

Abstract
NAD-dependent l-glutamate dehydrogenase (NAD-GDH) activity was detected in cell extract from the psychrophile Janthinobacterium lividum UTB1302, which was isolated from cold soil and purified to homogeneity. The native enzyme (1,065 kDa, determined by gel filtration) is a homohexamer composed of 170-kDa subunits (determined by sodium dodecyl sulfate-polyacrylamide gel electrophoresis). Consistent with these findings, gene cloning and sequencing enabled deduction of the amino acid sequence of the subunit, which proved to be comprised of 1,575 amino acids with a combined molecular mass of 169,360 Da. The enzyme from this psychrophile thus appears to belong to the GDH family characterized by very large subunits, like those expressed by Streptomyces clavuligerus and Pseudomonas aeruginosa (about 180 kDa). The entire amino acid sequence of the J. lividum enzyme showed about 40% identity with the sequences from S. clavuligerus and P. aeruginosa enzymes, but the central domains showed higher homology (about 65%). Within the central domain, the residues related to substrate and NAD binding were highly conserved, suggesting that this is the enzyme's catalytic domain. In the presence of NAD, but not in the presence of NADP, this GDH exclusively catalyzed the oxidative deamination of l-glutamate. The stereospecificity of the hydride transfer to NAD was pro-S, which is the same as that of the other known GDHs. Surprisingly, NAD-GDH activity was markedly enhanced by the addition of various amino acids, such as l-aspartate (1,735%) and l-arginine (936%), which strongly suggests that the N- and/or C-terminal domains play regulatory roles and are involved in the activation of the enzyme by these amino acids.
Glutamate dehydrogenases (GDHs) catalyze the reversible oxidative deamination of glutamate to α-ketoglutarate and ammonia using NAD(P) as a coenzyme (7, 11, 13, 21). The enzymatic activity of GDHs is well characterized, and GDHs have been classified into three groups based on their coenzyme specificity: NADP-dependent GDHs (EC 1.4.1.4) are involved in ammonia assimilation and are distributed in eukarya, bacteria, and archaea (7); NAD(P)-dependent GDHs (EC 1.4.1.3) are distributed in the mitochondria of mainly eukaryotic cells (22); and NAD-dependent GDHs (NAD-GDHs) (EC 1.4.1.2) are involved in glutamate catabolism and are distributed in animals, plants, and bacteria (11, 13). Almost all NADP- and NAD(P)-dependent GDHs from archaea to eukarya are known to be hexamers with subunit molecular masses of about 50 kDa. On the other hand, NAD-GDHs are classified into three subgroups with different molecular masses and oligomeric structures (5). Although most known types of NAD-GDHs from a variety of organisms occur as hexamers with subunit molecular masses of about 50 kDa (GDH-50s), those from several eukaryotic microorganisms, such as Neurospora crassa and Saccharomyces cerevisiae, exhibit a tetrameric structure with subunit molecular masses of about 115 kDa (5, 8, 10, 12, 18, 25). In addition, a third group of NAD-GDHs with subunit molecular masses of about 180 kDa (GDH-180s) have been found in three bacteria: Pseudomonas aeruginosa (17), Streptomyces clavuligerus (19), and Pscychrobacter sp. strain TAD1 (6). There is currently much less functional and structural information about GDH-180s than about the other two GDH groups.
Up to now we have been investigating the enzymological and structural characteristics of GDHs from hyperthermophiles such as Pyrococcus furiosus (21), Pyrobaculum islandicum (3, 14), and Aeropyrum pernix (2). These enzymes exhibit a high degree of activity at temperatures around 100°C, but the catalytic activity markedly declines as the temperature is reduced. By contrast, enzymes from psychrophiles are generally thermolabile and easily lose activity as the temperature is increased, but they often exhibit a high degree of catalytic activity at temperatures well below room temperature. We are therefore interested in the catalytic properties of GDHs from psychrophiles and in the differences in the catalytic and structural characteristics of the psychrophilic and thermophilic enzymes.
We isolated a psychrophile, Janthinobacterium lividum UTB1302, from low-temperature soil obtained from a mountain in Nagano, Japan, and found that it produced a GDH-180. In this paper, we describe the purification and characterization of this enzyme, as well as the cloning of the enzyme's gene. We also report that this enzyme is markedly different from other known GDH-180s with respect to its kinetics and the mechanism by which it is activated by amino acids.
MATERIALS AND METHODS
Materials.
A GenomicPrep cell and tissue DNA isolation kit and [γ-32P]ATP were purchased from Amersham Biosciences (Tokyo, Japan). A Quiafilter plasmid midi kit and a TKR MEGALABEL kit were obtained from QIAGEN (Tokyo, Japan) and Takara (Kyoto, Japan), respectively. Restriction endonucleases were purchased from New England Biolabs (Tokyo, Japan) and Takara. DEAE-Toyopearl and Butyl Toyopearl were obtained from Tosoh (Tokyo, Japan). All other chemicals were reagent grade.
Screening of the psychrophiles from soil and growth conditions for J. lividum UTB1302.
We obtained soil from a pond in Tsugaike kougen in the mountains of Nagano Prefecture, Japan, as a source from which to isolate psychrophiles. Notably, the temperature of this soil remains under 4°C throughout the year. One liter of the medium used for the isolation of psychrophiles contained 5 g of polypeptone, 2 g of yeast extract, 2 g of meat extract, 2 g of glycerol, 2 g of KH2PO4, 2 g of K2HPO4, 0.26 g of NH4Cl, and 0.1 g of MgSO4·7H2O, and the pH was adjusted to 7.2. Isolation was carried out at 4°C using 1.5% agar plates containing the medium components mentioned above. Of several colonies isolated, the one exhibiting the lowest upper-limit temperature (30°C) for growth was chosen as the candidate from which to isolate GDH. The cells were cultured at 10°C for 24 h and then collected by centrifugation (7,000 × g for 15 min at 4°C), washed with 0.85% NaCl, suspended with 10 mM potassium phosphate (pH 7.0) containing 1 mM EDTA, 0.1 mM dithiothreitol, and 10% glycerol (standard buffer), and stored at −30°C until they were used.
Assays for enzyme activity and protein concentration.
Oxidative deamination catalyzed by the isolated GDH was assayed spectrophotometrically at 35°C. The standard reaction mixture (total volume, 1 ml) contained 100 mM glycine/NaOH (pH 9.5), 20 mM glutamate, 5 mM NAD, and enzyme. The reaction was started by addition of the enzyme, and the change in absorbance caused by the formation of NADH was monitored at 340 nm (ɛ = 6.22 mM−1 cm−1). One unit of enzyme was defined as the amount catalyzing the formation of 1 μmol of NADH/min at 35°C during glutamate oxidation. The protein concentration was determined using the method of Bradford (4); bovine serum albumin served as the standard.
Purification of GDH.
All steps in the purification procedure were carried out at 4°C. Phenylmethylsulfonyl fluoride in dimethyl sulfoxide was added to the cell suspension (20 g [wet weight] in 100 ml of the standard buffer) to a final concentration of 1 mM, after which the cells were disrupted by sonication (TOMY UD-200; TOMY, Tokyo, Japan) using five cycles of 60-s pulses (35 W) followed by a 60-s rest at 10°C, and any remaining intact cells and the cell debris were removed by centrifugation (20,000 × g for 20 min at 4°C). The resultant supernatant was used as the crude extract and applied to a DEAE-Toyopearl column equilibrated with the standard buffer. The column was then washed with the standard buffer, and the enzyme was eluted with a linear 0 to 0.5 M NaCl gradient in the same buffer. The active fractions were pooled, and solid ammonium sulfate was added to 20% saturation while the pH was maintained at ∼7.0 by the addition of 14% NH4OH. The enzyme solution was applied to a Butyl Toyopearl column previously equilibrated with the standard buffer supplemented with 20% ammonium sulfate. After the column was washed with the same buffer, the enzyme was eluted with a linear 20 to 0% ammonium sulfate gradient in the same buffer. The active fractions were again pooled and then concentrated by ultrafiltration (Amicon Ultra YM-30). The enzyme solution was applied to a Superdex 200 column equilibrated with the standard buffer containing 0.2 M NaCl and then eluted with the same buffer. The active fractions were pooled and dialyzed against 10 mM potassium phosphate buffer (pH 6.5) containing 1 mM EDTA, 0.1 mM dithiothreitol, and 10% glycerol. The dialysate was applied to a Blue Sepharose CL-4B column equilibrated with the same buffer, after which the column was washed with the same buffer and the enzyme was eluted with the buffer containing 1.5 M NaCl. Active fractions were pooled and dialyzed against the standard buffer (pH 7.0).
Electrophoresis and determination of molecular mass.
Native polyacrylamide gel electrophoresis (PAGE) was carried out at 4°C on a 7.5% polyacrylamide gel using the method of Davis (9), after which the proteins were stained with Coomassie brilliant blue R-250. Active staining was performed at 25°C using a mixture containing 300 mM Tris-HCl (pH 8.0), 50 mM l-glutamate, 0.1 mM p-iodonitrotetrazolium violet, 0.04 mM phenazine methosulfate, and 0.25 mM NAD. Sodium dodecyl sulfate (SDS)-PAGE was carried out on a 10% polyacrylamide gel using the method of Laemmli (15). Maltose-binding protein-β-galactosidase (175 kDa), maltose-binding protein-paramyosin (83 kDa), glutamate dehydrogenase (62 kDa), aldolase (47.5 kDa), triose phosphate isomerase (32.5 kDa), β-lactoglobulin (25 kDa), lysozyme (16.5 kDa), and aprotinin (6.5 kDa) were used as the molecular standards (New England Biolabs). The molecular mass of the enzyme was determined by Superose 6 gel filtration chromatography using 10 mM potassium phosphate (pH 7.0) and 0.2 M NaCl as the elution buffer. Thyroglobulin (669 kDa), ferritin (440 kDa), catalase (232 kDa), and aldolase (158 kDa) were used as the molecular mass standards (Amersham Biosciences); bovine α-crystallin (about 810 kDa) also was used as a high-molecular-mass standard.
N-terminal amino acid sequence analysis.
The N-terminal amino acid sequence of the isolated enzyme was analyzed using an automated Edman degradation protein sequencer. The phenylthiohydantoin derivatives were separated and identified using a Shimadzu PPSQ-10 protein sequencer (Kyoto, Japan).
Cloning of the GDH gene.
Genomic DNA from J. lividum UTB1302 was prepared using a GenomicPrep cell and tissue DNA isolation kit according to the manufacturer's instructions. To clone the GDH gene, an oligonucleotide mixture probe (probe 1) (5′-ATGAA[T,C]CA[T,C]ACICCICA[A,G]GA[T,C]TT-3′; 23-mer; 16 kinds of sequence) was synthesized based on the N-terminal amino acid sequence of the enzyme (MNHTPQDL). The probe was labeled with [γ-32P]ATP using T4 polynucleotide kinase with MEGALABEL, purified using a ProbeQuant G-50 microcolumn, and used as a specific probe for Southern and colony hybridizations. The genomic DNA from J. lividum UTB1302 was digested with several restriction endonucleases and separated on a 0.8% agarose-Tris-acetate-EDTA gel. When the preparation was subjected to Southern hybridization, a positive signal was observed in the 2.5-kbp SphI fragment. A plasmid vector, pUC18, was therefore digested with SphI and then treated with alkaline phosphatase, after which the SphI fragments of genomic DNA were inserted into the vector, which was then used to transform Escherichia coli JM109 cells. The transformants were selected on a Luria-Bertani plate containing 0.01% ampicillin, 0.01% isopropyl-β-d-thiogalactopyranoside (IPTG), and 0.02% 5-bromo-4-chloro-3-indolyl-β-d-galactoside, after which the selected colonies were transferred and fixed onto nylon membranes. After the recombinant plasmids were screened by colony hybridization, a positive plasmid was isolated and used as the template for DNA sequencing. The sequencing was carried out using the dideoxynucleotide chain termination method with an Applied Biosystems PRISM model 377 DNA sequencer. A new probe for screening the GDH gene was then synthesized based on the 3′ sequence of the known GDH gene fragment, and the screening protocol was repeated until the entire length of the GDH gene was cloned. The sequence data were then analyzed using GENETYX-SV/RC9.0 software (Software Development, Tokyo, Japan).
Expression of the GDH gene and product purification.
The GDH gene was amplified by PCR using genomic DNA as the template with the following primers: 5′-TAATGGAGATGCCATATGAATCACACGCCA-3′ (forward) and 5′-GCGTTACTGCTTGGATCCTTACACCGCCGC-3′ (reverse). The forward primer introduced a unique NdeI restriction site that overlapped the 5′ initiation codon, and the reverse primer introduced a unique BamHI restriction site proximal to the 3′ end of the termination codon. After PCR using Pfu Turbo DNA polymerase, the amplified DNA fragment was confirmed from its sequence, digested with NdeI and BamHI, and inserted into pCold IV (Takara) to produce the expression plasmid pCold/JLGDH. E. coli TOP10 competent cells were then transformed with pCold/JLGDH, and the transformants were grown in Luria-Bertani medium containing 0.01% ampicillin at 37°C. After 6 h, the culture was cooled to 15°C, IPTG (1 mM) was added, and cultivation was continued for an additional 24 h at 15°C. Thereafter, the cells were collected by centrifugation (7,000 × g for 15 min at 4°C), suspended in the standard buffer containing 1 mM phenylmethylsulfonyl fluoride, and disrupted by sonication as described above. After centrifugation (20,000 × g for 15 min at 4°C), solid ammonium sulfate was added to the supernatant to 20% saturation (pH 7.0), and the recombinant GDH was fractionated with Butyl Toyopearl and concentrated by ultrafiltration as described above. The enzyme solution was applied to a Superdex 200 column equilibrated with the standard buffer containing 0.2 M NaCl and then eluted with the same buffer. The active fractions were pooled, dialyzed against the standard buffer, and used as the purified preparation.
Stereospecificity of hydrogen transfer to NAD.
The stereospecificity of the GDH-catalyzed hydrogen transfer to NAD was determined using 1H nuclear magnetic resonance (NMR). A reaction mixture (10 ml) containing 50 mM dl-glutamate (2,4,4-d3; Cambridge Isotope Laboratories, United States), 100 mM glycine/NaOH (pH 9.5), 1 mM NAD, and 2 U of J. lividum GDH was incubated at 25°C for 30 min. After the protein was removed by centrifugal filtration (Amicon Ultra YM-30), the solution was diluted with MilliQ water to obtain a volume of 200 ml and applied to a DEAE-Toyopearl column previously equilibrated with 10 mM NH4HCO3. The column was then washed with 10 mM NH4HCO3 to remove unreacted NAD, and the deuterated NADH was eluted with a linear 10 to 200 mM NH4HCO3 gradient. Fractions were monitored at 340 nm, and the fractions containing deuterated NADH were collected. The solution was lyophilized twice, after which the lyophilized material was dissolved in 99.9% 2H2O. Undeutrated NADH was also prepared using the same method with undeutrated l-glutamate as the substrate. The 1H NMR spectra at the C-4 position of the nicotinamide ring of NADH were recorded with a 400-MHz 1H NMR apparatus (JEOL, Japan).
Nucleotide sequence accession numbers.
The nucleotide sequences of the 16S rRNA and GDH genes have been submitted to DDBJ under accession numbers AB286844 and AB286655, respectively.
RESULTS
Screening of psychrophilic GDH.
By screening at 4°C, we were able to isolate 78 psychrophile strains from low-temperature soil collected from Tsugaike kougen in a mountainous region of Nagano Prefecture in Japan. These strains were then cultured in liquid medium at 4, 20, 25, 30, and 35°C, and the 16 strains that grew well at 4°C but did not grow at temperatures above 30°C were selected for a second screening. Crude extracts were prepared from these 16 strains, and GDH activity was detected using PAGE with active staining and with NAD or NADP as a coenzyme. NAD- or NADP-dependent GDH activity was observed in the crude extracts of seven strains. Of these, strain No. 68 was able to grow at 0°C but not at temperatures above 30°C and was chosen as a candidate producer of psychrophilic GDH. To identify strain No. 68, we determined its physiological characteristics (Table 1) and the sequence of its 16S rRNA gene (991 bp). We found that the sequence was completely identical to that of J. lividum and that the physiological properties were not contradictory to those of J. lividum as far as they are known (data not shown). Thus, strain No. 68 was identified as J. lividum UTB1302.
TABLE 1.
Physiological characteristics of J. lividum UTB1302
| Characteristic | Strain UTB1302 |
|---|---|
| Form | Rod |
| Gram staining | Negative |
| Spore formation | − |
| Mobility | + |
| Catalase activity | + |
| Oxidase activity | + |
| Urease activity | − |
| Production of dark-purple dye | + |
| Nitrate reduction | + |
| Indole production | − |
| Esculin hydrolysis | + |
| Oxidation/fermentation test | − |
| Glucose fermentation | + |
| Arginine dihydrolase | − |
| Gelatin hydrolysis | − |
| β-Galactosidase | − |
Purification of GDH from J. lividum UTB1302.
J. lividum UTB1302 cells (20 g, wet weight) were collected from 2 liters of culture medium, and about 2,000 mg of soluble protein was obtained in the crude extract after sonication and centrifugation. The purified enzyme was then obtained from the extract using the four-step procedure described in Materials and Methods. The purified GDH migrated as a single band on both native PAGE and SDS-PAGE gels and was homogeneous (Fig. 1). The subunit and native molecular masses of GDH were determined to be about 170 kDa (by SDS-PAGE) and about 1,065 kDa (by Superose 6 gel filtration chromatography), respectively, which indicates that the native enzyme occurs as a homohexamer. The N-terminal amino acid sequence of the enzyme was determined to be MNHTPQDLXTQXL (where X is an unidentified amino acid residue).
FIG. 1.
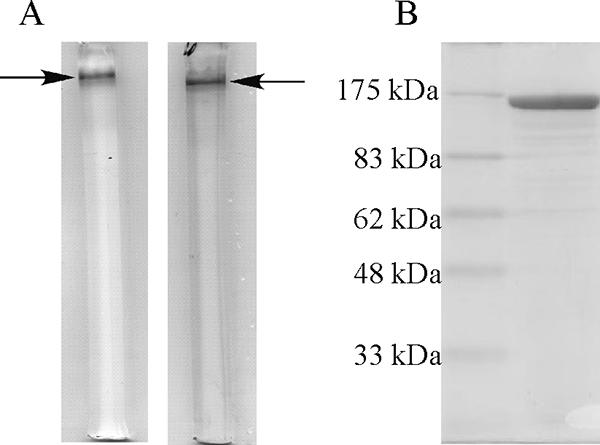
PAGE of purified GDH from J. lividum UTB1302. (A) Native PAGE on a 7.5% acrylamide gel was performed at 4°C. The left lane shows active staining, and the right lane shows protein staining. The arrows indicate the position of the native protein. (B) SDS-PAGE on a 10% acrylamide gel.
Cloning of the GDH gene from J. lividum UTB1302 genomic DNA.
To clone the GDH gene, we initially synthesized a degenerate oligonucleotide probe (probe 1) based on the N-terminal amino acid sequence of the enzyme. After Southern hybridization of the probe, a 2.5-kbp SphI fragment of genomic DNA showed a positive signal. Subsequent colony hybridization yielded a positive clone (pgdh1), and once inserted into pUC18, the DNA was sequenced. The fact that the amino acid sequence deduced from the nucleotide sequence included the known N-terminal amino acid sequence confirmed that we had determined part of the amino acid sequence (694 amino acids) of J. lividum GDH. Thereafter, a new probe (probe 2; 5′-GAACTCGAACGCGACATTCC-3′) was synthesized based on the nucleotide sequence of the known fragment, and cloning yielded pgdh2, in which a 0.5-kbp SalI fragment of genomic DNA was inserted. We then sequenced this fragment and deduced the amino acid sequence (175 amino acids). Using the same method, pgdh3 containing a 3.0-kbp SphI fragment of the genomic DNA was obtained, from which a 706-amino-acid sequence was deduced. In addition, a stop codon (TAA) was found within the nucleotide sequence.
Ultimately, we were able to clone the entire GDH gene using the three genome fragments (Fig. 2). The total length of the gene was 4,725 bp, and the deduced amino acid sequence contained 1,575 amino acids with a molecular mass of 169,360 Da, which corresponded well to the molecular mass determined by SDS-PAGE (170 kDa). A BLAST search revealed that the amino acid sequence of J. lividum GDH showed homology with the sequences of known NAD-dependent GDHs from S. clavuligerus (38%) and P. aeruginosa (38%) and with the sequences of putative GDHs from Chromobacterium violaceum ATCC 12472 (40%), Ralstonia metallidurans CH34 (41%), Shewanella putrefaciens CN-32 (40%), Burkholderia cenocepacia HI2424 (41%), Xanthomonas campestris pv. campestris str. 8004 (40%), Pseudoalteromonas atlantica T6c (39%), and Thermobifida fusca YX (40%).
FIG. 2.

Nucleotide and deduced amino acid sequences of J. lividum GDH. The N-terminal amino acid sequence determined by protein sequencing is underlined. The full nucleotide sequence was obtained by sequencing pgdh1, pgdh2, and pgdh3, which were comprised of genomic SphI (nucleotide positions 1 to 2295), SalI (nucleotide positions 2243 to 2821), and SphI (nucleotide positions 2296 to 5098) fragments, respectively. The amino acid sequence of the central domain (amino acids 719 to 1190) is shaded.
Purification and characterization of recombinant GDH expressed in E. coli.
After transformation of E. coli TOP10 cells with pCold/JLGDH, which harbored the recombinant GDH gene, high levels of GDH production were confirmed in the crude extract of the transformants (Fig. 3). The specific activity of the GDH in the crude extract of the transformants was 0.62 U/mg, which was about 25-fold higher than that in the crude extract of J. lividum UTB1302 cells. By contrast, no NAD-dependent GDH activity was detected in the crude extract of TOP10 cells transformed with empty pCold IV (negative control). The recombinant GDH was easily purified by successive Butyl Toyopearl and Superdex 200 chromatography steps, and 16 mg of purified protein was obtained from 3 g (wet weight) of E. coli cells (Table 2 and Fig. 3).
FIG. 3.
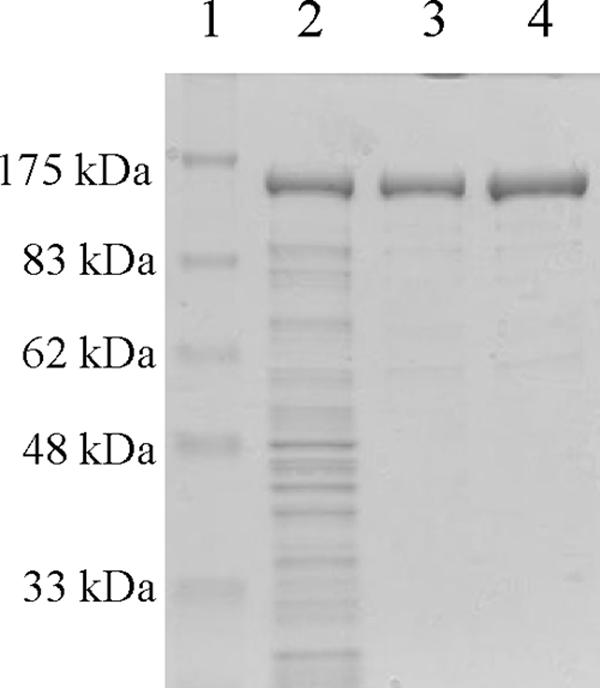
SDS-PAGE of recombinant GDH purified from E. coli. Lane 1, molecular markers; lane 2, crude extract; lane 3, Butyl Toyopearl fraction; lane 4, Superdex 200 fraction.
TABLE 2.
Purification of recombinant NAD-GDH from E. coli
| Step | Total activity (U) | Total protein (mg) | Sp act (U/mg) | Yield (%) |
|---|---|---|---|---|
| Crude extract | 163 | 264 | 0.62 | 100 |
| Butyl Toyopearl | 119 | 35 | 3.4 | 73 |
| Superdex 200 | 50 | 16 | 3.1 | 31 |
The activity of the purified recombinant GDH was maximal at 40°C in glycine-NaOH buffer at pH 9.0. The enzyme was stable for 10 min at temperatures up to 40°C but was completely inactivated by incubation for 10 min at 50°C.
To determine the amino acid specificity of the enzyme for oxidative deamination, the reactivities of the following amino acids (10 mM) were examined in the presence of NAD: l-glutamate, l-glutamine, l-aspartate, l-alanine, l-leucine, l-valine, l-lysine, l-2-aminobutyrate, l-methionine, l-ornithine, l-phenylalanine, l-arginine, l-tryptophan, l-methionine, l-histidine, d-glutamate, and d-aspartate. All of these amino acids except l-glutamate were inert. In addition, no l-glutamate deamination was detected when NAD was replaced with NADP.
To determine the kinetic parameters of the enzyme, the initial velocity was analyzed by varying the concentrations of l-glutamate and NAD. Typical substrate inhibition was observed at l-glutamate concentrations above 20 mM, but no such inhibition was seen with NAD. The apparent Km for NAD was determined to be 2.1 mM in the presence of 20 mM l-glutamate, while the Km for l-glutamate was 7.1 mM in the presence of 5 mM NAD. The Vmax was calculated to be 5.8 U/mg.
The effects of various other compounds on the enzyme activity also were examined (Table 3). Activation of GDH activity was observed when l-aspartate, l-arginine, l-tryptophan, l-methionine, l-lysine, l-histidine, or d-arginine was added to the reaction mixture to a final concentration of 10 mM. In particular, the addition of l-aspartate elicited a 1,735% increase in the reaction rate. This activation by l-aspartate was saturated at a concentration of 3 mM (Fig. 4), and in the presence of 5 mM l-aspartate the apparent Km values were 8.4 mM for l-glutamate and 0.50 mM for NAD; the Vmax was 102 U/mg. Inhibition of GDH activity was observed when citrate, isocitrate, fumarate, succinate, or malate (constituents of the tricarboxylic acid cycle) was added to the reaction mixture to a concentration of 10 mM. In addition, the GDH activity also was inhibited by the addition of 1 mM CoCl2, NiCl2, MgCl2, MnCl2, or HgCl2. Of these, HgCl2 completely blocked the enzyme's activity. By contrast, the GDH activity was increased to 224% of the control activity by the addition of 1 mM ZnCl2.
TABLE 3.
Additional effects of various compounds on the enzyme activity
| Effector (concn) | Activity (%) |
|---|---|
| None | 100 |
| l-Aspartate (10 mM) | 1,735 |
| l-Arginine (10 mM) | 936 |
| l-Tryptophan (10 mM) | 235 |
| l-Methionine (10 mM) | 206 |
| l-Lysine (10 mM) | 161 |
| l-Histidine (10 mM) | 155 |
| l-Asparagine (10 mM) | 118 |
| l-Glutamine (10 mM) | 89 |
| l-Ornithine (10 mM) | 83 |
| d-Arginine (10 mM) | 212 |
| d-Glutamine (10 mM) | 95 |
| d-Glutamate (10 mM) | 94 |
| d-Asparagine (10 mM) | 93 |
| d-Aspartate (10 mM) | 90 |
| Citrate (10 mM) | 67 |
| Malate (10 mM) | 65 |
| Isocitrate (10 mM) | 49 |
| Succinate (10 mM) | 36 |
| Fumarate (10 mM) | 32 |
| ZnCl2 (1 mM) | 224 |
| CoCl2 (1 mM) | 82 |
| NiCl2 (1 mM) | 77 |
| MgCl2 (1 mM) | 64 |
| MnCl2 (1 mM) | 52 |
| HgCl2 (1 mM) | 0 |
FIG. 4.

Saturation curve for enzyme activity elicited by addition of l-aspartate.
The stereospecificity of GDH-catalyzed hydrogen transfer to NAD at the C-4 position of the pyridine ring was determined by 1H NMR analysis. Mostad et al. have shown the specific spectra of the C-4 hydrogens of [4R-2H] and [4S-2H]NADHs (20). When unlabeled l-glutamate was used as the substrate in the presence of NAD, the 1H NMR spectrum of the proton at the C-4 position in the pyridine ring of the NADH produced showed detectable resonance peaks at 2.46, 2.51, 2.59, and 2.64 ppm (Fig. 5A). On the other hand, when deuterated dl-glutamate (2,4,4-d3) was used as the substrate, the 1H NMR spectrum of the NADH produced showed a resonance peak at 2.58 ppm (Fig. 5B). This means that the enzyme reaction produced [4R-1H, 4S-2H]NADH from NAD and deuterated l-glutamate. Thus, J. lividum GDH exhibits pro-S specificity in its hydrogen transfer to NAD (B-type stereospecificity).
FIG. 5.

Portion of the 1H NMR spectra for NADH produced by the enzyme reaction with l-glutamate (A) and dl-glutamate (2,4,4-d3) (B).
DISCUSSION
The first aim of the present study was to isolate psychrophiles and determine the characteristics of psychrophilic GDH. We succeeded in obtaining seven psychrophile strains that produced NAD- or NADP-dependent GDH. Of particular interest to us was strain No. 68. The mobility of the active GDH band detected when crude extract from this strain was run on a native PAGE gel was exceptionally low compared to the mobilities observed for the other strains. This suggested that the molecular mass of the GDH from this strain is much greater than those of the other GDHs, which prompted us to identify the strain (J. lividum) and to clone the gene and characterize the enzyme. As we anticipated, we found that J. lividum produces a very large NAD-dependent GDH, which has a homohexameric structure (1,065 kDa) with a subunit molecular mass of 170 kDa. Similar very large NAD-dependent GDHs with subunit molecular masses of around 180 kDa also have been found in S. clavuligerus, P. aeruginosa, and Psychrobacter sp. strain TAD1 (6, 17, 19). Interestingly, however, the GDHs from those three organisms exhibit hexameric, tetrameric, and dimeric structures, respectively. Judging from the subunit structure, we suggest that the hexameric J. lividum GDH belongs to the same group as the very large GDH from S. clavuligerus (1,100 kDa). On the other hand, the regulation of the catalytic activity of J. lividum GDH markedly differs from that of the S. clavuligerus enzyme and is more like the regulation of the P. aeruginosa and Psychrobacter GDHs. S. clavuligerus GDH is activated by AMP and is strongly inhibited by Tris (19). By contrast, the activity of J. lividum GDH does not appear to be affected by AMP, other nucleotides, or Tris.
One of the notable characteristics of J. lividum GDH is its marked activation by nonsubstrate amino acids, such as l-aspartate and l-arginine. In particular, l-aspartate enhanced the enzyme's activity by about 1,735% (Fig. 4). Although activation by several amino acids, including l-aspartate, l-asparagine, and l-arginine, is also observed in S. clavuligerus and P. aeruginosa GDHs, the degree to which their activity is increased is substantially smaller (no more than 492% of the control value) than the degree seen with J. lividum GDH. In addition, the reported activation of the S. clavuligerus and P. aeruginosa GDHs occurred when the substrate concentrations were unsaturated, suggesting that the enhanced activity reflected increased affinity of the substrate for its binding site on the enzyme. By contrast, activation of J. lividum GDH occurred when the substrate concentration was saturated, which suggests that the activation reflected an increase in the maximum reaction velocity. Differences in the manner of activation and in the activator specificity between these GDHs may be associated with differences in the sequences of their N-terminal and/or C-terminal domains, as the homology between these domains is rather low. We are presently working to clarify the details of the relationship between the activation and the structure of this enzyme.
We found that the entire amino acid sequence of J. lividum GDH shows about 40% homology with the sequences of S. clavuligerus and P. aeruginosa GDHs, as well as those of putative GDHs from C. violaceum, R. metallidurans, S. putrefaciens, B. cenocepacia, X. campestris, P. atlantica, and T. fusca. However, within the amino acid sequence of J. lividum GDH, the central domain (amino acid residues 719 to 1190) shows higher homology (60 to 65%) with the corresponding regions of these enzymes. Moreover, when we examined the sequence alignment of the J. lividum GDH central domain and the Clostridium symbiosum GDH (Fig. 6), which is a homohexamer comprised of 50-kDa subunits in which nine residues are known to be directly responsible for binding l-glutamate and expressing the catalytic activity (1, 23), six of the nine residues of C. symbiosum GDH, G91 (G786 in J. lividum GDH), K113 (K810), K125 (K820), D165 (D869), V377 (V1139), and S380 (S1142), are completely conserved in J. lividum GDH; however, K89, which interacts with the γ-carboxyl, A163, which interacts hydrophobically with the side chain, and G164, which interacts with the amino group of l-glutamate, are replaced by arginine (R784), proline (P867), and valine (V868), respectively, in J. lividum GDH. In addition, the classical dinucleotide binding motif GXGXXG is replaced by GXGXXS in the J. lividum enzyme. This motif is either GXGXXS or GXGXXA in the GDH-180 group. Upstream of this motif, T209, which interacts with the nicotinamide ring of NAD in the C. symbiosum enzyme, is conserved as T926 in the J. lividum enzyme. In addition, G376, V377, and S380 of the C. symbiosum enzyme, which are known to be responsible for the pro-S specificity (B-type stereospecificity) of hydrogen transfer to NADH, are completely conserved in the J. lividum enzyme as G1138, V1139, and S1142, respectively. In general, NAD- and NADP-dependent dehydrogenases show either A- or B-type stereospecificity for hydrogen transfer to the C-4 position of the nicotinamide ring of NAD(P), and it is known that GDH-50s show B-type stereospecificity (16). Here we show for the first time that the very large J. lividum GDH shows the same B-type specificity as GDH-50s (Fig. 5). These results suggest that the central domain of GDH-180s contains the active site where the enzymatic reaction occurs and that the central region of J. lividum GDH (amino acid residues 719 to 1190) strongly resembles that of C. symbiosum GDH. In addition, the N-terminal domain (amino acid residues 1 to 718) and C-terminal domain (amino acid residues 1191 to 1575) of J. lividum GDH may be responsible for regulating the enzyme activity, although their function is still unclear.
FIG. 6.

Amino acid sequence alignment of the central domains of P. aeruginosa, S. clavuligerus, and J. lividum GDHs (170 to 180 kDa) and C. symbiosum GDH-50. Sequences were aligned using Clustal W (24). Residues conserved in the three very large GDHs and in all the sequences are indicated by shading and asterisks, respectively. Residues that interact with l-glutamate and that are involved with the catalytic reaction are indicated by filled circles. Residues responsible for the pro-S specificity of the hydrogen transfer to NADH are enclosed in a box.
Acknowledgments
We are grateful to Hideyuki Nakatani and Takumi Toida for their excellent technical assistance.
This work was supported in part by the Pioneering Research Project in Biotechnology of the Ministry of Agriculture, Forestry and Fisheries of Japan and by the National Project on Protein Structural and Functional Analyses promoted by the Ministry of Education, Science, Sports, Culture and Technology of Japan. R.K. was supported in part by a Sasakawa Scientific Research Grant from the Japan Science Society.
Footnotes
Published ahead of print on 25 May 2007.
REFERENCES
- 1.Baker, P. J., K. L. Britton, P. C. Engel, G. W. Farrants, K. S. Lilley, D. W. Rice, and T. J. Stillman. 1992. Subunit assembly and active site location in the structure of glutamate dehydrogenase. Proteins 12:75-86. [DOI] [PubMed] [Google Scholar]
- 2.Bhuiya, M. W., H. Sakuraba, C. Kujo, N. Nunoura-Kominato, Y. Kawarabayasi, H. Kikuchi, and T. Ohshima. 2000. Glutamate dehydrogenase from the aerobic hyperthermophilic archaeon Aeropyrum pernix K1: enzymatic characterization, identification of the encoding gene, and phylogenetic implications. Extremophiles 4:333-341. [DOI] [PubMed] [Google Scholar]
- 3.Bhuiya, M. W., H. Sakuraba, T. Ohshima, T. Imagawa, N. Katunuma, and H. Tsuge. 2005. The first crystal structure of hyperthermostable NAD-dependent glutamate dehydrogenase from Pyrobaculum islandicum. J. Mol. Biol. 345:325-337. [DOI] [PubMed] [Google Scholar]
- 4.Bradford, M. M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248-254. [DOI] [PubMed] [Google Scholar]
- 5.Britton, K. L., P. J. Baker, D. W. Rice, and T. J. Stillman. 1992. Structural relationship between the hexameric and tetrameric family of glutamate dehydrogenases. Eur. J. Biochem. 209:851-859. [DOI] [PubMed] [Google Scholar]
- 6.Camardella, L., R. D. Fraia, A. Antignani, M. A. Ciardiello, G. di Prisco, J. K. Coleman, L. Buchon, J. Guespin, and N. J. Russell. 2002. The Antarctic Psychrobacter sp. TAD1 has two cold-active glutamate dehydrogenases with different cofactor specificities. Characterisation of the NAD+-dependent enzyme. Comp. Biochem. Physiol. Part A 131:559-567. [DOI] [PubMed] [Google Scholar]
- 7.Consalvi, V., R. Chiaraluce, L. Politi, R. Vaccaro, M. De Rosa, and R. Scandurra. 1991. Extremely thermostable glutamate dehydrogenase from the hyperthermophilic archaebacterium Pyrococcus furiosus. Eur. J. Biochem. 202:1189-1196. [DOI] [PubMed] [Google Scholar]
- 8.Coschigano, P. W., S. M. Miller, and B. Magasanik. 1991. Physiological and genetic analysis of the carbon regulation of the NAD-dependent glutamate dehydrogenase of Saccharomyces cerevisiae. Mol. Cell. Biol. 11:4455-4465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davis, B. J. 1964. Disc electrophoresis. 2. Method and application to human serum proteins. Ann. N. Y. Acad. Sci. 121:404-427. [DOI] [PubMed] [Google Scholar]
- 10.Degani, Y., F. M. Veronese, and E. L. Smith. 1974. Nicotinamide adenine dinucleotide-specific glutamate dehydrogenase of Neurospora. II. Selective chemical reactivity of amino and sulfhydryl groups. J. Biol. Chem. 249:7929-7935. [PubMed] [Google Scholar]
- 11.Gore, M. G. 1981. l-Glutamic acid deydrogenase. Int. J. Biochem. 13:879-886. [DOI] [PubMed] [Google Scholar]
- 12.Hemmings, B. A. 1980. Purification and properties of the phospho and dephospho forms of yeast NAD-dependent glutamate dehydrogenase. J. Biol. Chem. 255:7925-7932. [PubMed] [Google Scholar]
- 13.Hudson, R. C., and R. M. Daniel. 1993. l-Glutamate dehydrogenase: distribution, properties and mechanism. Comp. Biochem. Physiol. Part B 106:767-792. [DOI] [PubMed] [Google Scholar]
- 14.Kujo, C., and T. Ohshima. 1998. Enzymological characteristics of the hyperthermostable NAD-dependent glutamate dehydrogenase from the archaeon Pyrobaculum islandicum and effects of denaturants and organic solvents. Appl. Environ. Microbiol. 64:2152-2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head bacteriophage T4. Nature 227:680-685. [DOI] [PubMed] [Google Scholar]
- 16.Levy, H. R., and B. Vennesland. 1957. The stereospecificity of enzymatic hydrogen transfer from diphosphopyridine nucleotide. J. Biol. Chem. 228:85-96. [PubMed] [Google Scholar]
- 17.Lu, C.-D., and A. T. Abdelal. 2001. The gdhB gene of Psuedomonas aeruginosa encodes an arginine-inducible NAD+-dependent glutamate dehydrogenase which is subject to allosteric regulation. J. Bacteriol. 183:490-499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miller, S. M., and B. Magasanik. 1990. Role of NAD-linked glutamate dehydrogenase in nitrogen metabolism in Saccharomyces cerevisiae. J. Bacteriol. 172:4927-4935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miñambres, B., E. R. Olivera, R. A. Jensen, and J. M. Luengo. 2000. A new class of glutamate dehydrogenase (GDH). Biochemical and genetic characterization of the first member, the AMP-requiring NAD-specific GDH of Streptomyces clavuligerus. J. Biol. Chem. 275:39529-39542. [DOI] [PubMed] [Google Scholar]
- 20.Mostad, S. B., H. L. Helming, C. Groom, and A. Glasfeld. 1997. The stereospecificity of hydrogen transfer to NAD(P)+ catalyzed by lactol dehydrogenases. Biochem. Biophys. Res. Commun. 233:681-686. [DOI] [PubMed] [Google Scholar]
- 21.Ohshima, T., and N. Nishida. 1993. Purification and properties of extremely thermostable glutamate dehydrogenases from two hyperthermophilic archaebacteria, Pyrococcus woesei and Pyrococcus furiosus. Biosci. Biotechnol. Biochem. 57:945-951. [DOI] [PubMed] [Google Scholar]
- 22.Smith, E. L., B. M. Austen, K. M. Blumenthal, and J. F. Nyc. 1975. Glutamate dehydrogenase, p. 293-367. In P. D. Boyer (ed.), The enzymes, 3rd ed., vol. 11. Academic Press, New York, NY. [Google Scholar]
- 23.Stillman, T. J., P. J. Baker, K. L. Britton, and D. W. Rice. 1993. Conformational flexibility in glutamate dehydrogenase. Role of water in substrate recognition and catalysis. J. Mol. Biol. 234:1131-1139. [DOI] [PubMed] [Google Scholar]
- 24.Thompson, J. D., D. G. Higgins, and T. J. Gibson. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673-4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Veronese, F. M., J. F. Nyc, Y. Degani, D. M. Brown, and E. L. Smith. 1974. Nicotinamide adenine dinucleotide-specific glutamate dehydrogenase of Neurospora. I. Purification and molecular properties. J. Biol. Chem. 249:7922-7928. [PubMed] [Google Scholar]