

Abstract
The filamentous cyanobacterium Anabaena sp. strain PCC 7120 can fix N2 under oxic conditions, and the activity of nitrogen fixation occurs exclusively in heterocysts, cells differentiated from vegetative cells in response to a limitation of a combined-nitrogen source in the growth medium. At the late stages of heterocyst differentiation, an envelope layer composed of two glycolipids is formed to limit the entry of oxygen so that the oxygen-sensitive nitrogenase can function. The genome of Anabaena sp. strain PCC 7120 possesses a family of 13 genes (the hstK family), all encoding proteins with a putative Ser/Thr kinase domain at their N termini and a His-kinase domain at their C termini. In this study, we showed that the double mutant D4.3 strain, in which two members of this gene family, pkn44 (all1625) and pkn30 (all3691), were both inactivated, failed to fix N2 in the presence of oxygen (Fox−). In an environment without oxygen, a low level of nitrogenase activity was detectable (Fix+). Heterocyst development in the mutant D4.3 was delayed by 24 h and arrested at a relatively early stage without the formation of the glycolipid layer (Hgl−). Only the minor species of the two heterocyst-specific glycolipids (HGLs) was missing in the mutant. We propose that DevH, a putative transcription factor, coordinates the synthesis of both HGLs, while Pkn44/Pkn30 and the previously characterized PrpJ may represent two distinct regulatory pathways involved in the synthesis of the minor HGL and the major HGL, respectively.
Anabaena sp. strain PCC 7120 is a filamentous cyanobacterium capable of oxic N2 fixation upon limitation of a combined-nitrogen source in the growth medium (39, 42). In this organism, nitrogen fixation occurs exclusively in heterocysts, cells which are differentiated from vegetative cells in response to deprivation of combined nitrogen. Heterocysts, which account for 5 to 10% of the cells along each filament, provide a microoxic environment for nitrogenase that can be irreversibly inactivated by oxygen. Three mechanisms are known to contribute to the formation of a microoxic environment in heterocysts: formation of a thick cell envelope to limit oxygen penetration (23), absence of the oxygen-producing photosystem II, and increased respiration rate (for a review, see reference 39). Heterocyst development proceeds in stages and constitutes a complex process of morphogenesis (36, 37). From the proheterocyst stage, two tightly organized layers, an outer layer of polysaccharides and an inner layer of glycolipids, are deposited outside of the cells (39). At the end of heterocyst development, polar regions of the developing cells connected to vegetative cells are reorganized, becoming narrower and elongated. Three developmentally regulated genomic rearrangements occur, consisting of the excision of elements disrupting nifD, fdxN, or hupL (5, 12-14). The nif genes are expressed, and heterocysts then become mature and functional for nitrogen fixation.
Two heterocyst envelope glycolipids (HGLs), 1-(O-α-d-glucopyranosyl)-3,25-hexacosanediol and 1-(O-α-d-glucopyranosyl)-3-keto-25-hexacosanol (8, 11, 20), have been identified. 1-(O-α-d-Glucopyranosyl)-3-keto-25-hexacosanol (the minor HGL) is less abundant than 1-(O-α-d-glucopyranosyl)-3,25-hexacosanediol (the major HGL) and migrates faster on a plate of thin-layer chromatography with the solvent system as described previously (8, 11, 20). A number of genes, some of which are clustered as the Hgl island, are related to the synthesis or deposition of HGLs (8). The gene cluster hglEAFGDCAB (hglEA is also known as hglE) and all5341 are necessary for the synthesis of HGLs (1, 8). The devBCA cluster is presumably required for glycolipid export (10, 22), and the hglK gene is necessary for the localization of glycolipids (2). Inactivation of any of these genes affects either the synthesis or the localization of both HGLs. In addition, two genes that may play regulatory functions in the formation of the HGL layer have also been identified. One is devH (16, 28), which encodes a protein similar to NtcA (17), a transcription factor required for different steps of heterocyst development; the other is prpJ, which encodes a PP2C-type membrane protein phosphatase (18). In a devH mutant, the HGL layer is not formed, and neither of the two HGLs is detected. The inactivation of prpJ also leads to the absence of the HGL layer and the loss of only the major HGL while the minor HGL is still present. These results indicate that while devH may coordinate the synthesis of both HGLs, distinct regulatory pathways may exist for the synthesis of each of the two HGLs.
Anabaena sp. strain PCC 7120 has 53 proteins containing a catalytic domain of Ser/Thr kinases (19, 24, 34). Among them, members of the HstK (for His kinase and Ser/Thr kinase) family have a catalytic domain typical of Ser/Thr kinases at the N terminus and a catalytic domain of His kinases at the C terminus (6, 26, 34, 40). These proteins are all very large in size, with 1,777 to 2,121 amino acid residues. Two cotranscribed genes encoding members of the HstK family, pkn41 and pkn42, are involved in the cellular response to iron limitation and cell growth when both iron and combined nitrogen are limiting in the growth medium (6). The function of other members of the hstK family has not yet been characterized. We have constructed mutants with each of these genes inactivated by insertion of an antibiotic resistance cassette, and these mutants displayed little or very weak phenotypes under different conditions analyzed. We have therefore made several double mutants in which two members of the hstK gene family were inactivated. In this study, we report that two genes belonging to the hstK family, pkn44 (all1625) and pkn30 (all3691), are involved in the formation of the HGL layer. A mutant with both pkn44 and pkn30 inactivated simultaneously displayed a Het+ Fix+ Fox− Hgl− phenotype and the absence of the minor HGL, 1-(O-α-d-glucopyranosyl)-3-keto-25-hexacosanol.
MATERIALS AND METHODS
Strains and growth conditions.
Anabaena sp. strain PCC 7120 was grown in BG11 medium with nitrate or ammonium, or BG110 medium without fixed nitrogen (30, 41). When required, 5 μg/ml spectinomycin, 300 μg/ml neomycin, or 5 μg/ml erythromycin was added for the selection of exconjugants in Anabaena sp. strain PCC 7120 on plates. For the selection of double mutants, we used 300 μg/ml neomycin and 5 μg/ml spectinomycin for BG11 medium on plates and 50 μg/ml neomycin and 2.5 μg/ml spectinomycin in BG11 medium in liquid cultures.
Construction of mutants and complementation.
A DNA fragment corresponding to the 5′ coding regions of pkn44 (all1625) or pkn30 (all3691) was obtained by PCR amplification from chromosomal DNA of Anabaena sp. strain PCC 7120. The primers used for PCR are listed in Table 1. For pkn30, the corresponding PCR product was cloned into the PstI/XhoI sites of the integrative vector pRL271 (38), and interrupted by insertion of the Ω element Spr Smr (27) into a BstXI site. For pkn44, the corresponding PCR fragment was first cloned into the plasmid pBluescript SK(−), interrupted by insertion of the Ω element into an AccI site, and finally transferred into the PstI/XhoI sites of pRL271. All final constructs were conjugated into Anabaena sp. strain PCC 7120, and the selection of double recombinants was done as described previously (4). To construct double mutants, the Ω element in the constructed plasmid for pkn30 inactivation in pRL271 was removed by BamHI and replaced by a neomycin resistance gene cassette (33). The final construct was conjugated into the single mutant with pkn44 inactivated, to finally obtain the double mutant strain D4.3. All mutants used in this study were checked by Southern blotting. DNA probes were labeled with digoxigenin-dUTP (DIG High Primer DNA labeling and detection starter kit II; Roche Molecular Biochemicals) or [32P]dCTP.
TABLE 1.
DNA primers
| Target gene (primer name) | Sequence (5′-3′) | Purposea |
|---|---|---|
| pkn30 | CTTCTCGAGATGAATACGTATGTAGAT | Mutant construction |
| CTTCTGCAGCTTGTCCATTTTTACCTA | ||
| pkn44 | CTTCTGCAGATGGATACTGCAAATTCC | Mutant construction |
| CTTCTCGAGGTGTACAGGTGAAACTTC | ||
| rnpB | ATAGTACCACAGAAAAATACCG | RT-PCR |
| AAGCCGGGTTCTGTTCTCTG | ||
| hglC | TTGTTGCTAACAGTCTGG | RT-PCR |
| TTCACTCATTTGCTGACT | ||
| hglD | CACTATCTGTGGACAGGT | RT-PCR |
| GATGGGAGTTTGTACCAC | ||
| hglEA | CCGTAGTCAACCAAGTTG | RT-PCR |
| TATACTGACTGGCTGCCA | ||
| hglK | TGATGATGCGATCGCTGA | RT-PCR |
| CTACGGACAACGTGGATG | ||
| devA | GGTGTTGAGTTGTCAGGT | RT-PCR |
| GTTTGGGAATCACGAGCT | ||
| devB | TGACTGCGACTACTGCTA | RT-PCR |
| ATGTCAACCACTACCTTG | ||
| devC | CAGGAGTCCAGGAGAATT | RT-PCR |
| GGTCAGCAGAACGTAGTT | ||
| devH | TGCAATCTCCATCCTCCT | RT-PCR |
| CTGTTGATTGACTCTGCT | ||
| asr5349 | AACTAAATCTAGTGAGCATGG | RT-PCR |
| AAGCATATAAAAGGTGATGGT | ||
| asr5350 | ATGGCATTCATCAAGATACAG | RT-PCR |
| AACGCTTGACTCTTGATATTG | ||
| nifD (A1) | GTATCTCTCTACGCTTGCT | PCR |
| nifD (A2) | TTACGTGAAATCGCAGCTA | PCR |
| nifD (A3) | CCGTCTTCAGTCCATCCAA | PCR |
| rnpB | CCAGTTCCGCTATCAGAGAG | qRT-PCR |
| GAGGAGAGAGTTGGTGGTAAG | ||
| nifH | CTATTAGACGACGACACCAAG | qRT-PCR |
| GCTTCTGCGGGCTTACC | ||
| asr5349 | TAGTGAGCATGGCAGAAATAAC | qRT-PCR |
| AGGCAGGAGGCGGTAAG | ||
| asr5350 | GCTACTCCTCAGTTGCCATC | qRT-PCR |
| AACGCTTGACTCTTGATATTGTG |
qRT-PCR, real-time quantitative PCR.
For the complementation of the mutant D4.3, an EcoNI-Eco47III fragment (8,141 bp) from the plasmid anc1068 containing pkn30 with its own promoter region or an NruI-NheI fragment (7,047 bp) from the plasmid anp01211 (both anc1068 and anp01211 were kindly provided by C. Peter Wolk) containing pkn44 with its own promoter region was blunted and first cloned into the SmaI site of pBluescript SK(−) and then transferred into the PstI/SpeI sites of pRL271. The final construct was conjugated into the mutant D4.3 strain, and erythromycin-resistant colonies, with a single event of recombination between the plasmid and the chromosome, were selected to finally obtain complemented strains.
Nitrogenase assays and thin-layer chromatography.
Assays of nitrogenase activity were performed with the acetylene reduction method by gas chromatography as described previously (31). For oxic conditions, the nitrogenase activity was measured in the presence of air. For microoxic conditions, air was replaced by argon, and the filaments were kept in the presence of 3-(3′,4′-dichlorphenyl)-1,1-dimethylurea (DCMU) at a final concentration of 10 μM to block oxygen emission from photosystem II (31). Glycolipids were extracted and analyzed by thin-layer chromatography as described previously (18).
Light and electron microscopy.
An optical microscope (Nikon E400) was used to observe filaments and heterocysts of Anabaena sp. strain PCC 7120. In some cases, Alcian blue staining was used to color the polysaccharide layer of heterocysts (21). Samples for electron microscopic observation were prepared as reported previously (2).
Western blotting.
To detect the presence of NifH by immunoblotting, an antibody raised against NifH of Anabaena variabilis was used (29). Cells were broken with glass beads, and total proteins were extracted as described previously (43). Protein extracts were separated by electrophoresis in a 15% sodium dodecyl sulfate-polyacrylamide gel, transferred onto a nitrocellulose membrane, and incubated with antibodies as described previously (43). The primary and secondary antibodies were diluted 1,000- and 3,000-fold, respectively. The detection of the antibody-antigen complex was performed with an ECL kit from Amersham Biosciences.
PCR, reverse transcription-PCR (RT-PCR), and real-time quantitative PCR.
For the detection of DNA rearrangement of the 11-kb nifD element interrupting the nifD gene (13, 14), three PCR primers were designed (Table 1). Primer A1 is targeted to the 3′ coding region of nifD, primer A3 is targeted within the nifD element, and primer A2 is targeted to the 5′ coding region of nifD.
RNA was prepared using the hot-phenol method as described previously (43). For RT-PCR, 0.15 μg of total RNA was used for the synthesis of cDNA with reverse transcriptase using the sense primers shown in Table 1. For amplification of cDNA by PCR, both sense and antisense primers (Table 1) were added in the amplification system. The log phase of RT-PCR was determined by measuring the amounts of PCR products with different PCR cycles. For real-time quantitative PCR, a cDNA pool was synthesized using a PrimeScript RT reagent kit (Takara), with 400 ng RNA and 5 μM random 6-mer primer in a reaction volume of 10 μl. Reverse transcription was performed at 37°C for 30 min and then at 85°C for 5 s to inactivate the reverse transcriptase. For real-time PCR analysis, 0.2 μl of cDNA samples was used in a 25-μl mixture containing 0.2 μM specific primers (Table 1) and 12.5 μl of SYBR Premix Ex Taq (2×; Takara), using an iQ 5 System (Bio-Rad). A control without cDNA was also included. Amplification was initiated at 95°C for 1 min, followed by 40 cycles at 95°C for 10 s, 55°C for 15 s, and 60°C for 20 s. After amplification, melting-curve analysis was performed. The relative amount of transcript was calculated using the Bio-Rad iQ 5 Optical System software (Bio-Rad) based on the normalized-expression (ΔΔCT, where CT is threshold cycle) method. Transcript levels of target genes were normalized to those of the internal reference gene (rnpB) measured using the same samples.
RESULTS
The mutant D4.3 strain with both pkn44 and pkn30 inactivated was unable to grow diazotrophically.
pkn44 (all1625) and pkn30 (all3691) are two members of the hstK gene family, encoding protein kinases with a Ser/Thr kinase domain and a His kinase domain (Fig. 1). To investigate the function of pkn44 and pkn30, each of these two genes was inactivated by insertion of an antibiotic resistance cassette, and the mutants obtained were completely segregated (data not shown). Both mutants were able to grow normally compared to the wild type in the presence of different nitrogen sources (NH4+, NO3−, or N2), and no obvious morphological defects were identified for either vegetative cells or heterocysts (data not shown). We therefore made a double mutant in which both pkn44 and pkn30 were inactivated by the insertion of an Spr Smr cassette in the pkn44 locus and a neomycin resistance marker in the pkn30 locus (Fig. 1). After selection, several clones were obtained. The complete inactivation of these two genes was confirmed by PCR and Southern hybridization, and no transcripts of pkn44 or pkn30 were detected by RT-PCR (data not shown). This mutant was named D4.3. Although the mutant D4.3 grew at a similar rate as the wild type in nitrate-containing medium, it bleached rapidly and then died in medium lacking a source of combined nitrogen (Fig. 2A and B).
FIG. 1.

Schematic presentation of pkn44 and pkn30. The strain D4.3 was constructed by interrupting pkn44 by the insertion of the Ω element (Spr Smr) into an AccI site and by interrupting pkn30 by insertion of a neomycin resistance gene cassette into a BamHI site. Relevant domains of Pkn44 and Pkn30 are also shown.
FIG. 2.
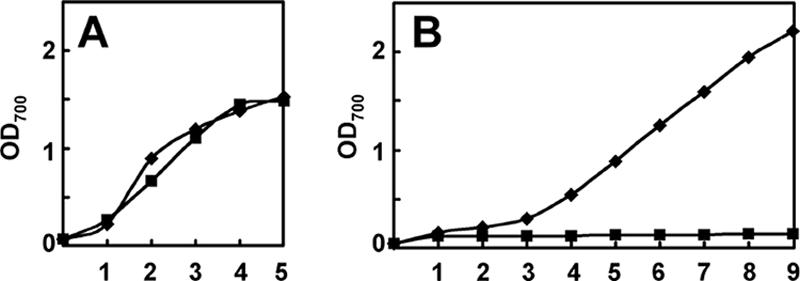
Growth curves of the wild type and D4.3 in the presence of fixed nitrogen (A) or under diazotrophic conditions (B). Growth was followed by measuring the optical density at 700 nm (OD700). Diamond, wild type; square, D4.3. Both strains were adjusted to an OD700 of 0.05 at the beginning of the growth measurement. Experiments were repeated twice, and a representative result is shown.
The mutant strain D4.3 was Fix+ Fox−.
Since the mutant strain D4.3 did not grow under diazotrophic conditions, we measured nitrogenase activity of the wild type and the mutant D4.3 at 24 and 48 h after a step-down of combined nitrogen using an acetylene reduction assay (31). Under oxic conditions, no acetylene reduction was detected in strain D4.3 at 24 h or at 48 h (Table 2). The ability of the mutant D4.3 to reduce acetylene was also examined under microoxic conditions where air was replaced by argon and the oxygen production by photosystem II was blocked by the presence of DCMU (31). Under such conditions, while no nitrogenase activity could be detected in samples of strain D4.3 at 24 h after the limitation of combined nitrogen, a low nitrogenase activity (9.7% of the level found in the wild type under similar conditions) was detected at 48 h (Table 2). According to the established criteria (7), the phenotype of D4.3 corresponds to Fix+ Fox− (showing the activity of acetylene reduction only under microoxic conditions), although the nitrogen-fixation activity was delayed and much lower compared to that found in the wild type.
TABLE 2.
Nitrogenase activity in wild-type and mutant D4.3 strains under different culture conditions
| Condition and time (h) in BG110 medium | Nitrogenase activity for indicated straina
|
|
|---|---|---|
| Wild type | D4.3 | |
| Oxic | ||
| 24 | 4.64 ± 0.15 | 0 |
| 48 | 1.53 ± 0.07 | 0 |
| Microoxic | ||
| 24 | 8.34 ± 0.32 | 0 |
| 48 | 3.17 ± 0.20 | 0.31 ± 0.02 |
The units of nitrogenase activity are micromoles of C2H4 per milligram of chlorophyll a per hour.
DNA rearrangement and expression of genes encoding nitrogenase in strain D4.3.
Since the mutant D4.3 could not fix N2 under oxic conditions, we examined whether DNA rearrangement that normally occurs during the late stages of heterocyst differentiation to remove the nifD element (13, 14) still took place in the mutant. Three PCR primers were designed for this purpose (Table 1): primer A2 was targeted to the upstream coding region of nifD, primer A3 was targeted within the nifD element, and primer A1 was targeted within the nifD coding region downstream of the nifD element. Primers A1 and A3 were used to detect nonrearranged DNA, while primers A1 and A2 were used to amplify rearranged DNA in heterocysts. The result is shown in Fig. 3A. A DNA fragment corresponding to a rearranged product could be amplified from both the wild type and the D4.3 strain at 18 h after the induction of heterocyst differentiation, while the same DNA fragment is absent in filaments cultured in the presence of ammonium.
FIG. 3.

Expression of nifH in the wild type and D4.3. (A) Rearrangement of the nifD element in the wild type (WT) and the strain D4.3. Primers A1 and A3 (A1A3) were used to amplify the DNA fragment corresponding to the nonrearranged DNA, and primers A1 and A2 (A1A2) were used to amplify the DNA fragment corresponding to the rearranged DNA. Cells were cultured for 3 days in the presence of ammonium (NH4+) and then incubated for 18 h in the absence of combined nitrogen (N2). DNA was extracted and used as template for PCR amplification. PCR products were loaded on an 0.8% agarose gel and separated by electrophoresis. (B) Immunodetection of NifH in the wild type (WT) and D4.3. Filaments were cultured in the absence of fixed nitrogen for 24 or 48 h and then incubated under four conditions: lanes 1 and 2, in the air; lanes 3 and 4, in the air and the presence of DCMU; lanes 5 and 6, in argon (Ar) instead of air; lanes 7 and 8, in argon instead of air and in the presence of DCMU. Fifty micrograms of total protein was separated on a 15% sodium dodecyl sulfate-polyacrylamide gel, transferred onto a nitrocellulose membrane, and incubated with antibodies against NifH. (C) The relative abundance of nifH transcripts determined by real-time quantitative PCR. The amounts of nifH transcripts were normalized to those of rnpB as an internal standard. The amount of the nifH transcripts measured under oxic conditions with wild-type cells at 24 h after the deprivation of combined nitrogen was set at 1, and all the other data were calculated relative to this value.
The impairment of the nitrogenase activity in D4.3 could result from defects in the expression of nifHDK encoding the nitrogenase. To test this possibility, cultures were starved for fixed nitrogen with or without DCMU in the presence or absence of oxygen, and then samples were harvested at 24 and 48 h. A polyclonal antibody against the dinitrogenase reductase (NifH) was used to investigate the expression of nifH. As shown in Fig. 3B, at 24 h after the step-down of combined nitrogen, no NifH was detected in strain D4.3 either under oxic (in the air) or microoxic (in the presence of DCMU and argon) conditions. However, a low level of NifH was detected at 48 h but only under microoxic conditions. The presence of DCMU, even without argon to replace air, was enough to allow the low-level expression of NifH in strain D4.3 at 48 h after the induction. We have also used real-time quantitative PCR to determine the transcript levels of nifH under oxic or microoxic conditions (Fig. 3C). The relative amounts of nifH transcripts found in the wild type and D4.3 mutant correlated well with the amounts of the NifH protein determined by Western blotting, and a low level of nifH transcripts was detected 48 h after the deprivation of combined nitrogen. These results demonstrated that providing artificial microoxic conditions could help to maintain a certain level of nifH expression and a low level of nitrogenase activity, and they suggest that the mutant D4.3 may have defects in heterocyst structures normally required to create a microoxic environment within these cells.
The mutant strain D4.3 was Hgl−.
Heterocyst differentiation of the wild type and D4.3 mutant was characterized after deprivation of combined nitrogen (Fig. 4). Whereas mature heterocysts were formed in the wild type within 24 h, none of the visually observed morphological changes were exhibited in D4.3. However, some regularly spaced cells with a morphology different from that of vegetative cells were found at 48 h after induction in the mutant (Fig. 4). These cells were not as much enlarged as mature heterocysts, and they lacked polar cyanophycin granules, which are normally present in mature wild-type heterocysts. However, similar to the wild type, they displayed a low level of fluorescence due to the degradation of photosynthetic pigments (Fig. 4), and they were stained with Alcian blue, which indicates the presence of the heterocyst-specific envelope polysaccharide (data not shown). From these results, we concluded that heterocyst differentiation was initiated in the mutant strain (but delayed compared to the wild type) and then arrested at a relatively early stage.
FIG. 4.

Heterocyst development after starvation for combined nitrogen in the wild type (A) and D4.3 (B). Strains were cultured for 48 h under diazotrophic conditions. Red fluorescent images reflecting photosynthetic pigments (right panels) or bright-field images (left panels) were pictured with the same filaments under a microscope. Arrows indicate the positions of heterocysts.
In order to confirm the phenotypes observed in the mutant D4.3, we performed a complementation assay. Two plasmids containing either pkn30 or pkn44 were transferred into strain D4.3, respectively, and then erythromycin-resistant colonies were selected, in which single recombination occurred between the plasmid and the chromosome. These strains contained both a normal copy and an interrupted copy of the same gene. Whereas no transcripts of either pkn30 or pkn44 could be detected in the double mutant D4.3 by RT-PCR, transcripts of pkn30 or pkn44 at a normal level could be found in D4.3 after complementation by pkn30 or pkn44, respectively (data not shown). Strains complemented by either pkn30 or pkn44 could differentiate heterocysts, distinguished by their enlargement and polar bodies resembling those found in the wild type or the single mutant with either pkn30 or pkn40 inactivated (data not shown). However, they displayed slower growth rates under conditions of combined-nitrogen deprivation than the wild type, whereas the mutant strain D4.3 was unable to grow at all under the same conditions (Fig. 2B).
The defect of nitrogen fixation under oxic conditions and the alteration of heterocyst structure displayed by the mutant D4.3 suggested that the process of heterocyst maturation could be affected, thus making the mutant unable to prevent the entry of oxygen into heterocysts. The ultrastructure of the mutant D4.3 was examined by electron microscopy and revealed that the HGL layer was absent, and only a thin layer of polysaccharides of the outer envelope was deposited (Fig. 5A), consistent with the weak staining by Alcian blue. The heterocyst envelope layers that were particularly thick at the junction of vegetative cells in the wild type were completely absent at the intercellular junction between heterocysts and vegetative cells in the mutant. These results were further confirmed by analysis of the lipid composition using thin-layer chromatography (Fig. 5B). The wild type exhibited two HGLs, 1-(O-α-d-glucopyranosyl)-3,25-hexacosanediol and 1-(O-α-d-glucopyranosyl)-3-keto-25-hexacosanol (8, 11, 20). However, the mutant strain D4.3 had only one HGL corresponding to 1-(O-α-d-glucopyranosyl)-3,25-hexacosanediol; the other one corresponding to 1-(O-α-d-glucopyranosyl)-3-keto-25-hexacosanol was undetectable.
FIG. 5.

The ultrastructure of heterocysts (A) and components of heterocyst glycolipids (B). (A) Electron microscopic images of the wild type (WT) and strain D4.3. Filaments were cultured in the absence of a fixed-nitrogen source. Boxed areas in the upper panels were enlarged with higher magnification in the lower panels. P, polysaccharide layer; GL, glycolipid layer. (B) Thin-layer chromatographic analysis of glycolipids in the wild type (WT) and D4.3. Cells were cultured in the presence of ammonium (NH4+) or in the absence of combined nitrogen (N2). The heterocyst-specific glycolipids are indicated by arrows; the upper one (the minor HGL) corresponds to 1-(O-α-d-glucopyranosyl)-3-keto-25-hexacosanol, and the lower one (the major HGL) corresponds to 1-(O-α-d-glucopyranosyl)-3,25-hexacosanediol.
Expression of genes involved in heterocyst-glycolipid synthesis in the mutant D4.3.
Because of the absence of the minor HGL in the mutant D4.3, we examined the expression pattern of several genes involved in HGL synthesis. The transcription of eight genes, namely hglEA, hglD, hglC, hglK, devBCA, devH, asr5349, and asr5350 (1, 8, 10, 16, 22, 28), was tested by RT-PCR (Fig. 6A). In the mutant D4.3, the transcripts of hglEA, hglC, and hglD were absent at 24 h after the deprivation of combined nitrogen but detectable at 48 h. Transcripts of hglK, devA, devB, devC, and devH were present in filaments at both 24 and 48 h after induction. Interestingly, transcripts of asr5349 and asr5350 were undetectable at either 24 h or 48 h after the deprivation of combined nitrogen. The expression pattern of asr5349 and asr5350 in the wild type and the mutant was further confirmed by real-time quantitative PCR (Fig. 6B). These results indicated that the expression of some of the genes involved in the synthesis or deposition of HGLs was affected in the mutant D4.3.
FIG. 6.

Analysis of transcripts of genes related to HGL synthesis or deposition. (A) Analyses by RT-PCR in the wild type (WT) and the mutant D4.3 strain. Total RNA was extracted from samples grown for 24 h and 48 h after deprivation of combined nitrogen. After cDNA synthesis, PCR was carried out for 20, 22, 24, and 26 cycles for rnpB; 24, 26, and 28 cycles for hglEACDK and devABC; 21, 23, and 25 cycles for devH; and 23, 25, and 27 cycles for asr5349 and asr5350. PCR products were loaded on a 1.2% agarose gel and separated by electrophoresis. For hglDEAK and devABC, the results are shown after 24 cycles; for rnpB, hglC, and devH, the results are shown after 22, 26, and 21 cycles, respectively; and for asr5349 and asr5350, results of PCR are shown after 27 cycles. The rnpB gene served as a control for the amount of RNA template. (B) Transcriptional analysis of asr5349 and asr5350 by real-time quantitative PCR in the wild type (gray bars) and D4.3 (open bars). The transcript levels were normalized to those of rnpB; the transcript level of wild-type cells at 24 h after the deprivation of combined nitrogen was set to 1, and other data were calculated according to this value.
DISCUSSION
In this study, we showed that a mutant strain D4.3 in which pkn44 (all1625) and pkn30 (all3691) were both inactivated had a Het+ Fix+ Fox− Hgl− phenotype, although single mutants with only one of these two genes inactivated had no detectable phenotype under similar conditions. pkn44 and pkn30 may have overlapping and redundant functions in heterocyst development. Several aspects of heterocyst development were affected in the mutant D4.3. First, heterocyst development was delayed. Secondly, the morphology of heterocysts appeared abnormal because of the absence of the glycolipid layer and a thin layer of polysaccharides that did not progress to envelop the junction between heterocysts and vegetative cells. Consequently, the mutant was unable to fix molecular nitrogen under oxic conditions (Fox−). The delayed appearance of morphologically discernible heterocysts in the mutant D4.3 was unlikely to be caused by a delayed initiation. Using antibodies against HetR, a protein crucial for the initiation of heterocyst differentiation (3), we found that HetR displayed similar expression patterns in both the wild type and the mutant at 0, 4, 24, and 48 h following induction (data not shown). It is likely that in the absence of pkn30 and pkn44, heterocyst differentiation was initiated but arrested at a stage when the heterocyst polysaccharide layer started to be deposited. The excision of the nifD element in the mutant took place 18 h after the deprivation of the combined-nitrogen source, a situation comparable to that occurring in the wild type. Some events of heterocyst differentiation are regulated by NtcA (BifA), and xisA encoding the site-specific recombinase responsible for the excision of the nifD element could be a target of NtcA regulation (25, 35), while other events of heterocyst differentiation are independent of NtcA (25). In the absence of Pkn30 and Pkn44, the morphogenesis of heterocyst differentiation was delayed while other events under the control of NtcA, such as the initiation of heterocyst differentiation, still occurred independently. DNA rearrangement and transcription of the nif genes are independent events in Anabaena sp. strain PCC 7120 (15). Consistent with this conclusion, we found that the expression of nifH was impaired in the mutant even under microoxic conditions, despite the fact that DNA rearrangement of the nifD element took place correctly. The expression of the nif genes in heterocysts could be at least partly controlled by oxygen (9, 32). When the mutant D4.3 was incubated under artificial microoxic conditions, a low level of NifH was detected, correlating with a low level of nitrogenase activity under similar conditions.
Only the minor HGL was absent in the mutant D4.3. Different classes of mutants affecting the synthesis or deposition of HGLs are known in Anabaena sp. strain PCC 7120. One class of mutants, such as the devH mutant, lacks both HGLs (28). The second class of mutants is represented by those with hglK inactivated. In these mutants, the HGLs are synthesized but not deposited outside the cells to form the HGL layer (2). The third class of mutants, like FQ1358 with asr5349 inactivated, still has both HGLs but at very much reduced levels compared to the wild type (8). The ΔprpJ mutant represents yet another class of mutants since it affects the synthesis of only the major HGL (18). The mutant D4.3 described in this study represents a distinct class of mutants in which the synthesis of only the minor HGL is affected. As previously pointed out (18), different regulatory pathways may be involved in the synthesis or deposition of the HGL layer, because genes related to these functions show different expression patterns (2, 10, 16, 18, 22, 28). Some of these genes are constitutive, while others are activated only during the process of heterocyst differentiation. We propose that DevH, a putative transcription factor similar to NtcA, is required for the synthesis of both HGLs (16, 28), while PrpJ represents a regulatory branch involved in the synthesis of the major HGL (18), and Pkn30 and Pkn44 correspond to another distinct regulatory pathway for the synthesis of the minor HGL. Consistent with this idea, the D4.3 mutant and the prpJ mutant each appeared to affect the expression of a subset of genes involved in HGL synthesis.
The process of HGL synthesis in Anabaena sp. strain PCC 7120 is still unclear, and the two HGLs differ only at the C-3 position of the aglycones, one with a hydroxyl and the other with a ketone (8, 11, 20). It is interesting that although both the mutant ΔprpJ and the mutant D4.3 lack only one HGL, the HGL layer is not formed, indicating that both HGLs must be present for the deposition and the formation of the HGL layer.
Acknowledgments
L.J.H. was supported by fellowships from the FRM foundation and the K.-C. Wong-C.N.R.S. foundation. We thank the Cheung-Kong scholarship program and the Natural Science Foundation of China (grant number 30500015) for support on works carried out in China.
We thank A. Janicki for technical assistance.
Footnotes
Published ahead of print on 18 May 2007.
REFERENCES
- 1.Awai, K., and C. P. Wolk. 2007. Identification of the glycosyl transferase required for synthesis of the principal glycolipid characteristic of heterocysts of Anabaena sp. strain PCC 7120. FEMS Microbiol. Lett. 266:98-102. [DOI] [PubMed] [Google Scholar]
- 2.Black, K., W. J. Buikema, and R. Haselkorn. 1995. The hglK gene is required for localization of heterocyst-specific glycolipids in the cyanobacterium Anabaena sp. strain PCC 7120. J. Bacteriol. 177:6440-6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Buikema, W. J., and R. Haselkorn. 2001. Expression of the Anabaena hetR gene from a copper-regulated promoter leads to heterocyst differentiation under repressing conditions. Proc. Natl. Acad. Sci. USA 98:2729-2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cai, Y. P., and C. P. Wolk. 1990. Use of a conditionally lethal gene in Anabaena sp. strain PCC 7120 to select for double recombinants and to entrap insertion sequences. J. Bacteriol. 172:3138-3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carrasco, C. D., J. A. Buettner, and J. W. Golden. 1995. Programmed DNA rearrangement of a cyanobacterial hupL gene in heterocysts. Proc. Natl. Acad. Sci. USA 92:791-795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheng, Y., J.-H. Li, L. Shi, L. Wang, A. Latifi, and C.-C. Zhang. 2006. A pair of iron-responsive genes encoding protein kinases with a Ser/Thr kinase domain and a His kinase domain are regulated by NtcA in the cyanobacterium Anabaena sp. strain PCC 7120. J. Bacteriol. 188:4822-4829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ernst, A., T. Black, Y. Cai, J. M. Panoff, D. N. Tiwari, and C. P. Wolk. 1992. Synthesis of nitrogenase in mutants of the cyanobacterium Anabaena sp. strain PCC 7120 affected in heterocyst development or metabolism. J. Bacteriol. 174:6025-6032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fan, Q., G. Huang, S. Lechno-Yossef, C. P. Wolk, T. Kaneko, and S. Tabata. 2005. Clustered genes required for synthesis and deposition of envelope glycolipids in Anabaena sp. strain PCC 7120. Mol. Microbiol. 58:227-243. [DOI] [PubMed] [Google Scholar]
- 9.Fay, P. 1992. Oxygen relations of nitrogen fixation in cyanobacteria. Microbiol. Rev. 56:340-373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fiedler, G., M. Arnold, S. Hannus, and I. Maldener. 1998. The DevBCA exporter is essential for envelope formation in heterocysts of the cyanobacterium Anabaena sp. strain PCC 7120. Mol. Microbiol. 27:1193-1202. [DOI] [PubMed] [Google Scholar]
- 11.Gambacorta, A., A. Soriente, A. Trincone, and G. Sodano. 1995. Biosynthesis of the heterocyst glycolipids in the cyanobacterium Anabaena cylindrica. Phytochemistry 39:771-774. [Google Scholar]
- 12.Golden, J. W., C. D. Carrasco, M. E. Mulligan, G. J. Schneider, and R. Haselkorn. 1988. Deletion of a 55-kilobase-pair DNA element from the chromosome during heterocyst differentiation of Anabaena sp. strain PCC 7120. J. Bacteriol. 170:5034-5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Golden, J. W., M. E. Mulligan, and R. Haselkorn. 1987. Different recombination site specificity of two developmentally regulated genome rearrangements. Nature 327:526-529. [DOI] [PubMed] [Google Scholar]
- 14.Golden, J. W., S. J. Robinson, and R. Haselkorn. 1985. Rearrangement of nitrogen fixation genes during heterocyst differentiation in the cyanobacterium Anabaena. Nature 314:419-423. [DOI] [PubMed] [Google Scholar]
- 15.Golden, J. W., L. L. Whorff, and D. R. Wiest. 1991. Independent regulation of nifHDK operon transcription and DNA rearrangement during heterocyst differentiation in the cyanobacterium Anabaena sp. strain PCC 7120. J. Bacteriol. 173:7098-7105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hebbar, P. B., and S. E. Curtis. 2000. Characterization of devH, a gene encoding a putative DNA binding protein required for heterocyst function in Anabaena sp. strain PCC 7120. J. Bacteriol. 182:3572-3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Herrero, A., A. M. Muro-Pastor, A. Valladares, and E. Flores. 2004. Cellular differentiation and the NtcA transcription factor in filamentous cyanobacteria. FEMS Microbiol. Rev. 28:469-487. [DOI] [PubMed] [Google Scholar]
- 18.Jang, J., L. Wang, R. Jeanjean, and C.-C. Zhang. 2007. PrpJ, a PP2C-type protein phosphatase located on the plasma membrane, is involved in heterocyst maturation in the cyanobacterium Anabaena sp. PCC 7120. Mol. Microbiol. 64:347-358. [DOI] [PubMed] [Google Scholar]
- 19.Kaneko, T., Y. Nakamura, C. P. Wolk, T. Kuritz, S. Sasamoto, A. Watanabe, M. Iriguchi, A. Ishikawa, K. Kawashima, T. Kimura, Y. Kishida, M. Kohara, M. Matsumoto, A. Matsuno, A. Muraki, N. Nakazaki, S. Shimpo, M. Sugimoto, M. Takazawa, M. Yamada, M. Yasuda, and S. Tabata. 2001. Complete genomic sequence of the filamentous nitrogen-fixing cyanobacterium Anabaena sp. strain PCC 7120. DNA Res. 8:205-213. [DOI] [PubMed] [Google Scholar]
- 20.Lambein, F., and C. P. Wolk. 1973. Structural studies on the glycolipids from the envelope of the heterocyst of Anabaena cylindrica. Biochemistry 12:791-798. [DOI] [PubMed] [Google Scholar]
- 21.Li, J.-H., S. Laurent, V. Konde, S. Bedu, and C.-C. Zhang. 2003. An increase in the level of 2-oxoglutarate promotes heterocyst development in the cyanobacterium Anabaena sp. strain PCC 7120. Microbiology 149:3257-3263. [DOI] [PubMed] [Google Scholar]
- 22.Maldener, I., G. Fiedler, A. Ernst, F. Fernandez-Pinas, and C. P. Wolk. 1994. Characterization of devA, a gene required for the maturation of proheterocysts in the cyanobacterium Anabaena sp. strain PCC 7120. J. Bacteriol. 176:7543-7549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Murry, M. A., and C. P. Wolk. 1989. Evidence that the barrier to the penetration of oxygen into heterocysts depends upon two layers of the cell envelope. Arch. Microbiol. 151:469-474. [Google Scholar]
- 24.Ohmori, M., M. Ikeuchi, N. Sato, P. Wolk, T. Kaneko, T. Ogawa, M. Kanehisa, S. Goto, S. Kawashima, S. Okamoto, H. Yoshimura, H. Katoh, T. Fujisawa, S. Ehira, A. Kamei, S. Yoshihara, R. Narikawa, and S. Tabat. 2001. Characterization of genes encoding multi-domain proteins in the genome of the filamentous nitrogen-fixing cyanobacterium Anabaena sp. strain PCC 7120. DNA Res. 8:271-284. [DOI] [PubMed] [Google Scholar]
- 25.Olmedo-Verd, E., E. Flores, A. Herrero, A. M., and Muro-Pastor. 2005. HetR-dependent and -independent expression of heterocyst-related genes in an Anabaena strain overproducing the NtcA transcription factor. J. Bacteriol. 187:1985-1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Phalip, V., J.-H. Li, and C.-C. Zhang. 2001. HstK, a cyanobacterial protein with both a serine/threonine kinase domain and a histidine kinase domain: implication for the mechanism of signal transduction. Biochem. J. 360:639-644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Prentki, P., and H. M. Krisch. 1984. In vitro insertional mutagenesis with a selectable DNA fragment. Gene 29:303-313. [DOI] [PubMed] [Google Scholar]
- 28.Ramirez, M. E., P. B. Hebbar, R. Zhou, C. P. Wolk, and S. E. Curtis. 2005. Anabaena sp. strain PCC 7120 gene devH is required for synthesis of the heterocyst glycolipid layer. J. Bacteriol. 187:2326-2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reich, S., and P. Böger. 1989. Regulation of nitrogenase activity in Anabaena variabilis by modification of the Fe protein. FEMS Microbiol. Lett. 58:81-86. [Google Scholar]
- 30.Rippka, R., J. Deruelles, J. B. Waterbury, M. Herdman, and R. Y. Stanier. 1979. Generic assignments, strain histories and properties of pure cultures of cyanobacteria. J. Gen. Microbiol. 111:1-61. [Google Scholar]
- 31.Rippka, R., and R. Y. Stanier. 1978. The effects of anaerobiosis on nitrogenase synthesis and heterocyst development by nostocacean cyanobacteria. J. Gen. Microbiol. 105:83-94. [Google Scholar]
- 32.Thiel, T., E. M. Lyons, J. C. Erker, and A. Ernst. 1995. A second nitrogenase in vegetative cells of a heterocyst-forming cyanobacterium. Proc. Natl. Acad. Sci. USA 92:9358-9362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vieira, J., and J. Messing. 1982. The pUC plasmids, an M13mp7-derived system for insertion mutagenesis and sequencing with synthetic universal primers. Gene 19:259-268. [DOI] [PubMed] [Google Scholar]
- 34.Wang, L., Y.-P. Sun, W.-L. Chen, J.-H. Li, and C.-C. Zhang. 2002. Genomic analysis of protein kinases, protein phosphatases and two-component regulatory systems of the cyanobacterium Anabaena sp. strain PCC 7120. FEMS Microbiol. Lett. 217:155-165. [DOI] [PubMed] [Google Scholar]
- 35.Wei, T.-F., T. S. Ramasubramanian, F. Pu, and J. W. Golden. 1993. Anabaena sp. strain PCC 7120 bifA gene encoding a sequence-specific DNA-binding protein cloned by in vivo transcriptional interference selection. J. Bacteriol. 175:4025-4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wilcox, M., G. J. Mitchison, and R. J. Smith. 1973. Pattern formation in the blue-green alga Anabaena. II. Controlled proheterocyst regression. J. Cell Sci. 13:637-649. [DOI] [PubMed] [Google Scholar]
- 37.Wilcox, M., G. J. Mitchison, and R. J. Smith. 1973. Pattern formation in the blue-green alga, Anabaena. I. Basic mechanisms. J. Cell Sci. 12:707-723. [DOI] [PubMed] [Google Scholar]
- 38.Wolk, C. P., Y. Cai, and J. M. Panoff. 1991. Use of a transposon with luciferase as a reporter to identify environmentally responsive genes in a cyanobacterium. Proc. Natl. Acad. Sci. USA 88:5355-5359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wolk, C. P., A. Ernst, and J. Elhai. 1994. Heterocyst metabolism and development, p. 769-823. In D. A. Bryant (ed.), The molecular biology of Cyanobacteria. Kluwer Academic Publishers, Dordrecht, The Netherlands.
- 40.Zhang, C.-C. 1996. Bacterial signalling involving eukaryotic-type protein kinases. Mol. Microbiol. 20:9-15. [DOI] [PubMed] [Google Scholar]
- 41.Zhang, C.-C. 1993. A gene encoding a protein related to eukaryotic protein kinases from the filamentous heterocystous cyanobacterium Anabaena PCC 7120. Proc. Natl. Acad. Sci. USA 90:11840-11844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang, C.-C., S. Laurent, S. Sakr, L. Peng, and S. Bedu. 2006. Heterocyst differentiation and pattern formation in cyanobacteria: a chorus of signals. Mol. Microbiol. 59:367-375. [DOI] [PubMed] [Google Scholar]
- 43.Zhang, C.-C., and L. Libs. 1998. Cloning and characterisation of the pknD gene encoding an eukaryotic-type protein kinase in the cyanobacterium Anabaena sp. PCC7120. Mol. Gen. Genet. 258:26-33. [DOI] [PubMed] [Google Scholar]