

Abstract
Forward genetic analysis is the most broadly applicable approach to discern gene functions. However, for some organisms like the filamentous ascomycete Neurospora crassa, genetic mapping frequently represents a limiting step in forward genetic approaches. We describe an efficient method for genetic mapping in N. crassa that makes use of a modified bulked segregant analysis and PCR-based molecular markers. This method enables mapping with progeny from a single cross, and requires only 90 PCR amplifications. Genetic distances between syntenic markers have been determined to ensure complete coverage of the genome and to allow interpolation of linkage data. As a result, most mutations should be mapped in less than one month to within 1–5 map units, a level of resolution sufficient to initiate map-based cloning efforts. This system also will facilitate analyses of recombination at a genome-wide level, and is applicable to other perfect fungi when suitable markers are available.
Keywords: Neurospora crassa, bulked segregant analysis, map-based cloning, genetic mapping, molecular marker; recombination
1. Introduction
Neurospora crassa has a long, rich history as a model organism due to its facile genetics, ease of culture and rapid growth rate (Davis and Perkins, 2002). In addition to these inherent attributes, many invaluable tools exist for genetic, molecular and biochemical analysis of N. crassa – a large collection of mutants housed at the Fungal Genetics Stock Center (McCluskey, 2003), a dense, well-ordered genetic map (Perkins, 2000), an RFLP map (Nelson and Perkins, 2000), efficient transformation (Margolin et al., 1997), a complete genome sequence (Galagan et al., 2003), commercially available DNA microarrays, and a rapidly growing number of targeted gene knock-out strains (Colot et al., 2006). While these tools are crucial for efforts to elucidate gene function, they are complementary and supplementary to novel forward genetic analysis, the primary experimental approach for a large majority of fungal researchers. However, forward genetics in N. crassa frequently is hampered by difficulties encountered in genetic mapping, which is a necessary step toward the identification of genes affected by mutation.
Mapping of mutations in N. crassa traditionally involves co-segregation analysis using phenotypic markers. In most cases multiply-marked tester strains are used to improve efficiency (Perkins, 1990; Perkins, 1991). However, new mutations often fail to show linkage to any of the markers in these strains (Perkins, 2006). When this occurs one must conduct co-segregation analysis using a series of marked strains that collectively test for linkage to regions within each arm of the seven linkage groups. In either case, even when linkage is clearly established, additional crosses are required to obtain sufficient resolution to identify the affected gene by candidate gene prediction and/or complementation. As a result the entire process requires a considerable investment of time, usually on the order of 3–12 months. For mutations that give rise to a phenotype that can be detected only via a complex screen or that require a defined genetic background, e.g. suppressor mutations, the time and effort needed for mapping is increased dramatically.
PCR-based molecular markers have been widely adopted for genetic mapping purposes (Elahi et al., 2004; Jenkins, 2003). The main advantages of molecular markers are that they seldom affect the fitness of an organism (selectively neutral) and are much more numerous than phenotypic markers. As a result, high-resolution mapping can be achieved using progeny from a single cross. On this basis, a mapping study in N. crassa could be completed in less than one month after a cross was initiated. As a first step toward this goal, Kotierk and Smith (2004) described a set of 18 PCR-based molecular markers that exploit the abundant sequence polymorphisms that exist between the laboratory standard Oak Ridge wild-type strain and the Mauriceville “exotic” wild-type strain (Kotierk and Smith, 2004; Metzenberg et al., 1984). In short, a mutant obtained in the Oak Ridge background is crossed to the Mauriceville strain and co-segregation analysis is conducted using the markers that distinguish polymorphic differences between the two parental backgrounds.
We have expanded upon the work of Kotierk and Smith (2004), and describe here a set of genetically defined molecular markers that provide complete map coverage. We also describe the use of these markers in an efficient genetic mapping strategy that employs bulked segregant analysis (Michelmore et al., 1991). Bulked segregant analysis is a widely used method to enhance the efficiency of mapping monogenic traits. Briefly, individual progeny from a single cross are pooled based on the segregating trait of interest. Within a bulk, all individuals have identical genotypes at the region related to the trait of interest (mutant or wildtype) but have random genotypes at all unlinked loci. Consequently, markers located near the region of interest will be in linkage disequilibrium and markers located further away will have a level of disequilibrium proportional to their distance. At far distances syntenic markers will display a maximum of 50% recombination, indistinguishable from unlinked loci. The primary advantage of bulked segregant analysis is that it greatly reduces the time and expense of mapping. For example, a standard co-segregation analysis using just 40 individual progeny from a segregating population and 30 different PCR-based molecular markers entails 1,200 amplifications. Alternatively, use of the same marker set but with bulked segregant analysis as described here requires only 90 amplifications, which can be accomplished in a single day using a 96-well plate.
Our mapping approach is designed to minimize effort and sample numbers, both as cost- and time-saving measures. Despite this minimalist approach, most mutations should be mapped in less than one month to within 1–5 map units, a resolution sufficient to proceed with map-based cloning efforts without the need for additional markers or analysis of large populations of individual segregating progeny.
2. Materials and Methods
2.1 Strains and culture conditions
Neurospora crassa strains used in this work included: wild type Oak Ridge 74-ORS-6a (FGSC 4200), wild type Mauriceville-1c (FGSC 2225), an albino (al-1) mutant (FGSC 3623) and where indicated, progeny from crosses between these parental strains. All strains were propagated in Vogel’s medium (Vogel, 1956) supplemented with 1% sucrose. Sexual crosses were conducted on agar plates containing Synthetic Crossing medium (Westergaard and Mitchell, 1947) supplemented with 1% sucrose.
2.2 Genomic DNA isolation
Mycelia were harvested by vacuum filtration from 1 ml stationary cultures after 2 days growth at 30°C. For DNA isolation from single cultures, mycelial pads were rinsed with water and vacuum filtered until just damp then transferred to 1.5 ml microcentrifuge tubes, frozen in liquid nitrogen and ground to a fine powder with plastic pestles. The freezing step could be omitted and mycelial pads instead lyophilized overnight. Equivalent yields and purity of genomic DNA were obtained regardless of which initial step was used, but most samples were frozen to expedite processing. To each ground sample 0.6 ml Extraction Buffer (100 mM Tris-HCl pH 8.0, 50 mM EDTA, 1% SDS) and 3 μl Proteinase K (20 mg/ml in 20 mM Tris-HCl pH 7.5, 50% glycerol) were added, mixed vigorously, and incubated at 65°C for 1 hr. When processing multiple samples the appropriate volumes of Extraction Buffer and Proteinase K solutions were combined immediately before use and added as a single reagent. After the 1 hr incubation, samples were again mixed thoroughly and 0.2 ml 7.5 M ammonium acetate was added followed by vigorous mixing then incubation on ice for 5 min. Samples were centrifuged for 3 min at 16,000 ×g, supernatants were transferred to fresh tubes, 3 μl RNase A (10 mg/ml in Tris-HCl, pH 8.0, 50% glycerol) was added, and samples were incubated at 37°C for 1 hr. Each sample was extracted once with 0.5 ml chloroform, then genomic DNA was precipitated with 0.65 ml isopropanol. DNA pellets were washed once with 70% ethanol before dissolving in 0.1 ml TE, pH 8.0. For bulked samples, 40 individual mycelial pads were combined, frozen with liquid nitrogen, ground with a mortar and pestle. DNA was then isolated as described but reagent volumes were increased 10-fold.
2.3 Primer design, PCR conditions and marker scoring
To ensure uniform amplification properties all PCR primers were designed using Web Primer (http://seq.yeastgenome.org/cgi-bin/web-primer) with the default parameters, and the output “best primers” were chosen. Primers were purchased from IDT, Inc. (Coralville, IA, USA). Criteria for selection of a primer pair for marker use included equivalent amplification efficiency and the same amplicon size when using genomic DNA isolated from either the Oak Ridge or the Mauriceville strains as PCR template. Amplicons were ideally 250–700 bp with a polymorphic restriction site located at the center.
A single PCR amplification scheme was used for all primer pairs: 50 ng genomic DNA, 0.5 μM each primer, 0.25 mM dNTPs, 1 unit Takara ExTaq polymerase (Fisher Scientific, Houston, TX, USA), and 1×ExTaq buffer were assembled in a 20 μl total volume on ice. Thermal cyclers were pre-heated to 95°C before tubes or 96-well plates were inserted. Samples were initially denatured for 3 min at 95°C then treated with 26 cycles of 15 sec at 95°C, 15 sec at 60°C, 1 min at 72°C, followed by 5 min at 72°C then stored at 4°C. The same amount of template DNA was used for analysis of individual strains and for bulked segregant analysis.
To score the genotype pattern for a given marker, 3 μl of the PCR mix was digested with 10 units of the appropriate restriction enzyme for 1.5 hr in a final volume of 20 μl then the entire volume was electrophoresed in a 1.5% agarose gel at 70 V. In all cases, the amplicon of one parental type was cleaved and the other parental type was uncleaved. The polymorphic pattern for each marker is listed in Tables 1 and 2. Restriction endonucleases were purchased from New England Biolabs (Ipswich, MA, USA).
Table 1.
Primary CAPS marker panel for bulked segregant analysis
| Marker name | Primers | Amplicon (Contig #: bp coordinates) | DNA fragment sizes (bp) | |
|---|---|---|---|---|
| Oak Ridge | Mauriceville | |||
| 1–23-MspI | CACAAGTTCACGCCTTGTCC
CTCGTTCATGGCGTAGAAGC |
7.39: 245326–245800 | 144/331 | 475 |
| 1–30-BamHI | TTCTTTCTCCGCTACTTCCGT
TCACAGAGTCCATGCCAGAAA |
7.59: 119075–119538 | 176/177 | 353 |
| 1–59-HaeIII | CACCGTCCCTAAACTCCATAA
TTGACTGTTCATCTGCCCACT |
7.3: 817717–818265 | 249/300 | 549 |
| 1–110-HaeIII | CCGTTGATCATGATGTGACA
ACAAAAAGAACGCTGCTCCC |
7.9: 362428–362948 | 228/293 | 521 |
| 1–185-TaqI | CGAGGCACATATCCGCAGA
AATAGCACCGCTCTTGCCA |
7.2: 274637–274940 | 148/156 | 304 |
| 1–226-DpnII | AATATCAATGGTCTGGCCCT
TCTCTACACTGTCAAGCACGG |
7.7: 802187–802427 | 110/131 | 241 |
| 2–36-PvuI | CTCCGGATGAGGTTGCCG
GTGCGGGCTTAACCGCTG |
7.8: 792192–793755 | 768/784 | 1552 |
| 2–60-DpnII | TCCTTCTCTACACCTCTGGCT
TGGGAGCAACGTAGAACTGA |
7.33: 287494–287790 | 297 | 148/149 |
| 2–88-TaqI | CCGACAAGCATCTGGCTCT
TTCTTCCTCGCACCCTCCTT |
7.5: 62054–62327 | 98/176 | 274 |
| 2–105-HhaI | TGCAACGAGATCCCAGACTAT
ACTGGTCCAAGGTCACCAAAT |
7.71: 66401–66681 | 132/149 | 281 |
| 3–17-AluI | CTTTGGGCGGTCAACTCCA
CAGGGCAACTTGTTTGGGC |
7.42: 202777–203079 | 96/207 | 303 |
| 3–52-EcoRI | GGGCGATGAGCAACAAATAA
ACAAAGTCTTACTGCCATGCG |
7.25: 33816–34195 | 189/191 | 380 |
| 3–102-Tsp509I | GAGCCAGAACTTGGTTGTGAT
ATTCTTCCATATTCCACCCCC |
7.67: 55700– 555988 | 152/137 | 289 |
| 4–2-MspI | CCATCCCCAAGCTTCTCAA
TGTGTGGTATCGCTTTCAACT |
7.79: 61171–61527 | 357 | 175/182 |
| 4–35-AluI | TGTCGATGGCACCCGTCT
TGAGGAGTTCGCCGTCAA |
7.18: 35189–35487 | 299 | 142/157 |
| 4–70-BamHI | TGGAGGGATTTGTGTCAAGGT
CGTATAGCTTGCCTCGTCGA |
7.53: 90510–90919 | 410 | 201/209 |
| 4–120-BglII | AATATCCTTCACCACCGTGGC
AATCCTTAAGCACCCCTTGG |
7.19: 771981–772470 | 242/248 | 490 |
| 5–27-TaqI | TCCTCTCCCTGTACTCGTCCA
CCCCCTTGTCCGTCAAGTA |
7.64: 47023–47419 | 198/199 | 397 |
| 5–98-Tsp509I | AAACCCATACCGAGGAGGA
GCGTGGTCTGTGACATCTACA |
7.11: 6101398–610423 | 113/173 | 286 |
| 5–135-HpaI | ATCCCTCCATCTAAAAGCTCA
CCCCTGATCATCGATCTCGT |
7.31: 259764–260117 | 170/184 | 354 |
| 5–175-DpnII | CCGAGCCAGGATTGTCA
ATCTGCATGTTGCCATCCGT |
7.13: 40616–40913 | 298 | 88/210 |
| 5–220-MspI | TAAGCCCAACGGCACTGT
AAAAAAGACCGTTGACGCCA |
7.48: 153273–153700 | 181/247 | 428 |
| 6–12-XhoI | GGTCCGCAGGTCTTACTTTAA
TCCGACAATGTTCAAACGCT |
7.16: 154977–155546 | 283/287 | 570 |
| 6–39-HaeIII | CCCGCTCCAAGTTCTACTCTT
GGTCTTCGGTCTGGACGTG |
7.22: 187185–187397 | 213 | 92/121 |
| 6–68-MspI | ATGTCTTGGGTGTTTGGCAT
TCCTCAAGATCGTCACTCAGC |
7.4: 334316–335039 | 724 | 216/508 |
| 6–95-KpnI | GAAAATGTCAACTTTGTTGCG
ATGAACGAATCAATGCCTCC |
7.4: 1131921–1132292 | 163/209 | 372 |
| 7–15-HaeIII | TAGCCATACCTGGTTTGAGGG
AACAGGTCCTTCCAGCGAGAT |
7.66: 11810–12098 | 289 | 125/164 |
| 7–40-DpnII | AAAGCTTGCGAGACGTCGGAT
TTAGAGCGCCAAGACACCAA |
7.21: 577678–578059 | 155/227 | 382 |
| 7–68-Tsp509I | ATGTTGCCTTTAAGCCCCA
GACCCATAGCCGCAAGGAT |
7.10: 232320–232919 | 600 | 295/305 |
| 7–94-HhaI | ATTGTCTCCTTGCATCCCGTT
CGAGGAGGAGGAGTCCAAGTA |
7.23: 348305–348727 | 68/355 | 423 |
Table 2.
Supplementary CAPS markers
| Marker name | Primers | Amplicon (Contig #: bp coordinates) | DNA fragment sizes (bp) | |
|---|---|---|---|---|
| Oak Ridge | Mauriceville | |||
| 1–85-HaeIII | TATTGATCTTCCGCTCTTTGG
AAGAAGGTCGAGTTCCTCGC |
7.6: 213347–213745 | 399 | 67/332 |
| 1–150-RsaI | CACCGTCCCTAAACTCCATAA
TTGACTGTTCATCTGCCCACT |
7.2: 1385454–1386006 | 553 | 252/301 |
| 1–184-HaeIII | GGTTATGTAGGAAGGGGGAGA
GGATGACACTGTCGTGGCTAA |
7.2: 315838–316741 | 904 | 320/584 |
| 3–76-AluI | CAAAGCATGATTGCAAGCCA
ACGGACATAGACAAGGGAAAA |
7.1: 304659–304923 | 130/135 | 265 |
| 5–55-MspI | ATTCCTCCTTCTCGTCCTCCT
TTGTCTGGTCGAACCATACCT |
7.24: 31028–31473 | 446 | 189/257 |
| 5–106-TaqI | TACGTTGATGCCGGAAGC
AGAAGGGAATCACCAACTCCA |
7.14: 465795–466056 | 262 | 62/200 |
| 5–127-Tsp509I | AGCCGAACCAGAAATACAAGA
TGCCGACTCCAAGATCAA |
7.15: 145012–145283 | 272 | 122/150 |
| 7–2-KpnI | ACTTGATAGCAGCCAAGCTCT
CGAGGAGGAGGAGTCCAAGTA |
7.78: 59602–60075 | 224/250 | 474 |
| 7–88-HindIII | TGTTTTCCGTGTTCTGGGTT
AGGAGCAAATCCAGGTTCTCA |
7.52: 168261–268752 | 244/248 | 492 |
3. Results and Discussion
3.1 Marker development
To facilitate single cross mapping, we initially intended to simply supplement the Kotierk and Smith (2004) marker collection as needed to achieve complete map coverage, and to use these markers for bulked segregant analysis to enhance efficiency. However, amplification conditions varied widely between the markers, which complicated high throughput, concurrent use. In addition, some markers were not suitable for bulked segregant analysis because the genetic contribution of only one parent was detected. To maximize efficiency we chose to utilize a single type of molecular marker that was readily amenable to bulked segregant analysis. Similarly, we defined parameters for PCR primer design to enable use of a single amplification scheme for all markers.
We chose to use cleaved amplified polymorphic sequence (CAPS) markers for our studies. A CAPS marker is a PCR-amplified sequence that corresponds to a defined genomic region and includes a polymorphic restriction endonuclease site, which allows one to distinguish the parent of origin. Because CAPS markers lack bias in the amplification step (both parental genotypes are amplified), these markers are readily amenable to bulked segregant analysis (Elahi et al., 2004; Jenkins, 2003; Michelmore et al., 1991; Vignal et al., 2002). For our purposes, CAPS markers distinguish between Oak Ridge- and Mauriceville-inherited sequences. This is the same combination of genetic backgrounds that are widely used for RFLP mapping (Metzenberg et al., 1984; Nelson and Perkins, 2000), and the use of a CAPS marker is fundamentally the same as an RFLP probe. The key advantage of CAPS markers is that analyses can be completed much more quickly and do not require an extensive series of hybridizations.
CAPS markers used in this study are listed in Tables 1 and 2. The approximate chromosomal locations of these markers relative to multiple known mutations are illustrated in Fig. 1. Markers were named based on the following convention: linkage group number – predicted genetic location in map units starting from the left telomere (Perkins, 2000; Radford and Parish, 1997) – restriction enzyme used to reveal a polymorphism. For example, the marker named 1–23-MspI denotes a marker located on Linkage Group I with a predicted map position 23 map units from the left telomere, and the restriction enzyme MspI. For clarity, marker names are abbreviated hereafter with the designated restriction enzyme omitted, e.g., 1–23. The values for the predicted genetic location serve solely as identifiers to orient the order of one marker relative to another on a chromosome; as discussed later they do not denote absolute map positions.
Fig. 1.
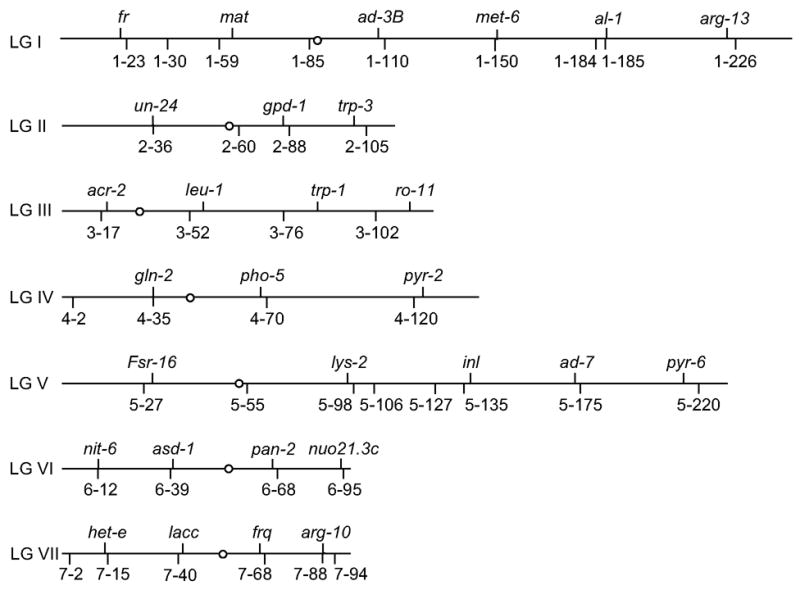
Positions of CAPS markers on the N. crassa genetic map. The positions of 39 CAPS markers relative to known mutations are shown with the overall scale based on Perkins (2000). Only the 30 markers listed in Table 1 are required for complete map coverage.
Our approach to PCR primer design was simplistic but effective. In short, candidate genomic sequences were selected from the complete genome sequence of the N. crassa Oak Ridge wild-type strain (Galagan et al., 2003) (http://www.broad.mit.edu/annotation/genome/neurospora/Home.html). Suitable primers to amplify the candidate regions were identified using the freely available Web Primer program (http://seq.yeastgenome.org/cgi-bin/web-primer) using the default parameters. All primer pairs were tested in separate PCR amplifications containing either Oak Ridge- or Mauriceville-derived genomic DNA as template to ensure that amplicons were of the same size and that amplification efficiency (yield) was comparable. In several cases, we were unable to amplify Mauriceville DNA, or only poorly so, presumably due to polymorphisms relative to the Oak Ridge-based primer sequences. In most of these cases, amplifications were successful when new primers were designed to flank the initial primer sites.
We used three different approaches to identify restriction site polymorphisms suitable for CAPS markers. We first evaluated the marker set described by Kotierk and Smith (2004). Our primary criterion for marker development was that a restriction site polymorphism would be flanked by at least 125 bp without an identical site to allow detection of both the cleaved and uncleaved amplicons by standard agarose gel electrophoresis. We set a more flexible upper limit for the ideal amplicon size at 600 bp to ensure that all amplifications could be conducted with a single protocol and resolved on a single gel. Although arbitrary, for economical reasons we limited our selection of polymorphisms to those that made use of readily available restriction endonucleases from a single vendor. Based on these criteria we adopted and/or developed five CAPS markers from the Kotierk and Smith data set (2–36, 4–120, 6–12, 6–95 and 7–88; see Tables 1 and 2). Amplicons were not sequenced and may contain additional polymorphisms, some of which could be small insertions/deletions that were undetected by standard agarose gel electrophoresis. The existence of such polymorphisms would not affect marker utility. Thus for simplicity, the sequence coordinates given for this marker set in Tables 1 and 2 are based on the reference Oak Ridge sequence.
Our second approach to identify restriction site polymorphisms was to mine available EST sequences derived from the Mauriceville strain. A set of 7,566 Mauriceville EST sequences prepared by M. A. Nelson (University of New Mexico) was obtained from the Broad Institute (Cambridge, MA, USA; http://www.broad.mit.edu/annotation/). Assembly of these ESTs using the StackPACK clustering system (Electric Genetics Corp., Reston, VA, USA; http://www.egenetics.com) yielded 649 singletons and 413 contigs (1,062 unigenes). Batch BlastN alignments (Altschul et al., 1990) were prepared for the unigene set using the complete Oak Ridge-derived genome sequence as the reference (Galagan et al., 2003). Unfortunately, automated search techniques were unable to reliably distinguish polymorphisms from random sequence errors. Therefore we manually screened the alignments to focus on regions with near identity. Due to this restriction it is likely that many useful polymorphisms were missed. Regardless, we identified 70 potential restriction site polymorphisms, of which 33 were confirmed and subsequently developed as a CAPS marker (Tables 1 and 2).
Our final strategy to identify restriction site polymorphisms was to amplify and screen defined target sequences from the Oak Ridge and Mauriceville genomes directly, a process we refer to as a de novo screen. For this strategy, a target sequence was selected on the assumption that the chromosomal location would serve as a useful genetic marker. Primers were designed to amplify a 5 kb region in three to five segments. Intergenic regions were preferred since non-coding sequence is likely to have greater numbers of random polymorphisms than coding sequence. The screen was conducted with four different restriction enzymes with 4 bp recognition sequences (4-bp cutters). We chose to use the enzymes HaeIII, MspI, TaqI and Tsp509I primarily because these enzymes were robust and among the least expensive 4-bp cutters. The rationale for this screen was that single nucleotide polymorphisms would exist in non-coding regions at a frequency of about 1% (double our crude estimate from analysis of coding regions in ESTs). On average, four 4-bp cutters would sample 320 bp in the target 5 kb region so the probability of failure to detect a polymorphism with four different enzymes would be (1-0.01)320, or 0.04. Thus one would have a better than 95% chance of successfully identifying at least one restriction site polymorphism in a specified 5 kb target region. We tested the de novo screen for a region near the al-2+ gene (albino-2) on Linkage Group I and found two polymorphisms, one of which was developed as marker 1–184 (Table 2). Even though 1–184 was the only marker we developed with this strategy, the successful outcome indicated that it is a tenable approach to screen user-defined regions.
3.2 Map coverage
Although we had developed markers with an eye toward uniform map distribution (Fig. 1) we could not be certain of the minimum number of markers or their placement on the map to achieve complete map coverage. This uncertainty was due to the fact that recombination frequencies in different genetic backgrounds can be highly variable (Perkins and Barry, 1977). An underestimate of the genetic distance between two markers could result in a gap whereby we would fail to detect linkage to a mutation located in the interstitial region. To identify potential gaps in map coverage and to assess genetic distances between markers we genotyped 96 randomly chosen progeny from a cross between the Oak Ridge and Mauriceville wild type strains with 34 of the CAPS markers. In essence, this was a 34-point cross with the ability to add markers as needed without repeating the entire process. The genetic distances were calculated as two-point data, e.g., from the percent recombination between two markers among the population of 96 individuals (Table 3). We made no correction for double crossover events in our estimates of map distances. However, we did assess the occurrence of these events for five different loci (Table 3) and found that for three of the loci, double crossovers reduced the apparent recombination frequency as might be expected (15% in the regions between markers 1–23 to 1–59 and 1–110 to 1–185, and 30% for the region between markers 5–27 to 5–98). In contrast, no double crossovers were detected for the distal right arms of Linkage groups III and VII, which was especially surprising for Linkage Group VII since the map length for this region (32 map units) was significantly greater than those regions where double crossovers were detected in Linkage Groups I and V (24, 17 and 9 map units).
Table 3.
Genetic distances between marker pairs
| Marker 1 | Marker 2 | Map unitsa | Marker 1 | Marker 2 | Map unitsa |
|---|---|---|---|---|---|
| 1–23-MspI | 1–30-BamHI | 10 | 5–27-TaqI | 5–55-MspI | 4 |
| 1–30-BamHI | 1–59-HaeIII | 18 | 5–55-MspI | 5–98-Tsp509I | 9 |
| 1–23-MspI | 1–59-HaeIII | 24 | 5–27-TaqI | 5–98-Tsp509I | 9 |
| 1–59-HaeIII | 1–110-HaeIII | 20 | 5–98-Tsp509I | 5–135-HpaI | 10 |
| 1–110-HaeIII | 1–150-RsaI | 15 | 5–135-HpaI | 5–175-DpnII | 9 |
| 1–150-RsaI | 1–185-TaqI | 5 | 5–175-DpnII | 5–220-MspI | 18 |
| 1–110-HaeIII | 1–185-TaqI | 17 | |||
| 1–185-TaqI | 1–226-DpnII | 25 | 6–12-XhoI | 6–39-HaeIII | 20 |
| 6–39-HaeIII | 6–68-MspI | 11 | |||
| 2–36-PvuI | 2–60-DpnII | 34 | 6–68-MspI | 6–95-KpnI | 32 |
| 2–60-DpnII | 2–88-TaqI | 8 | |||
| 2–88-TaqI | 2–105-HhaI | 26 | 7–15-HaeIII | 7–40-DpnII | 26 |
| 7–40-DpnII | 7–68-Tsp509I | 7 | |||
| 3–17-AluI | 3–52-EcoRI | 9 | 7–68-Tsp509I | 7–88-HindIII | 14 |
| 3–52-EcoRI | 3–76-AluI | 2 | 7–88-HindIII | 7–94-HhaI | 18 |
| 3–76-AluI | 3–102-Tsp509I | 13 | 7–68-Tsp509I | 7–94-HhaI | 32 |
| 3–52-EcoRI | 3–102-Tsp509I | 15 | |||
| 4–2-MspI | 4–35-AluI | 22 | |||
| 4–35-AluI | 4–70-BamHI | 24 | |||
| 4–70-BamHI | 4–120-BglII | 29 |
A map unit is defined as 1 percent recombination without correction for double crossovers. Map distances were determined as two-point data from the genotypes of 96 random progeny of a cross between the Oak Ridge mat a strain (FGSC 4200) and the Mauriceville mat A strain (FGSC 2225).
The N. crassa total map length is unknown, but estimates based on cytological chiasma counts and regional recombination frequencies suggest a value of about 1000 map units (Perkins and Barry, 1977; Radford and Parish, 1997). The sum of the regional map lengths determined from our 34-point cross (Table 3), however, suggest that the total map length for this combination of genetic backgrounds is likely to be less than 500 map units. Although the genetic length of each linkage group was less than anticipated from previous map data (Perkins, 2000; Radford and Parish, 1997), the reduction was not uniform. Map lengths of Linkage Groups III and V were each about 25% of the expected values, and Linkage Group I was about 45% of the expected value. In contrast, the genetic lengths of the left arm of Linkage group II and distal segments of the right arms of Linkage Groups II, VI and VII were all greater than the expected values. The presence of recombination hotspots and the nonrandom distribution of recombination events in general have been well documented in N. crassa (Bowring and Catcheside, 1999b; Catcheside, 1981; Catcheside, 1975), and attributed to modulators of recombination that include rec genes, cog and spo11 (Angel et al., 1970; Bowring et al., 2006; Catcheside, 1981; Yeadon et al., 2004). Allelic differences in these genes may also contribute to the distribution of recombination events that we detected. Regardless of mechanism, variation in the relationship between physical and genetic distances throughout the genome strongly supports the need for an empirical approach when investigating genetic map distances and meiotic recombination.
The greatest map distance between any two adjacent markers was 34 map units (markers 2–36 and 2–60). It should be noted that based on the physical map, marker 2–36 is located much closer to the left telomere than was estimated from previous genetic map data (Radford et al., 2006). Regardless, if we assumed that double crossovers occur within this region at high frequency, similar to that of the region between markers 5–27 to 5–98 (see above), we would estimate the corrected map distance at close to 50 map units. Given this conservative value, a mutation located midway between marker 2–36 and 2–60 would be roughly 25 map units from either marker, and this would represent the greatest distance any mutation would be found from a marker in our collection. Even at this distance, linkage would be confirmed from co-segregation analysis using only a modest number (40) of random progeny (95% confidence level, Chi square). Consequently, the set of 30 markers listed in Table 1 are more than sufficient to provide complete map coverage. Furthermore, most mutations will be linked to more than one marker when using this set of 30, adding precision to the estimate of genomic location. The supplementary markers listed in Table 2 may be useful to further refine some map positions although it is important to note that when markers are located very close to each other, the population size needed to detect an informative recombinant must be increased.
3.3 Bulked segregant analysis: sensitivity and limit of detection
Map distance data indicated that our 30-marker set (Table 1) would provide complete map coverage if co-segregation analysis were performed using individual progeny. The limiting factor was the ability to establish linkage at a maximum distance of 25 map units. To determine if this level of sensitivity could be achieved using bulked segregant analysis we tested three unlinked CAPS markers in a mock analysis. Genomic DNA from the Oak Ridge strain was mixed in different proportions with genomic DNA from the Mauriceville strain to mimic recombination and the related genetic distances. As shown in Fig. 2, each marker yielded band patterns that correlate with the “parental” DNA proportion, and we could easily distinguish mixes indicative of 25 map unit distances (25% and 75% of each parental DNA) from a mix representing an unlinked sample (50% each parent).
Fig. 2.
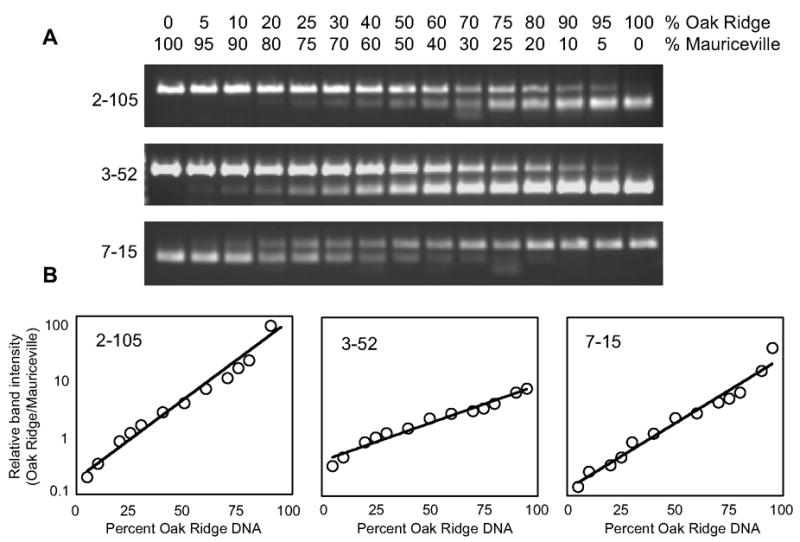
Quantitative assessment of sensitivity and detection limits for bulked segregant analysis. Mock bulks were prepared from proportions of Oak Ridge and Mauriceville-1c genomic DNA to mimic recombination and related genetic distances. (A) Representative band patterns from analysis with three CAPS markers. Note that for 2–105 and 3–52 it is the Oak Ridge “allele” that is cleaved but for 7–15 the Mauriceville allele is cleaved. (B) Band intensities for the parental alleles were determined by densitometry, and for each marker the ratio of Oak Ridge to Mauriceville allele is plotted vs. the percent Oak Ridge DNA in the bulk.
The mock bulk segregant analyses shown in Fig. 2 also established a working limit of detection for the presence of the rarer allele when linkage is tight. About 5% of the bulk must be composed of recombinants to distinguish the pattern from either of the parental, monomorphic patterns. For some markers detection at this 5-map unit level required examination of the equivalent, reciprocal mix, presumably due to subtle differences in the amplification efficiencies for specific target sequences. Quantitative analysis of band patterns using densitometry confirmed the congruence between reciprocal mixes and furthermore, indicated that the mock analyses yield remarkably consistent standard curves that could be used to estimate map distance to an unknown mutation.
3.4 Efficacy of bulked segregant analysis for genetic mapping
To test the overall utility of our mapping approach (outlined in Fig. 3), bulks were prepared from a population segregating a known mutation, al-1 (albino-1), and were analyzed with selected CAPS markers. In short, the al-1 mutant (Oak Ridge background) was crossed to the Mauriceville strain and random progeny were isolated. Progeny that carried the al-1 mutation were readily distinguished from wild-type progeny by their lack of the usual orange pigment. We pooled mycelia from 40 individuals of each phenotypic class to form two bulks, and isolated genomic DNA from both bulks. We chose 40 individuals as an ideal bulk size because, as discussed in section 3.2, this is the minimum number required to establish linkage if 25% recombination is observed (95% confidence level, Chi square). Although we could have analyzed only the albino class, which would be the norm for bulked segregant analysis in diploid organisms (Martinez-Morales et al., 2004; Michelmore et al., 1991; Rawls et al., 2003; Rymarquis et al., 2005), we also included the wild-type class for confirmation, and a 1:1 mix of the two pooled DNA samples as a control. In retrospect, the value of these extra samples in terms of increased confidence in the scoring far outweighs the modest increase in workload. The al-1+ gene is located on the right arm of Linkage group I (Fig. 1). As shown in Fig. 4, markers located on different chromosomes (4–120 and 7–88) displayed equivalent band patterns in all three bulks, indicative of unlinked loci. In contrast, markers located near the al-1+ gene (1–150 and 1–185) displayed reciprocal band patterns in the albino and wild-type bulks that clearly deviate from those in the 1:1 non-linkage control bulk, indicating that these markers are indeed linked to the al-1+ gene.
Fig. 3.
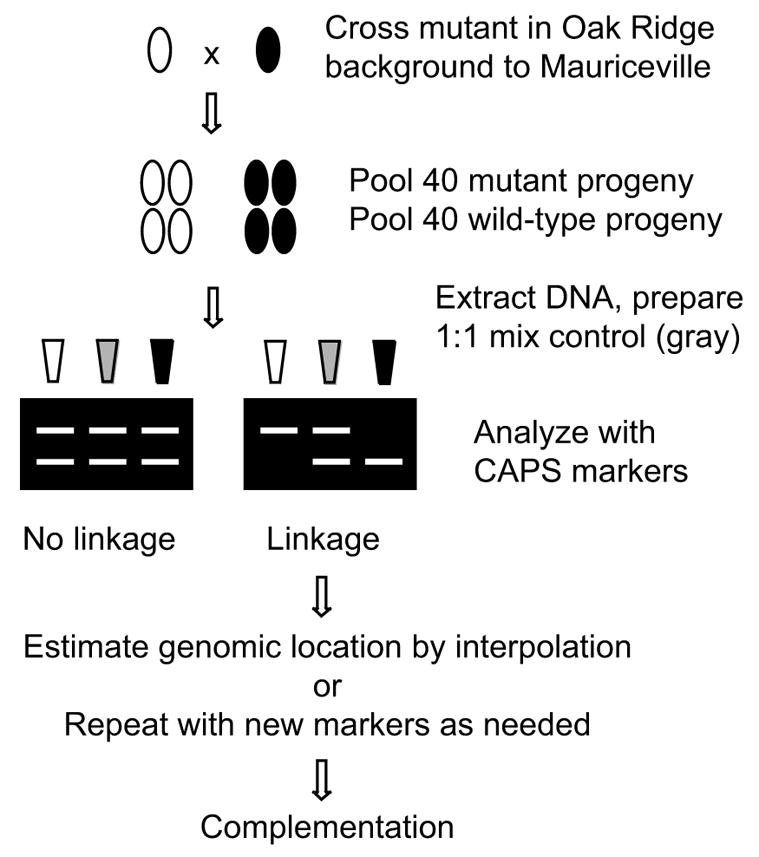
Flow chart for map-based cloning in N. crassa using bulked segregant analysis.
Fig. 4.

Bulked segregant analysis confirms the map location of the al-1 mutation at 1–185. The al-1 strain was crossed to Mauriceville-1c and genomic DNA bulks were prepared from progeny based on phenotype. Each DNA bulk as well as a 1:1 mixture of the two phenotypic bulks was analyzed with the indicated CAPS markers. Reciprocal patterns in the two phenotypic bulks that differ from those of the mix are indicative of linkage. The albino bulk contains only the Oak Ridge polymorphism at 1–185 while the wild-type bulk contains only the Mauriceville polymorphism, indicating strong linkage of the al-1 mutation to this locus. Significant linkage to the nearby marker 1–150 is also apparent. Unlinked markers 4–120 and 7–88 show equal parental polymorphisms.
The patterns detected for marker 1–185 (Fig. 4) from the segregated bulks are monomorphic suggesting that this marker is tightly linked to the al-1+ gene. Because our limit of detection is about 5% recombinants, we would estimate that the al-1+ gene is located at a position less than 5 map units from marker 1–185. However, this marker would give the same pattern regardless of whether it was located to the left or right of al-1+ so the gene must lie within a 10-map unit region. For some purposes this level of resolution may be sufficient but it is unlikely to be suitable for map-based cloning. To improve resolution we made use of the fact that marker 1–150 did undergo recombination with the al-1+ gene (Fig. 4). From a standard curve prepared the same as those shown in Fig. 2 (data not shown), we estimated that marker 1–150 contains 5% recombinants, and therefore is located approximately 5 map units from the al-1+ gene. Genotyping of individual progeny confirmed this estimate (4 recombinants out of 80 total progeny). Furthermore, our map distance data (Table 3) indicated that marker 1–150 is located 5 map units from marker 1–185. Through interpolation of these map distance data we estimate that the al-1+ gene is located within 1 map unit of marker 1–185. To verify this estimate we compared the physical and genetic distances between each of the markers and the al-1+ gene. Markers 1–150 and 1–185 are physically separated by about 1,110 kb and genetically by 5 map units. Thus for this region there is an average of 220 kb per map unit. Marker 1–185 is physically separated from the al-1+ gene by 75 kb, well within our estimated 1 map unit genetic distance at 220 kb per map unit. At this level of resolution we could easily have undertaken complementation studies to confirm the gene affected by mutation.
As a further validation of this technique, we mapped a trait for which the causative mutation was not known. The Oak Ridge derived strain bd, COP1–2 (Vitalini et al., 2004), has altered expression of the circadian clock-controlled genes ccg-1 and ccg-2, and conidia from the mutant strain are dark in color and wet-looking. When COP1–2 was backcrossed, the conidial phenotype segregated 1:1 in random progeny, and cosegregated with altered ccg-1 expression levels, indicating that a single gene was responsible for the COP1–2 mutant phenotypes. We crossed the bd, COP1–2 strain with the Mauriceville-1c strain, and assembled pools of wild-type and mutant segregants as described above. Using the CAPS markers, we located the mutation on the right arm of Linkage group IV, with linkage to markers 4–70 and 4–120. Recombination values derived from genotyping individual progeny (data not shown) suggested that the mutation was located within a 1 map unit region. Based on this genetic map location, a single candidate gene was identified and sequenced from the COP1–2 strain. While the identity of the gene and its characterization with respect to circadian rhythmicity will be published elsewhere, the sequence analysis identified a mutation in an open reading frame that would result in early termination, and confirmed the map data.
3.5 Method summary
Based on our investigations of map coverage by a set of defined molecular markers and the sensitivity and detection limits of bulked segregant analysis, we have devised an efficient method for rapid genetic mapping of mutations in N. crassa. The overall scheme for this procedure is illustrated in Fig. 3. In summary, a mutant isolated in the Oak Ridge background, i.e., FGSC 4200 (mat a) is crossed to the polymorphism-rich wild-type Mauriceville-1c (mat A) strain (FGSC 2225) to obtain a segregating population. It is important to note that the Mauriceville-1d strain (FGSC 2226), which is mat a, cannot be substituted in this method as a crossing partner with an Oak Ridge strain of opposite mating type. This is because the Mauriceville-1d strain is an independent isolate (D.D. Perkins, personal communication) that possesses mostly Oak Ridge-type polymorphisms (data not shown). Use of the Mauriceville-1c strain as the sole crossing partner is not a significant limitation because the routine backcrosses required for analysis of any new mutant would yield both mating types unless the mutation is tightly linked to the mat locus.
Genomic DNA bulks are assembled from wild-type and mutant progeny. The two bulk DNA samples, as well as a sample prepared from an equal mixture of the two bulks (non-linkage control) are analyzed with the 30 CAPS markers listed in Table 1. The analysis consists of PCR amplification, restriction digestion and gel electrophoresis. Although we describe additional markers (Table 2), there are two important considerations for our decision to limit the number of markers included in an initial screen. First, the distribution of the 30 markers listed in Table 1 provides complete map coverage so the location of any new mutation can quickly be defined to a relatively small region within a linkage group. Second, by limiting the number of markers to 32 or less, the PCR amplifications required for our strategy (two bulks plus a 1:1 control analyzed with each marker) can be accomplished in parallel using a single 96-well plate. Inclusion of the wild-type bulk provides immediate confirmation of map position determined in the mutant bulk; the mutation should be linked to the Oak Ridge polymorphism and the wild-type allele should be linked to the Mauriceville polymorphism at the same locus.
When linkage is detected to one or more markers, the genetic distance between a marker and the mutation can be estimated from a standard curve prepared from parental DNA, similar to those in Fig. 2, or can be calculated from two-point data in which recombination is scored from individual genotypes. Although the latter approach is more labor intensive, scoring individual progeny with two or three markers is likely to yield more consistent data. When the mutation lies between two markers, one can interpolate the position of the mutation to within 1–5 map units. If the linked markers are within 10 map units of the mutation one can, in most cases, safely assume that the map distances are not skewed by double crossover events. Thus the position of the mutation can be estimated to within a single map unit.
The level of resolution deemed sufficient to proceed with gene identification via complementation tests will undoubtedly depend on the nature of the mutant phenotype and whether complementation can be detected via a selection or requires a screen. For example, if a mutation that could be complemented by direct selection was mapped within 5 map units and this genetic distance corresponded to 1000 kb, one could immediately proceed with a relatively straightforward complementation strategy. That is, one could either test 25–50 cosmids from an ordered collection individually, or assemble binary pools (e.g., left half, right half, odd, even etc.) of the same cosmids to reduce the number of candidate clones to just a few individuals. This approach also could be applied for a complementation screen. However, if the screen is arduous it may be more expedient to first refine the map resolution. To refine the map location of a mutation one could of course repeat the analysis with additional, more closely spaced markers. This would be warranted if linkage were detected for only one marker in the initial screen, which might occur for mutations located near the telomeres. However, it is important, but perhaps counterintuitive, that closely spaced markers may not yield useful information unless very large numbers of individual progeny are analyzed. This simply relates to the fact that the probability for a crossover occurring within a small region is low. In contrast, with moderately spaced markers, the level of recombination is meaningful and the interpolation of genetic distances provides high map resolution. Ultimately, the method described here allows one to map any new mutation using progeny from a single cross, and the molecular analyses can be completed in parallel using a 96-well plate.
3.6 Applications
Although our motivation for this work was to facilitate map-based cloning, this is not the sole potential application for our genetic mapping method. One intriguing example is the analysis of genome-wide recombination. Since the genetic backgrounds are defined in our method, only the nature of the mutation one investigates would be expected to alter recombination. Thus it will be possible to investigate the effect of specific mutations on recombination and/or its control and to test whether the effects are global or localized to distinct chromosomal regions. Interesting candidates would include those genes previously identified through classical and related molecular approaches to modulate recombination (Angel et al., 1970; Bowring et al., 2006; Catcheside, 1981; Kato et al., 2004; Suzuki et al., 2005; Yeadon et al., 2004). Similarly, one could quantitatively assess chiasma interference at a genome-wide level (Bowring and Catcheside, 1999a; Foss et al., 1993).
The same mapping methodology described here also could be applied to other fungi both for the purpose of map-based cloning and for investigations of meiotic recombination. Key requirements would be that the fungus has a sexual cycle, a complete or nearly so genome sequence, and the availability of a polymorphism-rich out-crossing partner strain. There are currently 65 fungal species listed at the NCBI that have completed or initiated genome-sequencing projects. However, identifying a suitable polymorphic out-crossing strain may require screening of wild isolates. Once a suitable polymorphic out-crossing strain is identified, the de novo screen method to identify restriction site polymorphisms could be applied. Alternatively, adaptation of the method would be nearly trivial if genomic sequence also is available from the polymorphic strain.
Acknowledgments
We thank Tom McKnight, Deb Bell-Pedersen and Kathy Ryan for critical discussions and for comments on this manuscript. We also are grateful to Dan Ebbole for assistance with bioinformatic analyses. This work was supported by start-up funds from Texas A&M University and in part by the National Institutes of Health (GM58529).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–10. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Angel T, Austin B, Catcheside DG. Regulation of recombination at the his-3 locus in Neurospora crassa. Aust J Biol Sci. 1970;23:1229–1240. doi: 10.1071/bi9701229. [DOI] [PubMed] [Google Scholar]
- Bowring FJ, Catcheside DEA. Evidence for negative interference: clustering of crossovers close to the am locus in Neurospora crassa among am recombinants. Genetics. 1999a;152:965–969. doi: 10.1093/genetics/152.3.965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowring FJ, Catcheside DEA. Recombinational landscape across a 650-kb contig on the right arm of linkage group V in Neurospora crassa. Curr Genet. 1999b;36:270–274. doi: 10.1007/s002940050500. [DOI] [PubMed] [Google Scholar]
- Bowring FJ, Yeadon PJ, Stainer RG, Catcheside DEA. Chromosome pairing and meiotic recombination in Neurospora crassa spo11 mutants. Curr Genet. 2006;50:115–123. doi: 10.1007/s00294-006-0066-1. [DOI] [PubMed] [Google Scholar]
- Catcheside DEA. Genes in Neurospora that suppress recombination when they are heterozygous. Genetics. 1981;98:55–76. doi: 10.1093/genetics/98.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catcheside DG. Regulation of recombination. Neurospora crassa. In: Peacock WJ, Brock RD, editors. The eukaryotic chromosome. Australian National University Press; Canberra: 1975. pp. 301–312. [Google Scholar]
- Colot HV, Park G, Turner GE, Ringelberg C, Crew CM, Litvinkova L, Weiss RL, Borkovich KA, Dunlap JC. A high-throughput gene knockout procedure for Neurospora reveals functions for multiple transcription factors. Proc Natl Acad Sci USA. 2006;103:10352–7. doi: 10.1073/pnas.0601456103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RH, Perkins DD. Neurospora: a model of model microbes. Nat Rev Genet. 2002;3:397–403. doi: 10.1038/nrg797. [DOI] [PubMed] [Google Scholar]
- Elahi E, Kumm J, Ronaghi M. Global genetic analysis. J Biochem Mol Biol. 2004;37:11–27. doi: 10.5483/bmbrep.2004.37.1.011. [DOI] [PubMed] [Google Scholar]
- Foss E, Lande R, Stahl FW, Steinberg CM. Chiasma interference as a function of genetic distance. Genetics. 1993;133:681–91. doi: 10.1093/genetics/133.3.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galagan JE, Calvo SE, Borkovich KA, Selker EU, Read ND, Jaffe D, FitzHugh W, Ma LJ, Smirnov S, Purcell S, Rehman B, Elkins T, Engels R, Wang S, Nielsen CB, Butler J, Endrizzi M, Qui D, Ianakiev P, Bell-Pedersen D, Nelson MA, Werner-Washburne M, Selitrennikoff CP, Kinsey JA, Braun EL, Zelter A, Schulte U, Kothe GO, Jedd G, Mewes W, Staben C, Marcotte E, Greenberg D, Roy A, Foley K, Naylor J, Stange-Thomann N, Barrett R, Gnerre S, Kamal M, Kamvysselis M, Mauceli E, Bielke C, Rudd S, Frishman D, Krystofova S, Rasmussen C, Metzenberg RL, Perkins DD, Kroken S, Cogoni C, Macino G, Catcheside D, Li W, Pratt RJ, Osmani SA, DeSouza CP, Glass L, Orbach MJ, Berglund JA, Voelker R, Yarden O, Plamann M, Seiler S, Dunlap J, Radford A, Aramayo R, Natvig DO, Alex LA, Mannhaupt G, Ebbole DJ, Freitag M, Paulsen I, Sachs MS, Lander ES, Nusbaum C, Birren B. The genome sequence of the filamentous fungus Neurospora crassa. Nature. 2003;422:859–68. doi: 10.1038/nature01554. [DOI] [PubMed] [Google Scholar]
- Jenkins G. Unfolding large-scale maps. Genome. 2003;46:947–52. doi: 10.1139/g03-113. [DOI] [PubMed] [Google Scholar]
- Kato A, Akamatsu Y, Sakuraba Y, Inoue H. The Neurospora crassa mus-19 gene is identical to the qde-3 gene, which encodes a RecQ homologue and is involved in recombination repair and postreplication repair. Curr Genet. 2004;45:37–44. doi: 10.1007/s00294-003-0459-3. [DOI] [PubMed] [Google Scholar]
- Kotierk M, Smith ML. PCR-based markers for genetic mapping in Neurospora crassa. Fungal Genet Newsl. 2004;51:26–29. [Google Scholar]
- Margolin BS, Freitag M, Selker EU. Improved plasmids for gene targeting at the his-3 locus of Neurospora crassa by electroporation. Fungal Genet Newsl. 1997;44:34–36. [Google Scholar]
- Martinez-Morales JR, Naruse K, Mitani H, Shima A, Wittbrodt J. Rapid chromosomal assignment of medaka mutants by bulked segregant analysis. Gene. 2004;329:159–65. doi: 10.1016/j.gene.2003.12.028. [DOI] [PubMed] [Google Scholar]
- McCluskey K. The Fungal Genetics Stock Center: from molds to molecules. Adv Appl Microbiol. 2003;52:245–262. doi: 10.1016/s0065-2164(03)01010-4. [DOI] [PubMed] [Google Scholar]
- Metzenberg RL, Stevens JN, Selker EU, Morzycka-Wroblewska E. A method for finding the genetic map position of cloned DNA fragments. Fungal Genet Newsl. 1984;31:35–40. [Google Scholar]
- Michelmore RW, Paran I, Kesseli RV. Identification of markers linked to disease-resistance genes by bulked segregant analysis: A rapid method to detect markers in specific genomic regions by using segregating populations. Proc Natl Acad Sci USA. 1991;88:9828–9832. doi: 10.1073/pnas.88.21.9828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson MA, Perkins DD. Restriction polymorphism maps of Neurospora crassa: 2000 update. Fungal Genet Newsl. 2000;47:25–39. [Google Scholar]
- Perkins DD. New multicent linkage testers for centromere-linked genes and rearrangements in Neurospora. Fungal Genet Newsl. 1990;37:31–32. [Google Scholar]
- Perkins DD. Neurospora alcoy linkage tester stocks with group VII marked, and their use for mapping translocations. Fungal Genet Newsl. 1991;38:83. [Google Scholar]
- Perkins DD. Neurospora crassa gentic maps and mapped loci. Fungal Genet Newsl. 2000;47:40–58. [Google Scholar]
- Perkins DD. How to use alcoy. 2006 http://www.fgsc.net/Neurospora/NeurosporaProtocolGuide.htm.
- Perkins DD, Barry EG. The cytogenetics of Neurospora. Adv Genet. 1977;19:133–285. doi: 10.1016/s0065-2660(08)60246-1. [DOI] [PubMed] [Google Scholar]
- Radford A, Navarro-Coy N, Davies JR, Beck T. The Neurospora crassa e-Compendium. 2006 http://www.bioinf.leeds.ac.uk/~gen6ar/newgenelist/genes/gene_list.htm.
- Radford A, Parish JH. The genome and genes of Neurospora crassa. Fungal Genet Biol. 1997;21:258–66. doi: 10.1006/fgbi.1997.0979. [DOI] [PubMed] [Google Scholar]
- Rawls JF, Frieda MR, McAdow AR, Gross JP, Clayton CM, Heyen CK, Johnson SL. Coupled mutagenesis screens and genetic mapping in zebrafish. Genetics. 2003;163:997–1009. doi: 10.1093/genetics/163.3.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rymarquis LA, Handley JM, Thomas M, Stern DB. Beyond complementation. Map-based cloning in Chlamydomonas reinhardtii. Plant Physiol. 2005;137:557–66. doi: 10.1104/pp.104.054221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, Kato A, Sakuraba Y, Inoue H. Srs2 and RecQ homologs cooperate in mei-3-mediated homologous recombination repair of Neurospora crassa. Nucleic Acids Res. 2005;33:1848–58. doi: 10.1093/nar/gki326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vignal A, Milan D, SanCristobal M, Eggen A. A review on SNP and other types of molecular markers and their use in animal genetics. Genet Sel Evol. 2002;34:275–305. doi: 10.1186/1297-9686-34-3-275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitalini MW, Morgan LW, March IJ, Bell-Pedersen D. A genetic selection for circadian output pathway mutations in Neurospora crassa. Genetics. 2004;167:119–29. doi: 10.1534/genetics.167.1.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel HJ. A convenient growth medium for Neurospora (medium N) Microbiol Genet Bull. 1956;13:42–43. [Google Scholar]
- Westergaard M, Mitchell HK. Neurospora V. A synthetic medium favoring sexual reproduction. Am J Botany. 1947;34:573–577. [Google Scholar]
- Yeadon PJ, Bowring FJ, Catcheside DEA. Alleles of the hotspot cog are codominant in effect on recombination in the his-3 region of Neurospora. Genetics. 2004;167:1143–1153. doi: 10.1534/genetics.103.025080. [DOI] [PMC free article] [PubMed] [Google Scholar]