

Abstract
Certain penicillin binding protein mutants of Escherichia coli grow with spirillum-like morphologies when the FtsZ protein is inhibited, suggesting that FtsZ might govern aspects of cell wall growth other than those strictly associated with septation. While investigating the mechanism of spiral cell formation, we discovered conditions for visualizing this second function of FtsZ. Normally, inhibiting the cytoskeleton protein MreB forces E. coli cells to grow as smoothly enlarging spheres from which the poles disappear, yielding coccoid or lemon-shaped forms. However, when FtsZ and MreB were inhibited simultaneously in a strain lacking PBP 5 and PBP 7, the resulting cells ballooned outward but retained conspicuous rod-shaped extensions at sites representing the original poles. This visual phenotype was paralleled by the biochemistry of sacculus growth. Muropeptides are usually inserted homogeneously into the lateral cell walls, but when FtsZ polymerization was inhibited, the incorporation of new material occurred mainly in the central regions of cells and was significantly lower in those portions of side walls abutting a pole. Thus, reduced precursor incorporation into side walls near the poles explained why these regions retained their rod-like morphology while the rest of the cell grew spherically. Also, inhibiting FtsZ increased the amount of pentapeptides in sacculi by about one-third. Finally, the MreB protein directed the helical or diagonal incorporation of new peptidoglycan into the wall, but the location of that incorporation depended on whether FtsZ was active. In sum, the results indicate that in addition to nucleating cell septation in E. coli, FtsZ can direct the insertion of new peptidoglycan into portions of the lateral wall.
Growth of the cell wall in most gram-negative rod-shaped bacteria is believed to proceed by alternating between two independent modes of peptidoglycan insertion. Inserting new material into the cylindrical side wall drives cell elongation, whereas septation leading to division is accomplished by inserting new material into the leading edge of a ring that constricts and bisects the cell (17, 27, 37). The two processes are thought to be sufficiently independent that they constitute separate systems, sometimes described as “two competing sites” (4, 16, 31). In this view, switching between elongation and division occurs via a periodic back-and-forth tussle between mechanisms that direct peptidoglycan synthesis either into the lateral wall or into the septum, respectively. Although this idea has a long history (16, 31, 32), the nature of the competing entities was mostly a mystery. Recently, the general composition of the two systems has come into sharper focus. In Escherichia coli, peptidoglycan elongation of the side wall requires RodA and PBP 2 (17, 27), whose activities may be directed in whole or in part by the actin homologue MreB (22). The second mechanism, driving cell division, is nucleated by the tubulin homologue FtsZ, which recruits and organizes the activities of a cascade of septation proteins that includes FtsW and the putative transpeptidase PBP 3 (3). Similar proteins drive the two processes in other organisms, albeit with complicated variations (6, 12, 15, 19, 24, 34).
Although FtsZ is the focal point for initiating bacterial cell division (3, 24), there are hints that this protein may also play a role outside division per se by helping create or maintain the natural rod-shaped morphology of E. coli (36, 38). In particular, when FtsZ is inhibited, mutants lacking certain low-molecular-weight penicillin binding proteins (PBPs) grow as spiral filaments instead of as long straight rods (36). FtsZ collaborates in creating this phenomenon because the inhibition of PBP 3 or FtsA produces filamentous cells that do not spiral (36). Since changes in cellular morphology must be coordinated with changes in peptidoglycan structure or geometry, these results imply that FtsZ is involved in modulating, directly or indirectly, aspects of cell wall synthesis other than those that accompany the latter stages of cell division. Previous clues imply that this function may be related to penicillin-insensitive peptidoglycan synthesis (PIPS) (26, 28, 37), a process that produces an FtsZ-dependent but PBP 3-independent ring of inert peptidoglycan at sites of future septa (8). In any case, the general nature and molecular components of this second FtsZ-triggered pathway remain very much unknown.
We report here that this second role of FtsZ can be visualized more clearly by examining cell wall synthesis in an E. coli PBP mutant that retains elevated amounts of pentapeptide side chains in its cell wall. Inhibiting FtsZ under these circumstances substantially reduces peptidoglycan incorporation into those portions of the side wall near the poles and also alters the cell's muropeptide composition. These effects are made more apparent by the simultaneous inhibition of FtsZ and MreB. Instead of growing as smoothly enlarging spheres, the cells retain short tubular extensions that represent preexisting poles and their associated near-polar side walls. This observation provides visual confirmation of an underlying deficit of peptidoglycan insertion. Thus, in this strain, biochemical and physiological measures indicate that peptidoglycan is inserted into near-polar side walls only when FtsZ is active, suggesting that FtsZ influences peptidoglycan synthesis beyond its classical role during cell division. We also find that MreB is required for the homogeneous insertion of new material leading to the elaboration of spiral cells. Overall, the results imply that the two ostensibly separate mechanisms governing peptidoglycan synthesis are probably more intertwined than independent.
MATERIALS AND METHODS
Bacteria, plasmids, and growth conditions.
E. coli MG1655 (ilvG rfb-50 rph-1) served as the parent strain for E. coli AV15-1K (pbpG::Kan) (ΔPBP 7), AV21-10 (dacA::res) (ΔPBP 5), AV23-1 (dacA::res pbpG::Kan) (ΔPBPs 5 and 7), and AV65-1 [dacA::res pbpG::Kan ftsA12(Ts)] (36). Plasmid pFAD38 was supplied by J. Lutkenhaus and carries the sulA gene under the control of an arabinose promoter (2). Plasmid pLE7 (Plac-yfp::mreB) was supplied by L. Rothfield and expresses yellow fluorescent protein (YFP)-MreB from a gene under the control of the lac promoter (33). A22 (14) was a gift of A. Novikov. Strains were maintained in Luria-Bertani (LB) broth or on LB agar plates supplemented with ampicillin (50 μg/ml) or kanamycin (50 μg/ml) to maintain plasmids or with glucose (0.4%, wt/vol) to inhibit sulA expression from pFAD38. Cultures grown overnight were washed to remove glucose and then diluted 1:50 into fresh LB medium containing 0.5% NaCl. Arabinose (0.2% final concentration) was added to induce sulA expression from pFAD38, IPTG (isopropyl-β-d-thiogalactopyranoside) was added to a final concentration of 50 μM, and the cultures were incubated for 2 h at 42°C to induce the production of YFP-MreB from plasmid pLE7. When required, aztreonam (1 or 2 μg/ml) was added to block cell division, A22 (2 or 5 μg/ml) was added to inhibit MreB, and amdinocillin (2 μg/ml) was added to inhibit PBP 2. Different combinations of these procedures were implemented as diagrammed in Fig. 1A.
FIG. 1.

Experimental outlines. (A) Protocols for examining the effects of A22 on cell morphology. (B) d-Cysteine labeling experiments. (C) d-Cysteine labeling of the E. coli ftsA12(Ts) mutant AV65-1. The addition of 0.2% arabinose induced the expression of the sulA gene, MreB was inhibited by adding compound A22, and PBP 3 was inhibited by adding aztreonam. O/N, overnight.
d-Cysteine labeling of peptidoglycan.
d-Cysteine labeling was performed as described previously (8, 10) and as diagrammed in Fig. 1B. Briefly, cultures grown overnight were washed to remove glucose and diluted 1:50 in LB broth containing 0.5% salt and the appropriate antibiotics. The mixture was incubated for 10 min at 30°C, d-cysteine (200 μg/ml) was added and incubated for 30 min at 30°C, and arabinose (0.2%) and/or aztreonam (1 or 2 μg/ml) was then added and incubated for 1 h at 30°C. The cells were collected by centrifugation at 30°C (4 min at 8,000 × g) and resuspended in d-cysteine-free medium prewarmed to 42°C. For strain AV65-1 [MG1655 ftsA12(Ts) leu::Tn10] (36), d-cysteine labeling was performed as outlined in Fig. 1C.
Purification and immunolabeling of sacculi.
Cells were harvested, sacculi were isolated, and incorporated d-cysteine was biotinylated as described previously (8, 10). Biotin residues were visualized by immunolabeling followed by confocal microscopy. For confocal microscopy, multiple drops (1 to 2 μl) of appropriately diluted sacculi were placed onto glass coverslips and processed for immunolabeling (10). Biotin was detected by double labeling by adding mouse anti-biotin monoclonal antibody (Molecular Probes, Eugene, OR) diluted 1:250 into PBG medium (0.2% gelatin and 0.4% bovine serum albumin in phosphate-buffered saline [PBS] [pH 7.0]), which was followed by adding Alexa Fluor 488-conjugated anti-mouse secondary antibody (Molecular Probes) diluted 1:250 in PBG medium. To visualize the entire peptidoglycan surface of sacculi, samples were labeled with rabbit anti-murein antiserum (M. de Pedro, unpublished data), which was followed by staining with rhodamine red anti-rabbit antibody (Molecular Probes), each diluted 1:250 in PBG medium.
Vancomycin labeling of sacculi.
Areas enriched in muropeptides with pentapeptide side chains are likely to represent sites where new precursors are being inserted into the cell wall (6, 35). We labeled these areas by using vancomycin (Sigma Chemical Co., St. Louis, MO) as a specific pentapeptide-binding reagent and then imaged sacculi-bound vancomycin by immunofluorescence by staining with rabbit anti-vancomycin antibody (GTX19968; GeneTex, Inc., San Antonio, TX), followed by staining with Alexa Fluor 488-labeled goat anti-rabbit antibody (1:250) in PBG. This approach was preferable to the use of a fluorescent vancomycin-Bodipy-FL conjugate (Molecular Probes), mostly because the latter is subject to rapid photobleaching. However, unlabeled vancomycin also permitted the use of saturating concentrations of vancomycin and avoided artifacts that may be associated with the binding properties of derivatized vancomycin (35; de Pedro, unpublished). The following protocol was developed to optimize binding site saturation and specificity. A detailed account of our studies on the interaction between vancomycin and isolated E. coli sacculi will be reported elsewhere (M. de Pedro et al., unpublished data). Briefly, droplets of biotinylated sacculus suspensions were spotted onto glass cover slides and allowed to air dry. The immobilized sacculi were washed twice with water and incubated with 150 μl of vancomycin (100 μM) in 0.1 M NaHCO3-Na2CO3 buffer (pH 10.2) at room temperature for 15 min. The high pH prevents the nonspecific binding of vancomycin to sacculi devoid of pentapeptides that is observed at neutral pH, and the elevated concentration of vancomycin ensures binding site saturation. Samples were washed four times with 0.1 M NaHCO3-Na2CO3 buffer (pH 10.2), twice with PBS, and twice with PBG medium. After 5 min of incubation in PBG medium, sacculi were incubated for 1 h at room temperature with a mixture of rabbit anti-vancomycin antibody (1:250 in PBG) (to label pentapeptides) and mouse anti-biotin monoclonal antibody (1:250 in PBG) (to label biotinylated d-cysteine residues). Samples were washed thoroughly with PBG and incubated for 1 h with a mixture of Alexa Fluor 488-labeled goat anti-rabbit antibody and rhodamine-labeled goat anti-mouse antibody (1:250 each in PBG). Samples were washed, sequentially and extensively, with PBG medium, PBS, and water, respectively, after which the samples were air dried, mounted in Pro-Long Gold mounting medium (Molecular Probes), and cured overnight in the dark at room temperature.
Microscopy.
Confocal microscopy was performed with a Zeiss META microscope using a Zeiss 63× 1.4-numerical-aperture objective lens and parameters appropriate for the Nyquist criteria for deconvolution. Images were deconvolved using Huygens software (Scientific Volume Imaging B.V., Hilversum, The Netherlands) and then assembled with Adobe Photoshop and Image J.
RESULTS
MreB and PBP 2 are required for the formation of spiral cells.
When FtsZ polymerization is inhibited, a large fraction of E. coli AV23-1 (ΔPBPs 5 and 7) grows as spiral-shaped filaments (36). Purified sacculi isolated from these cells retain this spiral morphology, illustrating once again that it is the cell wall that determines bacterial size and shape. Since both FtsZ and MreB are homologues of eukaryotic cytoskeletal proteins (5, 15), it seemed likely that these two proteins might work in concert to create or maintain the spiral nature of this mutant. To see if this was true, we inhibited MreB by adding the compound A22 (14) to cells with active FtsZ and to cells in which FtsZ polymerization had been inhibited by SulA (25). The simultaneous inhibition of FtsZ and MreB in E. coli AV23-1 blocked the formation of spiral filaments and caused cells to become lemon shaped while retaining short tubular extensions at the poles (Fig. 2A). When cells were exposed transiently to A22 and the antibiotic was removed by washing, E. coli cells regained the ability to form coiled filaments (Fig. 2B), indicating that the effect of MreB inhibition was reversible and that continuous MreB function was required for the formation of spiral cells.
FIG. 2.
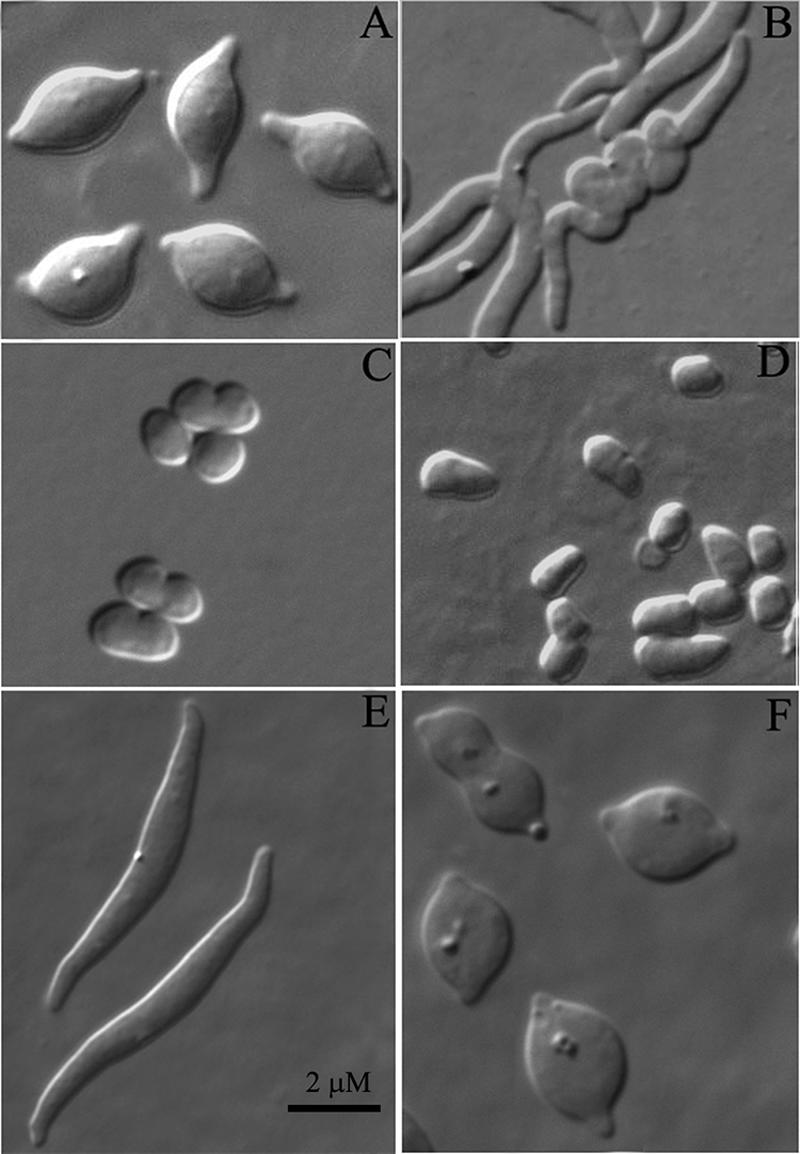
Morphological effects of inhibiting FtsZ, MreB, or PBP 2. E. coli MG1655 or AV23-1 was treated to inhibit one or more of the following proteins: FtsZ was inhibited by inducing SulA from pFAD38 by adding 0.2% arabinose, MreB was inhibited by adding A22 at the indicated concentrations, and PBP 2 was inhibited by adding amdinocillin (2 μg/ml). (A) AV23-1, with inhibition of both FtsZ and MreB (A22, 5 μg/ml). (B) AV23-1. Conditions were as described above (A), but A22 was removed before a shift to growth at 42°C. (C) MG1655, with inhibition of MreB (A22, 5 μg/ml). (D) AV23-1, with inhibition of MreB (A22, 5 μg/ml). (E) AV23-1, with inhibition of both FtsZ and MreB (A22, 2 μg/ml). (F) AV23-1, with inhibition of FtsZ and PBP 2.
A trivial explanation for the above-described results could have been that inhibiting MreB provoked the lysis of any preexisting coiled cells so that only uncoiled cells survived to adopt the observed morphologies. To rule out this possibility, we used time-lapse microscopy to observe how MreB inhibition affected preexisting coiled filaments. When exposed to A22, spiral cells progressively widened into lemon-shaped cells, proving that the disappearance of coiled cells was not the result of selective lysis (Fig. 3). Thus, both the generation and continued existence of spiral shapes depended on active MreB.
FIG. 3.

Effect of inhibiting MreB in a preformed spiral cell. Spiral cells were induced by inhibiting FtsZ in AV23-1 by expressing SulA from pFAD38. The cells were transferred onto agar wells containing A22 (5 μg/ml) (to inhibit MreB) and 0.2% arabinose (to inhibit FtsZ) and incubated at 42°C on a heated microscope stage. Individual spiral cells were selected for microscopic observation, and one typical experiment is shown. Numbers refer to minutes of incubation.
A more likely possibility for the failure of E. coli AV23-1 to coil was that inactivating MreB forced the cells to grow as spheres, which, due to simple geometry, could not adopt a spiral morphology. We tested this by adding A22 at a concentration (2 μg/ml) that took longer to produce coccoid cells (>1 h). Cells treated this way formed long and broadened straight filaments that did not coil (Fig. 2E), suggesting that this concentration of A22 inhibited only part of the MreB complement within the cells. Thus, although the disappearance of spiral forms could be due to geometric constraints (because sphere formation precluded spiraling), it was clear that partial inhibition of MreB was sufficient to block spiral formation. Furthermore, cells preincubated in the presence of A22 did not form spirals when FtsZ was inhibited at a later time, and spiral cells that grew in the presence of active MreB became unwound when A22 was added later (see, e.g., Fig. 3). Therefore, although a geometrical effect may have prevented cells from forming spirals in the first instance, the second situation indicates that peptidoglycan synthesis or remodeling remained active within the coiled areas and that these regions responded to A22 as would a normally growing cell. In summary, a fully functional MreB-driven complex was required to create a spiral sacculus.
Since MreB is cytoplasmic, it seemed unlikely that this protein would be involved directly in peptidoglycan synthesis. Current models envision a chain of connections between MreB and the synthetic machinery in the periplasm (22), including one periplasmic PBP, PBP 2, that is required for lateral cell wall synthesis. If this were true, then inhibiting PBP 2 and FtsZ should give a phenotype similar to that produced by inhibiting MreB and FtsZ. As predicted by this hypothesis, spiral formation was blocked after inhibiting FtsZ with SulA while also inactivating PBP 2 with the antibiotic amdinocillin, and the resulting spheroidal cells exhibited tubular projections at their poles (Fig. 2F). These results were the same as those obtained by inhibiting MreB and FtsZ and are consistent with MreB and PBP 2 being members of a common morphological pathway.
FtsZ is required for the expansion of near-polar side walls.
The most surprising aspect of the above-described experiments was the curious morphology adopted by E. coli AV23-1 when MreB and FtsZ were inhibited simultaneously. When both FtsZ and MreB were inhibited in AV23-1 cells, the cells bloated as expected, but there was little or no expansion of those portions of the side wall immediately adjacent to the original poles (Fig. 2A). Instead of smooth lemon shapes, these cells retained tubular extensions that persisted while the cells continued to enlarge, indicating that new peptidoglycan was not being inserted equally over the entire surface of the cell. In particular, there was little or no new cell wall growth in the vicinity of preexisting poles.
At first, we thought that this phenomenon was confined to the PBP mutant AV23-1. However, when we quantified the numbers of cells exhibiting these morphologies, it became clear that the mutant had magnified a phenotype present at a lower frequency in the parental strain MG1655 (Table 1). When both FtsZ and MreB were inhibited, MG1655 produced a range of morphotypes, including lemon-shaped cells without tubular poles (27% ± 3% of the population), cells with a single tubular pole (37% ± 6%), and cells with both poles extended (36% ± 3%) (Table 1). Deletion of either PBP 5 or PBP 7 alone did not change these frequencies appreciably (strains AV21-10 and AV15-1K) (Table 1). On the other hand, the absence of both of these PBPs in strain AV23-1 significantly decreased the percentage of lemon-shaped cells without tubular poles (down to 15% ± 5%) and increased the percentage of cells with tubular extensions at both poles (up to 47% ± 0.6%) (Table 1). When present, these rod-like protrusions were 1.0 to 1.5 μm long (average of 1.4 μm), and the lengths were comparable for parent strain MG1655 (measurements from 90 cells), for the single mutants AV21-10 (ΔPBP 5) (55 cells) and AV15-1K (ΔPBP 7) (77 cells), and for the double mutant AV23-1 (ΔPBPs 5 and 7) (36 cells).
TABLE 1.
Distribution of tubular polar extensions in PBP mutants after inhibition of MreB and FtsZa
 |
Most importantly, regardless of whether the strain was wild type or mutated, polar extensions were present only when FtsZ and MreB were inhibited simultaneously. Inhibiting FtsZ alone caused every cell to grow as a long filament (data not shown) (Fig. 2B), whereas inhibiting only MreB caused every cell to round up into a sphere (e.g., MG1655) (Fig. 2C) or into a quasilemon or squarish shape (e.g., AV23-1) (Fig. 2D). Inhibiting PBP 2 duplicated the results of MreB inhibition (Fig. 2F).
The results indicated that inhibiting polymerization of FtsZ prevented the expansion of side walls adjacent to preexisting polar regions. This situation, therefore, modified the morphological effects that normally accompany the inhibition of MreB or PBP 2. In contrast, when MreB alone was inhibited, cell wall expansion occurred over the entire surface, including those regions near the poles. The implication is that FtsZ plays a role in the disposition of new peptidoglycan into the near-polar domains of the lateral wall in E. coli and that this function is somewhat more prominent in a mutant lacking PBP 5 and PBP 7.
Murein is inserted transversely into the E. coli cell wall.
The simplest explanation for the persistence of tubular extensions in cells lacking active FtsZ and MreB was that FtsZ influenced the insertion of new peptidoglycan into near-polar side walls. Because evidence for the geometry of insertion of new murein into the wall of E. coli is indirect and incomplete (9, 13), we examined this question first. To facilitate the visualization of the cell wall structure in sacculi, we forced E. coli cells to grow as filaments by adding aztreonam, which inhibits the essential septation protein PBP 3 (FtsI) and blocks cell division. This procedure produced cells long enough that the extent and localization of new wall growth could be monitored more easily. The pattern of peptidoglycan insertion was tracked by using the d-cysteine pulse-chase technique (8, 10), and fluorescently labeled d-cysteine residues were visualized by acquiring a series of z-plane images by confocal microscopy, followed by deconvolution. These techniques substantially enhanced the quality and resolution of the resulting images compared to those previously obtained with the d-cysteine labeling method, thus expanding the amount of detail that could be observed.
Consistent with a roughly helical or diagonal incorporation of muropeptides into the side walls of E. coli MG1655 and AV23-1, new (d-cysteine-unlabeled) peptidoglycan was arrayed as a transverse lattice, visualized as short dark arcs separating the bright fluorescent fibers of older (d-cysteine-labeled) peptidoglycan. Both new and old material were distributed homogeneously in a criss-crossing meshwork that extended the length of each filament (Fig. 4A, panels 1 to 4 and 9). Although new murein was not inserted with a clearly defined periodicity, the images were compatible with the planar projection of a roughly helical distribution of insertion sites, as proposed previously for Bacillus subtilis (6, 35). We could not determine whether the diagonal incorporation pattern was continuous or if it arose from multiple overlapping synthetic events. The cells also had sharply defined, highly labeled polar regions (Fig. 4A, panels 9, 11, and 12), consistent with the presence of circumscribed domains of inert peptidoglycan known to exist at the poles of wild-type E. coli (8, 10). None of these peptidoglycan arrangements were artifacts of antibiotic treatment because similar results were obtained with strains in which filamentation was induced by inactivating temperature-sensitive variants of the FtsA or PBP 3 (FtsI) protein. More importantly, identical insertion patterns were observed in both the parental strain and PBP mutants, indicating that the mutations did not alter the general nature of peptidoglycan insertion. Thus, the overall incorporation of new peptidoglycan into the cell wall of E. coli occurs along diagonal lines, lending experimental credence to the notion that this organism and B. subtilis share an underlying helical mechanism that directs cell wall synthesis.
FIG. 4.

Peptidoglycan synthesis in E. coli detected by d-cysteine labeling. (A) Cells were filamented by adding aztreonam at either 2 μg/ml (for wild-type [WT] MG1655) or 1 μg/ml (for the AV23-1) (panels 1, 2, 5, 6, 9, 11, and 12). Alternatively, cells were filamented by inhibiting FtsZ by adding 0.2% arabinose to induce the expression of the SulA protein from plasmid pFAD38 (panels 3, 4, 7, 8, 10, and 13). Panels 1 to 4, 9, and 10, MG1655; panels 5 to 8 and 11 to 13, AV23-1 (ΔPBPs 5 and 7). Fluorescent fibers represent older (labeled) peptidoglycan, and dark areas represent sites of newly incorporated peptidoglycan during the chase period. Arrowheads in panels 9, 11, and 12 indicate the locations of dark transverse bands that represent new peptidoglycan incorporation at incipient sites of septation. (B) Differences in peptidoglycan insertion quantified by gray-level profiles of d-cysteine-labeled sacculi. Panel 1, MG1655 filamented with aztreonam (FtsZ+); panel 2, MG1655 filamented with SulA (FtsZ−); panel 3, AV23-1 filamented with aztreonam (FtsZ+); panel 4, AV23-1 filamented with SulA (FtsZ−).
Inhibiting MreB produces uneven cell wall synthesis.
When treated with A22 to inhibit MreB, many of the resulting lemon-shaped cells bulged asymmetrically (Fig. 2A, C, and D), suggesting that peptidoglycan incorporation was so uneven that new material was often inserted more frequently into one area of the cell than into another. To determine if this inference was supported on the molecular level, we visualized the location of newly incorporated peptidoglycan in strain AV23-1 in which FtsZ and MreB had been inhibited (Fig. 5). Confocal microscopy of sacculi from cells labeled with d-cysteine and then incubated in its absence (pulse-chase) revealed that the bulging regions were composed mostly of newly inserted peptidoglycan (dark zones) flanked by areas enriched for fibers of older peptidoglycan (Fig. 5A, green label). This was consistent with the impression that peptidoglycan insertion was unevenly distributed in the absence of MreB.
FIG. 5.

Inhibiting MreB alters the pattern of peptidoglycan insertion into the cell wall. AV23-1 was filamented by inhibiting FtsZ with SulA expressed from pFAD38, the cells were labeled with d-cysteine for 90 min and chased for 30 min at 42°C in the presence of A22 (5 μg/ml) and in the absence of d-cysteine, and sacculi were prepared and immunolabeled. (A) d-Cysteine pulse-chase-labeled sacculi. The left-hand cell in each panel displays older peptidoglycan (green) with uneven, localized incorporation of new material (dark areas). The right-hand cell in each panel is immunostained to detect all peptidoglycan; uniform labeling indicates that the dark areas are not the result of murein degradation. (B) Vancomycin labeling of a typical d-cysteine-labeled sacculus. The left-hand figure shows the location of vancomycin-labeled peptidoglycan (green), the middle figure shows the location of older d-cysteine-labeled peptidoglycan (red) and the right-most figure is a merged image. Note that the location of vancomycin-labeled material corresponds to the d-cysteine-free central dark area.
The unlabeled patches in sacculi synthesized in the absence of MreB could represent either the localized degradation of older peptidoglycan (resulting in the removal of d-cysteine) or the incorporation of new muropeptides (resulting in the insertion of new, unlabeled material). To distinguish between these two possibilities, we labeled sacculi with antibodies directed against total murein. Sacculi stained in this way showed a smooth and uniform labeling, indicating that the murein contained no large holes or thinned patches and suggesting that the large patches devoid of d-cysteine did not arise by localized degradation (Fig. 5A). To confirm that these darkened areas represented the incorporation of new material, we visualized pentapeptide-enriched regions, representing sites of active synthesis, in sacculi labeled with d-cysteine and chased for 30 min in its absence. The d-Ala-d-Ala termini of newly inserted pentapeptide chains were labeled with vancomycin and detected with anti-vancomycin antibodies. This method avoided artifacts that accompany the use of fluorescent derivatives (35; de Pedro et al., unpublished). Patches of vancomycin (Fig. 5B, left, green) were concentrated in regions of newly incorporated peptidoglycan (Fig. 5B, center, dark areas) and were surrounded by older wall material (Fig. 5B, center, red). Conversely, regions rich in d-cysteine (old peptidoglycan) showed reduced binding of vancomycin (Fig. 5B, center, red). As usual, peptidoglycan at the poles showed little or no turnover of old material or incorporation of new material so that these sites gave the strongest signal for d-cysteine (red) and the weakest signal for vancomycin (green) (Fig. 5B, center and right). Thus, the asymmetric bulges in sacculi were formed by redirecting the incorporation of new wall material rather than being due to murein degradation. These results were also reproduced in an E. coli mutant lacking only PBP 5 (not shown), meaning that the loss of PBP 7 did not contribute to the redistribution of cell wall synthesis.
Altogether, the results indicated that when MreB was inhibited, peptidoglycan synthesis continued but was no longer distributed uniformly over the entire cell surface. Instead, large amorphous patches of new wall material were inserted mainly into the central regions of the lateral wall in the previously rod-shaped cells.
FtsZ directs peptidoglycan insertion into near-polar side walls.
The retention of tubular polar extensions in E. coli AV23-1 cells in which both FtsZ and MreB were inhibited indicated that cell wall synthesis had been rearranged. However, the restricted and patchy insertion of peptidoglycan when MreB was inhibited did not explain why the poles did not expand when FtsZ was also inhibited. One alternative was that this phenotype depended on a novel function of FtsZ. Therefore, we examined how peptidoglycan incorporation was altered when FtsZ was inhibited in strain AV23-1. Cells were forced to grow as filaments either by overexpressing the FtsZ antagonist SulA (FtsZ− condition) (36) or by adding aztreonam to inhibit PBP 3 (FtsZ+ condition). The filaments were labeled with d-cysteine and incubated for 30 min in its absence (chased), and sacculi were prepared, immunolabeled, and observed by confocal fluorescence microscopy to visualize the insertion patterns of new precursors.
As described above, d-cysteine-labeled peptidoglycan appeared as a network of transverse fibers in both the parent strain and PBP mutant strain AV23-1 (Fig. 4A, panels 1 to 8). In both strains, cells with active FtsZ displayed prominent dark zones of newly incorporated material at sites that would normally develop into septa (Fig. 4A, panels 9, 11, and 12, arrowheads), whereas the striations disappeared when FtsZ was inhibited (Fig. 4A, panels 10 and 13). These FtsZ-dependent, septum-associated bands corresponded to the well-attested PIPS phenomenon, which represents an FtsZ-directed stage of peptidoglycan synthesis that precedes PBP 3-dependent synthesis during constriction and division (8, 26, 28, 37). Other than this, in the parental strain, there was no other FtsZ-dependent insertion of new cell wall material (dark regions) or distribution of older peptidoglycan (fluorescent fibers). Instead, similar patterns of side wall incorporation were observed when FtsZ was active or when the protein was inhibited (Fig. 4A, compare panels 9 and 10, and B, compare panels 1 and 2).
In contrast to the parental strain, inhibiting FtsZ in E. coli AV23-1 altered the pattern of peptidoglycan incorporation into the side wall. We quantified this effect by measuring the d-cysteine distribution along the length of individual sacculi and recording the insertion of new material from pole to pole for individual sacculi (Fig. 4B). As expected, when FtsZ was active, new material was incorporated homogenously along a cell's length in both the parent strain (Fig. 4B, panel 1) and the mutant (Fig. 4B, panel 3), with only a very small region at each pole being refractory to the insertion of new peptidoglycan (note the steep rise in incorporation at the extreme left and right of each graph in panels 1 and 3 of Fig. 4B). However, when FtsZ was inhibited, unlabeled peptidoglycan was inserted primarily into the central portion of each cell filament, with much less incorporation into the cylindrical walls near the poles (Fig. 4B, panel 4, note the gradual rise in incorporation at either end of the graph). Normally, only the hemispherical ends of the poles are distinguished by a small and distinct cap of inert peptidoglycan, the edges of which are sharply demarcated from the lateral walls, where active insertion occurs (7, 8, 10). This was true for the parent strain regardless of whether or not FtsZ was active (Fig. 4A, compare panels 9 and 10) and in PBP mutant strain AV23-1 when FtsZ was active (Fig. 4A, panels 11 and 12). However, when FtsZ was inhibited in AV23-1, the refractory polar domains were expanded significantly beyond their normal range (Fig. 4A, panel 13), and the cells exhibited a gradient of peptidoglycan incorporation that increased gradually from either pole towards the center of the cell (Fig. 4B, panel 4).
We quantified the extent of this alteration by measuring the slope of incorporation beginning from the tip of the pole (no incorporation) to the point where incorporation reached its maximum rate (defined as the boundary between the inert polar domain and the lateral wall) (Table 2). A short domain distance gave a larger value for the slope (incorporation rose quickly to its maximum), whereas a more extended domain gave a smaller value for the slope (incorporation rose slowly to its maximum). The incorporation slope was large and constant in the parent strain MG1655 whether or not FtsZ was active (Table 2), indicating that FtsZ had little or no effect on peptidoglycan insertion into the polar region. The same was true for strain AV23-1 (ΔPBPs 5 and 7) when FtsZ was active (Table 2). However, when FtsZ was inhibited in AV23-1, the value of the slope was significantly less than those under the other conditions (P < 0.001) (Table 2). These measurements confirmed the visual microscopic observations. Overall, when FtsZ was inhibited in AV23-1, peptidoglycan insertion was reduced in a region comprising ∼20 to 25% of the length of the filament nearest either pole, meaning that wild-type levels of incorporation occurred predominantly in the central 50% of each filament (Fig. 4B, panel 4, and data not shown). This was in contrast to the situation in the parent strain or in AV23-1 when FtsZ was active, where reduced incorporation was observed for only 5 to 8% of the cell length near each pole (Fig. 4B, panels 1 to 3). Thus, there was an FtsZ-dependent increase in the insertion of peptidoglycan into the lateral wall near cell poles, and this phenomenon was especially easy to measure in a mutant lacking PBP 5 and PBP 7 (although equivalent results were also obtained for a mutant lacking only PBP 5) (not shown).
TABLE 2.
Peptidoglycan incorporation into side walls near poles of E. coli
| Strain | Treatmenta (protein inhibited) | Avg slopeb (arbitrary units/pixel ± SD) | No. of measurements |
|---|---|---|---|
| MG1655 | Aztreonam (PBP 3) | 4.1 ± 2.4 | 12 |
| MG1655 | SulA (FtsZ) | 3.8 ± 1.7 | 12 |
| AV23-1 | Aztreonam (PBP 3) | 3.7 ± 1.5 | 13 |
| AV23-1 | SulA (FtsZ) | 1.2 ± 0.7c | 9 |
Aztreonam (1 to 2 μg/ml) was added to inhibit the essential septation protein PBP 3 while leaving the function of FtsZ intact. The SulA protein was produced by arabinose induction of the sulA gene on plasmid pFAD38, thereby inhibiting FtsZ.
E. coli sacculi were isolated from strains labeled with d-cysteine and then incubated (chased) in its absence. Peptidoglycan incorporation was quantified by measuring the gray-level profiles of 100 midline pixels along the length of each sacculus, beginning at each pole, moving toward the center of the cell.
The slopes for incorporation of peptidoglycan into E. coli treated with SulA were significantly different (according to Student's t test) between AV23-1 and MG1655 (P < 0.001) and between AV23-1 and AV23-1 treated with aztreonam (P < 0.001).
In addition to a quantitative difference in peptidoglycan incorporation at the poles, inhibiting FtsZ changed the microscale pattern of incorporation within PBP mutant strain AV23-1. Instead of numerous, narrow, sharp peaks of new material (Fig. 4B, panel 3), incorporation was distributed among fewer, broadened, blunted peaks (Fig. 4B, panel 4). This more coarse pattern of muropeptide insertion would be expected if the rate of incorporation was lower near the poles, which was the case. For total peptidoglycan insertion to remain approximately constant, the rate of insertion must rise in the central region to compensate for the decreased rate in the apical areas. More new murein must be accommodated at each insertion site, which gives the slight, but noticeable, widening of the unlabeled bands (Fig. 4B, panel 4).
In summary, the results indicate that in addition to directing the synthesis of septal murein, FtsZ is also required to distribute peptidoglycan incorporation properly in portions of the lateral cell wall and most especially in a mutant lacking PBP 5 and PBP 7.
Inhibiting FtsZ affects the muropeptide composition of E. coli AV23-1.
Because inhibiting FtsZ changed the pattern of peptidoglycan incorporation, it was possible that FtsZ might also influence the structure of the murein product. To evaluate this, muropeptide compositions were determined for sacculi from cells in which FtsZ was active or that had been inhibited by SulA.
Compared to E. coli MG1655, deleting PBP 5 and PBP 7 to create E. coli AV23-1 resulted in three major changes (Table 3). First, AV23-1 contained substantially more pentapeptides (22% of total muropeptides) than did the parent. This was expected because of the absence of PBP 5, the principal dd-carboxypeptidase that removes the terminal d-alanine from pentapeptide side chains. In MG1655, with active PBP 5, the amount of this component is undetectable (<0.2%). Second, the percentage of cross-linked muropeptides (dimers plus trimers) rose by 28%, increasing from 29.3% of all muropeptides in MG1655 to 36.1% in AV23-1. This was most likely due to the combined effects of a greater number of pentapeptides available for cross-linking plus the impairment of PBP 7, a dd-endopeptidase, that would normally remove some of these cross-links. The third change was that the cell wall contained 43% fewer muropeptides covalently linked to Braun's lipoprotein, decreasing from 9.4% of all muropeptides in MG1655 to 5.4% in AV23-1. Braun's lipoprotein is attached to the free amino group of diaminopimelic acid (DAP) via the same amino group required for cross-linking adjacent peptide side chains. Thus, the increased number of cross-links may have reduced a subset of DAP residues available for lipoprotein attachment.
TABLE 3.
Murein composition of wild-type E. coli and a PBP mutanta
| Composition | Mol%
|
|||||||
|---|---|---|---|---|---|---|---|---|
| MG1655
|
AV23-1
|
|||||||
| +FtsZ, +MreB | −FtsZ, +MreB | +FtsZ, −MreB | −FtsZ, −MreB | +FtsZ, +MreB | −FtsZ, +MreB | +FtsZ, −MreB | −FtsZ, −MreB | |
| Pentapeptide | 22.0 | 29.5 | 8.1 | 10.6 | ||||
| Monomers | 69.5 | 69.3 | 64.0 | 61.9 | 63.4 | 65.4 | 59.9 | 60.0 |
| Dimers | 27.8 | 28.6 | 31.8 | 32.9 | 33.8 | 32.5 | 35.8 | 34.5 |
| Trimers | 1.5 | 1.2 | 3.6 | 4.0 | 2.3 | 1.7 | 3.6 | 4.5 |
| Lipoprotein | 9.4 | 8.2 | 11.8 | 10.5 | 5.4 | 4.5 | 9.2 | 10.2 |
| Anhydro | 2.0 | 1.9 | 4.7 | 5.3 | 1.9 | 1.1 | 4.5 | 4.8 |
| DAP-DAP | 2.9 | 2.8 | 5.9 | 7.3 | 2.0 | 1.2 | 4.0 | 5.1 |
Peptidoglycan from E. coli strains MG1655 (parental) and AV23-1 (ΔPBPs 5 and 7) was isolated and treated as described in Materials and Methods. The FtsZ-dependent increases in pentapeptide levels were consistent in two independent experiments and four independent samples (not shown).
Most interestingly, when FtsZ was inhibited in AV23-1, the level of pentapeptides rose by 34% (from 22% to 29.5%) when MreB was active and by 35% (from 8.1% to 10.9%) when MreB was inactive (Table 3). The magnitude of this FtsZ-dependent increase in pentapeptides was surprising and unprecedented. The only other comparable result occurs when the dd-carboxypeptidase PBP 5 is deleted from E. coli, since this eliminates the enzyme responsible for removing the terminal d-alanine in the first place. The fact that inhibiting FtsZ produces an additional 35% increase in this component supports the existence of an FtsZ-dependent peptidoglycan-centered reaction that consumes pentapeptides. This reaction must operate independently of MreB since inhibiting FtsZ caused an increase in murein pentapeptides regardless of whether or not MreB was active. Also, the alteration in pentapeptide levels was not due to shifting the culture from growth in glucose (where SulA production was inhibited) to growth in arabinose (where SulA was produced) because pentapeptide composition under either of these conditions was 23%, which was equivalent to that in AV23-1 when both FtsZ and MreB were active (Table 3).
Inhibiting MreB changes the muropeptide composition of peptidoglycan.
Because inhibiting MreB had a strong effect on the general pattern of peptidoglycan insertion, we wondered if the treatment also altered the chemical nature of the material synthesized. Inhibiting MreB with A22 changed the murein composition in four ways in both the parent strain and PBP mutant strain AV23-1. First, the percentage of cross-linked muropeptides (dimers plus trimers) increased moderately, regardless of the state of FtsZ or the presence or absence of PBP 5 and PBP 7. In the parent strain MG1655, cross-linking increased 21% if FtsZ was active and 24% if FtsZ was inhibited by SulA, and in mutant strain AV23-1, cross-linking increased by 5.3% if FtsZ was active and by 15% if FtsZ was inhibited (Table 3). Inhibiting FtsZ in any of the pairings had no significant effect on cross-linking percentages. Second, the proportions of lipoprotein-bound muropeptides and DAP-DAP cross-links increased by ∼20 to 50% and 75 to 300%, respectively (Table 3). The third change was that the ratios of (1-6)-anhydromuramyl muropeptides increased by ∼135 to 400% depending on the strain pairings (Table 3). Because these anhydro derivatives terminate glycan strands, these increases reflect a rather significant shortening of the average glycan strand length from ∼50 to ∼20 disaccharides. These changes were qualitatively similar to those reported for other conditions that produce rounded cells, such as the inhibition of PBP 2 or RodA, and are therefore in agreement with a proposed relationship between MreB and PBP 2. Finally, the fourth and most substantial change was that when MreB was inhibited, the proportion of pentapeptides plummeted to roughly one-third of that in cells containing active MreB (Table 3). This decrease occurred in concert with the increases in cross-linking and lipoprotein-containing muropeptides, suggesting that the two pools may interchange with one another.
FtsZ does not affect lateral wall synthesis via classic divisome components.
Because FtsZ nucleates and drives septation during cell division, one explanation for the above-described results could be that FtsZ triggers an alternate peptidoglycan insertion pathway via components present in the classical septal ring. The ring itself or any element that assembles into the divisome after FtsZ could modulate this insertion pathway, with FtsZ acting as an indirect effector. If so, the behaviors noted above would depend on the operation of at least a partially assembled divisome. This would mean that interrupting divisome assembly at a stage after FtsZ polymerization should re-create the physiological and biochemical phenotypes that accompany FtsZ inhibition. To test this, we prevented septal ring formation in E. coli AV23-1 by inactivating the FtsA(Ts) protein by shifting the culture to 42°C. Unlike FtsZ inhibition, inactivating FtsA did not produce the same phenotypes. In the absence of FtsA, the cells elongated to form straight filaments without spirals, and the incorporation of new peptidoglycan was not inhibited in regions near the cell poles (not shown). Therefore, because inactivating FtsA prevents the assembly of the septal ring at an early stage, the complete divisome was not required to drive FtsZ-dependent peptidoglycan synthesis in the lateral wall.
A second possible explanation for the FtsZ dependence of lateral cell wall synthesis was that by inhibiting FtsZ, the mode of side wall incorporation was altered indirectly by changing the distribution or activity of MreB. Since MreB is thought to direct the insertion of new precursors into the side wall by interacting with PBP 2 (22), varying the distribution of MreB could affect the geometry of peptidoglycan synthesis. For example, the periodic distribution of Z rings along the length of the cell may stabilize helically arranged MreB filaments. If so, inhibiting Z-ring formation might lead to a rearrangement of MreB that is unfavorable to the polar regions, causing these areas to become impoverished for active insertion. Alternately, if FtsZ were required to anchor MreB fibers at the cell poles, inhibiting FtsZ might cause MreB to pull away from the poles so that peptidoglycan synthesis was decreased in these regions. In either case, domains near the poles would incorporate less new material during subsequent cell growth.
To see if any of the above-mentioned scenarios might be true, we determined if inhibiting FtsZ changed the distribution of MreB in E. coli AV23-1. When visualized by fluorescence microscopy, no change in the distribution of YFP-MreB was observed (Fig. 6). Instead, YFP-MreB cables extended from pole to pole both when FtsZ was active and when it was inactivated by SulA (Fig. 6). Specifically, there were no signs of a weakened YFP-MreB fluorescent signal in subpolar regions when FtsZ was inhibited. Thus, the gross localization pattern of MreB was not disrupted by SulA inhibition of FtsZ, even though the pattern of peptidoglycan insertion had changed. This indicated that the distribution of MreB must not, by itself, be solely responsible for directing the uniform insertion of muropeptides into the side wall near the poles.
FIG. 6.
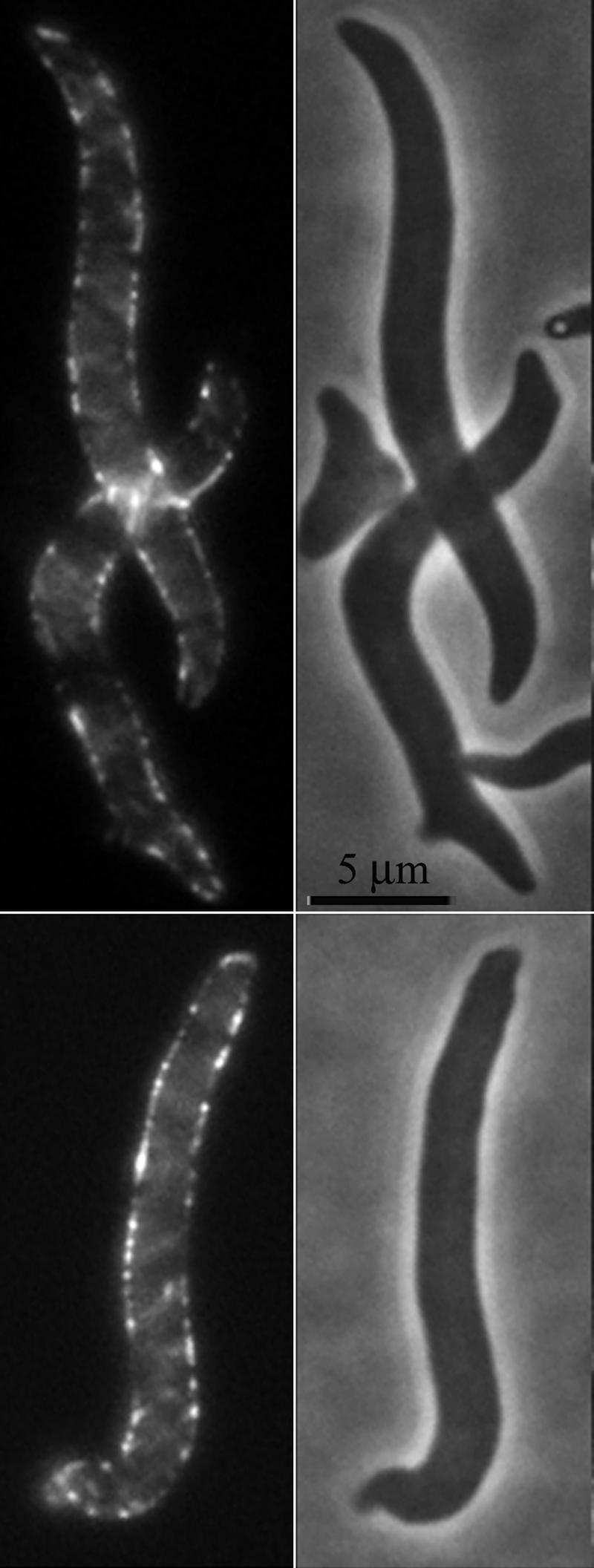
YFP-MreB is localized evenly throughout E. coli when FtsZ is inhibited. E. coli AV23-1 carrying plasmids pFAD38 (sulA gene under arabinose control) and pLE7 (yfp-mreB under the control of the lac promoter) was incubated for 2 h at 42°C in the presence of 0.2% arabinose (to induce SulA production and inhibit FtsZ) and 50 μM IPTG (to induce YFP-MreB). Fluorescent images (left) indicated that YFP-MreB was distributed throughout the cells (phase images are on the right), including at the poles and in regions near the poles.
By diminishing the likelihood of the two known possibilities by which FtsZ might influence peptidoglycan synthesis, the results reinforce the idea that FtsZ regulates peptidoglycan synthesis near the poles via a novel mechanism. The evidence supports the existence of a new pathway for peptidoglycan insertion that depends on FtsZ and requires some protein(s) other than or in addition to FtsA and MreB.
DISCUSSION
A new task for FtsZ.
Cell wall synthesis has been viewed as an either-or decision that the cell makes between elongation and cell division, but the results reported here suggest that these two processes are supplemented by yet another mechanism governed by FtsZ. Evidence for this third peptidoglycan insertion pathway can be observed in wild-type cells when MreB is nonfunctional but is more apparent in an E. coli mutant lacking PBP 5 and PBP 7. In these situations, FtsZ initiates or interacts with a third murein synthetic pathway and directs (or at least strongly influences) the incorporation of new peptidoglycan precursors into the side walls near each pole, perhaps with the concomitant utilization of pentapeptide-containing muropeptides as a principal substrate. Apparently, these conditions allow the visualization of an FtsZ-dependent activity that is normally invisible in wild-type cells. We speculate that the phenotype has not been observed in wild-type E. coli because the substrates (pentapeptides) for this third pathway are removed so rapidly that the effect is confined to an extremely narrow region near the incipient septum. In essence, inactivating MreB in these mutants allows us to see the results of an FtsZ-initiated reaction that probably occurs naturally near the septum but which is observed more easily in mutants that contain an overabundance of pentapeptides. In any event, these genetic backgrounds provide the opportunity to study this FtsZ function in more detail.
Previous results foreshadow some such additional role for FtsZ (38). The earliest indication of a third mode of muropeptide incorporation was the discovery of PIPS (26, 28, 37). During PIPS, a small amount of peptidoglycan is produced at the beginning of cell division by a process that is refractory to β-lactam inhibition (26, 28, 37). PIPS requires the activity of FtsZ but not the septation-specific protein PBP 3 (FtsI), which distinguishes PIPS from normal constriction (26, 28, 37). Furthermore, glycan chain lengths of this “preseptal murein” are longer, suggesting that this FtsZ-dependent process affects the chemical composition of the sacculus (18). In agreement with these results, de Pedro et al. previously observed the FtsZ-dependent creation of a ring of inert peptidoglycan at sites of future septa (8), which may be the visible product of the preseptal murein synthesized during PIPS (30). Interestingly, the R174D FtsZ mutant localizes to septal sites but fails to drive cell division to completion, and the resulting cell filaments contain shallow constrictions that may represent a very early stage of FtsZ-initiated peptidoglycan synthesis (21). All of this fits very nicely with recent results showing that the divisome matures in two stages, an early assembly of FtsZ with FtsA, ZipA, and ZapA (during which time PIPS would most likely occur), followed ∼17 min later by a second stage in which the FtsQWIN proteins complete the ring and initiate constriction (1). Overall, then, there are experimental precedents for an FtsZ-dependent, constriction-independent form of peptidoglycan synthesis. It is not clear whether the phenomenon reported here is equivalent to PIPS or represents yet another pathway.
Other experiments also intimate and extend the idea that FtsZ helps create or maintain the integrity of bacterial side walls. Instead of growing as filaments, certain E. coli PBP-FtsZ84 mutants lyse when shifted to the nonpermissive temperature (36). The phenotype depends on an impaired function of FtsZ and the deletion of PBP 5, which suggests a relationship between FtsZ activity and pentapeptide levels (36). Most interestingly, the form of lysis differs from the typical response elicited by β-lactams or by antibiotics that interfere with peptidoglycan synthesis. Cells treated with these latter treatments lyse by breaking open at septa or at poles, which extrude spherical bubbles that enlarge and burst. In contrast, lysis in the PBP-FtsZ84 mutant is peculiar: the cells disintegrate without intermediate blebbing, and the process affects the whole cell instead of circumscribed regions (36). The implication is that the impaired activity of FtsZ84 changes the synthesis or structural integrity of large regions of the wall and that it does so in the absence of cell division and at sites other than future septa.
Side wall insertion.
Cytoskeletal proteins are believed to organize peptidoglycan insertion by anchoring periplasmic synthases to the inner membrane so that they track along MreB- or Mbl-derived polymers to synthesize the wall as a set of interlocking helical fibers (6, 22, 23). If true, the absence of MreB or Mbl would result in an “untethered” synthetic apparatus that would be expected to insert muropeptide precursors unevenly. Our results show this clearly for E. coli. In the absence of FtsZ and MreB, E. coli peptidoglycan synthesis is patchy and not dispersed uniformly around the cell's circumference or along its length. Instead, the synthases seem to move irregularly, producing localized blooms of wall expansion.
Similar phenomena are observed in other organisms, thereby generalizing the concept that FtsZ and MreB tether peptidoglycan synthases (11, 12, 29, 35). When FtsZ is absent from Staphylococcus aureus, peptidoglycan synthesis is not confined to the septum but spreads nonuniformly over the entire wall, as though the relevant PBP has lost its anchor so that insertion is dispersed (29). In Caulobacter crescentus, the PBP synthases are organized as stripes or helices around the perimeter of the cell, but in the absence of MreB, the banding pattern disappears, and the PBP is found in spots, consistent with the haphazard operation of an untethered synthetic complex (12). Likewise, in B. subtilis, side wall synthesis continues when the cytoskeletal Mbl protein is absent, but insertion occurs in spots rather than as normal helical fibers (35). Cytoskeleton proteins may also compete for binding peptidoglycan synthases. In C. crescentus, the PBP 2 synthase condenses to the septum in an FtsZ-dependent manner when MreB or MreC is absent, implying that FtsZ captures PBP 2 released from the Mre proteins (11). However, if FtsZ is also depleted, PBP 2 returns to the side wall and inserts peptidoglycan randomly (11). In total, the results support the view that MreB (or Mbl) and FtsZ tether and direct the synthetic apparatus but that undirected synthesis can occur in the absence of these cytoskeletal proteins.
Overall, therefore, the partitioning and proper localization of cell wall synthesis can be viewed as a competition between FtsZ and MreB for otherwise independent peptidoglycan synthetic complexes. In its simplest form, such a contest can be envisioned as being the mechanism underlying the “two-competing-sites” hypothesis (31). However, our results suggest that the effects of this competition may be more complicated than a simple two-site decision. Thus, when FtsZ is inactivated in a PBP 5 mutant, the primary physiological effect may be that the cell cannot direct peptidoglycan synthetic complexes to regions close to the poles. Instead, the synthases would be recruited by MreB to insert peptidoglycan more frequently in the central zone of the cell body. In the absence of MreB, the distribution of peptidoglycan incorporation becomes even more random, leading to patchy insertion. Interestingly, these phenomena occur only when FtsZ is inhibited in a strain lacking PBP 5, suggesting that the vagaries of insertion are caused either by an excess of pentapeptides in the cell wall or by the absence of the PBP 5 protein itself.
Spiral cells, diagonal peptidoglycan.
There are at least four requirements for making E. coli adopt a spiral morphology: PBP 5 must be absent, one additional PBP (e.g., an endopeptidase) must be deleted, FtsZ must be inhibited by SulA, and MreB must be active (36; this work). The requirement for MreB may be the simplest to explain: in its absence, the cell incorporates muropeptides in an undirected way to give bulging, spherical cells. This suggests that a uniform elongation of the sacculus is necessary to form spiral cells. The other requirements are more complicated. Most likely, the removal of PBP 5 increases the level of pentapeptides in the sacculus, which leads to the biochemical events that accompany the inactivation of the FtsZ-dependent insertion of new peptidoglycan into the side wall. A “gradient” of MreB-directed wall expansion ensues, with much lower incorporation near the poles. The simplest explanation is that this failure to expand the near-polar side walls drives the formation of spiral cells, since they appear only under these conditions. A plausible scenario is that residual MreB-directed synthesis generates a torsional stress that the cell accommodates by twisting in the opposite direction, thereby relieving mechanical stress in the same way that superhelical twists are introduced into closed circular DNA. If true, this strengthens the interpretation that peptidoglycan is inserted into the side wall in a helical (or at least diagonal) manner. Also, because all twisted cells (so far as we have observed) take the form of left-handed spirals (36), peptidoglycan would have to be inserted into the existing wall in a right-handed fashion.
If MreB-directed cell wall growth is helical and does indeed create torsional stress, then in wild-type E. coli, FtsZ must somehow act as a “swivel” to counteract this force so that the cells do not bend but grow as straight rods. In the absence of FtsZ, then, the fixed nature of the poles might predispose the cells to grow as spirals. Some such function might account for an observation that is otherwise difficult to explain; that is, when FtsZ is inhibited in the PBP mutant, only about 33% of cell filaments adopt a spiral morphology (36). But if FtsZ serves as a “swivel,” then perhaps its function can sometimes be replaced by another protein or by a limited amount of residual MreB-directed peptidoglycan insertion into the polar zones.
Implications.
The present observations have several repercussions for general models of peptidoglycan synthesis. First, it is not essential that new peptidoglycan be inserted between the oldest preexisting peptidoglycan strands before it is inserted between more recently synthesized strands (17). An MreB-dependent process, not the overall chemical composition of the wall, determines where insertion occurs. In the absence of MreB, precursor muropeptides are inserted repeatedly into a restricted locale composed mainly of newly synthesized material, indicating that murein synthesis does not automatically copy the existing sacculus as though it were an exact template (17). Thus, when examined on a small enough scale, old and new murein do not form a perfectly homogeneous mixture; instead, new material is inserted as microdomains that leave fibers of older peptidoglycan arrayed in a net-like fashion. We cannot make the dogmatic claim that these microdomains are inserted as perfect helices, but the diagonal strands of peptidoglycan are compatible with a helical arrangement.
A second implication is that cell elongation and septation may not be as mutually exclusive as depicted in the classical “two-competing-sites” theory (31). Instead, these reactions may overlap and affect one another, or they may be joined by or interact with a second type of FtsZ-directed mechanism. The precise form of the interactions is impossible to state with assurance at this point, but three general possibilities present themselves. First, FtsZ may direct a completely independent mode of peptidoglycan incorporation (FtsZ only); second, FtsZ and MreB may collaborate (FtsZ plus MreB); and third, FtsZ might physically bind and secure MreB so that the latter protein can direct cell wall synthesis near the poles (FtsZ anchoring). Any of these possibilities might explain why, in the absence of FtsZ and PBP 5, MreB has difficulty directing muropeptide incorporation in the vicinity of the poles. One result in favor of the FtsZ-only mechanism is that FtsZ directs some subpolar side wall insertion in the absence of MreB, suggesting that FtsZ may not have to work with or anchor MreB. Suggestively, a similar phenomenon may occur in B. subtilis. When the Mbl protein is depleted, B. subtilis grows and retains its rod shape only when FtsZ is active, suggesting that in this organism, FtsZ may also direct the growth of the side wall in the absence of one of the actin homologues (6).
A third implication is that PBP 5 helps determine where new peptidoglycan is inserted. The presence of PBP 5 apparently restricts FtsZ-directed peptidoglycan incorporation to the septal area during division, whereas the absence of PBP 5 makes it difficult for MreB to direct peptidoglycan insertion into subpolar regions. One possibility is that MreB-directed muropeptide insertion prefers a different substrate than does FtsZ-directed synthesis. If, as suggested in this work and implied by previous observations (38), the FtsZ-associated reaction utilizes pentapeptide-containing substrates, then PBP 5-mediated destruction of pentapeptides may confine the FtsZ reaction to a narrow zone adjoining newly forming septa. In the absence of PBP 5, the extent of this zone may expand, thereby producing a visible, in vivo phenotype for this activity of FtsZ. Conversely, by reducing pentapeptide levels, active PBP 5 may extend the area into which MreB can direct peptidoglycan insertion.
Finally, a fourth implication is that cell shape and diameter are not determined solely by the geometry of the poles, as predicted by the surface stress theory (20), because cell poles can retain their shapes while growth in the central region of the cell becomes unconstrained.
Summary and questions.
In E. coli, FtsZ governs or participates in at least one other facet of cell wall synthesis that precedes the classical incorporation of peptidoglycan into the dividing septum and which modifies the growth of the cell's side walls near the poles. The principal substrate for this reaction seems to be composed of pentapeptide-containing muropeptides, because inactivating FtsZ changes the chemical composition of the sacculus. Of course, many questions remain. Do the same synthetic enzymes participate in MreB- and FtsZ-directed peptidoglycan insertion, or are there new enzymes to be discovered? How do the geometries of muropeptide insertion and cross-linking combine to create spiral cells? What is the nature of the competition between the cytoskeletal proteins? And what are the signals that ensure the orderly progression among the modes of peptidoglycan synthesis? The answers to these questions surely promise more mechanistic surprises.
ADDENDUM
After this article was accepted for publication, Aaron et al. reported similar findings regarding the function of FtsZ in Caulobacter crescentus (M. Aaron, G. Charbon, H. Lam, H. Schwarz, W. Vollmer, and C. Jacobs-Wagner, Mol. Microbiol. 64:938-952, 2007).
Acknowledgments
We especially thank the following people for their assistance: the Confocal Core Facility (UNDSM), Federico Acosta (CSIC-UAM) for deconvolution, Said Taimani (CSIC-UAM) for performing high-performance liquid chromatography analyses of muropeptides, and Alexei Novikov (UND) for synthesizing compound A22.
This work was supported by grant GM61019 from the National Institutes of Health (to K.D.Y.) and by grant PR2005-0024 of the Programa de Estancias de Profesores de Universidad e Investigadores del CSIC en Centros de Enseñanza Superior e Investigación Extranjeros y Españoles from the Spanish Ministry for Education and Science (to M.D.P.).
Footnotes
Published ahead of print on 18 May 2007.
REFERENCES
- 1.Aarsman, M. E., A. Piette, C. Fraipont, T. M. Vinkenvleugel, M. Nguyen-Disteche, and T. den Blaauwen. 2005. Maturation of the Escherichia coli divisome occurs in two steps. Mol. Microbiol. 55:1631-1645. [DOI] [PubMed] [Google Scholar]
- 2.Addinall, S. G., and J. Lutkenhaus. 1996. FtsA is localized to the septum in an FtsZ-dependent manner. J. Bacteriol. 178:7167-7172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Buddelmeijer, N., and J. Beckwith. 2002. Assembly of cell division proteins at the E. coli cell center. Curr. Opin. Microbiol. 5:553-557. [DOI] [PubMed] [Google Scholar]
- 4.Canepari, P., C. Signoretto, M. Boaretti, and M. M. Lleo. 1997. Cell elongation and septation are two mutually exclusive processes in Escherichia coli. Arch. Microbiol. 168:152-159. [DOI] [PubMed] [Google Scholar]
- 5.Carballido-Lopez, R. 2006. The bacterial actin-like cytoskeleton. Microbiol. Mol. Biol. Rev. 70:888-909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Daniel, R. A., and J. Errington. 2003. Control of cell morphogenesis in bacteria. Two distinct ways to make a rod-shaped cell. Cell 113:767-776. [DOI] [PubMed] [Google Scholar]
- 7.de Pedro, M. A., C. G. Grünfelder, and H. Schwarz. 2004. Restricted mobility of cell surface proteins in the polar regions of Escherichia coli. J. Bacteriol. 186:2594-2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Pedro, M. A., J. C. Quintela, J.-V. Höltje, and H. Schwarz. 1997. Murein segregation in Escherichia coli. J. Bacteriol. 179:2823-2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Pedro, M. A., H. Schwarz, and A. L. Koch. 2003. Patchiness of murein insertion into the sidewall of Escherichia coli. Microbiology 149:1753-1761. [DOI] [PubMed] [Google Scholar]
- 10.de Pedro, M. A., K. D. Young, J. V. Höltje, and H. Schwarz. 2003. Branching of Escherichia coli cells arises from multiple sites of inert peptidoglycan. J. Bacteriol. 185:1147-1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dye, N. A., Z. Pincus, J. A. Theriot, L. Shapiro, and Z. Gitai. 2005. Two independent spiral structures control cell shape in Caulobacter. Proc. Natl. Acad. Sci. USA 102:18608-18613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Figge, R. M., A. V. Divakaruni, and J. W. Gober. 2004. MreB, the cell shape-determining bacterial actin homologue, co-ordinates cell wall morphogenesis in Caulobacter crescentus. Mol. Microbiol. 51:1321-1332. [DOI] [PubMed] [Google Scholar]
- 13.Ghosh, A. S., and K. D. Young. 2005. Helical disposition of proteins and lipopolysaccharide in the outer membrane of Escherichia coli. J. Bacteriol. 187:1913-1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gitai, Z., N. A. Dye, A. Reisenauer, M. Wachi, and L. Shapiro. 2005. MreB actin-mediated segregation of a specific region of a bacterial chromosome. Cell 120:329-341. [DOI] [PubMed] [Google Scholar]
- 15.Graumann, P. L. 2004. Cytoskeletal elements in bacteria. Curr. Opin. Microbiol. 7:565-571. [DOI] [PubMed] [Google Scholar]
- 16.Higgins, M. L., and G. D. Shockman. 1971. Procaryotic cell division with respect to wall and membranes. CRC Crit. Rev. Microbiol. 1:29-72. [DOI] [PubMed] [Google Scholar]
- 17.Höltje, J.-V. 1998. Growth of the stress-bearing and shape-maintaining murein sacculus of Escherichia coli. Microbiol. Mol. Biol. Rev. 62:181-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ishidate, K., A. Ursinus, J.-V. Höltje, and L. Rothfield. 1998. Analysis of the length distribution of murein glycan strands in ftsZ and ftsI mutants of E. coli. FEMS Microbiol. Lett. 168:71-75. [DOI] [PubMed] [Google Scholar]
- 19.Jones, L. J., R. Carballido-Lopez, and J. Errington. 2001. Control of cell shape in bacteria: helical, actin-like filaments in Bacillus subtilis. Cell 104:913-922. [DOI] [PubMed] [Google Scholar]
- 20.Koch, A. L. 1988. Biophysics of bacterial wall viewed as a stress-bearing fabric. Microbiol. Rev. 52:337-353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koppelman, C. M., M. E. Aarsman, J. Postmus, E. Pas, A. O. Muijsers, D. J. Scheffers, N. Nanninga, and T. den Blaauwen. 2004. R174 of Escherichia coli FtsZ is involved in membrane interaction and protofilament bundling, and is essential for cell division. Mol. Microbiol. 51:645-657. [DOI] [PubMed] [Google Scholar]
- 22.Kruse, T., J. Bork-Jensen, and K. Gerdes. 2005. The morphogenetic MreBCD proteins of Escherichia coli form an essential membrane-bound complex. Mol. Microbiol. 55:78-89. [DOI] [PubMed] [Google Scholar]
- 23.Leaver, M., and J. Errington. 2005. Roles for MreC and MreD proteins in helical growth of the cylindrical cell wall in Bacillus subtilis. Mol. Microbiol. 57:1196-1209. [DOI] [PubMed] [Google Scholar]
- 24.Lowe, J., F. van den Ent, and L. A. Amos. 2004. Molecules of the bacterial cytoskeleton. Annu. Rev. Biophys. Biomol. Struct. 33:177-198. [DOI] [PubMed] [Google Scholar]
- 25.Mukherjee, A., C. N. Cao, and J. Lutkenhaus. 1998. Inhibition of FtsZ polymerization by SulA, an inhibitor of septation in Escherichia coli. Proc. Natl. Acad. Sci. USA 95:2885-2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nanninga, N. 1991. Cell division and peptidoglycan assembly in Escherichia coli. Mol. Microbiol. 5:791-795. [DOI] [PubMed] [Google Scholar]
- 27.Nanninga, N. 1998. Morphogenesis of Escherichia coli. Microbiol. Mol. Biol. Rev. 62:110-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nanninga, N., F. B. Wientjes, B. L. M. Dejonge, and C. L. Woldringh. 1990. Polar cap formation during cell division in Escherichia coli. Res. Microbiol. 141:103-118. [DOI] [PubMed] [Google Scholar]
- 29.Pinho, M. G., and J. Errington. 2003. Dispersed mode of Staphylococcus aureus cell wall synthesis in the absence of the division machinery. Mol. Microbiol. 50:871-881. [DOI] [PubMed] [Google Scholar]
- 30.Rothfield, L. 2003. New insights into the developmental history of the bacterial cell division site. J. Bacteriol. 185:1125-1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Satta, G., R. Fontana, and P. Canepari. 1994. The two-competing site (TCS) model for cell shape regulation in bacteria: the envelope as an integration point for the regulatory circuits of essential physiological events. Adv. Microb. Physiol. 36:181-245. [DOI] [PubMed] [Google Scholar]
- 32.Schwarz, U., A. Asmus, and H. Frank. 1969. Autolytic enzymes and cell division of Escherichia coli. J. Mol. Biol. 41:419-429. [DOI] [PubMed] [Google Scholar]
- 33.Shih, Y. L., T. Le, and L. Rothfield. 2003. Division site selection in Escherichia coli involves dynamic redistribution of Min proteins within coiled structures that extend between the two cell poles. Proc. Natl. Acad. Sci. USA 100:7865-7870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Slovak, P. M., S. L. Porter, and J. P. Armitage. 2006. Differential localization of Mre proteins with PBP2 in Rhodobacter sphaeroides. J. Bacteriol. 188:1691-1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tiyanont, K., T. Doan, M. B. Lazarus, X. Fang, D. Z. Rudner, and S. Walker. 2006. Imaging peptidoglycan biosynthesis in Bacillus subtilis with fluorescent antibiotics. Proc. Natl. Acad. Sci. USA 103:11033-11038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Varma, A., and K. D. Young. 2004. FtsZ collaborates with penicillin binding proteins to generate bacterial cell shape in Escherichia coli. J. Bacteriol. 186:6768-6774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wientjes, F. B., and N. Nanninga. 1989. Rate and topography of peptidoglycan synthesis during cell division in Escherichia coli: concept of a leading edge. J. Bacteriol. 171:3412-3419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Young, K. D. 2003. Bacterial shape. Mol. Microbiol. 49:571-580. [DOI] [PubMed] [Google Scholar]