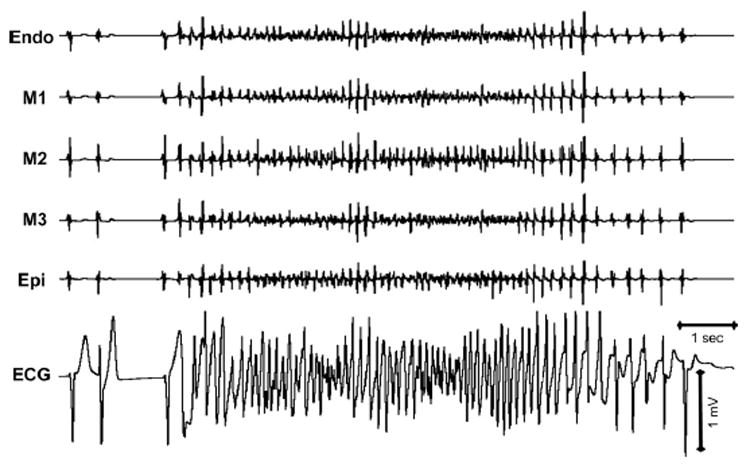
Electrograms recorded from endocardial (Endo), midmyocardial (M1–M3), and epicardial (Epi) sites together with transmural ECG. The episode of torsades de pointes that lasted 10 seconds terminates after progressive slowing.
Figure 5. Ranolazine suppression of BayK8644–induced torsades de pointes in a left ventricular wedge preparation.
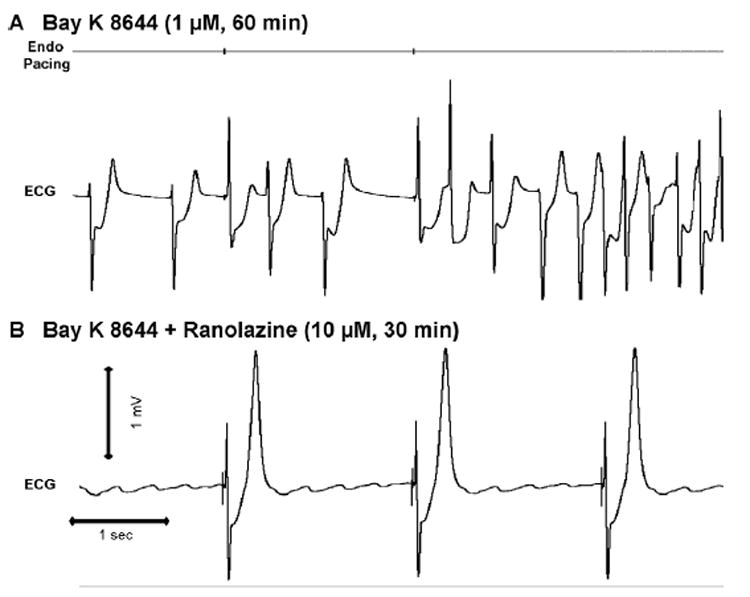
A: BayK8644 (1 μM, 60 minutes of exposure) induced spontaneous polymorphic ventricular tachycardia resembling torsades de pointes. B: Ranolazine (1 μM, 30 minutes of exposure) in the continued presence of BayK8644 abolishes ectopy and episodes of torsades de pointes.
BayK8644-induced ST-T alternans and its suppression by ranolazine
At relatively fast stimulation rates (BCL ≤500 ms), BayK8644 (1 μM, 60 minutes of exposure) induced marked ST-T wave alternans on the ECG of the wedge preparation (Figure 6). ST-segment alternans was largely due to alternation of plateau voltage and duration of the M cell AP. Ranolazine (10 μM) eliminated ST-T alternans in four of four preparations in which this activity was observed.
Figure 6. BayK8644–induced ST-T wave alternans in a left ventricular wedge preparation and its suppression by ranolazine.
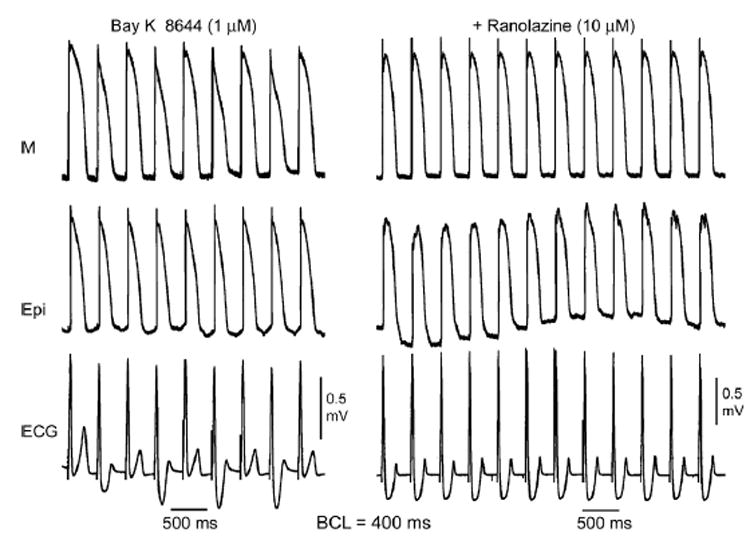
Traces were simultaneously recorded from M region (upper trace) and epicardial (middle trace) sites, together with a transmural ECG (lower trace). Left: BayK8644 (1 μM, 60 minutes of exposure) induces marked ST-T wave alternans largely due to alternation of plateau voltage and action potential duration of the M cell. Right: Ranolazine (10 μM, 30 minutes of exposure) in the continued presence of BayK8644 abolishes ST-T wave alternans and alternation of M cell action potential duration. Basic cycle length (BCL) = 400 ms.
BayK8644-induced DADs in LV wedge preparations
DADs were recorded in epicardial as well as subendocardial M region of LV wedge preparations pretreated with BayK8644 and paced at BCL ranging between 250 and 500 ms. Distinct DADs were observed at discrete sites uncovered only after careful mapping of the wedge preparations. Figure 7A shows an example of DADs simultaneously recorded form both surface epicardial and subendocardial M cells in an LV wedge preparation stimulated at BCL = 300 ms. The ST-segment alternans observed at this stimulation rate once again is associated with alternans of the plateau of the M cell AP.
Figure 7. BayK8644 (1 μM)-induced delayed afterdepolarizations (DADs) and its suppression by ranolazine.
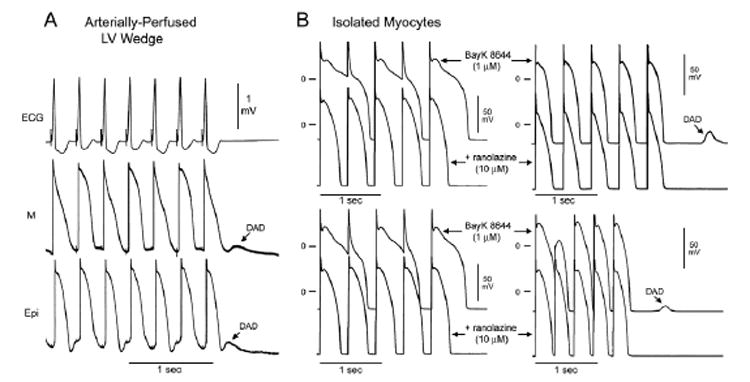
A: DADs simultaneously recorded from epicardial and subendocardial M cells in a left ventricular (LV), arterially perfused wedge preparation stimulated at basic cycle length (BCL) = 300 ms. Alternans of ST segment on the ECG correlates with alternans of the plateau of the M cell. B: BayK8644–induced action potential duration (APD) alternans and DADs in isolated myocytes (M cells). Left: Two cells in which pacing at BCL = 450 ms in the presence of 1 μM BayK8644 caused incomplete repolarization and alternans of APD. Addition of 10 μM ranolazine reduced duration and eliminates APD alternans in these cells. Right: Two cells in which rapid pacing in the presence of 1 μM BayK8644 caused DADs, which were abolished by 10 μM ranolazine.
BayK8644-induced DADs and AP alternans in isolated LV myocytes and suppression of this activity by ranolazine
In the presence of 1 μM BayK8644, a change in BCL from 800 to 290–450 ms induced DADs and/or major alternans of AP morphology due to interruption of repolarization of alternate beats. Addition of 10 μM ranolazine in the continued presence of 1 μM BayK8644 eliminated the DADs in four of four M cells as well as the alternans of APD. Figure 7B shows representative traces from two cells in which APD variations present in BayK8644 alone were eliminated by addition of ranolazine (left panels) and two cells in which DADs present with BayK8644 alone were abolished by ranolazine (right panels).
Like the clinical syndrome, the manifestation of the experimental model of the Timothy syndrome was variable. Prolongation of the QT interval together with the development of triggered activity following rapid pacing and the presence of ST-T wave alternans were constant features of the model, observed in 9 of 9 preparations. Discrete DADs were observed only occasionally (3/9 preparations), EADs were observed in only 1 preparation, TdP was seen in 2 of 9 preparations, monomorphic VT in 9/9 preparations, and bidirectional VT in 3 of 9 experiments.
Earlier studies have suggested that BayK8644, in addition to its agonist actions on ICa,L, may affect INa.8 In another series of experiments, we sought to establish the effects of BayK8644 on late and peak INa in canine ventricular myocytes. Both late INa and peak INa were unaffected by BayK8644 at membrane potentials below the threshold for activation of ICa,L. Figure 8, top left panel, shows traces recorded at −40 mV in control solution, 4 minutes after addition of 1 μM BayK8644, and immediately after the addition of 10 μM tetrodotoxin in the presence of BayK8644. Summary data for late INa before and 4 minutes after addition of BayK8644 are shown in Figure 8, lower left panel. At −50 mV, integrated late INa was −24,347 ± 1,200 pA*ms in control solution and −23,239 ± 1,983 pA*ms after BayK8644 (n = 10, P = .24). Figure 8, top right panel, shows traces recorded in reduced external sodium at −40 mV before and 4 minutes after addition of 1 μM BayK8644. Summary data for peak INa before and 4 minutes after addition of BayK8644 are shown in Figure 8, lower right panel. At −50 mV, peak INa was −2,609 ± 343 pA in control solution and −2,688 ± 330 pA after BayK8644 (n = 11, P = .38).
Figure 8. Peak and late INa were unaffected by 1 μM BayK8644.
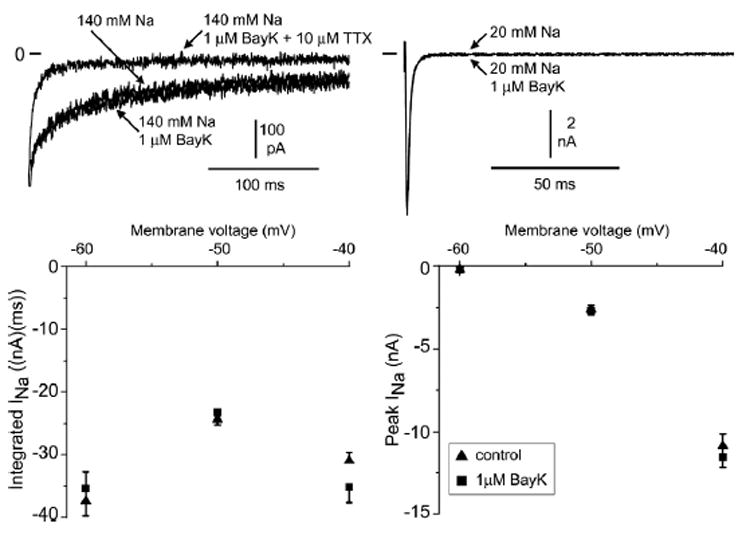
Representative traces of currents recorded at −40 mV after a depolarizing pulse step from a holding potential of −120 mV. Top left: Late INa recorded in 140 mM external sodium, after addition of 1 μM BayK8644, and in the presence of 1 μM BayK8644 and 10 μM tetrodotoxin. Top right: Peak INa recorded in 20 mM external sodium and after addition of 1 μM Bay K. Summary current–voltage plot for integrated current in 140 mM external sodium (bottom left; 7–10 cells) and peak INa in 20 mM external sodium (bottom right; 11 cells). Values are given as average ± SEM.
Decay of the whole-cell sodium current was unaffected by BayK8644. A biexponential fit was made to the decay of INa in both full and reduced external sodium at a voltage of −60 mV. In 140 mM sodium, τslow was 105 ± 15.6 ms in control solution and 110 ± 16.4 ms after BayK8644 (n = 10, P = .37). τfast was 26 ± 4.7 ms in control solution and 24 ± 3.3 ms after BayK8644 (n = 10, P = .26). In 20 mM sodium, τslow was 46 ± 5.3 ms in control solution and 45 ± 7.8 ms after BayK8644 (n = 7, P = .44). τfast was 20 ± 3.7 ms in control solution and 24 ± 6.7 ms after BayK8644 (n = 7, P = .32).
Discussion
Timothy syndrome, or syndactyly-associated LQTS or LQT8, is characterized by multisystem dysfunction, including prolongation of the QT interval, development of ventricular arrhythmias, and sudden cardiac death. Splawski et al1,9 reported linkage of Timothy syndrome to missense mutations in the CaV1.2 gene, resulting in a gain of function of the L-type calcium current (ICa,L). A second variant of the Timothy syndrome is not associated with syndactyly but is linked to a missense mutation in CaV1.2.9
Bradycardia is a characteristic feature of patients with Timothy syndrome.1,10 Zhang et al11 reported that 2:1 block resulting in a heart rate of 66 bpm developed in more than half of children affected with Timothy syndrome. Transient 2:1 block was observed in two patients in another study.12 Although bradycardia appears to be largely a consequence of 2:1 block, it also has been described in the absence of 2:1 block.
Levin et al13 reported “episodes of T-wave alternans” and, in the presence of sinus bradycardia (50–55 bpm), the development of phasic changes in T-wave configuration from upright to biphasic and finally inverted—without mention of accompanying 2:1 AV block. The dramatic QT prolongation in these patients (>LQT3 patients) suggest that the arrhythmia may be secondary to TdP episodes. Lo et al12 documented intermittent functional 2:1 AV block, frequent episodes TdP, T-wave alternans (TWA), and bi-geminy of ventricular extrasystolic or aberrantly conducted supraventricular beats. Marks et al10 reported the development of ventricular fibrillation.
In the present study, the calcium channel agonist BayK8644 was used to create a model of Timothy syndrome or LQT8. Preferential prolongation of the M cell AP was observed to lead to prolongation of the QT interval and an increase in TDR, the latter providing the substrate for development of TdP. Triggered activity in this model was found to originate from subendocardial, midmyocardial, as well as epicardial sites and appeared to be DAD mediated. The BayK8644–induced model of Timothy syndrome mimics the ECG and arrhythmic manifestations of Timothy syndrome, including prolonged QT intervals, rate-dependent ST-T wave alternans, and VT (monomorphic, bidirectional, and polymorphic).
Clinically relevant concentrations of ranolazine, an anti-anginal agent with antiarrhythmic properties, proved effective in reversing the abnormal ECG and arrhythmogenic manifestations induced by BayK8644 in this model of Timothy syndrome. Ranolazine was effective in reducing TDR and suppressing triggered activity, rate-dependent ST-T wave alternans, VT, and TdP. Ranolazine also was effective in suppressing AP alternans and DADs in canine ventricular myocytes isolated from the M region of the LV.
Models of LQTS and the effects of ranolazine
LQTS is characterized by the appearance of long QT intervals on the ECG, an atypical polymorphic VT known as TdP, and a high risk for sudden cardiac death.14 Models of the LQT1, LQT2, and LQT3 forms of the LQTS have been developed using the canine arterially perfused LV wedge preparation.15 LQT1 involves a loss of function in IKs and can be mimicked using an IKs blocker (chromanol 293B) together with a beta-adrenergic agonist (isoproterenol). LQT2 involves a defect in IKr and can be mimicked using an IKr blocker such as d-sotalol or dofetilide. LQT3 is associated with augmentation of late INa, which can be mimicked using ATX-II.16,17 In these three models of LQTS, as with the Timothy syndrome model, preferential prolongation of the M cell AP leads to an increase in the QT interval, as well as an increase in TDR, which provide the substrate for development of spontaneous as well as stimulation-induced TdP.15,18
The unique characteristics of the M cells are at the heart of LQTS. The hallmark of the M cell is the ability of its AP to prolong more than that of epicardium or endocardium in response to rate slowing.19 This feature of the M cell is due to weaker repolarizing current during phases 2 and 3 secondary to a smaller Iks and a larger late INa and INa-Ca20–22 compared with epicardial and endocardial cells. These ionic distinctions also sensitize the M cells to a variety of pharmacologic agents or genetic mutations that reduce IKr or IKs or increase late INa or ICa.
Ranolazine has been shown to be effective in reversing the arrhythmogenic substrate and in suppressing TdP in these three models of LQTS.3–5,23 By suppressing EAD and DAD activity and reducing TDR, ranolazine is capable of eliminating both the substrate (amplified dispersion of repolarization) and the trigger (EAD and DAD-induced extrasystoles) for initiation of reentry, leading to suppression of TdP. Similar actions of the drug may be responsible for its actions in the model of Timothy syndrome.
The effects of ranolazine on cardiac ion currents include inhibition of IKr, late INa, late ICa,L, peak ICa,L, INa-Ca, and IKs, with little or no inhibition of Itoor IK1.23 Multichannel inhibition, particularly the potent inhibition of late INa, has been suggested to explain the effect of IKr blockers to prolong QT without creating the substrate or trigger for development of TdP.24 This feature could contribute to suppression of EADs and reduction of spatial dispersion of repolarization, the trigger and substrate for TdP respectively.25 This is the case for amiodarone and sodium pentobarbital, as well as for high concentrations of quinidine and cisapride.26–30 The available data suggest that ranolazine fits this pharmacologic profile as well. Ranolazine has proven particularly effective in suppressing arrhythmogenesis in LQT3 models of LQTS.3,5 This effect of the drug is anticipated in light of its potent action in blocking late INa. Suppression of EAD and DAD is also secondary to the drug’s effects in reducing [Ca2+]i,which is known to modulate the manifestation of triggered activity.25,31
BayK8644, via its actions in augmenting ICa,L, contributes to cellular calcium loading and overloading. This action of the calcium agonist can lead to spontaneous release of calcium from the sarcoplasmic reticulum, promoting the development of both DAD and EAD activity32,33 and activating currents, including INa-Ca and calcium-activated chloride current ICl(Ca), which can contribute to beat-to-beat variability of APD.34,35 Both are harbingers of arrhythmia25 that may be antagonized by ranolazine.
T-wave alternans
TWA is an electrical phenomena characterized by beat-to-beat alternation of the morphology, amplitude, and/or polarity of the T wave. TWA often is associated with acquired as well as congenital LQTS. It is considered an important prognostic indicator in that it is commonly observed just before episodes of TdP. TWA as well as ST-T segment alternans has been described clinically in Timothy syndrome36 as well as in the wedge model of Timothy syndrome (Figures 6 and 7). ST-segment alternans is due largely to beat-to-beat alternation of plateau voltage and APD of the M cell. Previous studies on the wedge preparation have demonstrated that TWA observed at rapid rates under long QT conditions (LQT3 model) is due to alternation of M cell APD and that beat-to-beat alternans of [Ca2+] appears to be the underlying mechanism.37,38 Ranolazine has been shown to suppress TWA and TdP in this model of LQT3 (Fish and Antzelevitch, unpublished observations), as it does in the present model of Timothy syndrome (LQT8).
These findings suggest that arrhythmogenesis under long QT conditions may be suppressed via direct inhibition of late INa , as well as by preventing [Ca2+]i overload via sodium/calcium exchange as an indirect effect of late INa inhibition. This combined action of ranolazine may explain its effectiveness in our model of Timothy syndrome, where a gain of function in ICa,L is responsible for arrhythmogenesis. Ranolazine suppresses both the substrate (dispersion of repolarization) and the potential triggers (DADs) for development of TdP, with minimal effects on QT interval.
Of note, the calcium channel blocker verapamil has recently been shown to dramatically reduce the incidence of TdP in a patient with Timothy syndrome without significantly reducing the QT interval,39 thus paralleling the effects of ranolazine in the wedge preparation.
Our data suggest that ranolazine is a candidate for further investigation as a therapeutic agent in Timothy syndrome and provide further support for a therapeutic role of late sodium channel blockers in preventing or treating arrhythmias associated with all forms of LQTS.
Specificity of BayK8644 in augmenting ICa,L
A study by Yatani et al8 suggested that, in addition to ICa,L agonist activity, BayK8644 exerts an action on INa. The voltage clamp data illustrate that in the M cells studied, BayK8644 does not affect either late INaor peak INa at potentials below the threshold for activation of ICa,L. These data are different from those of Yatani et al, who reported that BayK8644 causes an increase in sodium current at these voltages in neonatal rat myocytes. The difference between our results and those of Yatani et al may be related to (1) the methodology used (i.e., substitution of calcium with cobalt in the bath solution may be responsible for the effect of BayK8644 on sodium currents observed by Yatani et al), (2) species differences, and (3) voltage clamp conditions. Our findings are consistent with the interpretation that the effects of BayK8644 in the present study are due to an increase in ICa,L rather than an increase in late INa.
Acknowledgments
Supported by Grant HL47678 from the National Heart, Lung, and Blood Institute to Dr. Antzelevitch and grants from CV Therapeutics and NYS and Florida Grand Lodges F. & A.M.
References
- 1.Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, Napolitano C, Schwartz PJ, Joseph RM, Condouris K, Tager-Flusberg H, Priori SG, Sanguinetti MC, Keating MT. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell. 2004;1:19–31. doi: 10.1016/j.cell.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 2.Chaitman BR, Skettino SL, Parker JO, Hanley P, Meluzin J, Kuch J, Pepine CJ, Wang W, Nelson JJ, Hebert DA, Wolff AA. Anti-ischemic effects and long-term survival during ranolazine monotherapy in patients with chronic severe angina. J Am Coll Cardiol. 2004;8:1375–1382. doi: 10.1016/j.jacc.2003.11.045. [DOI] [PubMed] [Google Scholar]
- 3.Wu L, Shryock JC, Song Y, Li Y, Antzelevitch C, Belardinelli L. Antiarrhythmic effects of ranolazine in a guinea pig in vitro model of long-QT syndrome. J Pharmacol Exp Ther. 2004;2:599–605. doi: 10.1124/jpet.104.066100. [DOI] [PubMed] [Google Scholar]
- 4.Antzelevitch C, Belardinelli L, Wu L, Fraser H, Zygmunt AC, Burashnikov A, Di Diego JM, Fish JM, Cordeiro JM, Goodrow RJ, Scornik FS, Perez GJ. Electrophysiologic properties and antiarrhythmic actions of a novel anti-anginal agent. J Cardiovasc Pharmacol Ther. 2004:S65–S83. doi: 10.1177/107424840400900106. [DOI] [PubMed] [Google Scholar]
- 5.Song Y, Shryock JC, Wu L, Belardinelli L. Antagonism by ranolazine of the pro-arrhythmic effects of increasing late INa in guinea pig ventricular myocytes. J Cardiovasc Pharmacol. 2004:192–199. doi: 10.1097/00005344-200408000-00008. [DOI] [PubMed] [Google Scholar]
- 6.Liu DW, Gintant GA, Antzelevitch C. Ionic bases for electrophysiological distinctions among epicardial, midmyocardial, and endocardial myocytes from the free wall of the canine left ventricle. Circ Res. 1993;3:671–687. doi: 10.1161/01.res.72.3.671. [DOI] [PubMed] [Google Scholar]
- 7.Zygmunt AC, Goodrow RJ, Weigel CM. INaCa and ICl(Ca) contribute to isoproterenol-induced delayed afterdepolarizations in midmyocardial cells. Am J Physiol. 1998;6(Pt 2):H1979–H1992. doi: 10.1152/ajpheart.1998.275.6.H1979. [DOI] [PubMed] [Google Scholar]
- 8.Yatani A, Kunze DL, Brown AM. Effects of dihydropyridine calcium channel modulators on cardiac sodium channels. Am J Physiol Heart Circ Physiol. 1988;1:H140–H147. doi: 10.1152/ajpheart.1988.254.1.H140. [DOI] [PubMed] [Google Scholar]
- 9.Splawski I, Timothy KW, Decher N, Kumar P, Sachse FB, Beggs AH, Sanguinetti MC, Keating MT. Severe arrhythmia disorder caused by cardiac L-type calcium channel mutations. Proc Natl Acad Sci U S A. 2005;23:8089–8096. doi: 10.1073/pnas.0502506102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marks ML, Whisler SL, Clericuzio C, Keating MT. A new form of long QT syndrome associated with syndactyly. J Am Coll Cardiol. 1995;1:59–64. doi: 10.1016/0735-1097(94)00318-k. [DOI] [PubMed] [Google Scholar]
- 11.Zhang L, Timothy KW, Antzelevitch C, Sicouri S, Vincent GM. Cardiac L-type channel mutations caused the longest QT interval compared to the common genotypes of long QT syndrome. Circulation. 2005:II-701. [Google Scholar]
- 12.Lo ANS, Wilde AA, van EL, Blom NA. Syndactyly and long QT syndrome (Ca(V)1.2 missense mutation G406R) is associated with hypertrophic cardiomyopathy. Heart Rhythm. 2005;2:1365–1368. doi: 10.1016/j.hrthm.2005.08.024. [DOI] [PubMed] [Google Scholar]
- 13.Levin SE, Harrisberg J, Govendrageloo K, du Plessis J. Idiopathic long QT syndrome in a black infant. Cardiovasc J S Afr. 1992:144–146. [Google Scholar]
- 14.Moss AJ, Liu JE, Gottlieb SS, Locati EH, Schwartz PJ, Robinson JL. Efficacy of permanent pacing in the management of high-risk patients with long QT syndrome. Circulation. 1991:1524–1529. doi: 10.1161/01.cir.84.4.1524. [DOI] [PubMed] [Google Scholar]
- 15.Antzelevitch C, Shimizu W. Cellular mechanisms underlying the long QT syndrome. Curr Opin Cardiol. 2002;1:43–51. doi: 10.1097/00001573-200201000-00007. [DOI] [PubMed] [Google Scholar]
- 16.Shimizu W, Antzelevitch C. Sodium channel block with mexiletine is effective in reducing dispersion of repolarization and preventing torsade de pointes in LQT2 and LQT3 models of the long-QT syndrome. Circulation. 1997:2038–2047. doi: 10.1161/01.cir.96.6.2038. [DOI] [PubMed] [Google Scholar]
- 17.Ueda N, Zipes DP, Wu J. Coronary occlusion and reperfusion promote early afterdepolarizations and ventricular tachycardia in a canine tissue model of long QT3. Am J Physiol Heart Circ Physiol. 2006;2:H607–H612. doi: 10.1152/ajpheart.00699.2005. [DOI] [PubMed] [Google Scholar]
- 18.Weiss JN, Qu Z, Chen PS, Lin SF, Karagueuzian HS, Hayashi H, Garfinkel A, Karma A. The dynamics of cardiac fibrillation. Circulation. 2005;8:1232–1240. doi: 10.1161/CIRCULATIONAHA.104.529545. [DOI] [PubMed] [Google Scholar]
- 19.Sicouri S, Antzelevitch C. A subpopulation of cells with unique electrophysiological properties in the deep subepicardium of the canine ventricle. The M cell Circ Res. 1991;6:1729–1741. doi: 10.1161/01.res.68.6.1729. [DOI] [PubMed] [Google Scholar]
- 20.Zygmunt AC, Goodrow RJ, Antzelevitch C. I(NaCa) contributes to electrical heterogeneity within the canine ventricle. Am J Physiol Heart Circ Physiol. 2000;5:H1671–H1678. doi: 10.1152/ajpheart.2000.278.5.H1671. [DOI] [PubMed] [Google Scholar]
- 21.Zygmunt AC, Eddlestone GT, Thomas GP, Nesterenko VV, Antzelevitch C. Larger late sodium conductance in M cells contributes to electrical heterogeneity in canine ventricle. Am J Physiol. 2001:H689–H697. doi: 10.1152/ajpheart.2001.281.2.H689. [DOI] [PubMed] [Google Scholar]
- 22.Liu DW, Antzelevitch C. Characteristics of the delayed rectifier current (IKr and IKs) in canine ventricular epicardial, midmyocardial, and endocardial myocytes. Circ Res. 1995;3:351–365. doi: 10.1161/01.res.76.3.351. [DOI] [PubMed] [Google Scholar]
- 23.Antzelevitch C, Belardinelli L, Zygmunt AC, Burashnikov A, Di Diego JM, Fish JM, Cordeiro JM, Thomas GP. Electrophysiologic effects of ranolazine: A novel anti-anginal agent with antiarrhythmic properties. Circulation. 2004:904–910. doi: 10.1161/01.CIR.0000139333.83620.5D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Antzelevitch C. Role of transmural dispersion of repolarization in the genesis of drug-induced torsades de pointes. Heart Rhythm. 2005;2(Suppl 2):S9–S15. doi: 10.1016/j.hrthm.2004.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Belardinelli L, Antzelevitch C, Vos MA. Assessing predictors of drug-induced torsade de pointes. Trends Pharmacol Sci. 2003:619–625. doi: 10.1016/j.tips.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 26.Antzelevitch C, Shimizu W, Yan GX, Sicouri S, Weissenburger J, Nesterenko VV, Burashnikov A, Di Diego JM, Saffitz J, Thomas GP. The M cell: its contribution to the ECG and to normal and abnormal electrical function of the heart. J Cardiovasc Electrophysiol. 1999;8:1124–1152. doi: 10.1111/j.1540-8167.1999.tb00287.x. [DOI] [PubMed] [Google Scholar]
- 27.Di Diego JM, Belardinelli L, Antzelevitch C. Cisapride-induced transmural dispersion of repolarization and torsade de pointes in the canine left ventricular wedge preparation during epicardial stimulation. Circulation. 2003;8:1027–1033. doi: 10.1161/01.CIR.0000085066.05180.40. [DOI] [PubMed] [Google Scholar]
- 28.Shimizu W, McMahon B, Antzelevitch C. Sodium pentobarbital reduces transmural dispersion of repolarization and prevents torsade de pointes in models of acquired and congenital long QT syndrome. J Cardiovasc Electrophysiol. 1999;8:156–164. doi: 10.1111/j.1540-8167.1999.tb00656.x. [DOI] [PubMed] [Google Scholar]
- 29.Weissenburger J, Nesterenko VV, Antzelevitch C. Transmural heterogeneity of ventricular repolarization under baseline and long QT conditions in the canine heart in vivo: torsades de Pointes develops with halothane but not pentobarbital anesthesia. J Cardiovasc Electrophysiol. 2000;9:290–304. doi: 10.1111/j.1540-8167.2000.tb01798.x. [DOI] [PubMed] [Google Scholar]
- 30.van Opstal JM, Schoenmakers M, Verduyn SC, De Groot SH, Leunissen JD, Der Hulst FF, Molenschot MM, Wellens HJ, Vos MA. Chronic Amiodarone evokes no torsade de pointes arrhythmias despite QT lengthening in an animal model of acquired long-QT syndrome. Circulation. 2001;22:2722–2727. doi: 10.1161/hc4701.099579. [DOI] [PubMed] [Google Scholar]
- 31.Belardinelli L, Antzelevitch C, Fraser H. Inhibition of late (sustained/persistent) sodium current: a potential drug target to reduce intracellular sodium-dependent calcium overload and its detrimental effects on cardiomyocyte function. Eur Heart J Suppl. 2004;(Suppl I):i3–i7. [Google Scholar]
- 32.Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;6868:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- 33.Katra RP, Laurita KR. Cellular mechanism of calcium-mediated triggered activity in the heart. Circ Res. 2005;5:535–542. doi: 10.1161/01.RES.0000159387.00749.3c. [DOI] [PubMed] [Google Scholar]
- 34.Thomsen MB, Verduyn SC, Stengl M, Beekman JD, de Pater G, van Opstal J, Volders PG, Vos MA. Increased short-term variability of repolarization predicts d-sotalol-induced torsades de pointes in dogs. Circulation. 2004:2453–2459. doi: 10.1161/01.CIR.0000145162.64183.C8. [DOI] [PubMed] [Google Scholar]
- 35.Pogwizd SM, Schlotthauer K, Li L, Yuan W, Bers DM. Arrhythmogenesis and contractile dysfunction in heart failure: Roles of sodium-calcium exchange, inward rectifier potassium current, and residual beta-adrenergic responsiveness. Circ Res. 2001;11:1159–1167. doi: 10.1161/hh1101.091193. [DOI] [PubMed] [Google Scholar]
- 36.Joseph-Reynolds AM, Auden SM, Sobczyzk WL. Perioperative considerations in a newly described subtype of congenital long QT syndrome. Paediatr Anaesth. 1997;3:237–241. [PubMed] [Google Scholar]
- 37.Shimizu W, Antzelevitch C. Cellular and ionic basis for T-wave alternans under long QT conditions. Circulation. 1999;11:1499–1507. doi: 10.1161/01.cir.99.11.1499. [DOI] [PubMed] [Google Scholar]
- 38.Sipido KR. Understanding cardiac alternans: the answer lies in the Ca2+ store. Circ Res. 2004;5:570–572. doi: 10.1161/01.RES.0000124606.14903.6F. [DOI] [PubMed] [Google Scholar]
- 39.Jacobs A, Knight BP, McDonald KT, Burke MC. Verapamil decreases ventricular tachyarrhythmias in a patient with Timothy syndrome (LQT8) Heart Rhythm. 2006;3:967–970. doi: 10.1016/j.hrthm.2006.04.024. [DOI] [PubMed] [Google Scholar]