

Abstract
The asynchronous secretion of gonadotrope LH and FSH under the control of GnRH is crucial for ovarian cyclicity but the underlying mechanism is not fully resolved. Because prostaglandins (PG) are autocrine regulators in many tissues, we determined whether they have this role in gonadotropes. We first demonstrated that GnRH stimulates PG synthesis by induction of cyclooxygenase-2, via the protein kinase C/c-Src/phosphatidylinositol 3′-kinase/MAPK pathway in the LβT2 gonadotrope cell line. We then demonstrated that PGF2α and PGI2, but not PGE2 inhibited GnRH receptor expression by inhibition of phosphoinositide turnover. PGF2α, but not PGI2 or PGE2, reduced GnRH-induction of LHβ gene expression, but not the α-gonadotropin subunit or the FSHβ subunit genes. The prostanoid receptors EP1, EP2, FP, and IP were expressed in rat gonadotropes. Incubations of rat pituitaries with PGF2α, but not PGI2 or PGE2, inhibited GnRH-induced LH secretion, whereas the cyclooxygenase inhibitor, indomethacin, stimulated GnRH-induced LH secretion. None of these treatments had any effect on GnRH-induced FSH secretion. The findings have thus elaborated a novel GnRH signaling pathway mediated by PGF2α-FP and PGI2-IP, which acts through an autocrine/paracrine modality to limit autoregulation of the GnRH receptor and differentially inhibit LH and FSH release. These findings provide a mechanism for asynchronous LH and FSH secretions and suggest the use of combination therapies of GnRH and prostanoid analogs to treat infertility, diseases with unbalanced LH and FSH secretion and in hormone-dependent diseases such as prostatic cancer.
GnRH IS THE central regulator of reproduction. GnRH alone, administered as hourly pulses, is sufficient to restore fertility in men and women with hypothalamic hypogonadism. GnRH binds and activates its cognate receptor in pituitary gonadotropes, resulting in the synthesis and release of LH and FSH, which control ovulation and spermatogenesis (1-4). The mechanisms mediating the differential synthesis and release of LH and FSH by GnRH during the ovarian cycle and the termination of the ovulatory LH surge are not well understood. Changes in GnRH pulse frequency have been implicated, but the ability to restore fertile reproductive cycles of normal length on administration of unvarying hourly pulses of GnRH to women with hypothalamic lesions (5-7) suggests that ovarian steroid hormones (estrogen and progesterone) and peptide hormone (inhibin, activin, and follistatin) mediate the differential LH and FSH responses to GnRH over the ovarian cycle (8).
In vivo, cells are exposed to numerous stimuli, which simultaneously activate multiple signaling pathways that potentially cross talk and affect the net biological outcome. Gonadotropes are simultaneously exposed to pulses of hypothalamic GnRH, neurotransmitters, melatonin, pituitary hormones, growth factors, and gonadal steroid and peptide hormones (9). Signaling of GPCRs is mainly studied by introducing a single ligand to target cells and analyzing the downstream events. To better mimic the in vivo situation, integrative signaling may be addressed by simultaneous activation of the cell by a particular ligand, together with effector molecules contributing to downstream pathway shared by GPCRs and growth factor receptors. Eicosanoids represent such a model downstream effector system modulated by multiple ligands. Eicosanoids are metabolites of arachidonic acid (AA) involved in the regulation of reproduction, cancer, and the inflammatory, neuronal, cardiovascular, gastrointestinal, respiratory, and immune systems (10, 11). We have previously shown that GnRH stimulates AA release from rat pituitary cells, followed by elevated expression of 12-lipoxygenase and formation of 5-lipoxygenase products, which participate in GnRH actions (3, 12). Here we demonstrate a novel GnRH signaling pathway mediated by PGF2α and the FP receptor and PGI2 and the IP receptor, which limit the homologous regulation of GnRH receptor (GnRHR), whereas PGF2α also exerts selective inhibition of LH release. This mechanism may underly the cyclical responsiveness of pituitary gonadotropes to GnRH and the asynchronous LH and FSH release during the mammalian ovarian cycle.
RESULTS
GnRH Stimulates Cycooxygnease (COX)-2 Induction
Because COX-2 is the key enzyme in the eicosanoid synthesis pathway, we looked for activation of COX-2 by GnRH. GnRH produced a marked time-dependent induction of COX-2 enzyme expression in the gonadotrope cell line LβT2 (13), as revealed by RT-PCR analysis (Fig. 1A). Preincubation of the cells with the selective MEK inhibitor PD98059 abolished the stimulatory effect of GnRH on COX-2 induction, indicating that activation of COX-2 by GnRH is mediated by ERK. Further quantitative PCR (Q-PCR) revealed a marked induction of COX-2 by GnRH (40-fold; basal and GnRH-stimulated levels were 0.12 and 4.8, respectively, n = 3) (Fig. 1B). This stimulatory effect was reduced by the selective inhibitors for MEK (PD98059), JNK (JNKI), p38 (SB203580), phosphatidylinositol 3′-kinase (PI3K) (wortmannin), protein kinase C (PKC) (GF109203X), and c-Src (PP2), with no significant effect on basal levels. The data implicate the known GnRH-activated PKC/c-Src/MAPK pathway (14-17) in GnRH induction of COX-2 (Fig. 1B). The substantial inhibition (50–75%) by all the inhibitors implies that the various MAPK cascades do not converge on the same signaling molecules, but rather act in parallel pathways to activate transcription factors, which act as a composite response element during activation of COX-2 gene expression. The epidermal growth factor (EGF) receptor kinase inhibitor, AG1478, had no significant effect on GnRH induced COX-2 expression. This accords with our observation that activation of MAPK by GnRH in gonadotropes is not mediated by transactivation of the EGF receptor (18, 19). The selective inhibitors for iPLA2 [bromoenol lactone (BEL)] and for sPLA2 [thioetheramide-PC (TE-PC)], but not that for cPLA2 (AACOCF3), reduced COX-2 induction by GnRH, suggesting that the two PLA2 isoforms may also be involved in GnRH induction of COX-2. To examine the effect of GnRH on COX-2 promoter activity, LβT2 cells were transiently transfected with the COX-2 promoter and treated with GnRH for various time periods (Fig. 1C). Activation was rapid and reached maximal activation after 8 h of stimulation with GnRH (3-fold, P < 0.01), with a decline to basal levels at 24 h. An increase in COX-2 mRNA stability by GnRH may explain the discrepancy between the degree of mRNA induction (40-fold) and the degree and kinetics of promoter activity (3-fold). It is also possible that the transfected promoter construct lacks the full complement of regulatory elements. COX-2 protein expression was similarly regulated by GnRH; maximal protein expression occurring at 8 h (2.5-fold; P < 0.05) (Fig. 1D). It is interesting to note that, whereas COX-2 protein levels have declined by 24 h of GnRH stimulation, mRNA levels remained high. This suggests that there is an increased rate of protein degradation and/or decreased translation.
Fig. 1. GnRH Induction of COX-2 Activity.
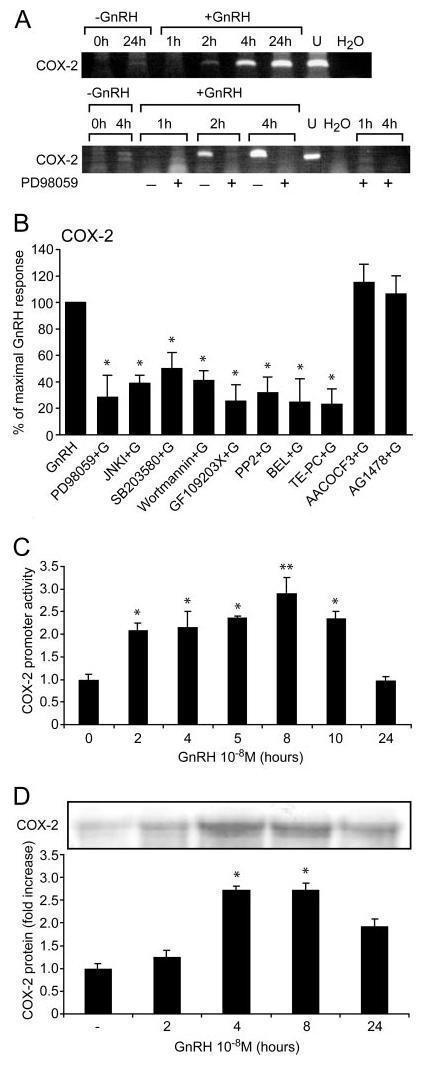
A, Effect of GnRH on COX-2 induction as revealed by RT-PCR. Subconfluent LβT2 cells were incubated and some groups were pretreated with the selective MEK inhibitor PD98059 (50 μm for 20 min) before the addition of GnRH (10 nm) for the time indicted. COX-2 was then determined by qualitative RT-PCR. Negative (H2O) and positive (U, uterus) controls are also shown. B, Quantitative-PCR for COX-2 induction by GnRH. Subconfluent LβT2 cells were pretreated for 20 min with the following selective inhibitors: for MEK (PD98059, 50 μm), for JNK (JNK inhibitory peptide, 2 μm), for p38 (SB203580, 10 μm), for PI3K (Wortmannin, 25 nm), for PKC (GF109203X, 3 μm), for c-Src (PP2, 5 μm), for iPLA2 (BEL, 20 μm), for sPLA2 (TE-PC, 25 μm), for cPLA2 (AACOCF3, 25 μm) and for EGF receptor kinase (AG1478, 5 μm). Cells were then stimulated with GnRH (100 nm for 8 h) and COX-2 was determined by Q-PCR. C, Effect of GnRH on COX-2 promoter activity. Subconfluent LβT2 cells were seeded into six-well plates and incubated overnight at 37 C before transfecting with 0.3 μg of −2307/+49 human COX-2 promoter. Renilla (33 ng) expression vector was also included as a measure of transfection efficiency and as an internal control. The transfected cells were serum starved for 18 h. GnRH (100 nm) was added for the indicated time points and cells were then harvested and assayed using a Dual-light Luciferase assay kit (Promega) in a FLUOStar Optima luminometer. Luciferase activity was normalized for Renilla to correct for transfection efficiency. Results for promoter activity are expressed as fold increase relative to untreated controls (n = 6). D, Effect of GnRH on COX-2 protein expression. LβT2 cells were grown in 4 × 12-well plates (5 × 105 cells/well). Cells were incubated in serum-free DMEM, 0.2% FCS overnight at 37 C. The cells were washed with DMEM and incubated with or without GnRH (100 nm) for various time periods and COX-2 was detected by Western blotting. The blot was re-run with antitotal ERK antibody to correct for equal loading of the samples. A representative gel is shown, and bars are the mean from triplicate samples (ANOVA: *, P < 0.05, **, P < 0.01 compared with control).
GnRH Stimulates PGE2, PGI2, and PGF2α Production
To determine whether gonadotropes could elaborate prostanoids, we incubated the LβT2 cells with GnRH for up to 24 h, with and without AA as substrate for the last 30 min of the incubation period. Incubation with GnRH alone for up to 24 h had no effect on PG production but addition of AA for the last 30 min of the incubation period resulted in a 2.5- to 3-fold increase (P < 0.05) in PGE2, PGI2, and PGF2α production (Fig. 2). Addition of AA alone had no effect on PG production (data not shown). Addition of AA had previously been shown to be required to demonstrate PG production (Ref. 20 and references therein).
Fig. 2. GnRH Stimulates PG Production.
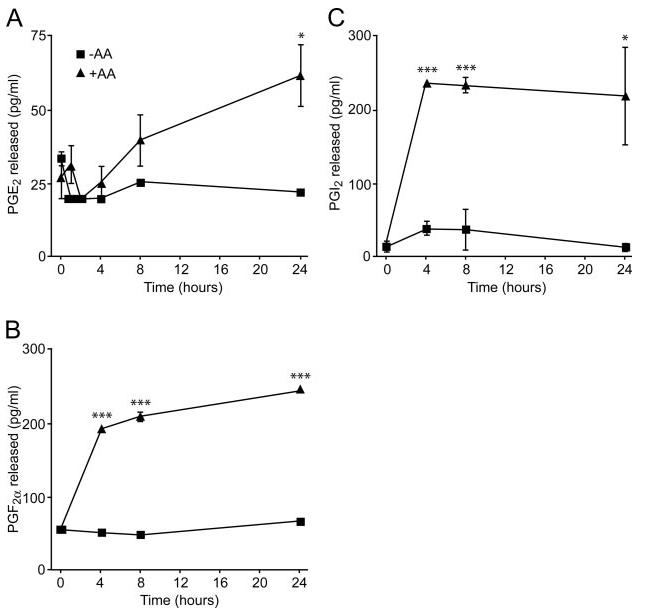
LβT2 cells were serum starved overnight washed with DMEM and incubated with GnRH (100 nm). Some of the wells received exogenous AA (1 μm) as substrate for the last 30 min of each incubation time. A, PGE2; B, PGF2α; and C, PGI2 levels were determined by ELISA. Addition of AA alone had no effect on PG production. One-way ANOVA established that *, P < 0.05 and ***, P < 0.001.
Expression of Prostanoid Receptors in LβT2 Cells
After biosynthesis, prostanoids are transported out of the cell and act in an autocrine/paracrine manner via cognate GPCRs (11). We therefore examined prostanoid receptor expression in the immortalized gonadotrope LβT2 cell line (13), which serve as a gonadotrope cell model to dissect the signaling of the GnRHR. Specific staining for EP1, EP2, EP3, EP4, FP, and IP was observed in LβT2 cells (Fig. 3A). Specificity was demonstrated by preabsorption of the antibodies with the specific peptides used for the immunization (C). The expression of EP1, EP2, EP3, EP4, FP, and IP in LβT2 cells was further demonstrated by RT-PCR (Fig. 3B).
Fig. 3. Prostanoids Receptor Expression in LβT2 Cells.
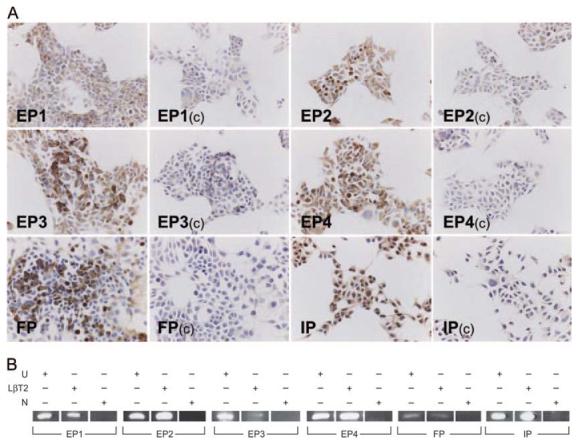
A, LβT2 cells were fixed for 10 min in 4% NBF and immunostained with antibodies to EP1, EP2, EP3, EP4, IP, and FP at a 1:200 dilution. To ensure specificity, the primary antibodies were also incubated with the immunization peptides at a 10-fold excess overnight at 4 C (control, C). After washes in TBS, sections were incubated for 30 min with ABC-HRP, and washed in TBS before visualization with diaminobenzidine (bar,50 μm). B, Qualitative RT-PCR of prostanoids receptors in LβT2 cells. Total RNA isolated from subconfluent LβT2 cells was reversely transcribed and aliquots of single-stranded cDNA were subjected to PCR (with negative controls, N) using appropriate oligonucleotide primers provided in Materials and Methods. PCR products were analyzed by UV fluorescence of ethidium bromide-stained DNA after agarose gel electrophoresis alongside DNA size markers. RNA isolated from rat uterus (U) served as a positive control.
Cross Talk between Prostanoid and GnRHRs
The known marked homologous induction of the GnRHR promoter construct by GnRH itself (21) was significantly reduced by PGF2α and the stable analog of PGI2, iloprost, but not by PGE2 (Fig. 4A). Furthermore, cotreatment of LβT2 cells with GnRH and PGF2α or iloprost for 6 h reduced GnRH binding [2180 ± 437 counts per minute (cpm) and 2587 ± 454 cpm respectively; sem, n = 3, P < 0.01] when compared with binding in cells incubated with GnRH alone (6134 ± 737 cpm; sem, n = 3), or with GnRH and PGE2 (6254 ± 1022 cpm; sem, n = 3). To elucidate the mechanisms involved in the inhibitory action of the PGs we first investigated whether they compete with the binding of GnRH for its cognate receptor. As shown in Fig. 4B, GnRH binding to LβT2 cells was not affected by the presence of PGE2, PGF2α or iloprost (each at 500 nm). We therefore proceeded to examine initial signaling events after receptor binding. The mechanism of GnRH stimulation of the GnRHR promoter is known to be via activation of its predominant signaling pathway (Gαq and PLC-β) (21). We confirmed this by demonstrating that GnRH stimulation of the GnRHR promoter was progressively inhibited by increasing concentrations of the PLC-β inhibitor (U73122) (Fig. 4C) and that this correlated well with the concomitant decrease in InsP production (Fig. 4D). Thus, it was feasible that the inhibitory effects of PGF2α and iloprost on the GnRHR promoter were mediated via an inhibition of InsP production. This notion is supported by our demonstration that PGF2α and iloprost inhibited GnRH stimulation of InsP formation but had no significant effect on basal levels, whereas PGE2 was ineffective (Fig. 4E).
Fig. 4. Effect of PG on GnRHR Regulation.
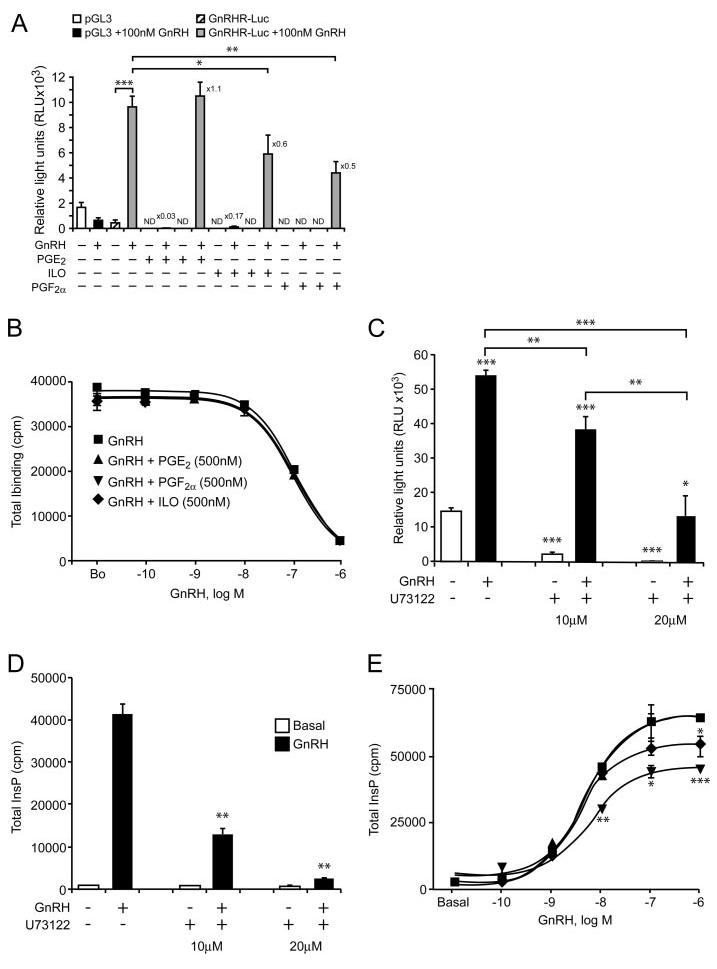
A, Effect of PG on GnRH-induced GnRHR promoter activity. LβT2 cells were transiently transfected with control pGL3 or GnRHR-Luc, pretreated for 30 min with PGE2, iloprost or PGF2α (500 nm) followed by addition of GnRH (100 nm) and continuous PG treatment for 4 h. Luciferase activity was measured, corrected for transfection efficiency and expressed as relative light units (RLU×103). Basal levels of pGL3 and GnRHR-Luc were not detectable (ND) after PG treatment, but there was no effect of PG treatment on the cotransfected control CMV-β-galactosidase plasmid. Fold stimulation was calculated relative to GnRH treated and a representative experiment of the three performed, each done in triplicate is shown. B, Effect of PG on GnRH binding. LβT2 cells were pretreated for 30 min with PGE2, iloprost or PGF2α (500 nm) followed by a competitive binding assay for GnRH in the presence and absence of PGE2, PGF2α or iloprost (500 nm) as detailed in Materials and Methods. A representative experiment of the three performed is shown. C and D, Effect of the phospholipase C inhibitor, U73122 on GnRH-induced GnRHR promoter activity and phosphoinositide turnover. C, LβT2 cells were transiently transfected with GnRHR-Luc, pretreated with U73122 for 30 min, followed by a further addition of GnRH (100 nm for 4 h). Luciferase activity was measured, corrected for transfection efficiency and expressed as relative light units (RLU×103). D, LβT2 cells were pretreated with U73122 for 30 min, followed by stimulation with GnRH (100 nm for 30 min) and total inositol phosphate (InsP) production was determined. A representative experiment of the three preformed, each done in triplicate is shown. E, Effect of PG on GnRH-induced phosphoinositide turnover. LβT2 cells were pretreated for 30 min with PGE2, iloprost or PGF2α (500 nm) followed by a 30-min incubation with increasing doses of GnRH in the presence and absence of PGE2, PGF2α or iloprost (500 nm), and total inositol phosphates (InsP) production was determined as detailed in Materials and Methods. A representative experiment of the three preformed, each done in triplicate is shown. One-way ANOVA determined that ***, P < 0.001; **, P < 0.01; and *, P < 0.05 were significantly different between treatment groups.
Effects of Prostanoids on GnRH-Stimulated α-Gonadotropin Subunit (αGSU), LHβ, and FSHβ Gene Expression
To determine whether prostanoids affect the GnRH transcriptional regulation of gonadotropin subunit genes (αGSU, LHβ, and FSHβ). LβT2 cells were transfected with αGSU, LHβ, or FSHβ promoters driving a luciferase reporter (Fig. 5). The exogenous addition of the PGs had no significant effect on GnRH induction of αGSU (Fig. 5A) and FSHβ (Fig. 5C) genes. However, PGF2α but not PGE2 or iloprost, reduced GnRH activation of the LHβ promoter (Fig. 5B; P < 0.05).
Fig. 5. Effect of PG on GnRH-Induced Gonadotropin Subunit Promoter Activities.
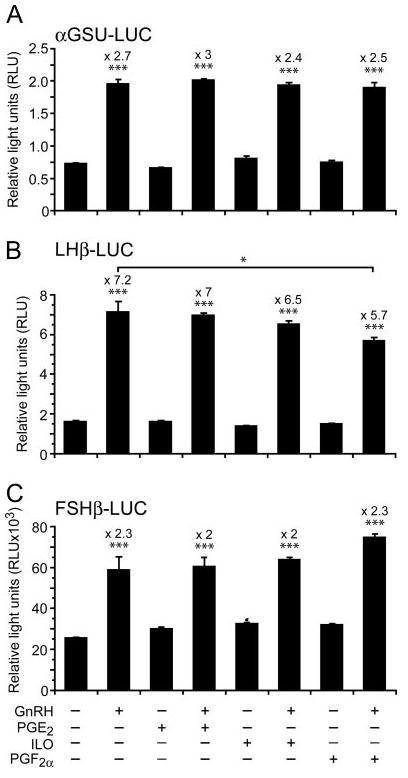
LβT2 cells were transiently transfected with αGSU-Luc (A), LHβ-Luc (B) and FSHβ-Luc (C) promoter constructs before being pretreated for 30 min with PGE2 or PGF2α or iloprost (500 nm) followed by addition of GnRH (100 nm for 6 h). Cells were harvested and assayed for an increase in reporter gene expression expressed as relative light units (RLU), after normalization of transfection efficiency with an internal control. GnRH significantly increased all three subunits (P < 0.001, one-way ANOVA). Note that only PGF2α had a significant inhibitory effect on GnRH induction of LHβ-Luc (*, P < 0.05).
Expression of Prostanoid Receptors in Rat Gonadotropes
We examined the expression of the various prostanoid receptors in rat pituitaries by immunohistochemistry and immunofluorescent microscopy using confocal microscopy (Fig. 6). Specific expression of a given receptor was followed by colocalization with pituitary hormones for potential cell-specific expression. Specific staining for receptors for PGE2 (EP1, EP2, EP3, and EP4), for PGF2α (FP) and PGI2 (IP) were demonstrated by immunohistochemistry. Negative controls included omission of the first antibody, pretreatment with preimmune serum and peptide immunogen competition. EP1 and EP2 staining colocalized to LH-containing gonadotropes, which comprise about 10–15% of the total cell population in the adult rat pituitary (22) (Fig. 6). EP3 and EP4 colocalized to prolactin-containing cells (mammotropes) and the GH-containing cells (somatotropes), respectively, which comprise about 20–30% and 40% of the total cell population in the adult rat pituitary (22) (data not shown). The PGI2 receptor IP and the PGF2α receptor FP were found to colocalize to the gonadotropes (Fig. 6).
Fig. 6. Expression of Prostanoid Receptors in Rat Pituitaries and Colocalization with LH.
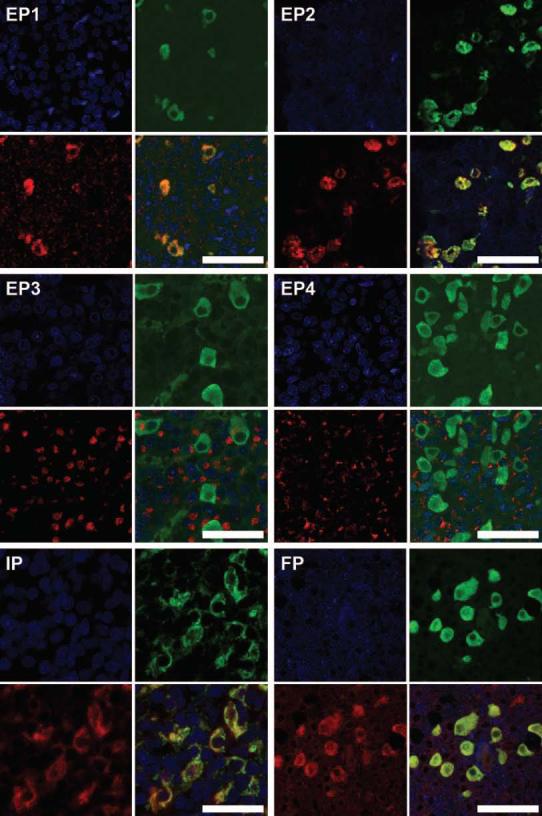
Rat pituitary sections were fixed and immunostained with antibodies to EP1 (1:40), EP2, (1:40), EP3 (1:40), EP4 (1:100), IP (1:100) and FP (1:100) (red). The sections were also stained for LHβ (green) and nuclear counterstained with TOPRO-3 (blue). Colocalization of prostanoids receptors and LH is examined by merge on the lower right figure for each receptor (bar, 50 μm). Note that EP1, EP2, IP, and FP colocalize to the gonadotropes.
Differential Inhibitory Effects of PGF2α on LH Release
Because prostanoids affect gonadal function (23) and GnRH secretion (24), it is not feasible to study their direct effects on pituitary function in vivo. We therefore sought to obtain direct evidence for a role for PGs on GnRH stimulation of gonadotropin secretion using an ex vivo approach. Rat pituitaries were stimulated with GnRH alone or in combination with indomethacin (COX enzyme inhibitor) or PGs. Indomethacin and the PGs had no significant effect on basal gonadotropin secretion (Fig. 7). COX enzyme inhibition by indomethacin treatment enhanced the GnRH-stimulated LH but not FSH secretion (Fig. 7, A and B; P < 0.05). Co-stimulation of pituitaries with GnRH and PGE2 or iloprost had no effect on the secretion of either gonadotropin in response to GnRH. However, PGF2α inhibited the GnRH-stimulated LH release, but had no effect on FSH release (Fig. 7, C and D; P < 0.01).
Fig. 7. Effect of the COX Inhibitor, Indomethacin and PG on GnRH-Induced Gonadotropin Secretion.
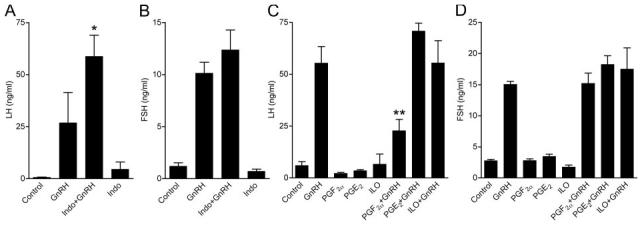
Rat pituitaries were pretreated with indomethacin (8 μm), PGE2, PGF2α, or iloprost (500 nm) for 4 h and further incubated with the drugs with or without GnRH (100 nm) for 3 h (Indomethacin + GnRH) and 4 h (PG + GnRH). The medium was collected and LH (A and C) and FSH (B and D) were determined by RIA. Note that indomethacin enhanced GnRH-stimulated LH, but not FSH release, whereas PGF2α, but not PGE2 or iloprost, inhibited GnRH-stimulated LH, but not FSH release. One-way ANOVA determined that **, P < 0.01 and *, P < 0.05 were significantly different between treatment groups.
DISCUSSION
GnRH is the central regulator of the reproductive system through the differential and asynchronous stimulation of LH and FSH (3, 4, 21, 25). The precise cyclical regulation of LH and FSH is crucial to normal reproduction and dysregulation results in conditions such as polycystic ovarian syndrome and amenorrhea anorexic patients (26). Feedback by ovarian steroid and peptide hormone at the pituitary gonadotrope and changes in GnRH pulse frequency and GnRHR expression have been proposed to contribute to this cyclical secretion (27, 28). GnRH auto-sensitizes the gonadotropes by up-regulating its own receptor preparatory to the onset of puberty and for the LH surge that triggers ovulation (1, 2, 29). GnRHR number increases from the evening of diestrus in rodents until late afternoon of proestrus, which culminates in the surge in LH secretion in late proestrus (2, 29). Conversely, potential signals to down-regulate GnRHR in mouse gonadotropes include the gonadal steroids and changes in pulse frequencies of GnRH (21, 30). In the sheep, the gonadotrope becomes refractory to GnRH after the LH surge (31). Despite these advances, the mechanisms involved have not been fully elucidated. We have now demonstrated that GnRH stimulates PG biosynthesis, which inhibits GnRHR and LH, but not FSH gene expression and hormone secretion. These findings thus provide a potential autocrine mechanism of refractoriness to GnRH after the LH surge and asynchronous gonadotropin secretion during the ovarian cycle (Fig. 8). Our results further provide a molecular mechanism to modulate GnRHR number and to mediate gonadotrope desensitization during prolonged GnRH stimulation. This is of particular importance because it provides a mechanism for the down-regulation of the GnRHR that is the only mammalian GPCR that lacks a cytoplasmic carboxyl-terminal tail and consequently does not undergo rapid desensitization and internalization as in other GPCRs (32-35). The loss of the carboxyl-terminal tail and lack of rapid desensitization is thought to have evolved to allow the prolonged LH surge, which is required for ovulation in women (32).
Fig. 8. Schematic Representation of the Inside-Out Prostanoid Signaling Pathway that Mediates GnRHR Autoregulation and LH Release.
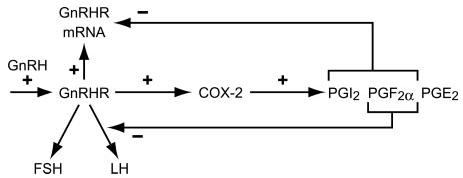
GnRH up-regulates the expression of its receptor (GnRHR) and COX-2 involved in the synthesis of prostaglandins. This is associated with elevated synthesis and release of various prostaglandins (PGI2, PGF2α, and PGE2). Prostaglandins (namely PGI2 and PGF2α, but not PGE2) limit the effect of GnRH on target cells by down-regulating the expression of the GnRHR. Moreover, elevated PGF2α synthesis (but not PGI2 or PGE2) can have diverging effects on the GnRH-induced release of gonadotropins from the pituitary, with negative effect on LH release but no effect on FSH release.
Multiple ligands interact with pituitary gonadotropes. In addition to GnRH, gonadal steroids and peptide hormones, growth factors, neurotransmitters, melatonin, and pituitary hormones have all been demonstrated to act as regulators (9). Hence, addition of a single ligand to cultured cells in vitro does not mimic the in vivo situation. Recognizing these caveats, attempts have been made to study the effect of multiple ligands simultaneously. Here we took another approach, which is based on the assumption that because many ligands stimulate PGs production, one can use these signaling molecules to elucidate the integrative signaling for a given ligand and better mimic the in vivo situation. Moreover, prostanoids may play a role in mediating the effects of ligands other than GnRH.
Eicosanoids are hydrolyzed from AA by PLA2, a super family of enzymes consisting of at least 14 groups (I–XIV) and at least 19 members (36, 37). Once released by PLA2, AA is converted to prostanoids (PG and thromboxanes) via COX-1, COX-2, and prostanoid synthases or undergoes dioxygenation to hydroperoxide derivatives by lipoxygenases or to epoxygenase products via cytochrome p450 epoxygenase (cyp2C11) (Ibid). We have previously shown that GnRH stimulates AA release from rat pituitaries (38) and have implicated 5-lipoxygenase and 12-lipoxygenase products of AA in GnRH actions (3, 12). Here, we have investigated the cross talk of GnRH and prostanoid receptors.
Because the inducible COX isoform (COX-2) is the key enzyme in the prostanoid synthesis pathway, we looked for activation of COX-2 by GnRH and showed a 40-fold increase in expression. A recent report documented that induction of COX-2 by angiotensin II reflects an immediate-early gene response (39). Our results support the findings because we show a rapid activation of the COX-2 promoter by GnRH (2 h), mRNA and protein production (4–8 h). The use of pharmacological inhibitors revealed a role for the PKC/c-Src/MAPK pathway, but not for EGF receptor kinase in the GnRH induction of COX-2. The results accord with previous studies suggesting that activation of MAPK by GnRH in gonadotropes is mediated mainly by PKC and c-Src but not via transactivation of the EGF receptor (14, 15, 18, 19, 40-46). MAPKs have previously been implicated in the induction of COX-2 (47, 48) and a role for PI3K and c-Src in COX-2 expression has been demonstrated (49). Indeed, GnRH activates c-Src and PI3K in LβT2 cells (19, 50). We also examined the role of PLA2 that has been implicated in COX-2 induction (51). Using specific inhibitors (36, 37), we found that iPLA2 and to a lesser degree sPLA2, but not cPLA2 as in Ref. 51, participate in GnRH induction of COX-2. Hence, iPLA2 and sPLA2 may also be involved in the induction of COX-2 possibly by a feed-forward mechanism via the newly formed PGs as described below. Thus, the signaling pathways and the various PLA2 and MAPK members are likely to be implicated in COX-2 induction in an agonist and cell context-dependent manner (39, 47, 48, 51) (present results). Activated ERK phosphorylates various transcription factors (e.g. c-Jun, c-Fos, ELK-1, and Sap-1) resulting in the activation of activator protein (AP)-1 (52), which binds and activates the COX-2 promoter via the CRE/ATF response element (53). ERK phosphorylation is also required for persistent activation of nuclear factor-κB, which is also involved in COX-2 transcription (54).
AA is converted to prostanoids via activated COX-2 and specific PG synthases (11). We therefore investigated whether GnRH activation of COX-2 results in PG synthesis. We first failed to detect production of PG after prolonged incubation of the cells with GnRH. We reasoned that although PLA2 activation by GnRH may be rapid (min), COX-2 induction is a slow process (4–8 h) and might be a limiting step in PG production. Furthermore, due to rapid uptake, the availability of free AA is a rate-limiting step in PG synthesis. Therefore, the two events, namely induction of COX-2 and availability of free AA, may not be appropriately and temporally coordinated during prolonged incubation periods in vitro. Indeed, exogenous AA was needed to demonstrate PGE2 production by IL-1 (20). Similarly, we could only detect stimulation of PG production by GnRH when exogenous AA was added for the last 30–60 min of the incubation period. The dose of AA used here (1 μm), is below the Michaelis-Menten constant (Km) values of 5-lipoxygenase and COX (55). In vivo, pituitary gonadotropes are exposed to GnRH pulses every 30–90 min (according to the species) thus, COX-2 induced in previous pulses may act on AA generated by PLA2 in the current pulse resulting in rapid formation of PGs. Once formed, PGF2α and PGI2 are now capable of inhibiting GnRH-stimulated phosphoinositide turnover (30 min), GnRHR promoter activity (4 h), GnRH binding sites and LHβ promoter activity (6 h) and LH release (8 h). Provided COX-2 was activated by GnRH for 2–4 h, once AA was added for 30 min (at any preincubation with GnRH for up to 24 h), we could detect the formation of the PGs at the end of the 30 min exposure to AA (Fig. 2). These temporal differences in activation of enzymes and substrate generation for PG production, which in turn regulate GnRHR and gonadotropin provides an elegant mechanism for translating changes in GnRH pulse frequency into physiological outputs.
Prostanoids are known to act in an autocrine/paracrine manner via cognate GPCRs (11). We therefore examined the expression of the various prostanoid receptors in rat pituitaries. Specific expression of receptors for PGE2 (EP1, EP2, EP3, and EP4), for PGF2α (FP) and PGI2 (IP) were found. EP1, EP2, IP, and FP colocalize to rat gonadotropes (Fig. 6), which comprise about 10–15% of the total cell population in the adult rat pituitary (22). EP3 and EP4 are present in the prolactin and GH producing cells, the mammotropes (20–30% of the total cell population) and somatotropes (∼40% of the cells), respectively (data not shown). The prostanoid receptors EP1, EP2, EP3, EP4, IP, and FP were detected in the embryonic-derived mouse LβT2 gonadotrope cells. The staining of the various prostanoid receptors was observed in the cytoplasmic and perinuclear membrane regions, and in some cases in the nucleus (Fig. 3). Because prostanoid receptor signaling is rapidly terminated, it is likely that only a small fraction of the receptor pool is at the plasma membrane under basal conditions and prostanoid receptors may rapidly cycle between the cell surface and the intracellular pool, with a relatively short half-life at the plasma membrane, as for many GPCRs. The predominantly intracellular staining observed here was also reported by others for different cell models. EP1 has been described to reside in the cytoplasm (56). The thromboxane A2/prostaglandin H2 receptor was found in the endoplasmic reticulum (57) and EP3 and EP4 were found in the nuclear envelope (58, 59). It is interesting to note that the dogma that PLA2 liberates AA from the sn-2 position of plasma membrane phospholipids has been challenged by the observations that both cPLA2α and COX-1/2 reside in the endoplasmic reticulum and the nuclear envelope membrane, where PGs are formed (60). It is therefore tempting to suggest that, aside from PG secretion and binding to receptors on the plasma membrane, PGs may also act intracellularly in the vicinity of their site of origin. The finding that EP3 and EP4 are present in LβT2 cells but not in rat pituitary gonadotropes may reflect incomplete differentiation of this gonadotrope-derived cell line and that maturation of murine gonadotropes may involve the loss of expression of both EP3 and EP4.
These studies begged the question as to whether the newly formed PGs affect the kernal physiological outcome of GnRH actions, namely the transcriptional regulation of gonadotropin subunits (αGSU, LHβ, and FSHβ) and gonadotropin secretion (21). Treatment with PGs had no significant effect on the induction of αGSU and FSHβ genes by GnRH. However, PGF2α, but not PGE2 or PGI2, significantly reduced the activation of the LHβ promoter by GnRH. We also investigated whether the exogenous addition of PGs affected the homologous induction of the GnRHR (21). The induction of GnRHR by GnRH was markedly reduced by PGF2α and PGI2, but not by PGE2. Homologous activation of the mouse GnRHR identified a role for PKC/MAPK/AP-1 signaling and CRE elements (21). Phosphoinositide turnover is tightly coupled to the PKC/MAPK/AP-1/CREB pathways (14, 52, 61). We therefore targeted phosphoinositide turnover as the first potential upstream signaling module for PG action. We found inhibition of GnRH-stimulated InsP formation by PGF2α and PGI2, but not PGE2, in line with their inhibition of the GnRHR. The link of phosphoinositide turnover to GnRHR induction was revealed by the use of the PLC-β inhibitor U73122, which produced a dose-related inhibition of GnRH-induced InsP formation and GnRHR promoter activity. Thus, inhibition of GnRHR by PGF2α and PGI2 appears to be mediated by an inhibition of phosphoinositide turnover, perhaps leading to an alteration of protein complexes at the AP-1 sites in the promoter (21).
Our findings present an enigma in that these ligands bind the FP and IP receptors, which stimulate InsP (11). Thus, in gonadotropes expressing GnRH, FP, and IP receptors (which are all believed to stimulate lnsP), EP1 and EP2 (which are believed to stimulate lnsP and cAMP, respectively), a more complex picture emerges and activation of the FP and IP receptors actually inhibits GnRHR stimulation of InsP. Hence, the presence of multiple receptors in the same cell may result in a different signaling profile from that obtained by over expressing a single prostanoid receptor. The dogma that PG receptors have discrete signaling pathways (11) should therefore be revisited because our results suggest that cell context and expression of multiple prostanoid receptors in the same cell can dictate the flavor of the signaling. The mechanism involved in PG inhibition of GnRH-stimulated InsP formation may include potentiating GnRH-stimulated PKC isoforms activation (62), which can feed back and inhibit PLC-β, or activation of specific InsP phosphatases.
Our demonstration of EP1, EP2, FP, and IP receptor expression in rat gonadotropes by double fluorescent confocal microscopy suggested that PGE2, PGF2α, and PGI2 might have direct effects on gonadotrope function. In vivo support for our in vitro cell line findings on inhibitory effects of endogenous PG on GnRHRs and LHβ expression was obtained by using an ex vivo approach of rat pituitaries incubated with GnRH alone or in the presence of the COX inhibitor, indomethacin or various PGs. Inhibition of COX enzyme activity by indomethacin treatment enhanced GnRH-stimulated LH but not FSH secretion. Furthermore, PGF2α, but not PGE2 or PGI2, inhibited GnRH-stimulated LH, but had no effect on FSH release. Thus, only the FP receptor in gonadotropes appears to regulate LH in this nondynamic system. Whereas the FP receptor therefore appears to play a significant role in differential gonadotropin secretion in response to GnRH, the findings do not rule out more subtle roles for the IP and EP receptors expressed in gonadotropes.
The data suggest that the reduction of GnRHR expression is insufficient to reduce LH secretion as evident from the lack of effect of PGI2, which like PGF2α reduced GnRHR expression but had no effect on LH secretion. Hence, the reduction of both GnRHR and LHβ expression, as observed with PGF2α are required to affect the LH exocytotic apparatus. The data also suggest that FSH secretion is less sensitive to the reduction of GnRHRs because it was not affected by PGI2 and PGF2α. The results are in line with a report that found that at high GnRHR concentrations, GnRH activates the αGSU and LHβ genes and exerts a selective inhibition of the FSHβ gene, whereas the FSHβ gene is optimally activated at lower number of GnRHRs (27).
In summary, our results provide a novel inside-out molecular mechanism to regulate GnRHR number and to mediate the differential LH and FSH secretion during GnRH stimulation (Fig. 8). These findings provide a basis for the use of combination therapies of GnRH and prostanoid analogs to treat infertility and diseases with unbalanced LH and FSH secretion such as polycystic ovarian syndrome. Combination therapy may also be contemplated in hormone-dependent disease such as prostatic cancer and endometriosis. These diseases are currently treated with GnRH analogs for inhibition of gonadotropin and hence sex steroids. Because PGs affect GnRHR and LHβ, modulation of PG input on gonadotropes has the potential to increase the efficacy of the GnRH analogs.
MATERIALS AND METHODS
Materials
Culture medium was from Invitrogen Inc. (Paisley, Scotland, UK). Penicillin-streptomycin and fetal calf serum (FCS) were from PAA Laboratories Ltd. (Middlesex, UK). GnRH was from Peninsula (St. Helens, UK). Prostaglandin E2 (PGE2), the stable PGI2 analog Iloprost, prostaglandin F2α (PGF2α), rabbit polyclonal antibodies to the various PG receptors and respective peptides, arachidonyl trifluoromethyl ketone (AACOCF3), TEPC, BEL, and ELISA kits for the PG were from Cayman Chemical Co. (Alexis Corp., Nottingham, UK). Rabbit antirat antibodies for LHβ and prolactin and guinea pig antirat GH were from National Hormone and Peptide Program [National Institutes of Health (NIH)]. Secondary horseradish peroxidase (HRP)-conjugated goat antimouse antibodies or goat antirabbit antibodies were from Jackson ImmunoResearch Laboratories (West Grove, PA). PD980595, a peptide JNK inhibitor (JNKI; Calbiochem 420116), SB203580, GF109203X, wortmannin, PP2, AG1478, and U73122 were from Calbiochem (Nottingham, UK).
Cell Culture
LβT2 cells were kindly obtained from P. Mellon (University of California, San Diego, San Diego, CA). The cells were cultured in matrigel (Becton Dickinson, Oxford, UK)-coated plastic flasks in DMEM/10% FCS/glutamine medium (Sigma, Poole, UK), with 1% antibiotics (stock 500 IU/ml penicillin and 500 μg/ml streptomycin) at 37 C and 5% CO2 (vol/vol) (19).
Immunohistochemistry
Adult rat pituitaries were fixed for either 6 or 24 h in Bouins, 4% neutral buffered formaldehyde (NBF) or modified Davidson's fixative. Pituitaries were processed to paraffin using standard procedures and 5μm sections were mounted on superfrost plus slides (BDH, Poole, Dorset, UK). Cells were cultured on chamber slides (Lab Tek, Naperville, IL) rinsed for 5 min in PBS followed by 10 min fixation. Each antiserum was tested using a range of dilutions on paraffin sections of rat pituitary fixed as above. They were evaluated with and without heat induced antigen retrieval using a pressure cooker (Tefal, Cambridge, UK) and 0.01 m citrate. For all antisera tested, antigen retrieval was not required. Negative controls were either omission of primary antisera or peptide absorption of antisera using a 10-fold excess of peptide incubated at 4 C overnight. Antisera were evaluated on LβT2 cells after 10 min fixation in 4% NBF. Sections were dewaxed in xylene before being rehydrated in graded ethanol. Sections were blocked in 3% hydrogen peroxide in Methanol for 30 min (HRP and Tyramide detections only), rinsed in running tap water then washed for 5 min in Tris-buffered saline (TBS, 0.05 m Tris; and 0.85% NaCl, pH 7.4). Sections were blocked in 20% normal swine serum in TBS (TBS/NSS) for 30 min at room temperature. Slide were drained and incubated with Primary antiserum diluted in TBS/NSS overnight at 4 C. After two 5-min washes in TBS, sections were incubated for 30 min with Swine anti Rabbit Biotinylated at 1:500 dilution in TBS/NSS for 30 min at room temperature. After two 5-min washes in TBS, sections were incubated for 30 min with HRP-conjugated avidin-biotin complex (ABC-HRP) (Dako, Cambridgeshire, UK). Sections were washed for a further two 5-min washes in TBS before visualizing with diaminobenzidine (Dako).
Immunofluorescence (LH, Prolactin, and GH)
After dewaxing and methanol/hydrogen peroxide blocking (if necessary) as described above. Sections were incubated in 20% normal goat serum in PBS (NGS/PBS). Slides were drained and incubated with Primary antiserum diluted in NGS/PBS overnight at 4 C. After two 5-min washes in PBS, sections were incubated for 30 min with either Goat antiguinea pig biotinylated at 1:500 dilution in PBS/NGS for 30 min at room temperature (prolactin, GH), or goat antimouse Alexa 488 at 1:200 dilution in PBS/NGS (LH). For GH or prolactin staining, after two 5-min washes in PBS, sections were incubated for 30 min with Streptavidin Alexa 488. All sections were washed for a further two 5-min washes in PBS before being counterstained in TOPRO-3 at 1:2000 dilution and coverslipped using permaflour (Coulter, Buckinghamshire, UK).
Fluorescent Tyramide Detection
After dewaxing and methanol/hydrogen peroxide blocking as described above. Sections were incubated in 20% NGS/PBS. Slides were drained and incubated with primary antiserum diluted in NGS/PBS overnight at 4 C. After two 5-min washes in PBS, sections were incubated for 30 min with goat antirabbit peroxidase at a 1:200 dilution in PBS/NGS for 30 min at room temperature. After two 5-min washes in PBS, sections were incubated for 10 min with Tyramide Cy3 Sections were washed for a further two 5-min washes in PBS before being counterstained in TOPRO-3 at 1:2000, and cover slipped using permafluour (Coulter).
Immunofluorescent Colocalization
After standard immunofluorescent detection as described above (prolactin, GH, and LH) sections were blocked in peroxide block for 10 min (Dako) before detection of EP1–4, IP, or FP using Fluorescent Tyramide Detection as described above. After two 5-min washes in PBS sections were counterstained in TOPRO-3 at 1:2000 and cover slipped using permaflour. Bright-field images were captured using an Olympus (Center Valley, PA) Provis microscope fitted with a Kodak (Rochester, NY) DCS 330 digital camera Fluorescent images were captured using a Zeiss (Oberkochen, Germany) LSM 510 confocal microscope.
PCR
Total RNA was isolated from cells using Tri-reagent (Sigma) according to the manufacturer's instructions. The RNA yield was calculated using ultraviolet spectrophotometry and samples were stored as an ethanol precipitate at −80 C before RT-PCR analysis. RNA samples (5 μg) were reverse-transcribed using deoxynucleotide triphosphate (dNTPs) (0.2 mm each), random primers (200 ng), ribonuclease inhibitor (2 U/μl) and SuperScript reverse transcriptase (10 U/μl; Invitrogen). The reverse transcription product (200 ng cDNA) was then amplified by PCR using homologous primers designed from the mRNA sequences of the various EP receptors and the COX enzymes. The sequence of the primers (forward and reverse primers respectively) was as follows: EP1, 5′-CGCTCCTTGCGGCATTAGTGTGC, 5′-CCAACACCACCAACACCAGCAGG; EP2, 5′-TAGGGCAGGTGAGGCACAGAAGC, 5′-GAAAGGAGCCACTGACGACTCTTGC; EP3, 5′-ATGTGTGTGCTGTCCGTCTGTTGG, 5′-CAACCAGACTCTCAGATTATCC; EP4, 5′-AGACACCACCTCGCTGAGAACTTTGC, 5′-CTTCAAGCCTGGGCACTCAAGGACC; FP, 5′-TAGGGAGGAAAGAGAGGTGGAACC, 5′-TGACTTCTGTCTAAATCTCTGG; IP, 5′-GATGCCGAAGGTTCTATGGC, 5′-TGTGTCCAGCAATGTCACCTGC; COX-2, 5′-TGCCACCTCTGCGATGCTCTTCC, 5′-CAGACTCCCTTGAAGTGGGTCAGG. Primers were selected to match sequences located in separate exons, enabling detection of spliced transcripts. The PCR mix consisted of 1× reaction buffer containing 1.5 mm MgCl2, 0.2 mm dNTPs, 0.2 μm of each primer and 1.25 U Taq DNA polymerase (Abgene, Epsom, Surrey, UK). Samples were denatured at 94 C for 5min, and then amplified by 35–40 cycles of 94 C for 1 min, 60 C for 30 sec and 72 C for 40 sec with a final extension of 72 C for 10 min. After amplification, 10 μl of each sample was visualized on a 1% agarose gel. Normal mouse uterus was used as a positive control (63).
Taqman Quantitative RT-PCR
RNA samples were quantified and reverse-transcribed using 5.5 mm MgCl2, 0.5 mm of each dNTP, 2.5 μm random hexamers, ribonuclease inhibitor (0.4 U/μl) and 1.25 U/μl Multiscribe reverse transcriptase (all from Applied Biosystems, Warrington, Cheshire, UK). RNA (200 ng) was added to each 10 μl reverse transcription reaction and samples were incubated for 60 min at 25 C, 45min at 48 C and 5 min at 95 C. The reaction mix for the PCR consisted of 1× mastermix, ribosomal 18S forward and reverse primers, ribosomal 18S probe (50 nm; all from Applied Biosystems), forward and reverse primers for COX-2 (300 nm) and COX-2 probe (200 nm) (all from Biosource UK, Nivelles, Belgium). The reaction mix (48 μl) was aliquoted into tubes and 2 μl cDNA was added. Duplicate 24-μl samples plus positive and negative controls were placed in a PCR plate and wells were sealed with optical caps. The PCRs were carried out using an ABI Prism 7700 (Applied Biosystems). All primers and probes were designed using the PRIMER express program (Applied Biosystems). The sequences of the COX-2 primers (forward, reverse, and probe, 6-carboxy fluoroscein labeled) were: COX-2, 5′-GCTTCGGGAGCACAACAGA-3′; 5′-TGGTTTGGAATAGTTGCTCATCAC-3′; 5′-TGTGCGACATACTCAAGCAGGAGCATC-3′. Data were analyzed and processed using Sequence Detector version 1.6.3 (Applied Biosystems) according to the manufacturer's instructions. Results were expressed relative to an internal positive standard cDNA obtained from a single sample of mouse uterus (63).
PG Production
LβT2 cells were grown in 4 × 12-well plates (5 × 105 cells/well). Cells were incubated in serum-free DMEM, 0.2% FCS overnight at 37 C. The cells were washed with DMEM and incubated with or without GnRH for various time periods as detailed in the legends. Some of the wells received exogenous AA (1 μm) for the last 30 or 60 min of each incubation time. At the end of the incubation time, the media were removed and stored at −20 C for determination of PGE2, PGI2, and PGF2α by ELISA as described in Ref. 64.
Reporter Gene Assays
The reporter constructs are described in (65), except 2307/+49 human COX-2 promoter [pGL3.C2.2 (P6P5)], which was obtained from R. Newton (University of Warwick, Coventry, UK). Reporter gene assays were carried out as recently described (19, 65, 66).
Receptor Binding Assays
Whole cell receptor binding assays used the 125I-[His (5), d-Tyr (6)]GnRH analog as described (67).
Inositol Phosphates Assays
LβT2 cells were incubated with GnRH (100 nm) in the presence and absence of PGE2, iloprost or PGF2α (500 nm) for 30 min and total inositol phosphate production was determined as previously described (68).
Gonadotropin Secretion
Pituitaries from 23-d-old Wistar-derived female rats were preincubated with indomethacin (8 μm), PGE2, iloprost or PGF2α (500 nm) in Krebs-Ringer-bicarbonate for 4 h, washed twice and further incubated with the drugs with or without GnRH (100 nm) for 3 h (Indomethacin + GnRH) and 4 h (PG + GnRH). The medium was collected and LH and FSH were determined by RIA using the kit provided by the National Hormone and Peptide Program (NIH).
Data Analysis
Results from two or three experiments were expressed as mean ± sem. Where appropriate, data were subjected to statistical analysis withy one-way ANOVA and Fisher's protected least significant difference tests (Statview 5.0; Abacus Concepts Inc., Carpinteria, CA), and statistical significance accepted when P < 0.05.
Acknowledgments
We thank Alan McNeilly, Rodney Kelly, and Gerald Lincoln [all from the Medical Research Council (MRC)], Naftali Stern (Tel Aviv University), David Klein (National Institutes of Health), and Iain Clarke (Prince Henry's Institute of Medical Research, Clayton, Australia) for their interest and input. We also thank Nancy Nelson, Dimitra Karali, Vivian Grant, Sheila MacPherson, and Nicola Miller (all from MRC), Fiorenza Prezedecki (Tel Aviv University), and Robin Sellar (MRC) for excellent technical assistance and Pamela Mellon (University of California, San Diego, CA) for the gonadotrope cell lines.
This research was supported by the Medical Research Council (United Kingdom), Medical Research Council (South Africa), and The Israel Science Foundation (Grant No. 221/05).
Abbreviations
- AA
Arachidonic acid
- AACOCF3
arachidonyltrifluoromethyl ketone
- ABC
avidin-biotin complex
- AP
activator protein
- BEL
bromoenol lactone
- COX
cyclooxygenase
- cpm
counts per minute
- CRE
cAMP responsive element
- dNTP
deoxynucleotide triphosphate
- EGF
epidermal growth factor
- EP
PGE2 receptor
- FCS
fetal calf serum
- FP
PGF2α receptor
- GnRHR
GnRH receptor
- G protein
guanine nucleotide binding protein
- GPCR
G protein-coupled receptor
- αGSU
α-gonadotropin subunit
- HRP
horseradish peroxidase
- InsP
inositol-phosphate
- IP
PGI2 receptor
- JNKI
JNK inhibitory peptide
- NBF
neutral buffered formaldehyde
- NGS
normal goat serum
- PLA2,
phospholipase A2
- cPLA2,
cytosolic phospholipase A2
- iPLA2
Ca2+-independent phospholipase A2
- PLC-β
phospholipase C-β
- PKC
protein kinase C
- PKCs
PKC isoforms
- PG
prostaglandin
- PI3K
phosphatidylinositol 3′-kinase
- TE-PC
thioetheramide-PC
Footnotes
Disclosure Statement: Z.N., H.N.J., M.N., A.J.P., K.M., S.B., M.R.M., P.B. have nothing to disclose. R.P.M. consults for Ardana plc.
REFERENCES
- 1.Loumaye E, Catt KJ. Homologous regulation of gonadotropin-releasing hormone receptors in cultured pituitary cells. Science. 1982;215:983–985. doi: 10.1126/science.6296998. [DOI] [PubMed] [Google Scholar]
- 2.Savoy-Moore RT, Schwartz NB, Duncan JA, Marshall JC. Pituitary gonadotropin-releasing hormone receptors during the rat estrous cycle. Science. 1980;209:942–944. doi: 10.1126/science.6250218. [DOI] [PubMed] [Google Scholar]
- 3.Naor Z. Signal transduction mechanisms of Ca2+ mobilizing hormones: the case of gonadotropin-releasing hormone. Endocr Rev. 1990;11:326–353. doi: 10.1210/edrv-11-2-326. [DOI] [PubMed] [Google Scholar]
- 4.Millar RP, Lu ZL, Pawson AJ, Flanagan CA, Morgan K, Maudsley SR. Gonadotropin-releasing hormone receptors. Endocr Rev. 2004;25:235–275. doi: 10.1210/er.2003-0002. [DOI] [PubMed] [Google Scholar]
- 5.Filicori M, Flamigni C, Vizziello G, Dalpiaz C, Ferrari P, Flamia R, Santoro N, Crowley WF., Jr Hypothalamic control of gonadotropin secretion in the human menstrual cycle. Prog Clin Biol Res. 1986;225:55–74. [PubMed] [Google Scholar]
- 6.Santoro N, Filicori M, Crowley WF., Jr Hypogonadotropic disorders in men and women: diagnosis and therapy with pulsatile gonadotropin-releasing hormone. Endocr Rev. 1986;7:11–23. doi: 10.1210/edrv-7-1-11. [DOI] [PubMed] [Google Scholar]
- 7.Martin K, Santoro N, Hall J, Filicori M, Wierman M, Crowley WF., Jr Clinical review 15: management of ovulatory disorders with pulsatile gonadotropin-releasing hormone. J Clin Endocrinol Metab. 1990;71:1081A–1081G. doi: 10.1210/jcem-71-5-1081. [DOI] [PubMed] [Google Scholar]
- 8.Hayes FJ, Hall JE, Boepple PA, Crowley WF., Jr Clinical review 96: differential control of gonadotropin secretion in the human: endocrine role of inhibin. J Clin Endocrinol Metab. 1998;83:1835–1841. doi: 10.1210/jcem.83.6.4884. [DOI] [PubMed] [Google Scholar]
- 9.Evans JJ. Modulation of gonadotropin levels by peptides acting at the anterior pituitary gland. Endocr Rev. 1999;20:46–67. doi: 10.1210/edrv.20.1.0355. [DOI] [PubMed] [Google Scholar]
- 10.Mollace V, Muscoli C, Masini E, Cuzzocrea S, Salvemini D. Modulation of prostaglandin biosynthesis by nitric oxide and nitric oxide donors. Pharmacol Rev. 2005;57:217–252. doi: 10.1124/pr.57.2.1. [DOI] [PubMed] [Google Scholar]
- 11.Hata AN, Breyer RM. Pharmacology and signaling of prostaglandin receptors: multiple roles in inflammation and immune modulation. Pharmacol Ther. 2004;103:147–166. doi: 10.1016/j.pharmthera.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 12.Limor R, Naor Z, Weisinger G, Naidich M, Knoll E, Sharon O, Stern N. Gonadotropin-releasing hormone activates the 12-lipoxygenase pathway in the LβT2 gonadotrope cell line. Neuroendocrinology. 2003;77:291–297. doi: 10.1159/000070895. [DOI] [PubMed] [Google Scholar]
- 13.Turgeon JL, Kimura Y, Waring DW, Mellon PL. Steroid and pulsatile gonadotropin-releasing hormone (GnRH) regulation of luteinizing hormone and GnRH receptor in a novel gonadotrope cell line. Mol Endocrinol. 1996;10:439–450. doi: 10.1210/mend.10.4.8721988. [DOI] [PubMed] [Google Scholar]
- 14.Naor Z, Benard O, Seger R. Activation of MAPK cascades by G-protein-coupled receptors: the case of gonadotropin-releasing hormone receptor. Trends Endocrinol Metab. 2000;11:91–99. doi: 10.1016/s1043-2760(99)00232-5. [DOI] [PubMed] [Google Scholar]
- 15.Liu F, Austin DA, Mellon PL, Olefsky JM, Webster NJ. GnRH activates ERK1/2 leading to the induction of c-fos and LHβ protein expression in LβT2 cells. Mol Endocrinol. 2002;16:419–434. doi: 10.1210/mend.16.3.0791. [DOI] [PubMed] [Google Scholar]
- 16.Navratil AM, Bliss SP, Berghorn KA, Haughian JM, Farmerie TA, Graham JK, Clay CM, Roberson MS. Constitutive localization of the gonadotropin-releasing hormone (GnRH) receptor to low density membrane microdomains is necessary for GnRH signaling to ERK. J Biol Chem. 2003;278:31593–31602. doi: 10.1074/jbc.M304273200. [DOI] [PubMed] [Google Scholar]
- 17.Dobkin-Bekman M, Naidich M, Pawson AJ, Millar RP, Seger R, Naor Z. Activation of mitogen-activated protein kinase (MAPK) by GnRH is cell-context dependent. Mol Cell Endocrinol. 2006;252:184–190. doi: 10.1016/j.mce.2006.03.035. [DOI] [PubMed] [Google Scholar]
- 18.Benard O, Naor Z, Seger R. Role of dynamin, Src, and Ras in the protein kinase C-mediated activation of ERK by gonadotropin-releasing hormone. J Biol Chem. 2001;276:4554–4563. doi: 10.1074/jbc.M006995200. [DOI] [PubMed] [Google Scholar]
- 19.Bonfil D, Chuderland D, Kraus S, Shahbazian D, Friedberg I, Seger R, Naor Z. Extracellular signal-regulated kinase, Jun N-terminal kinase, p38, and c-Src are involved in gonadotropin-releasing hormone-stimulated activity of the glycoprotein hormone follicle-stimulating hormone β-subunit promoter. Endocrinology. 2004;145:2228–2244. doi: 10.1210/en.2003-1418. [DOI] [PubMed] [Google Scholar]
- 20.Chen QR, Miyaura C, Higashi S, Murakami M, Kudo I, Saito S, Hiraide T, Shibasaki Y, Suda T. Activation of cytosolic phospholipase A2 by platelet-derived growth factor is essential for cyclooxygenase-2-dependent prostaglandin E2 synthesis in mouse osteoblasts cultured with interleukin-1. J Biol Chem. 1997;272:5952–5958. doi: 10.1074/jbc.272.9.5952. [DOI] [PubMed] [Google Scholar]
- 21.Cheng CK, Leung PC. Molecular biology of gonadotropin-releasing hormone (GnRH)-I, GnRH-II and their receptors in humans. Endocr Rev. 2004;26:283–306. doi: 10.1210/er.2003-0039. [DOI] [PubMed] [Google Scholar]
- 22.Ibrahim SN, Moussa SM, Childs GV. Morphometric studies of rat anterior pituitary cells after gonadectomy: correlation of changes in gonadotropes with the serum levels of gonadotropins. Endocrinology. 1986;119:629–637. doi: 10.1210/endo-119-2-629. [DOI] [PubMed] [Google Scholar]
- 23.McCracken JA, Custer EE, Lamsa JC. Luteolysis: a neuroendocrine-mediated event. Physiol Rev. 1999;79:263–323. doi: 10.1152/physrev.1999.79.2.263. [DOI] [PubMed] [Google Scholar]
- 24.Ojeda SR, Naor Z, Negro-Vilar A. The role of prostaglandins in the control of gonadotropin and prolactin secretion. Prostaglandins Med. 1979;2:249–275. doi: 10.1016/0161-4630(79)90060-0. [DOI] [PubMed] [Google Scholar]
- 25.Brown P, McNeilly AS. Transcriptional regulation of pituitary gonadotrophin subunit genes. Rev Reprod. 1999;4:117–124. doi: 10.1530/ror.0.0040117. [DOI] [PubMed] [Google Scholar]
- 26.DeGroot LJ, Jameson JL, editors. Endocrinology. 5th ed. Philadelphia: Elsevier Saunders; 2004. [Google Scholar]
- 27.Kaiser UB, Sabbagh E, Katzenellenbogen RA, Conn PM, Chin WW. A mechanism for the differential regulation of gonadotropin subunit gene expression by gonadotropin-releasing hormone. Proc Natl Acad Sci USA. 1995;92:12280–12284. doi: 10.1073/pnas.92.26.12280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haisenleder DJ, Dalkin AC, Ortolano GA, Marshall JC, Shupnik MA. A pulsatile gonadotropin-releasing hormone stimulus is required to increase transcription of the gonadotropin subunit genes: evidence for differential regulation of transcription by pulse frequency in vivo. Endocrinology. 1991;128:509–517. doi: 10.1210/endo-128-1-509. [DOI] [PubMed] [Google Scholar]
- 29.Clayton RN, Catt KJ. Gonadotropin-releasing hormone receptors: characterization, physiological regulation, and relationship to reproductive function. Endocr Rev. 1981;2:186–209. doi: 10.1210/edrv-2-2-186. [DOI] [PubMed] [Google Scholar]
- 30.Kaiser UB, Conn PM, Chin WW. Studies of gonadotropin-releasing hormone (GnRH) action using GnRH receptor-expressing pituitary cell lines. Endocr Rev. 1997;18:46–70. doi: 10.1210/edrv.18.1.0289. [DOI] [PubMed] [Google Scholar]
- 31.Moenter SM, Caraty A, Locatelli A, Karsch FJ. Pattern of gonadotropin-releasing hormone (GnRH) secretion leading up to ovulation in the ewe: existence of a preovulatory GnRH surge. Endocrinology. 1991;129:1175–1182. doi: 10.1210/endo-129-3-1175. [DOI] [PubMed] [Google Scholar]
- 32.Pawson AJ, Katz A, Sun YM, Lopes J, Illing N, Millar RP, Davidson JS. Contrasting internalization kinetics of human and chicken gonadotropin-releasing hormone receptors mediated by C-terminal tail. J Endocrinol. 1998;156:R9–R12. doi: 10.1677/joe.0.156r009. [DOI] [PubMed] [Google Scholar]
- 33.Pierce KL, Lefkowitz RJ. Classical and new roles of β-arrestins in the regulation of G-protein-coupled receptors. Nat Rev Neurosci. 2001;2:727–733. doi: 10.1038/35094577. [DOI] [PubMed] [Google Scholar]
- 34.McArdle CA, Franklin J, Green L, Hislop JN. Signalling, cycling and desensitisation of gonadotrophin-releasing hormone receptors. J Endocrinol. 2002;173:1–11. doi: 10.1677/joe.0.1730001. [DOI] [PubMed] [Google Scholar]
- 35.Shacham S, Cheifetz MN, Fridkin M, Pawson AJ, Millar RP, Naor Z. Identification of Ser153 in ICL2 of the gonadotropin-releasing hormone (GnRH) receptor as a phosphorylation-independent site for inhibition of Gq coupling. J Biol Chem. 2005;280:28981–28988. doi: 10.1074/jbc.M500312200. [DOI] [PubMed] [Google Scholar]
- 36.Lucas KK, Dennis EA. The ABC's of group IV cytosolic phospholipase A2. Biochim Biophys Acta. 2004;1636:213–218. doi: 10.1016/j.bbalip.2003.12.009. [DOI] [PubMed] [Google Scholar]
- 37.Six DA, Dennis EA. The expanding superfamily of phospholipase A(2) enzymes: classification and characterization. Biochim Biophys Acta. 2000;1488:1–19. doi: 10.1016/s1388-1981(00)00105-0. [DOI] [PubMed] [Google Scholar]
- 38.Naor Z, Catt KJ. Mechanism of action of gonadotropin-releasing hormone. Involvement of phospholipid turnover in luteinizing hormone release. J Biol Chem. 1981;256:2226–2229. [PubMed] [Google Scholar]
- 39.Slice LW, Chiu T, Rozengurt E. Angiotensin II and epidermal growth factor induce cyclooxygenase-2 expression in intestinal epithelial cells through small GT-Pases using distinct signaling pathways. J Biol Chem. 2005;280:1582–1593. doi: 10.1074/jbc.M408172200. [DOI] [PubMed] [Google Scholar]
- 40.Roberson MS, Misra-Press A, Laurance ME, Stork PJ, Maurer RA. A role for mitogen-activated protein kinase in mediating activation of the glycoprotein hormone α-subunit promoter by gonadotropin-releasing hormone. Mol Cell Biol. 1995;15:3531–3539. doi: 10.1128/mcb.15.7.3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sundaresan S, Colin IM, Pestell RG, Jameson JL. Stimulation of mitogen-activated protein kinase by gonadotropin-releasing hormone: evidence for the involvement of protein kinase C. Endocrinology. 1996;137:304–311. doi: 10.1210/endo.137.1.8536629. [DOI] [PubMed] [Google Scholar]
- 42.Haisenleder DJ, Cox ME, Parsons SJ, Marshall JC. Gonadotropin-releasing hormone pulses are required to maintain activation of mitogen-activated protein kinase: role in stimulation of gonadotrope gene expression. Endocrinology. 1998;139:3104–3111. doi: 10.1210/endo.139.7.6091. [DOI] [PubMed] [Google Scholar]
- 43.Saunders BD, Sabbagh E, Chin WW, Kaiser UB. Differential use of signal transduction pathways in the gonadotropin-releasing hormone-mediated regulation of gonadotropin subunit gene expression. Endocrinology. 1998;139:1835–1843. doi: 10.1210/endo.139.4.5972. [DOI] [PubMed] [Google Scholar]
- 44.Weck J, Fallest PC, Pitt LK, Shupnik MA. Differential gonadotropin-releasing hormone stimulation of rat luteinizing hormone subunit gene transcription by calcium influx and mitogen-activated protein kinase-signaling pathways. Mol Endocrinol. 1998;12:451–457. doi: 10.1210/mend.12.3.0070. [DOI] [PubMed] [Google Scholar]
- 45.Mulvaney JM, Roberson MS. Divergent signaling pathways requiring discrete calcium signals mediate concurrent activation of two mitogen-activated protein kinases by gonadotropin-releasing hormone. J Biol Chem. 2000;275:14182–14189. doi: 10.1074/jbc.275.19.14182. [DOI] [PubMed] [Google Scholar]
- 46.Vasilyev VV, Pernasetti F, Rosenberg SB, Barsoum MJ, Austin DA, Webster NJ, Mellon PL. Transcriptional activation of the ovine follicle-stimulating hormone-β gene by gonadotropin-releasing hormone involves multiple signal transduction pathways. Endocrinology. 2002;143:1651–1659. doi: 10.1210/endo.143.5.8771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cheng HF, Wang JL, Zhang MZ, McKanna JA, Harris RC. Role of p38 in the regulation of renal cortical cyclooxygenase-2 expression by extracellular chloride. J Clin Invest. 2000;106:681–688. doi: 10.1172/JCI10318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang T, Huang Y, Heasley LE, Berl T, Schnermann JB, Briggs JP. MAPK mediation of hypertonicity-stimulated cyclooxygenase-2 expression in renal medullary collecting duct cells. J Biol Chem. 2000;275:23281–23286. doi: 10.1074/jbc.M910237199. [DOI] [PubMed] [Google Scholar]
- 49.Zaric J, Ruegg C. Integrin-mediated adhesion and soluble ligand binding stabilize COX-2 protein levels in endothelial cells by inducing expression and preventing degradation. J Biol Chem. 2005;280:1077–1085. doi: 10.1074/jbc.M410006200. [DOI] [PubMed] [Google Scholar]
- 50.Kanasaki H, Mutiara S, Harada T, Miyazaki K. Phosphatidylinositol 3-kinase is involved in the regulation of gonadotropin and FSH-subunit gene expressions. Program of the 88th Annual Meeting of The Endocrine Society; Boston, MA. 2006. Abstract P2-349. [Google Scholar]
- 51.Pawliczak R, Logun C, Madara P, Lawrence M, Woszczek G, Ptasinska A, Kowalski ML, Wu T, Shelhamer JH. Cytosolic phospholipase A2 Group IValpha but not secreted phospholipase A2 Group IIA, V, or X induces interleukin-8 and cyclooxygenase-2 gene and protein expression through peroxisome proliferator-activated receptors γ 1 and 2 in human lung cells. J Biol Chem. 2004;279:48550–48561. doi: 10.1074/jbc.M408926200. [DOI] [PubMed] [Google Scholar]
- 52.Karin M. The regulation of AP-1 activity by mitogen-activated protein kinases. J Biol Chem. 1995;270:16483–16486. doi: 10.1074/jbc.270.28.16483. [DOI] [PubMed] [Google Scholar]
- 53.Bradbury DA, Newton R, Zhu YM, El-Haroun H, Corbett L, Knox AJ. Cyclooxygenase-2 induction by brady-kinin in human pulmonary artery smooth muscle cells is mediated by the cyclic AMP response element through a novel autocrine loop involving endogenous prostaglandin E2, E-prostanoid 2 (EP2), and EP4 receptors. J Biol Chem. 2003;278:49954–49964. doi: 10.1074/jbc.M307964200. [DOI] [PubMed] [Google Scholar]
- 54.Jiang B, Xu S, Hou X, Pimentel DR, Brecher P, Cohen RA. Temporal control of NF-κB activation by ERK differentially regulates interleukin-1β-induced gene expression. J Biol Chem. 2004;279:1323–1329. doi: 10.1074/jbc.M307521200. [DOI] [PubMed] [Google Scholar]
- 55.Ochi K, Yoshimoto T, Yamamoto S, Taniguchi K, Miyamoto T. Arachidonate 5-lipoxygenase of guinea pig peritoneal polymorphonuclear leukocytes. Activation by adenosine 5′-triphosphate. J Biol Chem. 1983;258:5754–5758. [PubMed] [Google Scholar]
- 56.Gould SF, Spaziani EP, Benoit R, O'Brien WFs. Immunohistochemical localization of the prostaglandin E subtype-1 receptor in cytokine-stimulated and unstimulated amnion cells. Obstet Gynecol. 1999;94:1027–1032. doi: 10.1016/s0029-7844(99)00417-2. [DOI] [PubMed] [Google Scholar]
- 57.Saussy DL, Jr., Mais DE, Baron DA, Pepkowitz SH, Halushka PV. Subcellular localization of a thromboxane A2/prostaglandin H2 receptor antagonist binding site in human platelets. Biochem Pharmacol. 1988;37:647–654. doi: 10.1016/0006-2952(88)90138-4. [DOI] [PubMed] [Google Scholar]
- 58.Bhattacharya M, Peri K, Ribeiro-da-Silva A, Almazan G, Shichi H, Hou X, Varma DR, Chemtob S. Localization of functional prostaglandin E2 receptors EP3 and EP4 in the nuclear envelope. J Biol Chem. 1999;274:15719–15724. doi: 10.1074/jbc.274.22.15719. [DOI] [PubMed] [Google Scholar]
- 59.Bhattacharya M, Peri KG, Almazan G, Ribeiro-da-Silva A, Shichi H, Durocher Y, Abramovitz M, Hou X, Varma DR, Chemtob S. Nuclear localization of prostaglandin E2 receptors. Proc Natl Acad Sci USA. 1998;95:15792–15797. doi: 10.1073/pnas.95.26.15792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Leslie CC. Regulation of the specific release of arachidonic acid by cytosolic phospholipase A2. Prostaglandins Leukot Essent Fatty Acids. 2004;70:373–376. doi: 10.1016/j.plefa.2003.12.012. [DOI] [PubMed] [Google Scholar]
- 61.Ellsworth BS, White BR, Burns AT, Cherrington BD, Otis AM, Clay CM. c-Jun N-terminal kinase activation of activator protein-1 underlies homologous regulation of the gonadotropin-releasing hormone receptor gene in α T3-1 cells. Endocrinology. 2003;144:839–849. doi: 10.1210/en.2002-220784. [DOI] [PubMed] [Google Scholar]
- 62.Harris D, Reiss N, Naor Z. Differential activation of protein kinase C δ and ε gene expression by gonadotropin-releasing hormone in αT3-1 cells. Autoregulation by protein kinase C. J Biol Chem. 1997;272:13534–13540. doi: 10.1074/jbc.272.21.13534. [DOI] [PubMed] [Google Scholar]
- 63.Sales KJ, Maudsley S, Jabbour HN. Elevated prostaglandin EP2 receptor in endometrial adenocarcinoma cells promotes vascular endothelial growth factor expression via cyclic 3′,5′-adenosine monophosphate-mediated transactivation of the epidermal growth factor receptor and extracellular signal-regulated kinase 1/2 signaling pathways. Mol Endocrinol. 2004;18:1533–1545. doi: 10.1210/me.2004-0022. [DOI] [PubMed] [Google Scholar]
- 64.Denison FC, Grant VE, Calder AA, Kelly RW. Seminal plasma components stimulate interleukin-8 and interleukin-10 release. Mol Hum Reprod. 1999;5:220–226. doi: 10.1093/molehr/5.3.220. [DOI] [PubMed] [Google Scholar]
- 65.Larder R, Karali D, Nelson N, Brown P. Fanconi anemia a (FANCA) is a nucleocytoplasmic shuttling molecule required for gonadotropin releasing hormone (GnRH) transduction of the GnRH receptor. Endocrinology. 2006;147:5676–5689. doi: 10.1210/en.2006-0383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Quirk J, Brown P. Hesx1 homeodomain protein represses transcription as a monomer and antagonises transactivation of specific sites as a homodimer. J Mol Endocrinol. 2002;28:193–205. doi: 10.1677/jme.0.0280193. [DOI] [PubMed] [Google Scholar]
- 67.Flanagan CA, Fromme BJ, Davidson JS, Millar RP. A high affinity gonadotropin-releasing hormone (GnRH) tracer, radioiodinated at position 6, facilitates analysis of mutant GnRH receptors. Endocrinology. 1998;139:4115–4119. doi: 10.1210/endo.139.10.6260. [DOI] [PubMed] [Google Scholar]
- 68.Pawson AJ, Maudsley SR, Lopes J, Katz AA, Sun YM, Davidson JS, Millar RP. Multiple determinants for rapid agonist-induced internalization of a nonmammalian gonadotropin-releasing hormone receptor: a putative palmitoylation site and threonine doublet within the carboxyl-terminal tail are critical. Endocrinology. 2003;144:3860–3871. doi: 10.1210/en.2003-0028. [DOI] [PubMed] [Google Scholar]