

Abstract
The thermal dissipation of absorbed light energy by the light-harvesting apparatus of higher plants is important in protecting the photosynthetic machinery from the effects of excess illumination. A major mechanism for such photoprotection, known as trans-thylakoid ΔpH-dependent chlorophyll fluorescence quenching (qE), is induced by acidification of the lumen, is correlated with the interconversion of xanthophyll pigments, and is manifested as quenching of chlorophyll fluorescence. The mechanistic basis for qE remains unknown. The reagent N,N′-dicyclohexylcarbodiimide (DCCD) specifically inhibits qE and covalently binds to two minor light-harvesting pigment–protein complexes (LHCII), LHCIIa and LHCIIc. It is shown that DCCD treatment of isolated LHCIIc complexes reverses acid-induced chlorophyll fluorescence quenching in an in vitro system. Fingerprinting of [14C]DCCD-labeled LHCIIc demonstrates that there are two DCCD-sensitive amino acid residues on this complex, and these are shown to be glutamate residues, each of which is located near the lumen. In view of the effects of DCCD on the pattern of proton release from photosystem II during photosynthesis, we propose a model for the mechanism of the induction of qE—that these residues form part of a proton pathway, the lumen pH being sensed via its effects on the rate of proton release. One possibility is that the resulting changes in the protonation state of these carboxyl side chains may modulate the structural and energetic organization of LHCII.
Keywords: fluorescence quenching, photoprotection, photosynthesis, photosystem II, thylakoid membrane
The role of the light-harvesting complexes (LHCII) of photosystem II (PSII) in protecting higher plants against the deleterious effects of excess illumination is now well-established. The thermal dissipation of energy by LHCII, manifested as quenching of chlorophyll fluorescence (qE), is known to depend on the acidification of the thylakoid lumen, which results from light-induced proton translocation. It is inferred that a reorganization of the light-harvesting apparatus is prompted by the protonation of specific amino acid residues or pigments (1, 2).
The reagent N,N′-dicyclohexylcarbodiimide (DCCD) has been widely used to identify active sites in proton-translocating complexes; it covalently modifies carboxyl residues within hydrophobic domains. Treatment of thylakoids with DCCD inhibits qE while also inhibiting proton translocation from the site of water splitting to the lumen (3, 4). Labeling studies using [14C]DCCD have shown that the only PSII proteins bound by DCCD are two minor LHCII complexes, LHCIIa (also referred to as CP29) and LHCIIc (CP26), and that the major complexes (LHCIIb) are not modified (5).
Recent work indicates that the minor LHCII complexes may be directly implicated in qE. LHCIIa and LHCIIc are enriched in violaxanthin and zeaxanthin (6, 7), the pigments of the xanthophyll cycle; the deepoxidation of violaxanthin to form zeaxanthin is strongly correlated with the capacity for qE (8). The minor complexes also show strong pH-dependent quenching in vitro (9).
These findings suggest a hypothesis in which DCCD-reactive sites on LHCIIa and/or LHCIIc are directly involved in energy dissipation, perhaps acting to sense the pH of the lumen. In further investigating this hypothesis, we have focused on the interaction of DCCD with LHCIIc. We describe a comparison of the inhibitory effects of DCCD on chlorophyll fluorescence quenching in isolated LHCIIb and LHCIIc complexes and report the identification of two DCCD-binding sites on LHCIIc.
MATERIALS AND METHODS
Isolation and Treatment of LHCII Complexes.
LHCII complexes were isolated from light-treated, hydroponically grown spinach (Spinacia oleracea L.) by isoelectric focusing as described (6) and were eluted in 200 μM n-dodecyl β-maltoside/20 mM Hepes, pH 7.5. [14C]DCCD-labeled LHCIIc apoprotein was prepared as described (5) by treating LHCIIc (35 μg of chlorophyll per ml) with 50 μM [14C]DCCD (at 20°C for 15 min). DCCD and detergent then were removed, and the protein was precipitated by extraction with methanol-chloroform (10). Digestion of labeled protein with l-1-tosylamido-2-phenylethyl chloromethyl ketone-treated trypsin (10 μg/ml−1, 37°C overnight) was carried out in 1% octyl glucoside/0.1% SDS/1 mM n-dodecyl β-maltoside/50 mM Hepes, pH 7.5. Digested protein again was extracted with methanol-chloroform. For chemical cleavage, labeled protein was dissolved in 0.5 ml 70% (vol/vol) formic acid. Formic acid cleavage was carried out overnight at 37°C. Alternatively, a few crystals of CNBr were dissolved, and cleavage was carried out at room temperature for at least 4 h. Cleaved protein was freeze-dried, washed in distilled water, and freeze-dried again. For analysis of LHCIIc from DCCD-treated thylakoids, protein (200 μg chlorophyll equivalent) was separated by SDS-urea/PAGE (5) and electroeluted from gel slices in 25 mM Tris/192 mM glycine/0.1% SDS, pH 8.3. Formic acid was added to the eluate to a final concentration of 70% (vol/vol), and the protein was cleaved with CNBr as above.
Chlorophyll Fluorescence Quenching in LHCII Complexes.
Measurements of chlorophyll fluorescence were carried out using a PAM101 fluorometer (Heinz Walz, Effeltrich, Germany) as described (11) with the following minor alterations: LHCII complexes were rapidly diluted to 3 μM chlorophyll (final concentration) in 20 mM Hepes (pH 8.0), and the medium was acidified to pH 5.4 by the addition of HCl. DCCD (stock solution, 50 mM in ethanol) was then added to the stated concentration, and the relaxation of chlorophyll fluorescence quenching was monitored until a steady-state was reached (3–5 min). Quenching inhibition was calculated using the approach of Johnson et al. (12). A sample diluted in 200 μM n-dodecyl β-maltoside/20 mM Hepes, pH 8.0 was used to give the “maximum” fluorescence yield.
Identification and Sequencing of 14C-Labeled LHCIIc Fragments.
Protein fragments were solubilized, separated by Tris–tricine electrophoresis (13), and electroblotted onto polyvinylidene difluoride membrane (Problott, Applied Biosystems). After staining with Coomassie blue R and autoradiography of the membrane, labeled protein bands were excised and sequenced by using an Applied Biosystems Sequenator model 473A. Where appropriate, the eluate from each sequencing cycle was collected and freeze-dried. After dissolving in water and adding scintillant, the eluted label was counted for a period that was sufficient to register a minimum of 2000 counts. Data were summed from duplicate samples corresponding to a total of at least 75 pmol of the N-terminal amino acid from each fragment.
Protein Sequence Analysis.
Protein sequence analysis was carried out using the University of Wisconsin Genetics Computer Group suite of programs (14). LHCII available in protein/DNA sequence data bases were identified using fasta; pileup was used for the compilation of consensus polypeptide sequences; and sequence alignments were carried out using bestfit.
RESULTS
Inhibition by DCCD of Fluorescence Quenching in LHCIIc.
We previously have shown that DCCD treatment of spinach thylakoids inhibits the protective dissipation of absorbed excitation energy that is induced by a trans-membrane ΔpH and which is manifested as quenching of chlorophyll fluorescence (3, 5). Using an in vitro system developed to enable the investigation of such fluorescence quenching in isolated chlorophyll–protein complexes, it has been shown that DCCD also inhibits fluorescence quenching in LHCII complexes (9). We have used this protocol to compare the effects of DCCD on quenching in LHCIIb and LHCIIc. Quenching was induced in detergent-solubilized complexes by diluting in detergent-free medium and then lowering the pH. When DCCD was added to LHCII complexes in this state, there was an increase in fluorescence (Fig. 1). This is hypothesized to reflect inhibition of the same process(es) responsible for fluorescence quenching in vivo.
Figure 1.
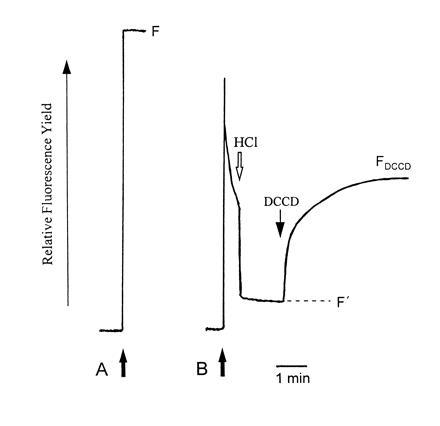
Inhibition by DCCD of chlorophyll fluorescence quenching in LHCII. Complexes isolated by preparative isoelectric focusing were diluted (solid arrows) either (A) in buffer containing detergent, or (B) in detergent-free buffer. For B, fluorescence quenching was induced by the immediate addition of HCl and then reversed by the addition of varying concentrations of DCCD. DCCD-reversible quenching was calculated as (F/F′) − (F/FDCCD). Data for LHCIIc treated with 100 μM DCCD are illustrated.
Fig. 2 shows the concentration dependence of inhibition of quenching by DCCD measured as kDrev, which is calculated in a manner directly analogous to that which has been used to calculate the Stern–Volmer rate constant for rapidly reversible energy dissipation (12). The reversal of quenching appears to saturate across the same DCCD concentration range, but the extent of quenching inhibition is substantially greater for LHCIIc compared with LHCIIb. This may reflect, in part, the greater quenching observed in LHCIIc in the absence of DCCD; however, only a small proportion of the difference between the two complexes can be accounted for in this way. In view of the finding that DCCD covalently binds to LHCIIc but not to LHCIIb, this adds weight to the hypothesis that LHCIIc has a particular role in protective energy dissipation. Furthermore, for both LHCIIb and LHCIIc, quenching inhibition nears saturation at concentrations of 100 μM. This concentration range is somewhat higher than (but of the same order of magnitude as) that used in thylakoid experiments to bring about inhibition of quenching in thylakoids and DCCD labeling of LHCIIc. However, it should be noted that direct comparisons of DCCD concentrations in aqueous- and membrane-based systems are somewhat complicated by the fact that DCCD partitions into lipid, perhaps increasing the local concentration in the latter experiments.
Figure 2.
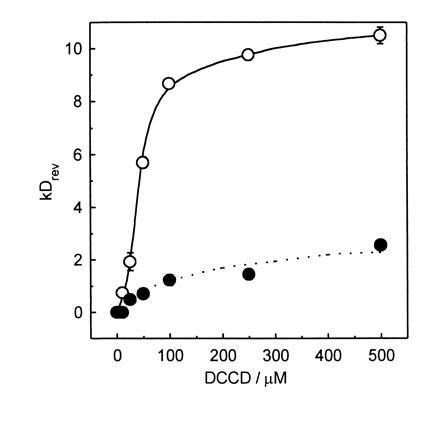
Concentration dependence of DCCD inhibition of chlorophyll fluorescence quenching in LHCIIb and LHCIIc. DCCD-reversible quenching of chlorophyll fluorescence (kDrev) was measured for LHCIIb (•) and LHCIIc (○) across a range of DCCD concentrations. Data are mean ± SE for measurements from two independent LHCII preparations.
Fingerprinting of [14C]DCCD-Labeled Peptide Fragments.
The site(s) at which DCCD binds to LHCIIc in vitro were investigated by treating isolated complex with [14C]DCCD and by cleaving the labeled apoprotein with trypsin, formic acid, or CNBr. The resulting fragments were separated by Tris-tricine electrophoresis, transferred to polyvinylidene difluoride membrane, and identified by autoradiography. Labeled fragments were characterized according to their approximate molecular weights (for the smallest fragments, such estimates were subject to significant error) and their N-terminal amino acid sequences, which were compared with published LHCIIc sequences. Since small fragments would be of greater use in locating binding sites to small regions of the protein, several CNBr digests were carried out overnight, so that formic acid-sensitive sites were also subject to cleavage. The relative strengths of the labeled bands produced in this way were somewhat variable, presumably as a consequence of different efficiencies for formic acid-mediated cleavage under these conditions.
The amino acid sequence information derived during this analysis of DCCD-labeled fragments can be combined into contiguous sequences for two regions of the LHCIIc apoprotein. Fig. 3 shows these aligned with previously published LHCIIc sequences. The sequenced regions have 91% sequence identity with the predicted sequences of LHCIIc from each of tomato, barley, and maize, and 84% with that from Scots pine, indicating that the labeled and sequenced polypeptides are indeed derived from the LHCIIc apoprotein.
Figure 3.

Sequence comparison of LHCIIc protein sequences. Contiguous stretches of amino acid sequence data, obtained during analysis of DCCD-labeled fragments of LHCIIc, were aligned with published sequences from LHCIIc from other species (refs. 15, 16, 17; GenBank accession nos. U23188U23188 and U23189U23189). Residues whose identification was doubtful or ambiguous are shown as queries. The Met residues (sites of CNBr cleavage) marking the N terminals of the fragments used to identify the DCCD-binding residues (see Figs. 4 and 5) are indicated with arrows. Residues are numbered according to ref. 17.
A large number of DCCD-labeled fragments were identified (Fig. 4). No single region of the protein was common to all of them; thus, it was clear that in vitro at least two separate DCCD-binding sites were present on LHCIIc. Their locations were identified as the regions of overlap common to several DCCD-binding fragments (indicated in Fig. 4). Inspection of their respective amino acid sequences showed the presence of at least two acidic amino acids in the region of each DCCD-binding site, any or all of which could be regarded as candidate binding sites. To further investigate the identity of the amino acids bound by DCCD, eluate was collected during sequence analysis of two LHCIIc fragments generated by CNBr cleavage (see Fig. 3). The released 14C label in each fraction was determined (counted for a period sufficient for analysis of the data to be statistically valid), in order to identify the positions at which the DCCD adduct was covalently attached.
Figure 4.
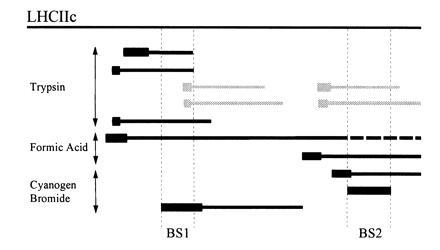
[14C]DCCD-labeled fragments of LHCIIc apoprotein. Labeled fragments of LHCIIc (solid bars) generated by cleavage with trypsin, formic acid, or CNBr were identified by estimation of their molecular weights (from their electrophoretic mobility), and comparison of their N-terminal amino acid sequences (thick bars) with the consensus sequence for LHCIIc apoprotein. Their positions are shown relative to the full LHCIIc apoprotein. In two cases, amino acid sequence data were ambiguous due to the presence of two comigrating fragments (gray bars). The regions of overlap identifying DCCD-binding sites (BS1, BS2) are bordered by dotted vertical lines, and begin at the Met residues identified in Fig. 3.
Fig. 5 shows the results for label release during sequence analysis of the two fragments. The yield of glutamate in each fraction is also shown, highlighting the points in the amino acid sequence at which glutamate was located. Repetitive yields of up to 95% were achieved, allowing unambiguous sequencing of 24 residues from each fragment, including all of the candidate DCCD-binding sites. Even with these high repetitive yields, it can be seen that the cumulative effect of low levels of noncleavage during each sequencing cycle was sufficient to cause glutamate peaks to “carry over” one or two fractions beyond those corresponding to Glu residues. The peaks observed at positions 214 and 220 during analysis of binding site 2 (BS2) are likely to be the result of degradation of the Gln residues at these positions.
Figure 5.
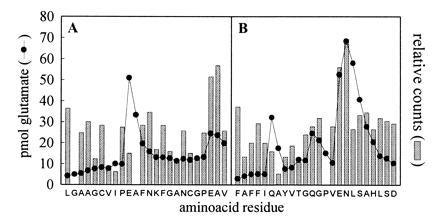
Label associated with fractions collected during sequence analysis of [14C]DCCD-labeled LHCIIc fragments. Eluate for each cycle during protein sequencing of the CNBr-generated LHCIIc fragments identified in Fig. 4 was collected and freeze-dried, and 14C content was determined by scintillation counting. Glu yields for each sequencing cycle are also shown. Amino acid sequences correspond to residues 95–118 (BS1) (A) and residues 209–232 (BS2) (B) of LHCIIc according to ref. 17.
14C label was released during every sequencing cycle. We presume that this is partly a result of protein degradation during sequencing; but it is also likely that the 14C-labeled N-acyl-urea residue formed by DCCD binding is susceptible to direct degradation. Similar observations have been reported elsewhere (18). Despite such nonspecific release, sufficient label remained bound to the protein to give rise to clear peaks. The amount of released label was appreciably higher in the fractions corresponding to residues 116–117 and 224–225. These positions were the same as where there were significant peaks in glutamate yield, indicating that Glu-116 and Glu-224 were bound by DCCD. Conversely, Glu-104 [in the region of binding site 1 (BS1)] was clearly not DCCD binding. It should be noted that the lag in the release of 14C at these positions mirrored the carry-over of glutamate release resulting from cumulative noncleavage during sequencing.
Comparison of DCCD-Labeling in Thylakoids and Isolated Complexes.
The above analysis required greater quantities of isolated LHCIIc than could be isolated from [14C]DCCD-labeled thylakoids. The possibility that the identified sites may not be the same as those bound during treatment of thylakoids was investigated by comparing the pattern of labeling in thylakoids with those in isolated LHCIIc complexes. LHCIIc was isolated from [14C]DCCD-labeled thylakoids by excision of the appropriate band from an SDS-urea/PAGE gel and electroelution. It was then cleaved with CNBr and analyzed simultaneously with similarly treated LHCIIc by Tris-tricine electrophoresis. The resulting autoradiograph is shown in Fig. 6.
Figure 6.
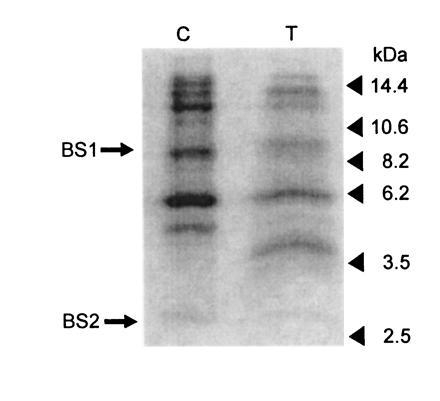
DCCD binding to LHCIIc in thylakoids and isolated complexes. [14C]DCCD-labeled LHCIIc apoprotein derived from either isolated complexes (lane C) or thylakoids (lane T) was cleaved with formic acid/CNBr, separated by Tris-tricine electrophoresis, and blotted onto polyvinylidene difluoride membrane. The resulting autoradiograph is shown. The positions of molecular weight markers (in kDa) are as shown. The fragments used for the identification of DCCD-binding sites in isolated LHCIIc (BS1, BS2) are also indicated.
There were marked differences in the relative strengths of some bands. Some bands could be visualized only after extended exposures. This is readily accounted for by the varying levels of formic acid-mediated cleavage noted above. For instance, although the 2.7-kDa band from isolated LHCIIc is barely visible in labeled thylakoids, this band appears to be a digestion product of the 4.0-kDa thylakoid band (arising from formic acid cleavage at Asp-232–Pro-233), which is barely visible in the isolated complex. After taking account of the variability in band strength, the banding pattern for CNBr-digested LHCIIc was very similar for the two samples. No bands were observed in LHCIIc from labeled thylakoids that were not present in the labeled isolated complex. The thylakoid-derived protein produced bands that corresponded to both identified binding sites. These data demonstrate that the same CNBr-generated fragments (i.e., the same regions of the protein) bind DCCD in detergent-solubilized LHCIIc as when the complex is located in the thylakoid membrane. Although not conclusive, these data suggest that the sites we have identified are the same as those bound during the inhibition of qE in thylakoids.
DISCUSSION
We have previously shown that DCCD inhibits the formation of photoprotective energy dissipation (qE) in thylakoids and that it binds to the minor complexes LHCIIa and LHCIIc but not to bulk light-harvesting complex LHCIIb. In this work, we have found that the effect of DCCD on LHCIIc is markedly greater than that on LHCIIb, and we have identified two Glu residues on LHCIIc that are bound covalently by DCCD in vitro. Comparison of labeled protein from thylakoids and detergent-solubilized complexes indicates that the identified sites correspond to DCCD-sensitive sites in thylakoids. Since the small effects of DCCD on fluorescence quenching in LHCIIb can be explained as being a consequence of the nonspecific effects of a hydrophobic reagent on proton–protein interactions, we conclude that differences in the fluorescence properties of LHCII complexes are related to their sensitivity to DCCD.
There is strong sequence homology between the light-harvesting proteins (17). In particular, trans-membrane helices identified in the crystal structure of LHCIIb (19) are heavily conserved, and it is inferred from this that the main features of the LHCIIb structure are also present in other light-harvesting complexes, including LHCIIc. Fig. 7 shows parts of a global alignment of LHCII consensus sequences, near the two LHCIIc DCCD-binding sites. Direct comparison of LHCIIb and LHCIIc shows that the DCCD-binding residues are located beyond the carboxy-terminal end of the B helix (BS1) and in a region with strong sequence homology to the amphiphilic D helix of LHCIIb (BS2). Both acidic residues are therefore likely to be located at or near the surface of the protein, on the lumen side of the membrane. DCCD-binding sites have previously been identified in amphiphilic helices in the cytochrome bc complexes (18).
Figure 7.

Sequence comparison of LHCII proteins near DCCD-binding sites. Consensus sequences compiled from published data for angiosperm LHCII proteins were aligned in the region of LHCIIc DCCD-binding sites (arrows). Conserved regions are shaded. Invariant residues (i.e., absolutely conserved across all species) are shown in uppercase letters. Regions corresponding to helices identified in the high-resolution structure of LHCIIb (19) are as indicated. Residues are numbered according to Jansson (17).
These observations are readily accounted for by suggesting that changes in the lumen pH are detected via changes in the protonation state of one or both of the DCCD-binding Glu residues. Such sensing of the lumen pH could be directly involved in the induction and maintenance of qE. Binding of DCCD at these sites would prevent this function from being fulfilled and would therefore inhibit qE. The question then arises: How does protonation of Glu residues lead to the induction of chlorophyll fluorescence quenching? It has been suggested that the residues bound by DCCD are involved in pigment coordination, so that Glu protonation directly affects pigment–pigment or pigment–protein interactions (20). However, this is unlikely since there is no indication from the crystal structure of LHCIIb that either of the residues that correspond to the DCCD-binding glutamates are involved in chlorophyll or carotenoid binding (19).
A more attractive hypothesis follows from the position of BS2. As noted above, this region has strong sequence similarity with LHCIIb and may likely form an amphiphilic helix similar to the LHCIIb D helix. A histidine residue that acts as a chlorophyll ligand is conserved in LHCIIc. Protonation of a lumen-exposed Glu residue possibly may alter the amphiphilic properties of this putative D helix, leading to movement of the helix, in turn affecting the conformation and energetics of the nearby chlorophyll. A similar hypothesis can be developed for BS1. Protonation of an acidic residue at the foot of the B helix near the lumenal surface may affect the position of the helix, thereby affecting one or more pigments bound by or otherwise interacting with that helix. It is worth noting that, although it might appear that single protonation events would only have minor effects on protein structure, small changes in chlorophyll environments are often sufficient to bring about large changes in chlorophyll energetics. There is extensive evidence that protonation of LHC complexes leads to dramatic changes in subunit–subunit interactions, resulting in aggregation of such complexes in vitro. The spectroscopic changes accompanying such aggregation indicate major changes in chlorophyll and carotenoid energetics and have the same principal features as those observed during the induction of qE in thylakoids, chloroplasts, and intact leaves (1, 2).
Further examination of the sequence alignment (Fig. 7) shows that the DCCD-binding sites on LHCIIc are missing from both LHCIIa and LHCIId, despite the observations that the former both binds DCCD (5) and exhibits strong DCCD reversibility of chlorophyll fluorescence quenching (9). The region surrounding BS1 is completely absent from both complexes, while the Glu residue at BS2 is replaced by conserved Asn and Gly residues (LHCIIa and LHCIId, respectively). The inevitable conclusion is that LHCIIa binds DCCD at sites different from those on LHCIIc. Conversely, the labeled glutamates are conserved in all three LHCIIb polypeptide types. It is unclear why none of them are bound by DCCD. One possibility is that the trimerisation of LHCIIb in some way prevents DCCD from gaining access to the sensitive sites, and that LHCIIa and LHCIIc are labeled simply because they are monomeric. It has been reported that LHCIIb is labeled if it is denatured in the presence of DCCD (21). An alternative is that differences in DCCD sensitivity arise from some particular function for the labeled residues or that some characteristic of the local environment alters their chemistry. For instance, more hydrophobic environments tend to favor the conversion of N-acyl-urea intermediates into stable covalent adducts.
As well as preventing the formation of qE, treatment of thylakoids with DCCD has been shown to inhibit the release into the lumen of protons derived from the water-splitting apparatus of PSII. The protons generated by PSII instead emerge on the stromal side of the membrane (4). This, in addition to the fact that DCCD is known to block proton channels by binding to acyl amino acids (see ref. 18), has led to the suggestion that this effect is due to the blocking of a proton-release pathway (21). A role for LHCII complexes in proton release is also implied by the fact that altered proton-release patterns are exhibited by LHCII-depleted PSII (22, 23, 24).
It has been suggested that LHCII (particularly LHCIIb) is important in the maintenance of localized proton domains that are involved in driving ATP synthesis and which are separated from and out of equilibrium with the lumen bulk phase (25). Proton pathways through the minor LHCII complexes may form part (possibly a key part) of such larger occluded domains. In this light, it is interesting to note that qE appears to show differential sensitivity to the pH of the lumen bulk phase and localized domains, as evidenced by its differential sensitivity to nigericin and the tertiary amine dibucaine (26, 27).
These effects of DCCD on both proton release and qE formation suggest a different interpretation of the effects of DCCD binding. We propose a model in which there are amino acid residues that are involved in both processes. DCCD-reactive residues form part of a proton channel. qE in vivo is formed as a result of the protonation of amino acid residues that form part of that channel and that may be (but are not necessarily) those bound by DCCD. The release of protons from this channel would be directly influenced by the local lumen pH, accounting for the response of qE to lumen pH. Conversely, in the absence of electron transport (i.e., where there is no proton flux away from PSII, such as in an in vitro system in which a ΔpH is developed by ATP hydrolysis), the transfer of protons onto these residues would depend on the external pH. Fig. 8 illustrates such a model, in which three DCCD-binding residues (two on LHCIIc plus at least one on LHCIIa) are on the proton-release pathway.
Figure 8.
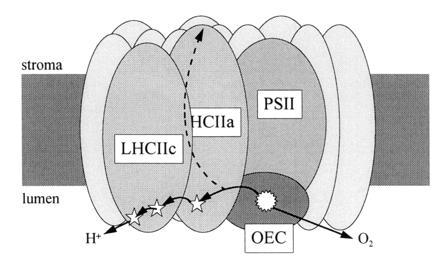
A model for the coregulation of proton channels and qE in the minor LHCII. The pathway for protons derived from the oxygen-evolving complex (OEC) passes through the light-harvesting complexes. DCCD-binding residues (⋆) in LHCIIa and LHCIIc are involved in proton release. Binding of DCCD to these residues, or inhibition of proton release by low lumen pH, activates an alternative proton pathway in which they are released into the stroma. At the same time, protonation of the active residues induces the formation of protective energy dissipation (qE) in LHCII.
According to this model, the release of protons via this channel becomes progressively inhibited as the lumen pH increases. In addition to inducing the formation of qE, we suggest that this has the effect of activating an alternative proton-release pathway (or making it energetically and kinetically favorable), leading to the release of protons into the stroma, as is observed when PSII is treated with DCCD. It has been suggested that donor side inhibition arising from low lumen pH is the major mechanism for high-light-induced photoinhibitory damage of PSII (28). “Gating” of proton flow would act to limit acidification of the lumen by water splitting, thereby providing additional protection for PSII.
Acknowledgments
We acknowledge the excellent technical assistance of Mrs. P. Scholes. We are grateful to Dr. A. Moir for his contribution to the protein sequencing, and to Dr. J. Keen (University of Leeds) for helpful discussions. This work benefited from use of the Biotechnology and Biological Sciences Research Council Seqnet facility, and was supported by grants from the Biotechnology and Biological Sciences Research Council.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: DCCD, N,N′-dicyclohexylcarbodiimide; qE, trans-thylakoid Δph-dependent chlorophyll fluorescence quenching; LHCIIa, -b, -c, and -d, light-harvesting chlorophyll–protein complexes of photosystem II; PSII, photosystem II; BS1 and BS2, binding sites 1 and 2.
References
- 1.Horton P, Ruban A V, Walters R G. Plant Physiol. 1994;106:415–420. doi: 10.1104/pp.106.2.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Horton P, Ruban A V, Walters R G. Annu Rev Plant Physiol Plant Mol Biol. 1996;47:655–684. doi: 10.1146/annurev.arplant.47.1.655. [DOI] [PubMed] [Google Scholar]
- 3.Ruban A V, Walters R G, Horton P. FEBS Lett. 1992;309:175–179. doi: 10.1016/0014-5793(92)81089-5. [DOI] [PubMed] [Google Scholar]
- 4.Jahns P, Polle A, Junge W. EMBO J. 1988;7:589–594. doi: 10.1002/j.1460-2075.1988.tb02851.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Walters R G, Ruban A V, Horton P. Eur J Biochem. 1994;226:1063–1069. doi: 10.1111/j.1432-1033.1994.01063.x. [DOI] [PubMed] [Google Scholar]
- 6.Ruban A V, Young A J, Pascal A A, Horton P. Plant Physiol. 1994;104:227–234. doi: 10.1104/pp.104.1.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bassi R, Dainese P. Eur J Biochem. 1992;204:317–326. doi: 10.1111/j.1432-1033.1992.tb16640.x. [DOI] [PubMed] [Google Scholar]
- 8.Demmig-Adams B, Adams W W. Annu Rev Plant Physiol Plant Mol Biol. 1992;43:599–626. [Google Scholar]
- 9.Ruban A V, Young A J, Horton P. Biochemistry. 1996;35:674–678. doi: 10.1021/bi9524878. [DOI] [PubMed] [Google Scholar]
- 10.Wessel D, Flügge U I. Anal Biochem. 1984;138:141–143. doi: 10.1016/0003-2697(84)90782-6. [DOI] [PubMed] [Google Scholar]
- 11.Ruban A V, Young A J, Horton P. Biochim Biophys Acta. 1994;1186:123–127. [Google Scholar]
- 12.Johnson G N, Young A J, Scholes J D, Horton P. Plant Cell Environ. 1993;16:673–679. [Google Scholar]
- 13.Schägger H, von Jagow G. Anal Biochem. 1987;166:368–379. doi: 10.1016/0003-2697(87)90587-2. [DOI] [PubMed] [Google Scholar]
- 14.Genetics Computer Group (1994) Program Manual for the Wisconsin Package (Genetics Computer Group, Madison, WI), Version 8.
- 15.Schwartz E, Stasys R, Aebersold R, McGrath J M, Pichersky E, Green B R. Plant Mol Biol. 1991;17:923–925. doi: 10.1007/BF00037074. [DOI] [PubMed] [Google Scholar]
- 16.Sørensen A B, Lauridsen B F, Gausing K. Plant Physiol. 1992;98:1538–1540. doi: 10.1104/pp.98.4.1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jansson S. Biochim Biophys Acta. 1994;1184:1–19. doi: 10.1016/0005-2728(94)90148-1. [DOI] [PubMed] [Google Scholar]
- 18.Beattie D S. J Bioenerg Biomembr. 1992;25:233–244. doi: 10.1007/BF00762585. [DOI] [PubMed] [Google Scholar]
- 19.Kühlbrandt W, Wang D N, Fujiyoshi Y. Nature (London) 1994;367:614–621. doi: 10.1038/367614a0. [DOI] [PubMed] [Google Scholar]
- 20.Crofts A R, Yerkes C T. FEBS Lett. 1994;352:265–270. doi: 10.1016/0014-5793(94)00976-7. [DOI] [PubMed] [Google Scholar]
- 21.Jahns P, Junge W. Eur J Biochem. 1990;193:731–736. doi: 10.1111/j.1432-1033.1990.tb19393.x. [DOI] [PubMed] [Google Scholar]
- 22.Jahns P, Junge W. Biochemistry. 1992;31:7398–7403. doi: 10.1021/bi00147a026. [DOI] [PubMed] [Google Scholar]
- 23.Wacker U, Haag E, Renger G. In: Current Research in Photosynthesis. Baltscheffsky M, editor. Vol. 1. Dordrecht, The Netherlands: Kluwer; 1990. pp. 869–872. [Google Scholar]
- 24.Lübbers K, Junge W. In: Current Research in Photosynthesis. Baltscheffsky M, editor. Vol. 1. Dordrecht, The Netherlands: Kluwer; 1990. pp. 877–880. [Google Scholar]
- 25.Renganathan M, Dilley R A. J Bioenerg Biomembr. 1994;26:117–125. doi: 10.1007/BF00763223. [DOI] [PubMed] [Google Scholar]
- 26.Laasch H, Weis E. Photosynth Res. 1989;22:137–146. doi: 10.1007/BF00035444. [DOI] [PubMed] [Google Scholar]
- 27.Noctor G D, Ruban A V, Horton P. Biochim Biophys Acta. 1993;1183:339–344. [Google Scholar]
- 28.Hurry V. In: Photosynthesis: From Light to Biosphere. Mathis P, editor. Vol. 4. Dordrecht, The Netherlands: Kluwer; 1995. pp. 417–420. [Google Scholar]