

Abstract
Biphenyl dioxygenase (BPDO) catalyzes the aerobic transformation of biphenyl and various polychlorinated biphenyls (PCBs). In three different assays, BPDOB356 from Pandoraea pnomenusa B-356 was a more potent PCB-degrading enzyme than BPDOLB400 from Burkholderia xenovorans LB400 (75% amino acid sequence identity), transforming nine congeners in the following order of preference: 2,3′,4-trichloro ∼ 2,3,4′-trichloro > 3,3′-dichloro > 2,4,4′-trichloro > 4,4′-dichloro ∼ 2,2′-dichloro > 2,6-dichloro > 2,2′,3,3′-tetrachloro ∼ 2,2′,5,5′-tetrachloro. Except for 2,2′,5,5′-tetrachlorobiphenyl, BPDOB356 transformed each congener at a higher rate than BPDOLB400. The assays used either whole cells or purified enzymes and either individual congeners or mixtures of congeners. Product analyses established previously unrecognized BPDOB356 activities, including the 3,4-dihydroxylation of 2,6-dichlorobiphenyl. BPDOLB400 had a greater apparent specificity for biphenyl than BPDOB356 (kcat/Km = 2.4 × 106 ± 0.7 × 106 M−1 s−1 versus kcat/Km = 0.21 × 106 ± 0.04 × 106 M−1 s−1). However, the latter transformed biphenyl at a higher maximal rate (kcat = 4.1 ± 0.2 s−1 versus kcat = 0.4 ± 0.1 s−1). A variant of BPDOLB400 containing four active site residues of BPDOB356 transformed para-substituted congeners better than BPDOLB400. Interestingly, a substitution remote from the active site, A267S, increased the enzyme's preference for meta-substituted congeners. Moreover, this substitution had a greater effect on the kinetics of biphenyl utilization than substitutions in the substrate-binding pocket. In all variants, the degree of coupling between congener depletion and O2 consumption was approximately proportional to congener depletion. At 2.4-Å resolution, the crystal structure of the BPDOB356-2,6-dichlorobiphenyl complex, the first crystal structure of a BPDO-PCB complex, provided additional insight into the reactivity of this isozyme with this congener, as well as into the differences in congener preferences of the BPDOs.
The microbial degradation of biphenyl has been well studied as a potential means of remediating soils contaminated with polychlorinated biphenyls (PCBs) (46). While the production of PCBs has been banned in industrial countries due to the adverse health effects that they cause in humans, these toxic pollutants are persistent and remain widespread in the environment (13). PCBs are aerobically transformed by the bph pathway, a pathway comprising four enzymes that initiates the catabolism of biphenyl. In most bacterial strains characterized to date, the pathway transforms up to tetrachlorobiphenyls, although some pathways can transform congeners containing up to six chlorine substituents (8, 44). A critical step in improving the microbial catabolic activities for the degradation of PCBs is understanding the reactivity of the four enzymes of the bph pathway for PCB metabolites.
Biphenyl dioxygenase (BPDO), the first enzyme of the bph pathway, is a typical three-component, ring-hydroxylating dioxygenase that catalyzes the insertion of molecular oxygen into an aromatic ring, forming cis-(2R,3S)-dihydroxy-1-phenylcyclohexa-4,6-diene (Fig. 1) (46). The oxygenase (BphAE) has an α3β3 composition. Each α subunit (BphA) contains a Rieske-type Fe2S2 cluster and a mononuclear iron center, located at the enzyme's active site. A reductase (BphG) and a ferredoxin (BphF) function to transfer electrons from NADH to the Rieske center of BphAE, where they are used in the hydroxylation of the biphenyl at the mononuclear iron center. The mechanism of dihydroxylation is thought to be very similar to that of the naphthalene dioxygenase from Pseudomonas sp. strain NCIB 9816-4, the best-characterized ring-hydroxylating dioxygenase (35). BPDO is a major determinant of a bacterium's PCB-transforming capabilities. Studies on BPDOs from different organisms have revealed significant differences in congener (substrate) preference and regiospecificity. For example, BPDOKF707 and BPDOLB00, from Pseudomonas alcaligenes KF707 and Burkholderia xenovorans LB400, respectively, show very different reactivities, although they share more than 95% sequence identity (21, 55). BPDOLB400 preferentially transforms ortho-substituted congeners containing up to six chlorines (10, 43). This enzyme has the relatively unusual ability to catalyze the 3,4-dihydroxylation of certain 2,5-substituted congeners, such as 2,2′,5,5′-tetrachlorobiphenyl (32). BPDOLB400 is also remarkable in that it catalyzes the dehalogenation of certain 2-Cl congeners, yielding 2,3-dihydroxybiphenyls (25, 50-52), the substrate of the third Bph pathway enzyme. By contrast, BPDOKF707 transforms a narrow range of PCB congeners and catalyzes neither 3,4-dihydroxylation nor o-dechlorination. Moreover, it preferentially transforms 4,4′-dichlorobiphenyl over either 3,3′- or 2,2′-dichlorobiphenyl (24). A third enzyme, BPDOB356 from Pandoraea pnomenusa (formerly Comamonas testosteroni [59]) B356, shares approximately 70% sequence identity with both BPDOKF707 and BPDOLB400. Previous studies have indicated this enzyme's preference for meta- > ortho- > para-substituted dichlorobiphenyls (33), as well as its higher specific activity against biphenyl than that of BPDOLB00.
FIG. 1.

Dihydroxylation of biphenyl by BPDO. The enzyme comprises a flavin adenine dinucleotide-containing reductase (BphG), a Rieske-type ferredoxin (BphF), and an oxygenase that contains a Rieske-type Fe2S2 cluster and a catalytic mononuclear iron center. Biphenyl is stereospecifically hydroxylated at positions 2 and 3, yielding cis-(2R,3S)-dihydroxy-2,3-dihydrobiphenyl. ox, oxidized; red, reduced.
Protein engineering efforts have yielded BPDO with improved PCB-degrading capabilities, as well as insights into the molecular basis of congener preference. Mutagenesis studies of BphALB400 and BphAKF707 identified four regions (regions I to IV) whose sequences influence the range of congeners attacked (44). All four regions occur in the C-terminal domain of BphA, consistent with the location of the oxygenase's active site. Substitution of individual residues in region III of BphALB400, comprising Thr335, Phe336, Asn338, and Ile341, improved the ability of the enzyme to transform 4,4′-dichlorobiphenyls, although the greatest improvements in activity were achieved by multiple substitutions in this region, suggesting that there is a cooperative or additive effect (4). Several studies have since confirmed the importance of these residues in determining the enzyme's congener preference and regiospecificity (3-5, 36, 54). For example, BphAEII9, a variant of BphAELB400 containing region III of BphAB356, is reported to possess better PCB-degrading properties than either parental enzyme (3). By contrast, BphAEII10, which differs from BphAEII9 by a single residue at position 267 (Ser, as in BphAEB356, instead of Ala, as in BphAELB400), was not able to transform any of the PCBs tested.
Here, we report the characterization of four variants of BPDO: BphAELB400, BphAEB356, and two variants of these enzymes generated via directed evolution, BphAEII9 and BphAEII10. The steady-state kinetic parameters for biphenyl of anaerobically purified non-His-tagged enzymes were determined, and the activities towards various PCB congeners were investigated. The degree of uncoupling between O2 utilization and congener transformation was also determined for the different enzymes using different congeners. To validate previous studies, activities determined using purified enzymes and whole cells were compared, as were activities determined using individual congeners and mixtures of congeners. Finally, a crystal structure of the BphAEB356-2,6-dichlorobiphenyl complex was determined. The results are discussed in terms of the proposed catalytic mechanism of ring-hydroxylating dioxygenases and the specificities of the different BPDOs.
MATERIALS AND METHODS
Materials.
Biphenyl was purchased from Aldrich (Mississauga, Ontario, Canada). PCB congeners either were purchased from Accustandard (New Haven, CT) or were kindly provided by Victor Snieckus (Queens University, Kingston, Ontario, Canada). Restriction enzymes and T4 DNA polymerase were purchased from New England Biolabs (Pickering, Ontario, Canada). Pwo DNA polymerase was purchased from Roche (Laval, Quebec, Canada). Synthetic oligonucleotides (see below) were purchased from the NAPS Service Unit at the University of British Columbia (Vancouver, British Columbia, Canada). Ferene S was purchased from ICN Biomedicals Inc. (Costa Mesa, CA). Acetonitrile, ethyl acetate, and hexane (Fisher Scientific, Mississauga, Ontario, Canada) were high-performance liquid chromatography (HPLC) grade. All other chemicals were reagent grade or better.
Strains, media, and growth.
Escherichia coli strains DH5α (27) and C41(DE3) (42) were used for DNA propagation and protein overexpression, respectively. The plasmids used in this study included pDB31-II9 and pDB31-II10 (3), pT7-7 (53), pT7-7AE3 (1), and pT7-6a (29). Strains harboring pDB31 or pT7 derivatives were grown in the presence of ampicillin (100 mg/ml) or carbenicillin (15 mg/ml). Strains harboring pPAISC-1 were grown in the presence of tetracycline (20 mg/ml). E. coli DH5α was cultured at 37°C and 250 rpm in Luria-Bertani broth with the appropriate antibiotics. For BPDO expression, E. coli C41(DE3) harboring the isc plasmid (1) and the appropriate pT7 vector was grown in Terrific broth (56) supplemented with 0.1 mg/ml ferric ammonium citrate. Culture media were inoculated with 1% (vol/vol) of an overnight culture and grown at 37°C until the optical density at 600 nm (OD600) reached 0.9 to 1.0. Expression of the bphAE genes was then induced by addition of isopropyl-1-thio-β-d-galactopyranoside (IPTG) to a final concentration of 1 mM, and each culture was transferred to 20°C (LB400, II9, and II10) or 25°C (B356) for an additional 18 h before it was harvested by centrifugation. The harvested cell pellet was washed twice with 500 ml of 25 mM HEPES (pH 7.3) containing 10% glycerol and frozen at −80°C until use.
DNA manipulation.
DNA was manipulated using standard protocols (49). Plasmid DNA was purified using a Quantum Prep kit (Bio-Rad, Mississauga, Ontario, Canada). To construct pT7II9 and pT7II10, a 2,064-bp DNA fragment containing bphAE was amplified from pDB31-II9 and pDB31-II10, respectively, using oligonucleotides II910F-NdeI (5′-TACGGAACATATGAGTTCAGCAATCAAAGAAG-3′) and II910F-HindIII (5′-CATGAAGCTTGTACCCCCTAGAAGAACTGC-3′) (introduced restriction sites are underlined) and Pwo polymerase according to the manufacturer's instructions (Roche, Laval, Quebec, Canada). Thirty reaction cycles were performed as follows: 95°C for 45 s, 45°C for 40 s, and 72°C for 2 min. The recovered amplicon was digested with NdeI/HindIII and ligated to similarly digested pT7-7. The sequences of the final constructs were verified at the NAPS Service Unit. To construct the pT7-6a derivatives, the 1,347-bp bphAE fragments from pT7II9 and pT7II10 were digested with EcoRV/SacII and ligated into the corresponding sites of pT7-6a.
Protein purification.
The BphAE terminal oxygenases were purified anaerobically as previously described (33). Accordingly, all preparations were manipulated under an N2 atmosphere (<2 ppm O2) using an MBraun Labmaster glove box (Stratham, United States). Chromatography was performed with an ÄKTA Explorer 100 (Amersham Pharmacia Biotech, Baie d'Urfé, Quebec, Canada) interfaced to the glove box to minimize the oxygen content of the purification buffers and protein fractions (58). The columns used were a Source 15Q anion-exchange column (2 by 9 cm) and a Phenyl-Sepharose column (1 by 9 cm) (Amersham Pharmacia Biotech). All buffers were prepared using water purified with a Barnstead NANOpure UV apparatus to a resistivity greater than 17 MΩ·cm. Buffers were sparged with N2 and equilibrated in the glove box for at least 24 h prior to use.
His-tagged ferredoxin from B. xenovorans LB400 (ht-BphFLB400) and His-tagged reductase from P. pnomenusa B-356 (ht-BphGB356) were prepared using the QIAexpress system from QIAGEN as described previously (18, 30).
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis was performed using a 12% resolving gel and a Bio-Rad mini-Protean II cell. Gels were stained with Coomassie blue according to standard protocols (38). Protein concentrations were determined using the Bradford reagent (11), with bovine serum albumin as a standard. For the purified enzymes, concentrations were determined spectrophotometrically using the appropriate extinction coefficients, as follows: for BphAELB400, ɛ455 = 10.1 mM−1 cm−1 (25); for BphAEB356, ɛ455 = 8.3 mM−1 cm−1 (31); for BphFLB400, ɛ326 = 9 mM−1 cm−1 (18); and for BphGB356, ɛ450 = 11.8 mM−1 cm−1 (30). The extinction coefficients for BphAEII9 and BphAEI10, determined in this study based on sulfur content, were ɛ455 = 8.4 mM−1 cm−1 and ɛ455 = 9.6 mM−1 cm−1, respectively. The sulfur content was determined colorimetrically using N,N-dimethyl-p-phenylenediamine and sodium sulfide as a standard (15). The value was used in the assays performed with purified enzymes.
Steady-state kinetic measurements and data analysis.
Enzymatic activity was measured by following the consumption of O2 using a Clark-type polarographic O2 electrode (model 5301; Yellow Springs Instruments, Yellow Springs, OH) (58). The activity assay was performed in a thermojacketed Cameron Instrument Co. model RCI respiration chamber (Port Aransas, TX) connected to a Lauda model RM6 circulating bath. Data were recorded every 0.1 s, and initial velocities were calculated from the slope of the progress curve for each consecutive 6-s interval.
The standard activity assay was performed in 1.4 ml (total volume) of air-saturated 50 mM morpholineethanesulfonic acid (MES) (pH 6.0) (25°C). The reaction mixture contained 150 μM biphenyl, 320 μM NADH, 3.6 μM ht-BphFLB400, 1.8 μM ht-BphGB356, and 0.6 μM BphAE. For PCBs, the biphenyl was replaced with 50 μM of the appropriate congener. The assay was initiated by adding the oxygenase after equilibration of the reaction mixture with all other components for 30 s. The reaction buffer and stock solutions used in the assay were prepared fresh daily. Stock solutions and protein samples were prepared anaerobically. The electrode was zeroed on the day of use by adding an excess of sodium hydrosulfite to the buffer in the reaction chamber. It was calibrated using standard concentrations of catechol and an excess of catechol 2,3-dioxygenase. Activity determinations were corrected for the consumption of O2 observed in the absence of oxygenase. One unit of enzyme activity is defined as the amount of enzyme required to consume 1 μmol of O2/min under the conditions described above. Apparent steady-state kinetic parameters for biphenyl were determined by measuring rates of oxygen uptake in the presence of concentrations of biphenyl ranging from 0.1 to 150 μM. The Michaelis-Menten equation was fitted to initial velocities determined at different substrate concentrations using the least-squares fitting and dynamic weighting options of LEONORA (17).
Coupling measurements.
Coupling experiments were carried out using 50 mM MES (pH 6.0) at 25°C, an excess of biphenyl or congener, 350 μM NADH, and the same concentrations of BPDO components used in the standard activity assay. Reactions were initiated by adding oxygenase and quenched 1 to 4 min later by adding acetonitrile (1:1, vol/vol). Oxygen consumption was monitored using the O2 electrode. The amount of hydrogen peroxide was estimated using catalase, 650 U of which was added to the reaction mixture at the time corresponding to the acetonitrile quench. The amount of oxygen detected upon addition of catalase was taken to represent 50% of the total hydrogen peroxide produced during biphenyl transformation. The consumption of biphenyl was determined by HPLC.
Depletion of PCB mixtures by purified BPDOs.
Depletion assays were performed in 12-ml glass vials sealed with Teflon caps in 1.0 ml (total volume) of air-saturated 50 mM MES (pH 6.0) (25°C). The reaction mixture contained the same concentrations of BPDO components as the standard oxygraph assay mixture, 350 μM NADH, and 10 μM each of 3,3′-dichlorobiphenyl, 4,4′-dichlorobiphenyl, 2,6-dichlorobiphenyl, 2,3,4′-trichlorobiphenyl, 2,3′,4-trichlorobiphenyl, 2,4,4′-trichlorobiphenyl, 2,2′,3,3′-tetrachlorobiphenyl, and 2,2′,5,5′-tetrachlorobiphenyl (3). Reactions were initiated by adding oxygenase and quenched immediately or 20 min later (each enzyme retained at least 60% of its activity after 20 min [data not shown]). After quenching, 2,2′,4,4′,6-pentachlorobiphenyl was added as an internal standard, and the reaction mixture was extracted twice with hexane. The hexane fractions were pooled, dried over anhydrous sodium sulfate, and analyzed for PCB congener content by gas chromatography (GC)-mass spectrometry (MS).
Whole-cell assays.
E. coli C41(DE3) cells freshly transformed with the isc plasmid and either pT7-6a, pT7II9a, or pT7II10a were grown at 37°C to mid-log phase, induced with 0.5 mM IPTG, and then grown at 22°C to an OD600 of 1.0. Cells were then harvested, washed twice with 50 mM sodium phosphate (pH 7.5), supplemented with 1 g/liter glucose, and resuspended in the same buffer at an OD600 of 2.0. One-milliliter portions of the suspension were distributed in 12-ml glass vials with Teflon caps. An aliquot of the PCB mixture described above was added to each vial such that the final concentration of each congener was 10 μM. The reaction mixtures were shaken at 25°C at 225 rpm, and the reactions were stopped after 0, 3, and 18 h by adding a drop of 1 N HCl. The reaction mixtures were then frozen at −80°C. Assays were performed in duplicate. Controls contained C41(DE3) cells without any plasmid and otherwise were treated the same. An internal standard, 2,2′,4,4′,6-pentachlorobiphenyl, was added to a final concentration of 10 μM. The samples were extracted twice with 1 ml of hexane, pooled, dried over sodium sulfate, and transferred to GC vials. The PCB content was analyzed as described above. Protein levels in the different strains were verified using Sypro Ruby-stained denaturing gels (sodium dodecyl sulfate-polyacrylamide) of whole cells. Band intensities were quantified with ImageQuant 5.2 (Amersham Pharmacia). The levels of BPDO components were comparable in all assays and corresponded to ∼0.5 μM BphAE, 0.6 μM BphG, and 0.6 μM BphF. The wild-type strains, B. xenovorans LB400 and P. pnomenusa B-356, were cultured to mid-log phase at 29°C and 250 rpm in M9 medium (28) containing 0.01% (wt/vol) biphenyl as the sole carbon source. The cultures were filtered on glass wool to get rid of residual biphenyl. The cells were harvested by centrifugation, washed once with M9 medium, and suspended in M9 medium to an OD600 of 2.0. One-milliliter portions of the suspensions were distributed into 20-ml glass vials with Teflon caps. Each tube received 10 μl of a synthetic mixture dissolved in acetone that added 1 μM of each of six congeners (2,6-dichlorobiphenyl, 2,4,4′-trichlorobiphenyl, 2,3′,4-trichlorobiphenyl, 2,3,4′-trichlorobiphenyl, 2,2′,5,5′-tetrachlorobiphenyl, and 2,2′,3,3′-tetrachlorobiphenyl), 5 μM of two congeners (3,3′-dichlorobiphenyl and 4,4′-dichlorobiphenyl), and 1 μM 2,2′,3,3′,4,5,5′,6,6′-nonachlorobiphenyl (an internal standard). Incubation was carried out at 250 rpm at 29°C for 18 h. PCBs were extracted with three 1-ml portions of hexane and processed similar to metabolites generated using E. coli cells. All values reported in the present study are averages from triplicate experiments.
Characterization of the transformation product of 2,4,4′-trichlorobiphenyl and 2,6-dichlorobiphenyl by BPDOB356.
To characterize the BPDOB356-catalyzed transformation product of 2,4,4′-trichlorobiphenyl, two reactions were carried out in parallel. Each mixture contained 600 μM of the substrate, 1.4 mM NADH, 5 μM ht-BphG, 15 μM ht-BphF, and 3 μM BphAE. After 30 min, reactions were quenched with acetonitrile, and the product was purified by HPLC and dried under a stream of nitrogen. The sample was resuspended in acetone-d6 and analyzed using a 500-MHz Varian nuclear magnetic resonance (NMR) spectrometer (Department of Chemistry, University of British Columbia). To characterize the products of the BPDOB356-catalyzed transformation of 2,6-dichlorobiphenyl, reactions were performed at 37°C in 200-ml mixtures that contained 10 μM BphAE, 10 μM ht-BphF, 5 μM ht-BphG, 100 μM substrate, and 1 mM NADH. Ethyl acetate was added after 5 min of incubation to stop the reaction and to extract the metabolites. The latter were derivatized using butylboronate and analyzed by GC-MS as previously described (3).
HPLC and GC-MS analyses.
HPLC analyses were performed using a Waters 2695 Separations module equipped with a Waters 2996 photodiode array detector and a C18 Waters Nova-Pak column (3.9 by 150 mm; Waters Limited, Mississauga, Ontario, Canada). The instrument was operated at a flow rate of 1 ml/min. Biphenyls were eluted with a 20-ml gradient of 50% to 90% acetonitrile in H2O. Samples (100 μl) were injected, and the amount of biphenyl was determined from the area of the absorbance peak at the appropriate wavelength using a standard curve. GC-MS was performed using an HP-5MS equipped with an Agilent column (19091S-433; 0.25 mm by 30 m by 0.25 mm; Agilent, Mississauga, Ontario, Canada). The instrument was run at a flow rate of 53.5 μl/min and a pressure of 10.7 lb/in2, with the oven temperature ramping from 40 to 280°C. Samples (2 μl) were injected in a splitless mode, and the amount of biphenyl was determined from the area of the corresponding peak at the appropriate m/z using a standard curve. For HPLC and GC-MS, standard curves for biphenyl were established by determining the peak areas of known amounts of the biphenyl. Samples for standard curves were treated in the same way as reaction mixtures to account for losses of biphenyl that may have occurred during sample manipulation. All standard curves had correlation factors higher than 0.99.
Crystallization of BphAEB356 and preparation of the BphAEB356-2,6-dichlorobiphenyl complex.
BphAEB356 was crystallized by the sitting-drop vapor diffusion method at 21°C under an N2 atmosphere in a glove box (Innovative Technologies, Newburyport, MA) maintained at ≤2 ppm O2. The typical protein sample contained 8 mg/ml BphAE B356, 25 mM HEPES (pH 7.3), 10% (vol/vol) glycerol, 10% (vol/vol) ethanol, 0.25 mM ferrous ammonium sulfate, and 2 mM dithiothreitol. Crystals of BphAEB356 were prepared by combining 3 μl of the protein sample with 3 μl of a reservoir solution containing 100 mM MES (pH 6.0) and 16% (vol/vol) 2-propanol and incubating the preparation with 1,000 μl of reservoir solution. Crystals had been grown previously from citrate-buffered solutions, but the presence of citrate tended to reduce the occupancy of Fe in the active site (16). The BphAEB356-2,6-dichlorobiphenyl complex was formed by adding 2,6-dichlorobiphenyl (solid) to protein crystals sitting in the original mother liquor droplet and incubating the preparation for 7 days. Longer incubation times apparently damage the crystals inasmuch as the diffraction patterns had lower resolution and overall quality.
Diffraction data measurement and processing.
Crystals were preserved by flash freezing in liquid nitrogen following a very short immersion in a solution similar to the reservoir solution except that the 2-propanol was replaced with 20% glycerol. Diffraction data were acquired from crystals cooled by an N2 gas stream at a nominal temperature of 100K. For preliminary experiments, the primary equipment included a Cu rotating anode generator equipped with double focusing mirrors or focusing multilayer optics and an R-axis IV++ imaging plate detector (Rigaku/MSC). Data were also measured using monochromatic synchrotron radiation (wavelength, 1.0 Å) and the facilities of SERCAT beamline 22-ID at the Advanced Photon Source, Argonne National Laboratory. The detector was a MarMosaic 300 charge-coupled device detector (Mar USA, Inc., Evanston, IL). The HKL2000 program suite was used to analyze the diffraction patterns, as well as to merge, scale, and reduce observed intensities. Analysis of the intensity distribution using the program DETWIN from the CCP4 software suite (16a) indicated approximately 20% twinning.
Structure determination and refinement.
The new crystals grown from MES-buffered solutions and the prior crystals from citrate-buffered solutions were essentially isomorphous. Thus, a previously refined model for the latter crystals (16) provided initial phases by straightforward molecular replacement using the program MOLREP (57) from the CCP4 v.4.2 software suite (16a). Restrained refinement was carried out with REFMAC5 (45) without adjusting intensities for twinning. Solvent molecules were added using ARP-wARP (39) and were manually inspected in O (34) and COOT (19). Coot and O were also used for calculation of density maps or for visualization of maps generated with REFMAC. The final model has an Rfactor of 20.5% and an Rfree value of 26.3%.
Modeling of BphAELB400.
The structure of the BphAELB400-biphenyl complex was modeled using the interactive mode of the 3D-JIGSAW protein comparative modeling server (7) and the crystallographic coordinates of the BphAERHA1-biphenyl complex (ProteinData Bank identifier 1ULJ) (23).
RESULTS
Purification and characterization of BPDO.
Relevant details of the anaerobic purification of the BPDOs are summarized in Table 1. The enzymes were greater than 90% pure as estimated from denaturing gels, comparable to previous preparations of aerobically and anaerobically purified BPDOB356 (30, 33). Anaerobically purified BphAELB400, BphAEB356, BphAEII9, and BphAEII10 had specific activities of 0.2, 4, 0.6, and 0.3 U/mg, respectively. The sulfur contents of the oxygenase preparations ranged from 5.7 to 5.9 mol of S per α3β3 hexamer. The iron contents ranged from 8.1 to 9.6 mol per hexamer. These values indicate that the BphAEs contained full complements of the Rieske-type [2Fe-2S] cluster and mononuclear Fe2+ center. Under aerobic conditions, the absorbance spectrum was characteristic of an oxidized Rieske-type [2Fe-2S] cluster, with maxima at 323 and 455 nm and a shoulder at 575 nm (data not shown). The ratios of the absorbance at 280 nm to the absorbance at 323 nm of the oxidized BphAEs ranged from 7 to 8.5.
TABLE 1.
Purification of oxygenase variantsa
| Enzyme | Purification step | Total protein (mg) | Total activity (U) | Sp act (U/mg) | Yield (%) |
|---|---|---|---|---|---|
| BphAELB400 | Crude extract | 1,069 | 32 | 0.03 (0.01) | 100 |
| Source Q | 72 | 7 | 0.10 (0.02) | 22 | |
| Phenyl-Sepharose | 15 | 3 | 0.22 (0.03) | 9 | |
| BphAEB356 | Crude extract | 1,200 | 600 | 0.5 (0.1) | 100 |
| Source Q | 153 | 230 | 1.5 (0.5) | 38 | |
| Phenyl-Sepharose | 41 | 164 | 4.0 (0.3) | 27 | |
| BphAEII9 | Crude extract | 2,196 | 198 | 0.09 (0.03) | 100 |
| Source Q | 105 | 32 | 0.3 (0.1) | 16 | |
| Phenyl-Sepharose | 41 | 25 | 0.6 (0.1) | 13 | |
| BphAEII10 | Crude extract | 1,314 | 40 | 0.03 (0.02) | 100 |
| Source Q | 174 | 17 | 0.12 (0.03) | 43 | |
| Phenyl-Sepharose | 28 | 8 | 0.30 (0.02) | 20 |
One unit of enzyme activity is defined as the amount of enzyme required to consume 1 μmol of O2/min. Standard deviations (n = 4) are indicated in parentheses.
Steady-state utilization of biphenyl.
Steady-state kinetic analyses of the initial rate of oxygen uptake as a function of biphenyl concentration (0.1 to 150 μM) indicated that each of the four BPDO variants exhibited Michaelis-Menten behavior. The apparent kinetic parameters for each of the four enzymes are shown in Table 2. Of the four variants, BPDOLB400 possessed the highest apparent specificity constant (kcat/Km) for biphenyl, which was approximately 10-fold greater than that of BPDOB356. By contrast, BPDOB356 exhibited the highest turnover number (kcat). The kinetic parameters for biphenyl reported here for BPDOB356 are similar in magnitude to those reported previously (33). However, direct comparison is not possible as the assay conditions were modified to facilitate comparison of all four isozymes. The steady-state parameters of BPDOII9 and BPDOII10 for biphenyl fell between those of the two parent enzymes. However, BPDOII9 was more similar to BPDOLB400. In each of the four enzymes, the consumption of O2 was well coupled to the consumption of biphenyl (Table 2). That is, in the presence of a saturating concentration of biphenyl (150 μM), the amount of biphenyl consumed corresponded to the amount of O2 utilized within experimental error. Moreover, no hydrogen peroxide, a possible uncoupling product, was detected upon the addition of catalase to reaction mixtures.
TABLE 2.
Apparent kinetic parameters and coupling constants of BPDOB356, BPDOLB400, BPDOII9, and BPDOII10 using biphenyl as the substratea
| Enzyme | Km (μM) | kcat (s−1) | kcat/Km (106 M−1 s−1) | Biphenyl/O2 | H2O2/O2 |
|---|---|---|---|---|---|
| BPDOB356 | 20 (4) | 4.1 (0.2) | 0.21 (0.04) | 1.0 (0.2) | ND |
| BPDOLB400 | 0.18 (0.03) | 0.4 (0.1) | 2.4 (0.7) | 1.1 (0.1) | ND |
| BPDOII9 | 0.3 (0.2) | 0.67 (0.08) | 2 (1) | 1.0 (0.1) | ND |
| BPDOII10 | 2 (1) | 1.03 (0.05) | 0.5 (0.3) | 0.9 (0.2) | ND |
Experiments were performed using 50 mM MES (pH 6) at 25°C. The values are means (n = 4 to 6), with standard deviations in parentheses. ND, not detected. Additional experimental details are provided in Materials and Methods.
Reactivity of BPDO variants with individual chlorinated biphenyls.
Steady-state kinetic parameters for chlorinated biphenyls were not determined in the current study due to the low solubilities of the congeners, the low activities of the isozymes, and the poor coupling of the transformation of the more chlorinated biphenyls to O2 consumption. For these reasons, the reactivity of each BPDO with each of five chlorinated biphenyls was examined using a single congener concentration (50 μM). The rates of congener and O2 depletion are summarized in Table 3. Of the four isozymes, BPDOB356 showed the best congener-transforming activity: it transformed each of the five congeners at rates that equaled or exceeded those of the other isozymes. Of particular note, BPDOB356 depleted 2,2′-dichlorobiphenyl as well as BPDOLB400. By contrast, BPDOII10 had the poorest ability to transform congeners, significantly depleting only 2,2′- and 3,3′-dichlorobiphenyls. The overall congener depletion activity of BPDOII9 was intermediate between those of the two parental enzymes, except for 4,4′-dichlorobiphenyl, which BPDOII9 transformed more slowly than either parental enzyme. None of the four enzymes transformed any of the congeners tested significantly faster than biphenyl. Except for the transformation of 2,2′-dichlorobiphenyl by BPDOII9 andBPDOLB400, congener and O2 consumption were uncoupled. In most cases, the O2 that was not incorporated into the PCB was detected as H2O2. However, not all uncoupling resulted in H2O2 production, suggesting that H2O was also produced in some cases.
TABLE 3.
Reactivities of purified BPDOs with individual congenersa
| Congener | BPDOB356
|
BPDOLB400
|
BPDOII9
|
BPDOII10
|
||||
|---|---|---|---|---|---|---|---|---|
| Activity (nmol biphenyl/min) | H2O2/O2 | Activity (nmol biphenyl/min) | H2O2/O2 | Activity (nmol biphenyl/min) | H2O2/O2 | Activity (nmol biphenyl/min) | H2O2/O2 | |
| Biphenyl | 55.3 (0.6) | ND | 12 (4) | ND | 44 (2) | ND | 25 (1) | ND |
| 3,3′-Dichloro | 32.2 (0.2) | 0.2 (0.04) | 6 (2) | 0.3 (0.1) | 6 (2) | 0.31 (0.01) | 10 (1) | 0.6 (0.3) |
| 2,4,4′-Trichloro | 20.5 (0.3) | 0.31 (0.02) | 1.1 (0.3) | 0.7 (0.2) | 8 (1) | 0.5 (0.1) | ND | ND |
| 2,2′-Dichloro | 15 (1) | 0.6 (0.2) | 15 (4) | ND | 12 (2) | ND | 16 (4) | 0.4 (0.1) |
| 4,4′-Dichloro | 9 (5) | 0.4 (0.2) | 3.9 (0.3) | 0.4 (0.2) | 1 (0.4) | 0.15 (0.05) | ND | ND |
| 2,6-Dichloro | 6 (1) | 0.4 (0.1) | 1.5 (0.6) | 0.9 (0.1) | 5 (1) | 0.4 (0.2) | ND | ND |
Experiments were performed using 50 mM MES (pH 6) at 25°C. Each substrate was tested individually using a final concentration of 50 μM. The values are means (n = 3 to 5), with standard deviations in parentheses. ND, not detected. Additional experimental details are provided in Materials and Methods. The activities obtained using biphenyl are not comparable to the parameters reported in Table 2 as depletion was determined after 1 min.
Transformation products of 2,4,4′-trichlorobiphenyl and 2,6-dichlorobiphenyl.
The relatively rapid transformation of 2,4,4′-trichlorobiphenyl by BPDOB356 was unexpected in light of previous reports that the enzyme transforms double para-substituted congeners poorly (1, 3). To better characterize this transformation, the reaction product was purified and found to absorb maximally at a λ of 293.5 nm, which is within the range of λmaxs of other dihydrodiols (2). When the transformation was performed in the presence of additional Bph enzymes, the bright yellow ring-cleaved product was observed when BphB and BphC were added to the reaction mixture but not when BphC alone was added. This result indicates that the BPDOB356-catalyzed dihydroxylation of 2,4,4′-trichlorobiphenyl did not involve a dehalogenation as this is expected to yield a catechol, the substrate of BphC. Analysis of the dihydrodiol by 1H NMR identified it as 2,3-dihydro-2,3-dihydroxy-2′,4,4′-trichlorobiphenyl (Fig. 2). In particular, the values of the chemical shifts and coupling constants [4.5 (1H, d, J = 6 Hz, H2′/H3′), 4.95 (1H, d, J = 6 Hz, H2′/H3′), 6.12 (1H, d, J = 6.1 Hz, H5′/H6′), and 6.43 (1H, d, J = 6.1 Hz, H5′/H6′] were consistent with the presence of four protons on the nonaromatic ring (excluding the hydroxyl protons), which are upfield of the aromatic ring due to the shielding effect of the —OH groups. Moreover, the chemical shifts were comparable to values predicted using ChemDraw Ultra 7.0 (CambridgeSoft, Cambridge, MA), as well as to those of similar chlorinated 2,3-dihydroxybiphenyls (5, 48). Overall, these results demonstrate that BPDOB356 catalyzes the 2,3-dihydroxylation of the monochlorinated ring of 2,4,4′-trichlorobiphenyl.
FIG. 2.

NMR spectrum of the product of 2,4,4′-trichlorobiphenyl dihydroxylation by BPDOB356. 1H NMR (300 MHz, acetone-d6, δ): 4.5 (1H, d, J = 6 Hz, H2′/H3′), 4.95 (1H, d, J = 6 Hz, H2′/H3′), 6.12 (1H, d, J = 6.1 Hz, H5′/H6′), 6.43 (1H, d, J = 6.1 Hz, H5′/H6′), 7.65 (2H, d, J = 2 Hz, H3/H5/H6), 7.5 (1H, d, J = 2 Hz, H3/H5/H6). The data collected were insufficient to assign protons 3, 5, 6, 5′, and 6′ to a specific chemical shift. The intensity of the peak for H2′/H3′ was affected by the water signal suppression specified in the instrument.
Transformation products of 2,6-dichlorobiphenyl were characterized by GC-MS (Fig. 3). BPDOB356, BPDOII9, and BPDOLB400 each transformed this congener to a mixture of two dichlorodihydrodihydroxybiphenyls. More specifically, the butylboronate derivatives of these compounds yielded mass spectra with the characteristic molecular ion (m/z 322) and a fragmentation pattern reflecting loss of a chlorine radical (m/z 287), the n-butyl moiety (m/z 265), the C4H9BO moiety (m/z 238), and the n-C4H9BO2 moiety (m/z 222) (40). The data did not identify the location of the hydroxyl groups. However, BPDOLB400 catalyzes the 2,3-dihydroxylation of 2-chlorobiphenyl, 3-chlorobiphenyl, and 2,5-dichlorobiphenyl on the unchlorinated ring (26), suggesting that the major metabolite produced from 2,6-dichlorobiphenyl (representing 85% of total dihydrodiol produced by BPDOB356) is 2,6-dichloro-2′,3′-dihydro-2′,3′-dihydroxybiphenyl. Moreover, no monochlorinated dihydroxybiphenyl, the expected product of 2,3-dihydroxylation of the chlorinated ring, was detected, indicating that the second metabolite results from 3,4-dihydroxylation of one of the rings.
FIG. 3.

GC spectra of butylboronate-derived metabolites of 2,6-dichlorobiphenyl produced by BPDOII9, BPDOB356, and BPDOLB400. The mass spectra of metabolites 1 and 2 are characterized by major ions at m/z 322, 287, 265, 238, and 222, which are characteristic features for dihydro-dihydroxy-dichlorobiphenyls (see Results fore more details). The exact location of the hydroxyl groups on the biphenyl ring cannot be determined precisely. However, one of the two metabolites must result from a meta-para oxygenation on either the chlorinated or nonchlorinated ring. The metabolites are thus tentatively identified as the 2,6-dichloro-3,4-dihydro-3,4-dihydroxybiphenyl and 2,6-dichloro-2′,3′-dihydro-2′,3′-dihydroxybiphenyl.
Depletion of PCB mixtures by purified BPDOs.
The activities of the purified BPDOs were investigated using a previously described mixture of eight congeners (3): 3,3′-dichlorobiphenyl, 4,4′-dichlorobiphenyl, 2,6-dichlorobiphenyl, 2,3,4′-trichlorobiphenyl, 2,3′,4-trichlorobiphenyl, 2,4,4′-trichlorobiphenyl, 2,2′,3,3′-tetrachlorobiphenyl, and 2,2′,5,5′-tetrachlorobiphenyl. The percent depletion of each congener in the mixture after 20 min is shown in Table 4. Consistent with the assays performed with individual congeners, BPDOB356 showed the best congener-transforming activity; it was the only variant that significantly depleted all congeners and, with the exception of 2,3,4′-trichlorobiphenyl and 2,2′,5,5′-tetrachlorobiphenyl, depleted each congener more completely than any of the other variants. Indeed, BPDOB356 was the only variant that detectably depleted 2,6-dichlorobiphenyl, 3,3′-dichlorobiphenyl, and 4,4′-dichlorobiphenyl. By contrast, BPDOLB400 detectably depleted only four of the congeners in the mixture. However, it depleted 2,3,4′-trichlorobiphenyl and 2,2′,5,5′-tetrachlorobiphenyl faster than BPDOB356. The activity of BPDOII9 was similar to that of BPDOLB400, while BPDOII10 had the lowest overall depletion activity. Nevertheless, both BPDOII9 and BPDOII10 depleted 2,3,4′-trichlorobiphenyl more efficiently than either parental enzyme.
TABLE 4.
Depletion of a PCB mixture by purified BPDOsa
| Congener | % Depletion with:
|
|||
|---|---|---|---|---|
| BPDOB356 | BPDOLB400 | BPDOII9 | BPDOII10 | |
| 2,6-Dichloro | 21 (11) | <10 | <10 | <10 |
| 3,3′-Dichloro | 29 (11) | <10 | <10 | <10 |
| 4,4′-Dichloro | 13 (5) | <10 | <10 | <10 |
| 2,3′,4-Trichloro | 80 (14) | 57 (5) | 35 (13) | 18 (11) |
| 2,4,4′-Trichlorol | 21 (5) | <10 | 10 (4) | <10 |
| 2,3,4′-Trichloro | 49 (5) | 65 (18) | 82 (9) | 73 (23) |
| 2,2′,5,5′-Tetrachloro | 19 (12) | 51 (13) | 25 (12) | 23 (14) |
| 2,2′,3,3′-Tetrachloro | 18 (12) | 16 (4) | 16 (6) | 10 (3) |
Assays were performed using 50 mM MES (pH 6) 25°C. The values are means, with standard deviations in parentheses (n = 4 to 6). Additional experimental details are provided in Materials and Methods.
Depletion of a PCB mixture by whole cells.
The abilities of BPDOLB400, BPDOII9, and BPDOII10 to deplete congeners in a mixture were also tested using whole cells. The bphAE genes were coexpressed with bphFGLB400 in E. coli C41(DE3) (29). Overall, the whole-cell assays mirrored the apparent substrate preference observed in the enzyme assays, but the levels of depletion were higher in the former (Table 5). Interestingly, cells expressing BPDOII9 depleted every congener tested and did so at higher rates than cells expressing BPDOLB400. Although the high activity of BPDOII9 in whole cells was similar to that of BPDOB356 in purified enzyme assays, the apparent substrate preference of these two enzymes was still different. Whole cells containing BPDOII10 did not degrade any of the congeners. The abilities of strains B. xenovorans LB400 and P. pnomenusa B-356 to deplete congeners in a mixture were similarly tested (Table 5). Thus, the substrate preference of BPDOLB400 with the mixture of congeners was 2,3′,4-trichlorobiphenyl > 2,2′,5,5′-tetrachlorobiphenyl ∼ 2,2′,3,3′-tetrachlorobiphenyl > 2,3,4′-trichlorobiphenyl ∼ 3,3′-dichlorobiphenyl > 2,4,4′-trichlorobiphenyl > 4,4′-dichlorobiphenyl ∼ 2,6-dichlorobiphenyl. The substrate preference of BPDOB356 with the mixture of congeners was 2,3′,4-trichlorobiphenyl ≥ 2,3,4′-trichlorobiphenyl > 3,3′-dichlorobiphenyl > 2,6-dichlorobiphenyl ≥ 2,4,4′-trichlorobiphenyl > 2,2′,5,5′-tetrachlorobiphenyl ∼ 2,2′,3,3′-tetrachlorobiphenyl ≥ 4,4′-dichlorobiphenyl. The reactivity of mutant BPDOII9 was closer to that of BPDOLB400: 2,3,4′-trichlorobiphenyl > 2,3′,4-trichlorobiphenyl > 2,2′,5,5′-tetrachlorobiphenyl ≥ 2,2′,3,3′-tetrachlorobiphenyl ∼ 2,4,4′-trichlorobiphenyl > 4,4′-dichlorobiphenyl ∼ 2,6-dichlorobiphenyl ∼ 3,3′-dichlorobiphenyl.
TABLE 5.
Depletion of a PCB mixture by E. coli cells expressing BPDOsa
| Congener | % Depletion with:
|
||||||
|---|---|---|---|---|---|---|---|
| BPDOLB400
|
BPDOII9
|
BPDOII10
|
Wild types
|
||||
| 3 h | 18 h | 3 h | 18 h | 18 h | LB400 | B356 | |
| 2,6-Dichloro | <10 | <10 | 39 | 100 | <10 | 0 | 89 (17) |
| 3,3′-Dichloro | 26 | 79 | 38 | 100 | <10 | 78 (11) | 98 |
| 4,4′-Dichloro | 11 | 18 | 39 | 100 | <10 | 11 | 88 (13) |
| 2,3′,4-Trichloro | 49 | 100 | 67 | 100 | <10 | 96 | 100 (5) |
| 2,4,4′-Trichloro | 18 | 37 | 36 | 99 | <10 | 45 (6) | 45 (6) |
| 2,3,4′-Trichloro | 26 | 80 | 89 | 100 | <10 | 89 (17) | 100 (5) |
| 2,2′,5,5′-Tetrachloro | 40 | 100 | 45 | 99 | <10 | 80 (8) | 32 (6) |
| 2,2′,3,3′-Tetrachloro | 39 | 100 | 35 | 96 | <10 | 92 (10) | 32 (5) |
The values are means (n = 3 to 5). Standard deviations were less than 5% except where indicated in parentheses. The results obtained for BPDOII10 were the same at 3 and 18 h. Additional experimental details are provided in Materials and Methods.
Structure of a BphAEB356-2,6-dichlorobiphenyl complex.
Crystals of the complex were in space group R3 with the hexagonal cell parameters a = 136.3 Å and c = 107.0 Å, and the asymmetric unit included one αβ protomer. Diffraction to 2.4-Å resolution was measured from the best crystal, with an overall completeness of 89% and an overall redundancy of 3.1 measurements per unique reflection; the completeness and redundancy were 64% and 1.6, respectively, in the in the last shell. Rsym was 14.2% overall and 44.9% in the last shell, where 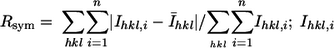 is the intensity of the ith observation of reflection hkl from the asymmetric unit in reciprocal space, and Īhkl is the mean of n observations. The mean values of I/σ(I) were 11.9 overall and 2.3 for the data in the last shell. The refined model includes residues 18 to 457 from the α subunit, residues 1 to 186 from the β subunit, one molecule of 2,6-dichlorobiphenyl, and 124 water molecules. An R value of 20.5% was obtained for the structure factors used in the refinement; Rfree was 26.3% for the data reserved for validation. The average B factor was 41 Å2 for all atoms.
is the intensity of the ith observation of reflection hkl from the asymmetric unit in reciprocal space, and Īhkl is the mean of n observations. The mean values of I/σ(I) were 11.9 overall and 2.3 for the data in the last shell. The refined model includes residues 18 to 457 from the α subunit, residues 1 to 186 from the β subunit, one molecule of 2,6-dichlorobiphenyl, and 124 water molecules. An R value of 20.5% was obtained for the structure factors used in the refinement; Rfree was 26.3% for the data reserved for validation. The average B factor was 41 Å2 for all atoms.
Following refinement of the protein and water atoms, 2,6-dichlorobiphenyl was clearly represented in the (Fo-Fc) map by positive density at 3.0σ. The electron density maps indicate a single binding mode for 2,6-dichlorobiphenyl, and this mode is consistent with hydroxylation of C-2′ and C-3′ on the nonchlorinated ring; in particular, (Fo-Fc) maps presented no evidence for an alternative binding mode that would support hydroxylation of the C-3′—C-4′ bond. Superposition of the BphAEB356-2,6-dichlorobiphenyl complex and the BphAERHA1-biphenyl complex (23) (either on the basis of the active site Fe and its ligands or on the basis of the Cα models) indicated that the C-2—C-3 bond of biphenyl and the C-2′—C-3′ bond of 2,6-dichlorobiphenyl are in similar locations relative to each other and to their respective Fe atoms. In the superposed models, the normals to the reactive ring planes are within 10° of each other, the C-2 and C-2′ positions are 0.4 Å apart, the C-3 and C-3′ atoms are 0.2 Å apart, and the distances from C-2/C-3 and C-2′/C-3′ to their respective active Fe atoms are all in the range from 4.3 to 4.6 Å. The angle between the planes of the two aromatic rings is 70° for 2,6-dichlorobiphenyl in the complex with BphAEB356 and 60° for biphenyl in the complex with BphAERHA1. The consistency in binding modes supports a conclusion that the ring presented to the Fe atom is in a productive location and orientation.
As illustrated in Fig. 4, the reactive ring is in nonbonded contact with the side chains of Gln226, Phe227, Met231, His321, Leu331, and His233 (one of the ligands to the Fe atom) and with the carbonyl groups of Gln226 and Asp230. Five of the six ring atoms are in nonbonded contact with multiple atoms. By contrast, only four of the ring atoms in the chlorinated ring are in contact with side chain atoms, each of the four ring atoms makes contact with only a single protein atom, and only three protein atoms are involved, Cγ2 of Ile283, Cδ1 of Ile334, and Cζ of Phe376 (two contacts). Thus, it seems that the distal ring pocket would place few restrictions on the binding of a nonchlorinated ring and presents sufficient space to accommodate chlorination at any position. One Cl atom lies close to the active Fe in contact with the Cβ atom of Ala234 as well as the Fe ligands, His233 and His239. The other Cl atom binds in contact with the backbone atoms of Gln320 and His321 and with the side chain of Leu331.
FIG. 4.

Crystal structure of the BphAEB356-2,6-dichlorobiphenyl complex. In this stereoscopic drawing carbon atoms from the protein and biphenyl are yellow and gray, respectively. Nitrogen, oxygen, sulfur, chlorine, and iron atoms are blue, red, orange, green, and magenta, respectively. Protein residues are identified by sequence numbers and the standard one-letter codes.
Model of the BphAELB400-biphenyl complex.
To better understand the molecular basis of the differences in congener specificity of the BPDOs, the structure of the BphAELB400-biphenyl complex was modeled on the basis of the crystallographic coordinates of the BphAERHA1-biphenyl complex (23). The two proteins share 70% sequence identity, and the modeling score was 3.8/4. The α carbons of BphALB400 had root mean square deviations of 0.74 Å and 0.64 Å with those of BphARHA1 and BphAB356 (16; C. L. Colbert and J. T. Bolin, personal communication), respectively. Overall, the biphenyl-binding pocket of the modeled BphAELB400 is expected to be smaller than that of BphAEB356 because it includes more aromatic residues. In the hypothetical model of BphAELB400, the first shell residues of the biphenyl-binding pocket include Gln226, Phe227, Met231, His323, Leu333, Phe336, and Phe384. Among the residues that are substituted in BphAEII9 and BphAEII10, Phe336 is expected to help define the biphenyl-binding pocket; residues 335, 338, and 341 may exert indirect effects. The model predicts that Phe336 is approximately 1 Å closer to C-4 of the distal ring of the bound biphenyl than the equivalent residue in BphAEB356, Ile334. Residue 267, which is also substituted in BphAEII10, is remote from the active site; it is located more than 20 Å from the closest mononuclear iron in the α3β3 oxygenase.
DISCUSSION
This study establishes that BPDOB356 transforms PCBs better than BPDOLB400, an enzyme that has been well studied for its potent PCB-transforming ability, and provides insight into the molecular basis for this activity. The PCB-transforming activities were compared using three different experimental designs: purified enzyme and individual congeners, purified enzyme and a mixture of congeners, and whole cells and a mixture of congeners. This comparison established the validity of each of the individual approaches. Moreover, the current study made use of anaerobically purified preparations of nontagged oxygenases to maximize the specific activity and stability of the oxygenases.
The higher apparent specificity constant for biphenyl of BPDOLB400 than of BPDOB356 is consistent with what has been previously reported (3). However, the current finding that the Km of BPDOB356 for biphenyl is 100-fold higher than that of BPDOLB400 contradicts previous results obtained with His-tagged enzymes indicating that the enzymes have similar Km values for biphenyl (3). This could reflect the influence of the His tag or the lower sensitivity of the assay used. Similarly, previous studies with aerobically prepared His-tagged oxygenases had indicated that BPDOII9 and BPDOII10 were more active than BPDOB356 (3). The kinetic parameters of mutants BPDOII9 and BPDOII10 for biphenyl are within the range of those of their parental enzymes. This is consistent with the observation that most engineered BPDOs described to date degrade biphenyl at lower rates than their parental enzymes (28, 54, 60) and likely reflects the optimization of the parental enzymes for their natural substrate.
The similar results obtained from the three congener depletion assays demonstrate that appropriately designed experiments may be compared. Small differences between the assays may be rationalized. Thus, the rate of depletion of the individual congeners was lower in the presence of competing congeners, consistent with previous studies (9) and the fact that multiple substrates compete for the same active site. Moreover, enzymes in whole cells transformed congeners faster than the reconstituted dioxygenases, consistent with previous reports (10, 24). It seems unlikely that these differences arise from a lower quality of the purified BPDO components compared with those in the cell as the former were prepared anaerobically and were highly active. Such differences may be due to the higher relative levels of BphAE inside the cells, which were estimated to be 3 orders of magnitude higher than those in the in vitro assay when the number of E. coli cells and their individual volume were considered. Overall, while data obtained with purified enzymes are essential for mechanistic and biochemical studies, assays performed using whole cells provide valuable insights into the physiological behavior of the enzyme.
A major finding of the current study is that BPDOB356 is a potent PCB-degrading enzyme: it was more active than the other purified BPDOs against all congeners tested with the exception of 2,2′,5,5′-tetrachlorobiphenyl (Tables 3 and 4). Characterization of the transformation products of 2,6-dichlororbiphenyl and 2,4,4′-trichlororbiphenyl confirmed the previously unreported ability of BPDOB356 to transform these congeners and also provided the first evidence that this enzyme catalyzes the 3,4-dihydroxylation of some congeners. The transformation of 2,2′,5,5′-tetrachlorobiphenyl by BPDOB356 may also involve 3,4-dihydroxylation. The current study is nevertheless consistent with previous reports of the apparent substrate preference of BPDOB356 (meta- > para- > ortho-dichlorobiphenyls) (33), as well as the enzyme's ability to degrade 2,3′,4-trichlorobiphenyl, 2,3,4′-trichlorobiphenyl, and 3,3′-dichlorobiphenyl (3). The low activity of BPDOB356 against other congeners reported in the latter study perhaps reflects the assay, which used whole cells expressing His-tagged enzyme. Recent studies using His-tagged preparations have since confirmed that BPDOB356 transformed 2,6-dichlorobiphenyl better than either BPDOLB400 or BPDOP4 (D. Barriault and M. Sylvestre, unpublished data), a variant of BPDOLB400 that degrades most congeners at a higher rate than BPDOLB400 (4). Although BPDOB356 shares similar sequence identity with BPDOLB400 (75%) and BPDOKF707 (76%), the reactivity of BPDOB356 appears to be closer to that of BPDOKF707. BPDOKF707 also shows a high level of depletion for both 2,4,4′-trichlorobiphenyl and 4,4′-dichlorobiphenyl, although it preferentially degrades para-substituted congeners over ortho-substituted congeners (12, 20, 24).
The congener preference of BPDOLB400 reported here (Tables 3 and 4) is consistent with data reported by others (1, 2, 6, 22, 44). Thus, the enzyme dihydroxylated biphenyl and 2,2′-dichlorobiphenyl at similar rates and showed an apparent preference for ortho- > meta- > para-dichlorobiphenyls. Overall, the current results substantiate and extend the conclusion that the introduction of ortho-chloro substituents into an otherwise meta- and/or para-chlorobiphenyl renders the congener more susceptible to attack by BPDOLB400 despite the increased level of chlorination (12). As discussed below, this is consistent with modeling experiments that indicate that the active site of BPDOLB400 better accommodates nonplanar congeners.
The overall substrate preference of BPDOII9 was closer to that of BPDOLB400 than to that of BPDOB356 (Tables 3 and 4). This is consistent with the steady-state parameters for biphenyl, which were also closer to those of BPDOLB400. Although BPDOII9 has PCB-transforming properties superior to those of BPDOLB400 in whole-cell assays (3; this study), the purified enzyme did not show superior activity against any individual congeners compared to purified BPDOB356. The second engineered variant, BPDOII10, was the least active enzyme in this study. The overall pattern of congener degradation by purified BPDOII10 in the mixed-congener assay was similar to that of BPDOLB400 and BPDOII9 (Tables 3 and 4), although BPDOII10 showed a slightly improved ability to degrade 3,3′-dichlorobiphenyl compared to the latter enzymes. On the other hand, the improved degradation of 2,4,4′-trichlorobiphenyl and 2,6-dichlorobiphenyl by BPDOII9 compared to that by BPDOLB400 was not observed for BPDOII10. These two findings may indicate a crucial role of Ala267 in substrate binding, particularly in meta-substituted congeners. Finally, it is not clear why whole cells expressing BPDOII10 did not degrade any of the congeners, although this is consistent with previous observations (3).
In all the variants studied, the degree of coupling between the transformation of congener and the consumption of O2 was approximately proportional to the rate of congener depletion. The results obtained with BPDOLB400 and BPDOB356 for 2,2′-, 3,3′-, and 4,4′-dichlorobiphenyls are consistent with previous results obtained in identical conditions (33, 41). Uncoupling has been better characterized in cytochrome P450 monooxygenases (47), in which the efficiency of hydroxylation is thought to depend on the position of the organic substrate with respect to the activated oxygen intermediate and the exclusion of solvent molecules from this environment. The current results are consistent with this notion and highlight the ability of PCBs to poison the degradation process and to stress the cell through the production of reactive oxygen species. This is consistent with a recent study demonstrating that PCB-degrading cells are oxidatively stressed (14).
The crystallographic data for the BphAEB356-2,6-dichlorobiphenyl complex are consistent with the observed preponderance of the 2′,3′-dihydroxylation product. While the structural data do not reveal a binding mode consistent with 3,4-dihydroxylation, the limited contact between the substrate and protein suggests that there is sufficient space to accommodate an appropriate binding mode. In this case, displacement of a short helix spanning residues 282 through 288 might be required. This appears to be an unhindered change in conformation inasmuch as the helix is solvent exposed and does not seem to be constrained by intramolecular contacts. Moreover, superposition of the BphAEB356-2,6-dichlorobiphenyl complex with the BphAERHA1-biphenyl complex revealed that the backbone atoms of the corresponding helix in the latter are displaced 1.9 Å further away from the Fe atom at the position corresponding to Ile283 of BphAEB356, the single residue from this helix in contact with the distal ring of 2,6-dichlorobiphenyl.
Comparison of the crystal structure of the BphAEB356-2,6-dichlorobiphenyl complex to that of the modeled BphAELB400-biphenyl complex revealed several features that explain the different reactivities of the two isozymes. Most strikingly, the increased space around C-4 of the distal ring in BphAEB356 likely explains the superior ability of BphAEB356 to transform para-substituted congeners. This conclusion is corroborated by BphAEII9 and BphAEKF707, which also have Ile at this position and which transform 4,4′-dichlorobiphenyl better than BphAELB400 (54). Some studies have further suggested an important role of residues 338 and 341 in the dihydroxylation of ortho-substituted congeners (12, 37). However, the levels of depletion of 2,2′-dichlorobiphenyl by BPDOB356 and BPDOII9 in this study were comparable to those by BPDOLB400. This result suggests that residues at positions 338 and 341 have a more subtle effect on congener binding, consistent with the model of the BphAELB400-biphenyl complex. Finally, it is not obvious how residue 267 influences the reactivity of BphAE to the extent that it does, due to its location remote from the active site. However, the different activities of BPDOII10 and BPDOB356, two enzymes that have Ser267, highlight the importance of sequence context for the influence of a specific residue.
Considerable effort has been invested in engineering BPDO to improve its PCB-degrading activities, as well as to improve our understanding of the enzyme's mechanism and the role of active site residues. While much of this effort has involved mutations of residues in region III, it is clear from the available structural data that other residues play an equally important role in determining the congener preference of the enzyme. Finally, most of the engineering efforts have involved BPDOLB400 and BPDOKF707. The high PCB-transforming activity of BPDOB356 highlights the importance of including this and other less-well-characterized enzymes in engineering efforts, particularly directed evolution.
Acknowledgments
This work was supported by Discovery and Strategic grants from the Natural Sciences and Engineering Research Council of Canada (NSERC). X-ray diffraction data were collected at the Southeast Regional Collaborative Access Team (SER-CAT) 22-ID beamline at the Advanced Photon Source, Argonne National Laboratory; supporting institutions may be found at www.ser-cat.org/members.html. Use of the Advanced Photon Source was supported by the U.S. Department of Energy Office of Science Office of Basic Energy Sciences under contract W-31-109-Eng-38.
Cheryl Whiting and Gordon Stewart provided valuable technical assistance. Geoff Horsman collected the NMR data. Shiva Bhowmik assisted in preparing Fig. 4. Elitza Tocheva assisted in interpreting modeled structures.
Footnotes
Published ahead of print on 25 May 2007.
REFERENCES
- 1.Agar, N. Y. R. 2002. Identification of molecular determinants of substrate specificity and activity for biphenyl dioxygenase from Comamonas testosteroni B-356. Concordia University, Montréal, Canada.
- 2.Arnett, C. M., J. V. Parales, and J. D. Haddock. 2000. Influence of chlorine substituents on rates of oxidation of chlorinated biphenyls by the biphenyl dioxygenase of Burkholderia sp. strain LB400. Appl. Environ. Microbiol. 66:2928-2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barriault, D., M. M. Plante, and M. Sylvestre. 2002. Family shuffling of a targeted bphA region to engineer biphenyl dioxygenase. J. Bacteriol. 184:3794-3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barriault, D., and M. Sylvestre. 2004. Evolution of the biphenyl dioxygenase BphA from Burkholderia xenovorans LB400 by random mutagenesis of multiple sites in region III. J. Biol. Chem. 279:47480-47488. [DOI] [PubMed] [Google Scholar]
- 5.Barriault, D., F. Lepin, M. Mohammadi, S. Milot, N. Leberre, and M. Sylvestre. 2004. Revisiting the regiospecificity of Burkholderia xenovorans LB400 biphenyl dioxygenase toward 2,2′-dichlorobiphenyl and 2,2′,3,3′-tetrachlorobiphenyl. J. Biol. Chem. 279:47489-47496. [DOI] [PubMed] [Google Scholar]
- 6.Barriault, D., C. Pelletier, Y. Hurtubise, and M. Sylvestre. 1997. Substrate selectivity pattern of Comamonas testosteroni strain B-356 towards dichlorobiphenyls. Int. Biodeterior. Biodegrad. 39:311-316. [Google Scholar]
- 7.Bates, P. A., L. A. Kelley, R. M. MacCallum, and M. J. E. Sternberg. 2001. Enhancement of protein modeling by human intervention in applying the automatic programs 3D-JIGSAW and 3D-PSSM. Proteins 5:39-46. [DOI] [PubMed] [Google Scholar]
- 8.Bedard, D., and M. Haberl. 1990. Influence of chlorine substitution pattern on the degradation of polychlorinated biphenyls by eight bacterial strains. Microb. Ecol. 20:87-102. [DOI] [PubMed] [Google Scholar]
- 9.Bedard, D. L., R. Unterman, L. H. Bopp, M. J. Brennan, M. L. Haberl, and C. Johnson. 1986. Rapid assay for screening and characterizing microorganisms for the ability to degrade polychlorinated biphenyls. Appl. Environ. Microbiol. 51:761-768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bopp, L. H. 1986. Degradation of highly chlorinated PCBs by Pseudomonas strain LB400. J. Ind. Microbiol. 1:23-29. [Google Scholar]
- 11.Brown R. E., J. L. Jarvis, and K. J. Hyland. 1989. Protein measurement using bicinchoninic acid: elimination of interfering substances. Anal. Biochem. 180:136-139. [DOI] [PubMed] [Google Scholar]
- 12.Bruhlmann, F., and W. Chen. 1999. Tuning biphenyl dioxygenase for extended substrate specificity. Biotechnol. Bioeng. 63:544-551. [DOI] [PubMed] [Google Scholar]
- 13.Carpenter, D. O. 2006. Polychlorinated biphenyls (PCBs): routes of exposure and effects on human health. Rev. Environ. Health 21:1-23. [DOI] [PubMed] [Google Scholar]
- 14.Chavez, F. P., H. Lunsdorf, and C. A. Jerez. 2004. Growth of polychlorinated-biphenyl-degrading bacteria in the presence of biphenyl and chlorobiphenyls generates oxidative stress and massive accumulation of inorganic polyphosphate. Appl. Environ. Microbiol. 70:3064-3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen, J. S., and L. E. Mortenson. 1977. Inhibition of methylene blue formation during determination of the acid-labile sulfide of iron-sulfur protein samples containing dithionite. Anal. Biochem. 79:157-165. [DOI] [PubMed] [Google Scholar]
- 16.Colbert, C. L. 2000. Initiating the biodegradation of PCBs: structural analysis of the biphenyl dioxygenase system. Purdue University, West Lafayette, IN.
- 16a.Collaborative Computational Project by number 4. 1994. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. Sect. D 50:760-763. [DOI] [PubMed] [Google Scholar]
- 17.Cornish-Bowden, A. 1995. Fundamentals of enzyme kinetics. Portland Press, London, United Kingdom.
- 18.Couture, M. M., C. L. Colbert, E. Babini, F. I. Rosell, A. G. Mauk, J. T. Bolin, and L. D. Eltis. 2001. Characterization of BphF, a Rieske-type ferredoxin with a low reduction potential. Biochemistry 40:84-92. [DOI] [PubMed] [Google Scholar]
- 19.Emsley, P., and K. Cowtan. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr. Sect. D 60:2126-2132. [DOI] [PubMed] [Google Scholar]
- 20.Erickson, B. D., and F. J. Mondello. 1993. Enhanced biodegradation of polychlorinated biphenyls after site-directed mutagenesis of a biphenyl dioxygenase gene. Appl. Environ. Microbiol. 59:3858-3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Erickson, B. D., and F. J. Mondello. 1992. Nucleotide sequencing and transcriptional mapping of the genes encoding biphenyl dioxygenase, a multicomponent polychlorinated-biphenyl-degrading enzyme in Pseudomonas strain LB400. J. Bacteriol. 174:2903-2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Furukawa, K. 2000. Engineering dioxygenases for efficient degradation of environmental pollutants. Curr. Opin. Biotechnol. 11:244-249. [DOI] [PubMed] [Google Scholar]
- 23.Furusawa, Y., V. Nagarajan, M. Tanokura, E. Masai, M. Fukuda, and T. Senda. 2004. Crystal structure of the terminal oxygenase component of biphenyl dioxygenase derived from Rhodococcus sp. strain RHA1. J. Mol. Biol. 342:1041-1052. [DOI] [PubMed] [Google Scholar]
- 24.Gibson, D. T., D. L. Cruden, J. D. Haddock, G. J. Zylstra, and J. M. Brand. 1993. Oxidation of polychlorinated biphenyls by Pseudomonas sp. strain LB400 and Pseudomonas pseudoalcaligenes KF707. J. Bacteriol. 175:4561-4564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Haddock, J. D., and D. T. Gibson. 1995. Purification and characterization of the oxygenase component of biphenyl 2,3-dioxygenase from Pseudomonas sp. strain LB400. J. Bacteriol. 177:5834-5839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haddock, J. D., J. R. Horton, and D. T. Gibson. 1995. Dihydroxylation and dechlorination of chlorinated biphenyls by purified biphenyl 2,3-dioxygenase from Pseudomonas sp. strain LB400. J. Bacteriol. 177:20-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hanahan, D. 1983. Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol. 166:557-580. [DOI] [PubMed] [Google Scholar]
- 28.Hirose, J., A. Suyama, S. Hayashida, and K. Furukawa. 1994. Construction of hybrid biphenyl (bph) and toluene (tod) genes for functional analysis of aromatic ring dioxygenases. Gene 138:27-33. [DOI] [PubMed] [Google Scholar]
- 29.Hofer, B., L. D. Eltis, D. N. Dowling, and K. N. Timmis. 1993. Genetic analysis of a Pseudomonas locus encoding a pathway for biphenyl/polychlorinated biphenyl degradation. Gene 130:47-55. [DOI] [PubMed] [Google Scholar]
- 30.Hurtubise, Y., D. Barriault, J. Powlowski, and M. Sylvestre. 1995. Purification and characterization of the Comamonas testosteroni B-356 biphenyl dioxygenase components. J. Bacteriol. 177:6610-6618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hurtubise, Y., D. Barriault, and M. Sylvestre. 1996. Characterization of active recombinant his-tagged oxygenase component of Comamonas testosteroni B-356 biphenyl dioxygenase. J. Biol. Chem. 271:8152-8156. [DOI] [PubMed] [Google Scholar]
- 32.Hurtubise, Y., D. Barriault, and M. Sylvestre. 1998. Involvement of the terminal oxygenase beta subunit in the biphenyl dioxygenase reactivity pattern toward chlorobiphenyls. J. Bacteriol. 180:5828-5835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Imbeault, N. Y., J. B. Powlowski, C. L. Colbert, J. T. Bolin, and L. D. Eltis. 2000. Steady-state kinetic characterization and crystallization of a polychlorinated biphenyl-transforming dioxygenase. J. Biol. Chem. 275:12430-12437. [DOI] [PubMed] [Google Scholar]
- 34.Jones, T. A., J. Y. Zou, S. W. Cowan, and M. Kjeldgaard. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. Sect. A 47:110-119. [DOI] [PubMed] [Google Scholar]
- 35.Karlsson, A., J. Parales, R. Parales, D. Gibson, H. Eklund, and S. Ramaswamy. 2003. Crystal structure of naphthalene dioxygenase: side-on binding of dioxygen to iron. Science 299:1039-1042. [DOI] [PubMed] [Google Scholar]
- 36.Kimura, N., A. Nishi, M. Goto, and K. Furukawa. 1997. Functional analyses of a variety of chimeric dioxygenases constructed from two biphenyl dioxygenases that are similar structurally but different functionally. J. Bacteriol. 179:3936-3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kumamaru, T., H. Suenaga, M. Mitsuoka, T. Watanabe, and K. Furukawa. 1998. Enhanced degradation of polychlorinated biphenyls by directed evolution of biphenyl dioxygenase. Nat. Biotechnol. 16:663-666. [DOI] [PubMed] [Google Scholar]
- 38.Laemmli, U. 1970. Cleavage of structural proteins during the assembly of the head of the bacteriophage T4. Nature 227:680-685. [DOI] [PubMed] [Google Scholar]
- 39.Lamzin, V. S., A. Perrakis, and K. S. Wilson. 2001. The ARP/WARP suite for automated construction and refinement of protein models, p. 720-722. In M. G. Rossmann and E. Arnold (ed.), International tables for crystallography, vol. F. Crystallography of biological macromolecules. Kluwer Academic Publishers, Dordrecht, The Netherlands. [Google Scholar]
- 40.Massé, R., F. Messier, C. Ayotte, M.-F. Lévesque, and M. Sylvestre. 1989. A comprehensive gas chromatographic/mass spectrometric analysis of 4-chlorobiphenyl bacterial degradation products. Biomed. Environ. Mass Spectrom. 18:27-47. [Google Scholar]
- 41.Master, E. 2002. Polychlorinated biphenyl (PCB) metabolism in psychrotolerant and mesophilic bacteria. University of British Columbia, Vancouver, Canada.
- 42.Miroux, B., and J. Walker. 1996. Over-production of proteins in Escherichia coli: mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J. Mol. Biol. 260:289-298. [DOI] [PubMed] [Google Scholar]
- 43.Mondello, F. J. 1989. Cloning and expression in Escherichia coli of Pseudomonas strain LB400 genes encoding polychlorinated biphenyl degradation. J. Bacteriol. 171:1725-1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mondello, F. J., M. P. Turcich, J. H. Lobos, and B. D. Erickson. 1997. Identification and modification of biphenyl dioxygenase sequences that determine the specificity of polychlorinated biphenyl degradation. Appl. Environ. Microbiol. 63:3096-3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Murshudov, G. N., A. A. Vagin, and E. J. Dodson. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. Sect. D 53:240-255. [DOI] [PubMed] [Google Scholar]
- 46.Pieper, D. H. 2005. Aerobic degradation of polychlorinated biphenyls. Appl. Microbiol. Biotechnol. 67:170-191. [DOI] [PubMed] [Google Scholar]
- 47.Raag, R., and T. L. Poulos. 1991. Crystal structures of cytochrome P-450CAM complexed with camphane, thiocamphor, and adamantane: factors controlling P-450 substrate hydroxylation. Biochemistry 30:2674-2684. [DOI] [PubMed] [Google Scholar]
- 48.Raschke H., M. Meier, J. G. Burken, R. Hany, M. D. Muller, J. R. Van Der Meer, and H. P. Kohler. 2001. Biotransformation of various substituted aromatic compounds to chiral dihydrodihydroxy derivatives. Appl. Environ. Microbiol. 67:3333-3339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY
- 50.Seeger, M., B. Camara, and B. Hofer. 2001. Dehalogenation, denitration, dehydroxylation, and angular attack on substituted biphenyls and related compounds by a biphenyl dioxygenase. J. Bacteriol. 183:3548-3555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Seeger, M., K. N. Timmis, and B. Hofer. 1995. Degradation of chlorobiphenyls catalyzed by the bph-encoded biphenyl-2,3-dioxygenase and biphenyl-2,3-dihydrodiol-2,3-dehydrogenase of Pseudomonas sp. LB400. FEMS Microbiol. Lett. 133:259-264. [DOI] [PubMed] [Google Scholar]
- 52.Seeger, M., M. Zielinski, K. N. Timmis, and B. Hofer. 1999. Regiospecificity of dioxygenation of di- to pentachlorobiphenyls and their degradation to chlorobenzoates by the bph-encoded catabolic pathway of Burkholderia sp. strain LB400. Appl. Environ. Microbiol. 65:3614-3621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Studier, F. W., A. H. Rosenberg, J. J. Dunn, and J. W. Dubendorff. 1990. Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol. 185:60-89. [DOI] [PubMed] [Google Scholar]
- 54.Suenaga, H., T. Watanabe, M. Sato, Ngadiman, and K. Furukawa. 2002. Alteration of regiospecificity in biphenyl dioxygenase by active-site engineering. J. Bacteriol. 184:3682-3688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Taira, K., J. Hirose, S. Hayashida, and K. Furukawa. 1992. Analysis of bph operon from the polychlorinated biphenyl-degrading strain of Pseudomonas pseudoalcaligenes KF707. J. Biol. Chem. 267:4844-4853. [PubMed] [Google Scholar]
- 56.Tartoff, K. D., and C. A. Hobbs. 1987. Improved media for growing plasmid and cosmid clones. Bethesda Res. Lab. Focus 9:12-14. [Google Scholar]
- 57.Vagin, A., and A. Teplyakov. 1997. MOLREP: an automated program for molecular replacement. J. Appl. Crystallogr. 30:1022-1025. [Google Scholar]
- 58.Vaillancourt, F. H., S. Han, P. D. Fortin, J. T. Bolin, and L. D. Eltis. 1998. Molecular basis for the stabilization and inhibition of 2,3-dihydroxybiphenyl 1,2-dioxygenase by t-butanol. J. Biol. Chem. 273:34887-34895. [DOI] [PubMed] [Google Scholar]
- 59.Vézina, J., D. Barriault, and M. Sylvestre. Diversity of the C-terminal portion of the biphenyl dioxygenase large subunit. J. Mol. Microbiol. Biotechnol., in press. [DOI] [PubMed]
- 60.Zielinski, M., S. Kahl, H. J. Hecht, and B. Hofer. 2003. Pinpointing biphenyl dioxygenase residues that are crucial for substrate interaction. J. Bacteriol. 185:6976-6980. [DOI] [PMC free article] [PubMed] [Google Scholar]