

Abstract
The genome sequences of several pseudomonads have revealed a gene cluster containing genes for a two-component heavy metal histidine sensor kinase and response regulator upstream of cinA and cinQ, which we show herein to encode a copper-containing azurin-like protein and a pre-Q0 reductase, respectively. In the presence of copper, Pseudomonas putida KT2440 produces the CinA and CinQ proteins from a bicistronic mRNA. UV-visible spectra of CinA show features at 439, 581, and 719 nm, which is typical of the plastocyanin family of proteins. The redox potential of the protein was shown to be 456 ± 4 mV by voltametric titrations. Surprisingly, CinQ is a pyridine nucleotide-dependent nitrile oxidoreductase that catalyzes the conversion of pre-Q0 to pre-Q1 in the nucleoside queuosine biosynthetic pathway. Gene disruptions of cinA and cinQ did not lead to a significant increase in the copper sensitivity of P. putida KT2440 under the conditions tested. Possible roles of CinA and CinQ to help pseudomonads adapt and survive under prolonged copper stress are discussed.
Copper is required as a cofactor for many enzymes, but in excess, it can have deleterious effects through metal-catalyzed protein oxidation and the generation of reactive oxygen species. Several mechanisms to handle and regulate intracellular levels of copper have been identified in bacteria (5, 10, 12, 31). In Escherichia coli, CopA is a P-type ATPase (8, 30) involved in ATP-dependent translocation of Cu(I) and Ag(I) from the cytoplasm to the periplasm, while CueO (12) is a multicopper oxidase that oxidizes Cu(I) to the less toxic Cu(II) in the periplasm. Both of the relevant genes, copA and cueO, are regulated by the MerR family-like activator CueR. In addition, E. coli contains the multicomponent Cus complex, which is responsible for expulsion of Cu(I) and Ag(I) from the periplasm across the outer membrane. CusA is localized in the cytoplasmic membrane; the essential energy-providing, proton-driven transporter CusB is also essential and belongs to the family of membrane fusion proteins; and CusC is an outer membrane factor. Another accessory protein, CusF, is believed to be a periplasmic copper chaperon delivering Cu(I) to the CusABC complex. In other prokaryotes, additional, often plasmid-encoded mechanisms have been shown to be involved in copper homeostasis. For Pseudomonas syringae copABCDRS (5) and its E. coli homolog, the plasmid-carried operon pcoABCDRSE (3), CopA is a multicopper oxidase that is thought to oxidize Cu(I) to Cu(II) in the periplasm, CopB is an outer membrane protein, CopC is a periplasmic protein, and CopD is probably an inner membrane protein.
Pseudomonas putida is a ubiquitous saprophytic bacterium that can be found in bulk soils, contaminated soils, waters, and the rhizosphere and has been studied extensively for its ability to metabolize aromatic compounds (37). Genomic analysis revealed that P. putida is equipped with a variety of genes involved in metal homeostasis, including two copA (a gene homologous to those encoding the multicopper oxidase CopA from P. syringae pv. syringae and PcoA from E. coli) and two copB (a gene homologous to that encoding the P. syringae pv. syringae CopB outer membrane factor) genes, a cusCBA system similar to E. coli cusCFBA, and two genes encoding putative Cu(I)- and Ag(I)-transporting P-type ATPases (4).
Sequence comparison revealed another putative operon involved in copper homeostasis conserved in P. putida KT2440, Pseudomonas fluorescens PFO-1 and PF-05, Pseudomonas aeruginosa PAO1, and Pseudomonas chlororaphis. The cinRS operon encodes a histidine sensor kinase (cinS) and a response regulator (cinR) in the opposite direction of cinAQ (copper induced). Sequence analysis has annotated cinA as encoding a putative azurin-plastocyanin-like protein and cinQ as encoding a putative GTP cyclohydrolase/pre-Q0 reductase (NCBI accession numbers NP_744308 and NP_744309, respectively). Known members of the azurin-plastocyanin family from bacteria are electron shuttles located in the periplasm.
CinA is similar to the P. fluorescens DF57 Cot protein, which is involved in copper tolerance (38). In addition, the P. aeruginosa homolog, cinA (PA2806), is also transcribed in copper-exposed cells, and its disruption leads to a slight reduction in copper tolerance (12-mm inhibition radius in a disk assay, compared to 8 mm for the wild type) (36).
A homolog of GTP cyclohydrolase I has been shown to catalyze the NADPH-dependent reduction of 7-cyano-7-deazaguanine (pre-Q0) to 7-aminomethyl-7-deazaguanine (pre-Q1) (39). The reductase in Bacillus, QueF, was found to be encoded in a cluster of genes that are thought to be involved in the biosynthesis of the hypermodified tRNA base queuosine (29).
In this paper, we report the analysis of the cinAQ operon in P. putida KT2440. The studies show that CinA is a copper-containing protein with a high redox potential and that CinQ catalyzes the conversion of pre-Q0 to pre-Q1 (Fig. 1). Disruption strains where cinA or cinQ was interrupted were examined to determine the role(s) of the encoded proteins in copper homeostasis.
FIG. 1.

Reaction catalyzed by CinQ. CinQ catalyzes the reduction of pre-Q0 to pre-Q1. The final product of the pathway is queuosine (Q-tRNA).
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The bacterial strains and plasmids used for this study are listed in Table 1. P. putida strain KT2440 and all derived strains were grown either in Luria-Bertani broth (LB; EMD), on LB agar (Difco), or in minimal salts medium (MM) (22) with 5 mM benzoate and 1% agar (Bacto agar; Difco) at 27°C. E. coli strains were grown in LB at 37°C. P. aeruginosa PAO1 and its derivative transposon mutants PA2806::ISphoA/hah, PA2807::ISlacZ/hah, PA2807::ISphoA/hah, PA2809::ISlacZ/hah, and PA2810::ISphoA/hah, obtained from the University of Washington Genome Center, were grown in LB at 37°C. E. coli DH5α was used to maintain engineered constructs. E. coli BL21(pLys) (Stratagene) was used for overexpression of recombinant proteins. For conjugative gene transfer, overnight cultures of donor strain E. coli S17-1 and recipient strain P. putida KT2440 were grown at 27°C in LB, mixed (3:1 and 1:3), and plated onto sterilized filters on LB agar plates. After overnight incubation, cells were suspended in saline solution (0.85% NaCl), diluted, and plated onto selective medium.
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Description or relevant phenotype | Reference or source |
|---|---|---|
| Strains | ||
| E. coli strains | ||
| DH5α | F2 end-1 hsdR17 supE44 thi-1 recA1 deoR gyrA96 relA1 Δ(argF-lacZYA)U169 | New England Biolabs |
| S-17 | thi pro hsdR hsdM1 recA tra-1 | 34 |
| BL21 | DE3(pLys) [E. coli B F−dcm ompT hsdSB(rB− mB−) gal λ(DE3) (pLysS Camr)] | Stratagene, Novagen |
| P. putida strains | ||
| KT2440 | Wild type | ATCC 47054 |
| KTΔcinA | KT2440 with deletion of cinA | This work |
| KTcinQ::kan | KT2440 with gene disruption of cinQ::kan | This work |
| P. aeruginosa strains | ||
| PAO1 | Wild type | University of Washington Genome Center |
| PA2806::ISphoA/hah | PAO with gene disruption of cinQ::Tetr | University of Washington Genome Center |
| PA2807::ISlacZ/hah | PAO with gene disruption of cinA::Tetr | University of Washington Genome Center |
| PA2807::ISphoA/hah | PAO with gene disruption of cinA::Tetr | University of Washington Genome Center |
| PA2809::ISlacZ/hah | PAO with gene disruption of cinR::Tetr | University of Washington Genome Center |
| PA2810::ISphoA/hah | PAO with gene disruption of cinS::Tetr | University of Washington Genome Center |
| Plasmids | ||
| PASK-IBA3 | Overexpression vector (Strep-TagII); Ampr | IBA, GmbH |
| pGEM-T-Easy | Cloning vector; Ampr | Promega, Madison, WI |
| pLO2 | Suicide vector; sacB Kanr | 18 |
| pET29 | Overexpression vector (S tag); Kanr | Novagen |
| pLO2NCinQ | Contains nt 45 to 786 of cinQ | This work |
| pLO2KTPCR4 | Contains deletion of cinA | This work |
| pASKP-12 | CinA in pASK-IBA3 with C-terminal Strep-TagII | This work |
| pASK-X4 | CinQ in pASK-IBA3 with C-terminal Strep-TagII | This work |
Antibiotics (kanamycin [25 mg/liter for E. coli and 50 mg/liter for P. putida], ampicillin [100 mg/liter], and chloramphenicol [25 mg/liter]) were added when appropriate.
Genetic techniques.
Standard genetic techniques were used (33). Whole genomic DNA was extracted according to the method of Marmur (21). DNAs were amplified by PCR, using Taq polymerase (Fermentas). P. putida KTΔcinA (P. putida KT2440 with an in-frame deletion of cinA) was constructed as follows. A 900-bp region upstream of the start codon and containing the first codons of the gene and a 500-bp region downstream of cinA were amplified using primers KTko1 and KTko2tag (PCR 1) and primers KTko3tag and KTko4 (PCR 2), respectively. These two flanking DNA fragments were joined together by overlap extension PCR (OE-PCR). For OE-PCR, 1 μl each of the PCR 1 and PCR 2 products was used as a template in a PCR mixture containing a 0.2 mM concentration of each deoxynucleoside triphosphate, 2.25 mM of MgCl2, 75 mM Tris-HCl (pH 8.8), 20 mM (NH4)2SO4, and 2.5 U of Taq polymerase. The PCR program consisted of 55 cycles of 94°C for 30 s, 56°C for 30 s, and 72°C for 30 s. The gene assembly reaction mixture (1 μl) was diluted in 50 μl of a PCR mixture containing a 0.2 mM concentration of each deoxynucleoside triphosphate, 2.25 mM of MgCl2, 75 mM Tris-HCl (pH 8.8), 20 mM (NH4)2SO4, and primers KTko1 and KTko4 and then amplified. The resulting PCR product (KTPCR4), containing a deletion of cinA, was first cloned into pGEM-T Easy (Promega) according to the manufacturer's protocol, and this construct was then digested using the SacI restriction enzyme and ligated into pLO2 (18) to obtain pLO2KTPCR4. Plasmid pLO2 contains a kanamycin cassette and sacB.
A gene deletion was obtained by transfer of pLO2KTPCR4 from E. coli S17-1 to P. putida KT2440 via parental mating. Transformants were first selected on MM with 5 mM benzoate and 50 μg/liter of kanamycin (single recombination event) and then counterselected on LB containing 10% sucrose to screen for the loss of sacB (double recombination event). Gene deletion was confirmed by a PCR using primers (KTDPOD and KTDXUU) external to the region used for the OE-PCR.
Strain KTcinQ::kan, containing a gene disruption of cinQ, was constructed as follows. The central part of the gene was amplified (using primers KTDXU and KTDXD) and cloned into plasmid pLO2, using the MluI and SacI restriction sites, generating plasmid pLO2NCinQ. Gene disruption was obtained after parental mating with E. coli S17-1(pLO2NCinQ) and selection of transformants growing on MM with 5 mM benzoate and 50 μg/liter of kanamycin. Gene disruption was confirmed by Southern blotting.
Construction of expression vectors.
To construct the cinA expression vector pASK-P12, cinA was amplified from the genomic DNA and cloned into the EcoRI and SalI restriction sites of pASK-IBA3 (IBA). Plasmid pASK-P12 expressed CinA as a C-terminal fusion protein with Strep-TagII (amino acid sequence, VDLQGDHGLSAWSHPQFEK), with an MGDRGPEL peptide at the N terminus.
To construct the cinQ expression vector pASK-X4, cinQ was amplified from the genomic DNA and cloned into the EcoRI and SalI restriction sites of pASK-IBA3. Plasmid pASK-X4 expressed CinQ as a C-terminal fusion protein with Strep-TagII (amino acid sequence, VDLQGDHGLSAWSHPQFEK), with an MGDRGPEL peptide at the N terminus.
The gene encoding QueF was amplified by PCR from B. subtilis genomic DNA (ATCC 23857), using forward primer VBD136 and reverse primer VBD137, with an Epicenter Technologies fail-safe PCR kit. The resulting fragment was cloned between the NdeI and HindIII sites of pET29 for expression of the native recombinant QueF protein. Constructs were introduced by electroporation into E. coli BL21(DE3) for protein expression (Novagen).
Protein expression and purification.
CinA and CinQ were purified using Strep-TagII technology. For protein overexpression, pASK-P12 and pASK-X4 were always transformed fresh into E. coli BL21(pLys) (Stratagene).
Cells were cultivated overnight at 37°C in LB with ampicillin and chloramphenicol, diluted 1:50 into 0.5 liter of fresh medium, and cultivated with shaking at 37°C to an optical density at 600 nm (OD600) of 0.5. Overexpression of recombinant protein was induced by the addition of anhydrotetracycline to a final concentration of 200 μg/liter, and incubation was continued for 3 to 5 h at 30°C. Cells were harvested by centrifugation at 5,500 × g for 12 min at 4°C. CinA was extracted from the periplasm by osmotic shock (12, 27). Briefly, cell pellets were suspended in buffer P (100 mM Tris-HCl, pH 8, 500 mM sucrose), kept on ice for 30 min, and then centrifuged at 15,000 × g for 15 min at 4°C. CinQ was extracted from the cytoplasm. The pellet was resuspended in buffer W (100 mM Tris-HCl, pH 8) containing DNase I (10 mg/liter) and a proteinase inhibitor cocktail (Sigma), lysed with a French press (1,260 lb/in2, cell type 20K; Amicon, SLM Instruments, Inc., Rochester, NY), and then centrifuged at 15,000 × g for 15 min at 4°C.
In both cases, after centrifugation (15,000 × g, 15 min, 4°C) the supernatant was applied to a streptactin-agarose column (1 ml/400 ml of culture) equilibrated with buffer W. The column was washed with buffer W, and the protein was then eluted in aliquots of 2.5 ml of buffer E (buffer W containing 2.5 mM desthiobiotin).
E. coli BL21(DE3) transformants containing the QueF expression plasmid were plated on an LB agarose plate containing 34 μg/ml kanamycin and incubated at 37°C. After 16 h, one colony from the plate was used to inoculate 50 ml LB containing 34 μg/ml kanamycin. The culture was allowed to grow overnight at 37°C. To inoculate cultures for protein expression, 10 ml of the overnight culture was added to flasks containing 1 liter of LB and 34 μg/ml kanamycin. Cells were grown to an OD600 of ∼0.5, and protein expression was induced by adding IPTG (isopropyl-β-d-thiogalactopyranoside) to a final concentration of 0.1 mM. After 6 h of growth at 37°C, cells were collected by centrifugation (20,500 × g) and frozen in liquid N2.
For protein purification, a cell paste (3 g) was suspended in a solution containing 0.05 M Tris-HCl (pH 8.0), 2 mM dithiothreitol (DTT), and 1 mM phenylmethylsulfonyl fluoride. Cell walls were disrupted using a Branson 450D sonifier at 50% power. Cell suspensions were then centrifuged at 26,500 × g for 30 min. The soluble extract was loaded on an 80-ml (19.3 × 2.3 cm) column of Q-Sepharose that had been equilibrated with buffer containing 0.05 M Tris-HCl (pH 8.0) and 2 mM DTT. Protein was eluted using a linear gradient from 0 to 0.5 M KCl in a total volume of 0.9 liter. Fractions containing QueF were identified by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), pooled, and concentrated in an Amicon pressure cell (YM-10 membrane). The resulting protein was desalted on a Sephacryl S-300 size-exclusion column (Amersham) which had been equilibrated with a buffer containing 0.05 M Tris-HCl (pH 8.0) and 2 mM DTT. Fractions containing QueF were identified by gel electrophoresis, pooled, and concentrated as described above. The protein was quantified by a BCA assay (Pierce), with bovine serum albumin as the standard.
RNA-DNA hybridization.
Regulation of transcription was examined by Northern blot analysis. P. putida KT2440 cells were grown on MM containing 5 mM benzoate to mid-log phase at 27°C, at which point either no metal (control), NiCl2, CuCl2, FeSO4 (with 10 mM ascorbic acid), or ZnCl2 was added to a final concentration of 100 μM. Incubation was continued with shaking at 27°C for 30 min, after which cells were harvested and total RNA was extracted using a RNeasy Plant Mini kit (QIAGEN).
Equal amounts of total RNA were loaded in a 1.2% agarose-formaldehyde gel and electrophoresed for 3 h at 70 V/cm. RNAs were transferred to a nylon membrane by using a vacuum blotter (Boekel Appligen) and then hybridized overnight with 32P-labeled probes at 42°C. 32P was purchased from Perkin-Elmer. The probes were prepared using the RadPrime DNA labeling system (Invitrogen). Probes for cinA and cinQ were obtained by PCR with the KTDPU-KTDPD and KTDXU-KTDXD primers, respectively, using P. putida KT2440 genomic DNA as the template.
RT-PCR analyses.
Total RNA was isolated from CuCl2-induced P. putida KT2440 as described above. The RNA sample was then digested with 1 U of DNase I (Sigma) per microgram of RNA for 2 h at 37°C. RNAs were then purified by phenol-chloroform extraction and resuspended in diethyl pyrocarbonate-treated water. Reverse transcription-PCR (RT-PCR) was performed using Superscript II reverse transcriptase (GIBCO BRL, Cambridge, MA) at 50°C for 1 h with primer RT-PX3′A (Table 2). The cDNA was PCR amplified using primers RT-PX3′A and RT-PX5′S and Taq DNA polymerase (Fermentas) for 30 cycles with an annealing temperature of 63°C and an extension time of 30 seconds. RT-PCR products were analyzed by electrophoresis on a 2% agarose gel stained with ethidium bromide.
TABLE 2.
Primers used in this study
| Primer | Sequence (5′-3′)a | Use | Restriction site |
|---|---|---|---|
| KTDPU | AAAACGCGTCATTACCGCCACCCTTGCCCTG | NBAb | MluI |
| KTDPD | AAAGAGCTCCTGGTAATGCCCCGGCACGTT | NBA | SacI |
| KTDXU | AAAACGCGTATACATCGCCACCTACTCCCG | Disruption of cinQ/NBA | MluI |
| KTDXD | AAAGAGCTCGCTGCGATACGGGTTGATGTCC | Disruption of cinQ/NBA | SacI |
| KTko1 | AAAGAGCTCAGCTTGTTGGGGTGCGGGTCG | cinA deletion | SacI |
| KTko2tag | cccatccactaaatttaaataGGTGGCGGTAATGAACAG | cinA deletion | |
| KTko3tag | tatttaaatttagtggatgggATTGAGTGACTGGCTCTG | cinA deletion | |
| KTko4 | GTGGAGCTCTTCTTCCACCACCCGCTCC | cinA deletion | SacI |
| KTNCinQ3′ | AGGGTCGACCTGCCGGACCAGCCGTTTGTTATCC | cinQ in pASK-IBA3 | SalI |
| KTNCinQ5′ | GTCCAATTGCATCCCGCTGCCGAACATTCC | cinQ in pASK-IBA3 | MunI |
| KTcinA5′ | GTCCAATTGAAACACCTGTTCATTACCGCCAC | cinA in pASK-IBA3 | MunI |
| KTcinA3′ | AGCGTCGACCTCATTGGTCAATGCCCCG | cinA in pASK-IBA3 | SalI |
| KTDPOD | AGGGAATTCATCGTGTCCTGGCTCGGGC | Verification of cinA deletion | EcoRI |
| KTDXUU | CCTGAGCTCCGGCTTGTGGACGGTAGGC | Verification of cinA deletion | SacI |
| RT-PX5′S | GCGCGCCGAACTGACCTGGAC | RT 5′ sense | |
| RT-PX3′A | TGCTCCGGGGAGTAGGTGGCG | RT 3′ antisense | |
| VBD136 | AAAGGAAGATGCATATGACGACAAGAAAAGAATCAG | queF in pET29 | NdeI |
| VBD137 | CACACAACGGAAAGCTTATTAACGATTATCAATTGTC | queF in pET29 | HindIII |
Lowercase indicates DNA tag used for overlap extension PCR.
NBA, Northern blot analysis.
Disk sensitivity assay.
P. putida KT2440, KTΔcinA, and KTcinQ::kan were grown overnight at 27°C. P. aeruginosa PAO1 and its derivative transposon mutants PA2806::ISphoA/hah, PA2807::ISlacZ/hah, PA2807::ISphoA/hah, PA2809::ISlacZ/hah, and PA2810::ISphoA/hah were grown, and 100 μl of cell suspension was plated on an LB agar plate. Ten microliters of 1 M CuSO4 or 1 M CuCl2 was added to each filter disk. Disks were air dried and placed at the center of the agar plates. Plates were incubated at room temperature for 24 (P. aeruginosa) or 48 (P. putida) hours, and the zones of clearance were measured.
Spectroscopic and spectroelectrochemical analysis of CinA.
UV-visible spectra of CinA were obtained with a Varian Cary 300 spectrophotometer. Before the spectrum was obtained, CinA was first loaded with 20 μM of CuCl2, oxidized with one crystal of ferricyanide, and then dialyzed in buffer W.
The redox potential was investigated by spectroelectrochemical techniques as described by Ding et al. (6), carried out using the same optically transparent thin-layer electrode cell (OTTLE) and the same Ag/AgCl reference electrode (−205 mV versus a standard hydrogen electrode [SHE]; Bioanalytical Systems, Inc.) (6).
A 150 μM solution of CinA was prepared in 0.1 M sodium phosphate buffer (pH 7.5). The solution was degassed for 24 h at 4°C by dialysis against argon-bubbled buffer. After the addition of 0.2 mM of the electrochemical mediators ferrocene 1,1-dicarboxylic acid (Aldrich) (E0′ = 644 mV versus SHE) and hexaammineruthenium(II) chloride (Aldrich) (E0′ = 51 mV versus SHE), the sample was loaded into the OTTLE cell under argon. Electrochemical titrations were carried out by setting the potential at a chosen value, using a computer-controlled BAS CV-50W voltametric analyzer, for a period of 5 to 10 min until the optical spectrum did not change significantly with time. An optical spectrum was then recorded, the potential was changed in steps of 20 to 50 mV, and the process was repeated at the new applied potential. The temperature of the chamber was 27 ± 1°C. The data were then fit to the Nernst equation, using the nonlinear least-squares fitting algorithm of Origin software. The Nernst equation describes the effect of applied potential on the ratio of the concentrations of oxidized and reduced forms of the complex and on the standard reduction potential, E0, as follows: Eapp = E0 + 2.303(RT/nF)·log10([Ox]/[Red]), where Eapp is the applied potential, E0 is the reduction potential determined from these data, [Ox] and [Red] are the concentrations of the protein in the oxidized and reduced states, respectively, R is the universal gas constant, T is the temperature in kelvin units, n is the number of electrons transferred in half reaction, and F is the Faraday constant. [Ox] and [Red] were calculated from the optical spectra by using Beer's law.
Enzymatic characterization of CinQ. (i) Synthesis of pre-Q0.
Methyl formate (3.1 ml) and chloroacetaldehyde (3.2 ml) were added to anhydrous toluene (25 ml) at 0°C. Sodium methoxide (2.9 g) was added slowly over the course of 5 min. The reaction was allowed to proceed for 2 h. The precipitate was collected and washed with 50 ml toluene and 25 ml anhydrous ethyl ether, yielding an off-white powder of chloro(formyl)acetonitrile. The product was placed in 10 ml water containing 2.9 ml of acetic acid at <10°C. The pH of the solution was adjusted to ∼4 to 5 prior to the solution being combined with 2,4-diamino-6-hydroxypyrimidine (6.3 g) in N,N-dimethylformamide (10 ml). The mixture was stirred for 2 h at room temperature. The solution was placed at 4°C overnight. The resulting precipitate was collected and washed with 0.4 liter of water and 0.2 liter of acetone. The resulting solid was refluxed for 1 h in 50 ml water. The precipitate that formed upon cooling was collected and washed with 0.4 liter water and 0.3 liter acetone. The nuclear magnetic resonance and mass spectra were consistent with those of the desired product (23).
(ii) Assays for GTP cyclohydrolase I activity.
Assays to analyze the GTP cyclohydrolase I activity of P. putida KT2440 CinQ were carried out with the following reagents: 20 mM Tris-HCl (pH 8.0), 100 μM GTP, 1 mM MgCl2, 1 mM DTT, and 10 μM CinQ protein. Reactions were carried out at 30°C and quenched by centrifugation through YM-10 Microcon centrifugal membranes to remove CinQ at 0, 30, 60, and 120 min. A control reaction mix lacking CinQ was also prepared in parallel. An aliquot (20 μl) obtained at each time point was loaded on a Zorbax SAX high-performance liquid chromatography (HPLC; Agilent) column (4.6 × 250 mm) that had been equilibrated with H2O (solution A). The column was eluted at a flow rate of 0.75 ml/min with increasing proportions of 0.75 M NH4H2PO4-2% acetonitrile (solution B) in water. The elution program was as follows: 5 min with 99% solution A, 25 min with 95% solution A, 30 min with 30% solution B, and 35 min with 30% solution B. An aliquot (5 μl) containing 0.8 mM (each) GTP, GDP, and GMP served as a standard. UV spectra were recorded from 190 to 500 nm on an Agilent 1100 series photodiode array detector.
(iii) Assays for pre-Q0 reductase activity.
The P. putida CinQ protein (18.2 nmol) was buffer exchanged into 0.05 M HEPES (pH 7.5) by repeated concentration and dilution in a Microcon centrifugal concentrator (YM-10 membrane). The assay mixtures contained 0.05 M HEPES (pH 7.5), 0.1 mM pre-Q0, 0.1 M KCl, 1 mM NADPH, 10 mM DTT, and 10 μM CinQ protein. A positive control reaction was carried out as described above, but with recombinant B. subtilis QueF protein, which was shown previously to catalyze the conversion of pre-Q0 to pre-Q1 (37). Reactions were allowed to proceed for 60 min at 30°C and then quenched by centrifugation for 8 min at 12,000 × g through YM-10 Microcon centrifugal membranes to remove CinQ prior to being analyzed by reverse-phase HPLC as described below for liquid chromatography-mass spectrometry (LC-MS) experiments.
For LC-MS analysis of the reaction, an aliquot (20 μl) of each reaction mix was injected onto a 4.6- by 250-mm Eclipse XDB-C18 column (Agilent) which had been preequilibrated in 20 mM ammonium acetate at pH 6.0 (solution A) at a flow rate of 0.3 ml/min. The column was developed with a linear gradient of 0 to 40% methanol over 40 min, followed with a gradient of 40 to 50% methanol over 5 min. The elution was monitored by UV-visible and MS detection. UV-visible spectra were obtained from 220 to 500 nm, using a ThermoFinnigan Surveyor photodiode array detector. Mass spectra were obtained in positive mode, scanning the m/z range of 90 to 500 atomic mass units (amu), using an electrospray ionization-equipped LCQ ThermoFinnigan Deca XP mass spectrometer. The instrument was set at a 42-V ionization energy and a 300°C ion source temperature.
To fully assign the identities of the species that were observed in the course of the CinQ studies, reactions were carried out in 61% 18O-labeled water, or aliquots of the reaction mixtures (30 μl) were treated with NaBH4 and NaBD4 at a 10 mM final concentration. The borohydride reduction was allowed to proceed for 1 h at room temperature prior to LC-MS analysis.
RESULTS
cinA and cinQ are induced by copper and cotranscribed.
Metal-dependent induction of transcription of cinA and cinQ was analyzed by RNA-DNA hybridization, using RNAs isolated from cells grown in the presence of no metal, nickel, copper, ferrous iron, or zinc at 0.1 mM. The RNA samples were probed with amplified DNAs corresponding to the cinA and cinQ genes. As shown in Fig. 2, induction was observed only in the presence of copper.
FIG. 2.

Northern blot analysis of cinA and cinQ. Northern blot analysis was performed with cinA (left) and cinQ (right) from P. putida strain KT2440. RNAs were isolated from KT2440 grown on MM containing 5 mM sodium benzoate, supplemented with metal as follows: lanes 1, no metal; lanes 2, 100 μM FeSO4 and 10 mM ascorbic acid; lanes 3, 10 mM ascorbic acid; lanes 4, 100 μM NiCl2; lanes 5, 100 μM ZnCl2; and lanes 6, 100 μM CuCl2.
RT-PCR was performed to investigate if cinA and cinQ are transcribed from a bicistronic mRNA. The translational stop codon of cinA and the start codon of cinQ are separated by 68 nucleotides. Primers were designed to amplify the region between the last 100 nucleotides of cinA and the first 100 nucleotides of cinQ, spanning the intragenic space. DNase I-treated RNA from copper-induced cells was used as a template for RT. The obtained single-stranded DNA was amplified by PCR. Figure 3 shows that RT-PCR revealed a 250-bp band that contains the intragenic region between cinA and cinQ.
FIG. 3.

RT-PCR analysis of expression of cinA and cinQ in P. putida KT2440. Electrophoresis was performed on a 2% agarose gel stained with ethidium bromide. Lane bp, 100-bp ladder; lane 1, RT-PCR product from RNAs extracted from CuCl2-induced cells (after DNase I treatment), using RT 5′ sense and RT 3′ antisense primers; lane 2, negative control for RT, obtained by PCR amplification of the RT product obtained when the reverse transcriptase was omitted, to verify that no genomic contamination was present in the RNA extract; lane 3, PCR negative control obtained by omitting the genomic DNA from the PCR; lane 4, PCR positive control obtained using genomic DNA from P. putida KT2440 as the template.
We not only demonstrated that cinA and cinQ are induced by the addition of copper (Fig. 2) but also detected the intragenic region (Fig. 3). These results, along with sequence analysis of the genomic region, strongly suggest that cinA and cinQ are transcribed as a bicistronic mRNA, not as two monocistronic mRNAs, in a copper-inducible manner.
Overexpression and purification of CinA and CinQ.
CinA with a C-terminal Strep-TagII was overexpressed in E. coli BL21(pLys), extracted from the periplasm by osmotic shock, and then purified using a streptactin-agarose column. Samples from the eluates containing the overexpressed protein were analyzed in an SDS-polyacrylamide gel and stained with Coomassie brilliant blue 250 (Fig. 4).
FIG. 4.

SDS-PAGE analysis of CinA. Typical elution profiles of purified CinA are shown. Elution samples from the affinity column were loaded into a 12.5% SDS-PAGE gel and stained with Coomassie brilliant blue 250. Proteins usually eluted in the second and third fractions. Fermentas SM0661 was used as a molecular size ladder.
The estimated molecular mass of CinA containing the C-terminal Strep-TagII, after cleavage of the N-terminal signal sequence, is 19.1 kDa. The purified protein preparation showed a band corresponding to the correct molecular mass (Fig. 4) in SDS-PAGE analysis. A band at 21.8 kDa would have been expected had the signal peptide not been processed. The UV-visible spectrum of the streptavidin-purified CinA protein revealed absorbance maxima at 439 nm and 581 nm and a shoulder at 719 nm; these spectral features are typical of members of the azurin-plastocyanin family (Fig. 5). The molar extinction coefficient at 581 nm (ɛ581) was calculated to be 3,092 M−1 cm−1.
FIG. 5.

UV-visible spectrum of CinA. The UV-visible spectrum of CinA was recorded with a Varian Cary 300 UV-visible spectrophotometer at room temperature. The CinA concentration was 0.5 mg/ml in 100 mM Tris-HCl, pH 8.
CinQ with a C-terminal Strep-TagII was overexpressed in E. coli BL21(DE3)pLys and purified by affinity chromatography.
CinA is an azurin-like protein of high electrochemical potential.
The redox potential of CinA was determined by spectroeletrochemical titrations. The protein sample was placed in an OTTLE cell equipped with a Ag/AgCl reference electrode. By changing the potential applied to the OTTLE cell and analyzing the concentrations of the oxidized and reduced forms of the sample, it was possible to calculate the reduction potential of the protein, using the Nernst equation. The potential in the cell was changed using a voltametric analyzer in steps of 20 to 50 mV; spectra were recorded after each change in the potential of the system. The data were fit to the Nernst equation, yielding a redox potential for CinA of 456 ± 4 mV (Fig. 6).
FIG. 6.

Voltametric titrations for redox potential of CinA. A 150 μM CinA solution was loaded into an OTTLE cell, and spectroelectrochemical titration was carried out by changing the potential in steps of 20 to 50 mV. Spectra were recorded 10 min after each change. Ferrocene 1,1-dicarboxylic acid (0.2 mM) and 0.2 mM hexaammineruthenium(III) chloride were used as mediators. (Left) Background-subtracted (205 mV) spectra taken at different potentials (255 to 605 mV versus SHE). (Right) Plot of absorbance at 439 nm versus equilibration potential. The red line shows the nonlinear least-squares fit with the Nernst equation (n = 1, with one electron transfer center), obtained using Origin (R2 = 0.98). From the fit, the reduction potential of the protein is 456 ± 4 mV (versus SHE).
CinQ possesses queuosine reductase activity.
To determine the enzymatic role of CinQ, the purified protein was assayed initially for turnover with GTP by HPLC. The traces (not shown) did not reveal conversion of GTP to dihydroneopterin triphosphate, as would have been expected. However, some GTP was converted to GDP in the presence of CinQ, to a greater extent than that in buffer solution alone (data not shown). Since PSI-BLAST analysis of the protein relative to the protein database had also revealed similarity to a recently discovered class of proteins that are involved in the biosynthesis of queuosine (39) (49% similarity to Bacillus subtilis QueF), CinQ was assayed for pre-Q0 reductase activity. In these experiments, 10 μM CinQ was incubated with pre-Q0 prior to being analyzed by reverse-phase HPLC. The chromatograms show that in the time course of the reaction (1 h), there was a virtually complete loss of the peak due to the substrate, pre-Q0 (24 min) (Fig. 7A), and the appearance of two new species, eluting at 19 and 21 min (Fig. 7B). The retention time and UV-visible spectrum of the 19-min peak correlate with those for pre-Q1 synthesized enzymatically from pre-Q0 by recombinant QueF from B. subtilis (Fig. 7C). In control experiments, we showed that pre-Q1 was not observed if pre-Q0, NADPH, or the enzyme was omitted from the incubation mixtures (data not shown).
FIG. 7.

UV chromatograms revealing the conversion of pre-Q0 to pre-Q1 by P. putida CinQ. The protein sampled (10 μM) was incubated with 50 mM HEPES (pH 7.5), 100 μM pre-Q0, 100 mM KCl, 1 mM NADPH, and 10 mM DTT for 60 min at 30°C. An aliquot (20 μl) of each of the reaction mixtures was analyzed by HPLC. Trace A shows unreacted pre-Q0 from a reaction mixture lacking CinQ and QueF. Trace B shows pre-Q1 and an additional unknown product (*) from a reaction mixture containing CinQ. Trace C is a control reaction in which pre-Q0 was converted to pre-Q1 by the B. subtilis nitrile reductase, QueF.
The presence of a minor product in the CinQ incubation mixtures prompted a more careful LC-MS analysis of the reactions carried out by CinQ. Extracted ion chromatograms for the reactions are shown in Fig. 8. Pre-Q0 (42 min) and pre-Q1 (32 min) exhibited the appropriate masses, 176 and 180 amu, respectively, expected for the protonated molecular ion (M + H+) (Fig. 8A and F). The pre-Q1 standard (Fig. 8F) was obtained enzymatically as described above (Fig. 7C). When a sample of the CinQ reaction mix was treated with NaBH4, we observed a peak at 181 amu, consistent with reduction of the minor product and incorporation of two protons (Fig. 8C). The small peak with the same retention time as that of pre-Q1, which is also observed in the chromatogram of the NaBH4-treated sample, is from the isotope envelope of pre-Q1. Consistent with the NaBH4-treated samples, when an aliquot of the CinQ reaction mix was treated with NaBD4, the peak due to the minor product at 179 amu disappeared and a new peak at 182 amu appeared. This is consistent with the incorporation of one deuterium molecule and one proton into the starting material. Finally, when the reaction was carried out in the presence of 61% 18O-enriched water, both the 16O- and 18O-labeled forms of the unknown were observed in the chromatograms (Fig. 8E), at 179 and 181 amu, respectively. We infer from these results that the minor product is a 7-deazaguanine-7-aldehyde. This molecule is unlikely to be a physiologically relevant product of CinQ and was not observed in the published B. subtilis QueF studies (39), nor was it observed in the QueF reactions shown in this study. The unusual product may result from the release of the two-electron reduced pre-Q0 from the active site of the protein prior to reduction with a second equivalent of NADPH. In solution, the imine would undergo spontaneous hydrolysis to the aldehyde.
FIG. 8.

Extracted ion chromatograms for a set of six CinQ reactions. Reactions were conducted as described in Materials and Methods. The traces were obtained as follows: A, a control lacking CinQ; B, a reaction mix containing CinQ; C, the CinQ reaction treated with 10 mM NaBH4; D, the CinQ reaction treated with 10 mM NaBD4; E, a reaction mix containing CinQ run in 61% H218O; and F, a B. subtilis QueF control reaction. Constituents resulting from chemistry on the cyano group of pre-Q0 are indicated next to each representative peak. The R groups in the figure represent the 7-deazaguanine base (see Fig. 1). There is a slight inconsistency in retention times for products eluting in panels E and F, but UV spectra of the peaks (not shown) confirmed that each one represents expected compounds. The retention times in this experiment are internally consistent but different from those shown in Fig. 7 due to the presence of the MS detector, which substantially changes the backpressures that are observed, altering the retention times.
Deletion of cinA or disruption of cinQ does not significantly influence copper sensitivity.
To investigate the role of cinA and cinQ in copper homeostasis, strains where cinA or cinQ was inactivated by deletion (cinA) (Fig. 9) or insertion (cinQ) (not shown) were prepared as described in Materials and Methods.
FIG. 9.
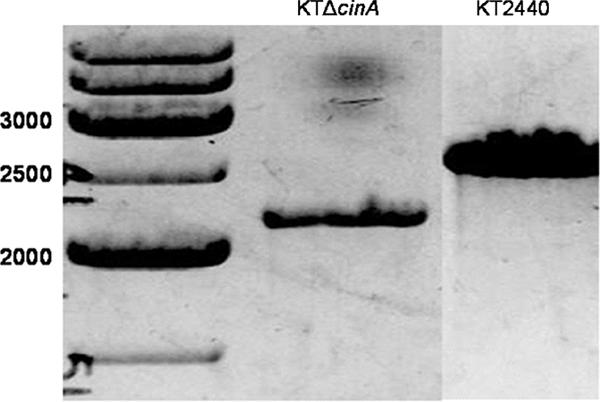
Gene deletion of cinA in strain KTΔcinA. PCR amplification of genomic DNA was done using primers KTDPOD and KTDXUU, which are external to the DNA sequence used for the gene deletion of cinA. The expected size of the PCR product from the deletion strain was 2,200 bp, while the expected size of the PCR product from KT2440 (wild type) was 2,640 bp.
The copper sensitivity of the mutated strains was investigated by exposing the strains to different concentrations of CuCl2. First, wild-type or mutant P. putida strains were streaked on LB agar plates containing different concentrations (2 to 4.6 mM) of CuCl2. P. putida KTcinQ::kan and KTΔcinA exhibited slightly smaller colony sizes than did the isogenic P. putida KT2440 wild type, both on LB agar and on LB agar containing concentrations of copper between 2.5 and 3.75 mM CuCl2. At concentrations above 4 mM CuCl2, all three strains presented very small colonies. Disk assays were performed to measure the inhibition radii with CuSO4 and CuCl2, but no significant difference was observed between P. putida KTcinQ::kan, KTΔcinA, and the wild type.
To analyze the possible roles of cinA and cinQ homologues in P. aeruginosa PAO1, disk inhibition assays were also carried out with P. aeruginosa PAO1 and strains carrying disruptions of the PA2806 (cinQ), PA2807 (cinA), PA2809 (cinR), and PA2810 (cinS) open reading frames (ORFs), obtained from the University of Washington Genome Center. The PA2807, PA2809, and PA2810 mutants presented larger inhibition radii than did the PAO1 wild type, as previously reported by Teitzel et al. (36), while the PA2806 (cinQ) mutant did not present any significant difference from the wild type in disk assays.
DISCUSSION
Sequence comparison revealed a gene cluster, characterized by a heavy metal sensor kinase gene (cinS), a response regulator gene (cinR), a gene encoding a copper protein of the plastocyanin/azurin family (cinA), and a gene encoding a pre-Q0 reductase (cinQ), conserved in several pseudomonads, including P. putida KT2440 and P. fluorescens Pf-5 and PfO-1. P. aeruginosa PAO1 contains a putative operon that is similar to the cin operon, but with an additional gene, the negative regulator of type III secretion gene ptrA (PA2808), preceding cinA (PA2807). CinA exhibits significant sequence similarity (67%) to the P. fluorescens DF57 Cot protein, which is involved in copper tolerance (38). Moreover, transcriptional analysis has shown that cinA from P. aeruginosa PAO1 (PA2807) is the most upregulated gene in copper-adapted cells (2,376-fold induction) (36). Transcriptional activation by copper was also demonstrated for PA2806, PA2809, and PA2810 (corresponding to the P. putida KT2440 cinQ, cinR, and cinS genes, respectively). Disruption of PA2807 resulted in a slight increase in copper sensitivity in disk assays, while disruption of PA2809 caused significant copper sensitivity (36). Our results showed that disruption of PA2806 (cinQ) did not result in any increase in copper sensitivity in disk assays.
In this paper, we show that cinA and cinQ from P. putida KT2440 are strongly induced by the presence of Cu2+ but not by Cd2+, Zn2+, or Fe2+. We also provide evidence that cinA and cinQ are transcribed as a bicistronic mRNA. In contrast to the analyses of copper sensitivity of a cinA disruptant in P. aeruginosa PAO1 performed by us and by Teitzel et al. (36), deletion of cinA in P. putida KT2440, although resulting in a smaller colony size for cells grown on LB, did not lead to an increase in copper sensitivity in disk assays (data not shown). Gene disruption of cinQ in either P. aeruginosa PAO1 or P. putida KT2440 did not result in an increase in copper sensitivity in disk assays (not shown). The different phenotypes in response to copper challenges for P. putida and P. aeruginosa could be the result of redundancy in copper resistance genes in P. putida. P. putida KT2440 possesses multiple genes thought to be involved in copper homeostasis that appear to be redundant. Genome analysis (4) revealed two copA genes, encoding multicopper oxidases, two copper heavy metal two-component systems, a putative cus system, and two putative copper-transporting P-type ATPases.
An indication of a possible role of CinA in conferring copper resistance comes from examining the promoter region of cinA. The promoter regions of proteins involved in copper homeostasis and resistance often contain a cop box, which is presumably the site of regulation of transcription in a copper-dependent fashion. cop boxes are found in promoters preceding P. syringae copA (24) and E. coli cusC (10) and pcoA (26). For P. aeruginosa, PA2808 (ptrA), which precedes cinA, has been reported to be induced by copper (14, 36) via a mechanism involving the two-component system cinRS (PA2809 and PA2810) and resulting in the repression of the type III secretion system (14).
cinA was upregulated by copper, and its gene product could be extracted from the periplasm after overexpression in E. coli. CinA contains a signal peptide, and CinA extracted from the periplasm had an estimated molecular mass of 19.1 kDa, corresponding to the cleaved mature peptide containing the C-terminal tag. CinA as a member of the plastocyanin/azurin family could function as an electron shuttle in the periplasm. The electrochemical potential was determined to be 456 ± 4 mV. However, members of this family of proteins usually have redox potentials that are lower than that found for CinA. For example, azurins and pseudoazurines have potentials of 260 to 310 and 260 to 275 mV, respectively, and the redox potentials of plastocyanins are ∼375 mV. These proteins are believed to be involved in electron transfer to nitrous oxide reductase, another copper-containing enzyme (pseudoazurins), or in transfer between photosystems I and II (plastocyanin). Azurins are thought to be involved in anaerobic electron transfer in P. aeruginosa and are known to exchange electrons with cytochrome c551. Rusticyanin, another member of this family, possesses a much higher redox potential (∼680 mV) and is involved in iron oxidation (for reviews, see references 9, 13, 15, and 20).
CinA contains higher percentages of methionine (8%) and histidine (6%) residues than those of azurins and pseudoazurins. A single T1 “blue copper” metal binding site is predicted to be formed by the side chains of H131, C158, H163, and M168. Furthermore, CinA presents a series of methionine- and histidine-rich motifs similar to P. syringae CopA's MXXMXHXXM motif. Methionine-rich regions can be found in many proteins involved in copper homeostasis, and it has been proposed that they might be involved in copper binding (5). Roberts et al. (32) suggested that although the methionine-rich region in CueO binds a labile Cu(II) molecule, this might not be the main role of this region, while Wernimont et al. (40) proposed that the role of the similar methionine-rich region in PcoC may be in protein recognition with PcoA.
Homologs of CinA can be found in other genomes, located in clusters containing other genes involved in copper homeostasis. Most CinA homologs are associated with a CopA multicopper oxidase, suggesting a possible role in electron transfer (Table 3). Genes encoding CinA can be found associated with copA, encoding putative multicopper oxidases; with copB, encoding putative outer membrane factors; and with two-component systems (Roseovarius nubinhibens ISM 334 and Alcanivorax borkumensis SK2) or without a two-component system (Sulfitobacter sp. strain NAS-14.1). In other cases, cinA can be found with copA and an ORF encoding a putative outer membrane protein of the TolC family (Fulvimarina pelagi HTCC2506, Agrobacterium tumefaciens, and Sinorhizobium meliloti 1021). In Hahella chejuensis KCTC 2396, cinA is found associated with a putative cus system. Finally, in Xanthomonas campestris pv. vesicatoria, cinA is associated with the multicopper oxidase, a TolC-like outer membrane protein, and a two-component heavy metal sensor kinase and response regulator. Basim et al. (1) reported that for X. campestris pv. vesicatoria, disruption of cinA (ORF5) resulted in a partial loss of copper resistance, while disruption of the sensor kinase gene (ORF1), a multicopper oxidase gene (ORF4), or the TolC family outer membrane factor gene (ORF3) had a much greater effect. These results suggest an as yet undefined accessory role for CinA homologs in copper homeostasis. However, the finding that CinA homologs are most often associated with a CopA multicopper oxidase suggests a possible role in electron transfer.
TABLE 3.
CinA homologues in selected genomes
| Bacterium | ORFa | NCBI accession no. |
|---|---|---|
| Roseovarius nubinhibens | CinA | ZP_00958990 |
| ISM 334 | CopA | ZP_00958992.1 |
| CopB | ZP_00958993.1 | |
| CopR | ZP_00958995.1 | |
| CopS | ZP_00958994.1 | |
| Alcanivorax borkumensis | CinA | YP_693084 |
| SK2 | CopA | YP_693083.1 |
| CopB | YP_693082.1 | |
| CopR | YP_693085.1 | |
| CopS | YP_693086.1 | |
| Sulfitobacter sp. strain | CinA | ZP_00964732 |
| NAS-14.1 | CopA | ZP_00964730.1 |
| CopB | ZP_00964731.1 | |
| ORF4 | ZP_00964733.1 | |
| CinA | ZP_00963604 | |
| CopA | ZP_00963605.1 | |
| TolC | ZP_00963606.1 | |
| Fulvimarina pelagi | CinA | ZP_01440306 |
| HTCC2506 | CopA | ZP_01440305.1 |
| TolC | ZP_01440304.1 | |
| CinA | ZP_01440770 | |
| CopA | ZP_01440771.1 | |
| TolC | ZP_01440772.1 | |
| Agrobacterium tumefaciens | CinA | NP_534476.1 |
| strain C58 | CopA | NP_534477.1 |
| TolC | NP_534478.1 | |
| Sinorhizobium meliloti | CinA | NP_384693 |
| 1021 | CopA | NP_384692.1 |
| TolC | NP_384691 | |
| CinA | NP_435810.1 | |
| CopA | NP_435809.1 | |
| TolC | NP_435808.1 | |
| Hahella chejuensis KCTC | CinA | YP_434213 |
| 2396 | CusA | YP_434212.1 |
| CusB | YP_434211.1 | |
| CusC | YP_434210.1 | |
| Xanthomonas campestris | CinA | AAP42068.1 |
| pv. vesicatoria | CopA | AAP42069 |
| TolC | AAP42070 | |
| CopR | AAP42072 | |
| CopS | AAP42071 |
CinA homologues and adjacent ORFs potentially involved in copper homeostasis in selected genomes.
In this report, we demonstrate that contrary to its database annotation, P. putida CinQ is an NADPH-dependent pre-Q0 reductase, not a GTP cyclohydrolase I. The activity of this protein is similar to that of the B. subtilis QueF protein, whose gene is colocalized in a cluster of genes involved in the biosynthesis of the hypermodified tRNA base queuosine (29, 39). Queuosine is found in the wobble position of several tRNAs and is known to prevent the readthrough of the stop codon UAG by tRNATyr (2, 11), but it does not seem to have a major influence on the translational efficiency of tRNA (7). A lack of queuosine was also found to strongly reduce the virulence of Shigella flexneri, possibly altering the efficiency of translation of the virF-encoded transcription activator of the AraC family (7). In contrast, E. coli mutants lacking queuosine exhibit reduced survival in stationary phase when competing with the isogenic wild-type strain (28) but are otherwise indistinguishable from the wild type.
While conversion of pre-Q0 to pre-Q1 was readily observed with CinQ, analysis of the reactions revealed the formation of a second product as well. Using LC-MS analysis and carrying out the reaction in 18O-labeled water or reducing the product mixture with NaBH4 or NaBD4, the product was assigned tentatively as a 7-deazaguanine-7-aldehyde. The pyridine nucleotide-dependent reduction of pre-Q0 presumably occurs in two half-reactions, one where the nitrile is reduced to an imine and the second where the imine is reduced to the amine of pre-Q1. Escape of the imine from the active site into bulk solvent could lead to conversion to the observed aldehyde. Recent homology modeling (35) suggested that B. subtilis QueF is a homodecameric protein with active sites located at intersubunit interfaces. It is possible that the affinity tag that was used in our studies to facilitate purification of CinQ interferes with the native quaternary structure of this protein and leads to the diffusion of intermediates from the active site.
A link between copper and tRNA modification is novel. The presence of CinQ in a copper-induced system was found only for pseudomonads. CinQ is upregulated sevenfold in P. aeruginosa PAO1 in copper-adapted cells (36). Several lines of evidence prompted us to hypothesize a possible role for CinQ as a regulator of biofilm formation. Environmental stresses are known to induce the expression of resistance factors. Copper enhances the production of the exopolysaccharide (EPS) alginate in P. syringae (17), and EPSs are known to contribute to metal resistance by a mechanism involving chelation (25). Kazy et al. (16) also reported that a copper-resistant strain of P. aeruginosa was found to adsorb more copper in its EPS than a copper-sensitive strain did. Furthermore, a homolog of pre-Q0 reductase (cinQ) from P. chlororaphis PCL1391 (ippA; NCBI accession number DQ339483) was reported to induce phenazine production. Phenazines were recently shown to be involved in biofilm formation by Pseudomonas chlororaphis (aureofaciens) strain 30-84 (19). The expression of a pre-Q0 reductase gene during copper stress could therefore be linked to copper tolerance of the organism; however, a more precise link between tRNA modification and copper must await more detailed analysis of the physiological role(s) of queuosine.
Further studies regarding the roles of cinA and cinQ in biofilm formation, copper resistance, and copper accumulation in the biofilm matrix are currently in progress in our laboratory.
Acknowledgments
We thank F. Ann Walker and Robert E. Berry for electropotential measurements of CinA. Furthermore, we acknowledge Terrance Meyer, John Fitch, and Michael Cusanovich for acquisition of the UV-visible spectra and Sylvia Franke for her suggestions.
Research in the lab of V.B. is supported by a Career Award in Biomedical Sciences from the Burroughs Wellcome Fund and by NIH grant GM72623.
Footnotes
Published ahead of print on 4 May 2007.
REFERENCES
- 1.Basim, H., G. V. Minsavage, R. E. Stall, J. F. Wang, S. Shanker, and J. B. Jones. 2005. Characterization of a unique chromosomal copper resistance gene cluster from Xanthomonas campestris pv. vesicatoria. Appl. Environ. Microbiol. 71:8284-8291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bienz, M., and E. Kubli. 1981. Wild-type tRNA GTyr reads the TMV RNA stop codon, but Q base-modified tRNAQTyr does not. Nature 294:188-190. [DOI] [PubMed] [Google Scholar]
- 3.Brown, N. L., S. R. Barrett, J. Camakaris, B. T. Lee, and D. A. Rouch. 1995. Molecular genetics and transport analysis of the copper-resistance determinant (pco) from Escherichia coli plasmid pRJ1004. Mol. Microbiol. 17:1153-1166. [DOI] [PubMed] [Google Scholar]
- 4.Cánovas, D., I. Cases, and V. de Lorenzo. 2003. Heavy metal tolerance and metal homeostasis in Pseudomonas putida as revealed by complete genome analysis. Environ. Microbiol. 12:1242-1256. [DOI] [PubMed] [Google Scholar]
- 5.Cha, J. S., and D. A. Cooksey. 1991. Copper resistance in Pseudomonas syringae mediated by periplasmic and outer membrane proteins. Proc. Natl. Acad. Sci. USA 88:8915-8919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ding, X. D., A. Weichsel, J. F. Andersen, T. K. Shokhireva, C. Balfour, A. J. Pierik, B. A. Averill, W. R. Montford, and F. A. Walker. 1999. Nitric oxide binding to the ferri- and ferroheme states of nitrophorin 1, a reversible NO-binding heme protein from the saliva of the blood sucking insect, Rhodnius prolixus. J. Am. Chem. Soc. 121:128-138. [Google Scholar]
- 7.Durand, J. M., B. Dagberg, B. E. Uhlin, and G. R. Bjork. 2000. Transfer RNA modification, temperature and DNA superhelicity have a common target in the regulatory network of the virulence of Shigella flexneri: the expression of the virF gene. Mol. Microbiol. 35:924-935. [DOI] [PubMed] [Google Scholar]
- 8.Fan, B., G. Grass, C. Rensing, and B. P. Rosen. 2001. Escherichia coli CopA N-terminal Cys(X)(2)Cys motifs are not required for copper resistance or transport. Biochem. Biophys. Res. Commun. 286:414-418. [DOI] [PubMed] [Google Scholar]
- 9.Farver, O., and I. Pecht. 1991. Electron transfer in proteins: in search of preferential pathways. FASEB J. 5:2554-2559. [DOI] [PubMed] [Google Scholar]
- 10.Franke, S., G. Grass, C. Rensing, and D. H. Nies. 2003. Molecular analysis of the copper-transporting efflux system CusCFBA of Escherichia coli. J. Bacteriol. 185:3804-3812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frey, B., G. Jänel, U. Michelsen, and H. Kersten. 1989. Mutations in the Escherichia coli fnr and tgt genes: control of molybdate reductase activity and the cytochrome d complex by fnr. J. Bacteriol. 171:1524-1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grass, G., and C. Rensing. 2001. CueO is a multi-copper oxidase that confers copper tolerance in Escherichia coli. Biochem. Biophys. Res. Commun. 286:902-908. [DOI] [PubMed] [Google Scholar]
- 13.Gray, H. B., B. G. Malmstrom, and R. K. P. Williams. 2000. Copper coordination in blue proteins. J. Biol. Inorg. Chem. 5:551-559. [DOI] [PubMed] [Google Scholar]
- 14.Ha, U. H., J. Kim, H. Badrane, J. Jia, H. V. Baker, D. Wu, and S. Jin. 2004. An in vivo inducible gene of Pseudomonas aeruginosa encodes an anti-ExsA to suppress the type III secretion system. Mol. Microbiol. 54:307-320. [DOI] [PubMed] [Google Scholar]
- 15.Holwerda, R. A., S. Wherland, and H. B. Gray. 1976. Electron transfer reactions of copper proteins. Annu. Rev. Biophys. Bioeng. 5:363-396. [DOI] [PubMed] [Google Scholar]
- 16.Kazy, S. K., S. Pinaki, S. P. Singh, A. K. Sen, and S. F. D'Souza. 2002. Extracellular polysaccharides of a copper-sensitive and a copper-resistant Pseudomonas aeruginosa strain: synthesis, chemical nature and copper binding. World J. Microbiol. Biotechnol. 18:583-588. [Google Scholar]
- 17.Kidambi, S. P., G. W. Sundin, D. A. Palmer, A. M. Chakrabarty, and C. L. Bender. 1995. Copper as a signal for alginate synthesis in Pseudomonas syringae pv. syringae. Appl. Environ. Microbiol. 61:2172-2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lenz, O., E. Schwartz, J. Dernedde, T. Eitinger, and B. Friedrich. 1994. The Alcaligenes eutrophus H16 hoxX gene participates in hydrogenase regulation. J. Bacteriol. 176:4385-4393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maddula, V. S. R. K., Z. Zhang, E. A. Pierson, and L. S. Pierson III. 2006. Quorum sensing and phenazines are involved in biofilm formation by Pseudomonas chlororaphis (aureofaciens) strain 30-84. Microb. Ecol. 52:289-301. [DOI] [PubMed] [Google Scholar]
- 20.Malmstrom, B. G., and J. Leckner. 1998. The chemical biology of copper. Curr. Opin. Chem. Biol. 2:286-292. [DOI] [PubMed] [Google Scholar]
- 21.Marmur, J. 1961. A procedure for the isolation of deoxyribonucleic acid from micro-organisms. J. Mol. Biol. 3:208-218. [Google Scholar]
- 22.Mergeay, M., D. H. Nies, H. G. Schlegel, J. Gerits, P. Charles, and F. van Gijsegen. 1985. Alcaligenes eutrophus CH34 is a facultative chemolithotroph with plasmid-bound resistance to heavy metals. J. Bacteriol. 162:328-334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Migawa, M. T., J. M. Hinkley, G. C. Hoops, and L. B. Townsend. 1996. A two step synthesis of the nucleoside Q precursor 2-amino-5-cyanopyrrolo2,3-dipyrimidin-4-one (preQ 0). Synth. Commun. 26:3317-3322. [Google Scholar]
- 24.Mills, S. D., C. K. Lim, and D. A. Cooksey. 1994. Purification and characterization of CopR, a transcriptional activator protein that binds to a conserved domain (cop box) in copper-inducible promoters of Pseudomonas syringae. Mol. Gen. Genet. 244:341-351. [DOI] [PubMed] [Google Scholar]
- 25.Mittelman, M. W., and G. G. Geesey. 1985. Copper-binding characteristics of exopolymers from a freshwater-sediment bacterium. Appl. Environ. Microbiol. 49:846-851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Munson, G. P., D. L. Lam, F. W. Outten, and T. V. O'Halloran. 2000. Identification of a copper-responsive two-component system on the chromosome of Escherichia coli K-12. J. Bacteriol. 182:5864-5871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Neu, H. C., and L. A. Heppel. 1965. The release of enzymes from Escherichia coli by osmotic shock and during the formation of spheroplasts. J. Biol. Chem. 240:3685-3692. [PubMed] [Google Scholar]
- 28.Noguchi, S., Y. Nishimura, Y. Hirota, and S. Nishimura. 1982. Isolation and characterization of an Escherichia coli mutant lacking tRNA-guanine transglycosylase. Function and biosynthesis of queuosine in tRNA. J. Biol. Chem. 257:6544-6550. [PubMed] [Google Scholar]
- 29.Reader, J. S., D. Metzgar, P. Schimmel, and V. de Crécy-Lagard. 2004. Identification of four genes necessary for biosynthesis of the modified nucleoside queuosine. J. Biol. Chem. 279:6280-6285. [DOI] [PubMed] [Google Scholar]
- 30.Rensing, C., B. Fan, R. Sharma, B. Mitra, and B. P. Rosen. 2000. CopA: an Escherichia coli Cu(I)-translocating P-type ATPase. Proc. Natl. Acad. Sci. USA 97:652-656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rensing, C., and G. Grass. 2003. Escherichia coli mechanisms of copper homeostasis in a changing environment. FEMS Microbiol. Rev. 27:197-213. [DOI] [PubMed] [Google Scholar]
- 32.Roberts, S. A., G. F. Wildner, G. Grass, A. Weichsel, A. Ambrus, C. Rensing, and W. R. Montford. 2003. A labile regulatory copper ion lies near the T1 copper site in the multicopper oxidase CueO. J. Biol. Chem. 278:31958-31963. [DOI] [PubMed] [Google Scholar]
- 33.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY.
- 34.Simon, R., U. Priefer, and A. Pühler. 1983. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in gram negative bacteria. Bio/Technology 1:784-791. [Google Scholar]
- 35.Swairjo, M. A., R. R. Reddy, B. Lee, S. G. Van Lanen, S. Brown, V. de Crecy-Lagard, D. Iwata-Reuyl, and P. Schimmel. 2005. Crystallization and preliminary X-ray characterization of the nitrile reductase QueF: a queuosine-biosynthesis enzyme. Acta Crystallogr. F 61:945-948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Teitzel, G. M., A. Geddie, S. K. De Long, M. J. Kirisits, M. Whiteley, and M. R. Parsek. 2006. Survival and growth in the presence of elevated copper: transcriptional profiling of copper stressed Pseudomonas aeruginosa. J. Bacteriol. 188:7242-7256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Timmis, K. N. 2002. Pseudomonas putida: a cosmopolitan opportunist par excellence. Environ. Microbiol. 4:779-781. [DOI] [PubMed] [Google Scholar]
- 38.Tom-Petersen, A., C. Hosbond, and O. Nybroe. 2001. Identification of copper-induced genes in Pseudomonas fluorescens and use of a reporter strain to monitor bioavailable copper in soil. FEMS Microbiol. Ecol. 38:59-67. [Google Scholar]
- 39.Van Lanen, S. G., J. S. Reader, M. A. Swairjo, V. de Crecy-Larard, and B. Lee. 2005. From cyclohydrolase to oxidoreductase: discovery of nitrile reductase activity in a common fold. Proc. Natl. Acad. Sci. USA 102:4264-4269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wernimont, A. K., D. L. Huffman, L. A. Finney, B. Demeler, T. V. O'Halloran, and A. C. Rosenzweig. 2003. Crystal structure and dimerization equilibria of PcoC, a methionine-rich copper resistance protein from Escherichia coli. J. Biol. Inorg. Chem. 8:185-194. [DOI] [PubMed] [Google Scholar]