

Abstract
Expression of the hurIR bhuRSTUV heme utilization locus in Bordetella bronchiseptica is coordinately controlled by the global iron-dependent regulator Fur and the extracytoplasmic function sigma factor HurI. Activation of HurI requires transduction of a heme-dependent signal via HurI, HurR, and BhuR, a three-component heme-dependent regulatory system. In silico searches of the B. bronchiseptica genome to identify other genes that encode additional participants in this heme-dependent regulatory cascade revealed hurP, an open reading frame encoding a polypeptide with homology to (i) RseP, a site 2 protease (S2P) of Escherichia coli required for modifying the cytoplasmic membrane protein RseA, and (ii) YaeL, an S2P of Vibrio cholerae required for modification of the cytoplasmic membrane protein TcpP. A mutant of B. bronchiseptica defective for hurP was incapable of regulating expression of BhuR in a heme-dependent manner. Furthermore, the hurP mutant was unable to utilize hemin as a sole source of nutrient Fe. These defects in hemin utilization and heme-dependent induction of BhuR were restored when recombinant hurP (or recombinant rseP) was introduced into the mutant. Introduction of hurP into a yaeL mutant of V. cholerae also complemented its S2P defect. These data provided strong evidence that protease activity and cleavage site recognition was conserved in HurP, RseP, and YaeL. The data are consistent with a model in which HurP functionally modifies HurR, a sigma factor regulator that is essential for heme-dependent induction of bhuR.
Iron is indispensable for survival of most bacteria. However, excessive amounts of Fe in the cytoplasm of bacteria usually is toxic due to Fe-dependent Fenton reactions that produce lethal hydroxyl radicals (30). Thus, Fe storage and acquisition systems in bacteria are tightly regulated in response to Fe availability. In a variety of bacteria, Fe-dependent regulation is conferred by Fur, a global Fe-dependent DNA binding protein that reversibly binds Fe (31). In most cases, regulation by Fur is sufficient to control expression of most Fe-regulated genes. In other cases, however, additional levels of regulation are required to more precisely control expression of genes that encode particular Fur-dependent Fe acquisition systems. These more complicated regulatory systems often operate in concert with Fur and activate specific Fur-repressed genes under explicit environmental circumstances. Multicomponent Fur-dependent regulatory systems include AraC-type regulators (5, 24, 48, 51, 61), LysR-type regulators (27, 41, 52, 60), and extracytoplasmic function (ECF) sigma factors. In each of these cases, the ancillary components are required to control the expression of specific Fe-dependent genes (1, 11, 39, 47, 54, 56, 58).
ECF sigma factors are a subfamily of σ70-type bacterial proteins that control responses to the local environment by regulating expression of genes that encode adaptive proteins (42). In Bordetella bronchiseptica, the heme utilization locus (hurIR bhuRSTUV) encodes an ECF sigma factor (HurI), an ECF sigma factor regulator (HurR), an outer membrane heme receptor (BhuR), and several other proteins (BhuSTUV) that are predicted to provide transport functions for acquisition of Fe in the form of heme (57, 58). This locus is composed of two operons that are expressed from two independent promoters (PhurI and PbhuR) (59). When Fe is abundant, expression of both operons is repressed in a Fur-dependent manner. Under Fe-limited conditions, however, Fur-dependent repression of PhurI is relieved, thus promoting expression of HurI and HurR. A low-level expression of BhuR ensues by infrequent read-through transcription from PhurI into the second operon (59). In the presence of heme or hemoproteins, high-level expression of the entire downstream operon (bhuRSTUV) is promoted by activation of PbhuR by the three-component signal transduction complex comprised of HurI, HurR, and BhuR. While heme induction in B. bronchiseptica requires coordination between HurI, HurR, and BhuR, other ancillary factors are likely involved in heme-dependent signal transduction in the bacterium.
RseP, also known as YaeL (2, 34) or EcfE (19), is a member of the site 2 protease (S2P) class of membrane metalloproteases that are present in most bacterial genomes (3). The S2P appears to cleave within or near transmembrane segments of its respective substrate. Cleavage releases the resulting polypeptides from the membrane (14). Substrates for S2Ps have been described for RseP homologues in Vibrio cholerae (46), Caulobacter crescentus (17), Bacillus subtilis (10, 43, 55), and Escherichia coli (4, 35). In E. coli, σE, an ECF sigma factor encoded by rpoE, regulates the bacterium's extracytoplasmic stress response (20). σE is negatively controlled by RseA, a membrane-bound anti-sigma factor that binds to σE, thus rendering the sigma factor inactive. Release of σE from RseA is modulated via a mechanism of regulated intramembrane proteolysis, in which RseA is initially cleaved by the successive proteolytic actions of DegS and RseP (3). In a final regulatory step, ClpXP and SspB, which acts as an adaptor protein for ClpX, perform the final cleavage of RseA, thus freeing σE for binding to core RNA polymerase and directing the holoenzyme to σE-dependent promoters (26). In V. cholerae, TcpP is a membrane-localized transcriptional regulator that is required for virulence activation. TcpP activity requires TcpH, a membrane protein that protects TcpP from degradation (7). The TcpP/TcpH transcription complex is required for activation of toxT, the direct modulator for expression of the genes (ctxAB) encoding cholera toxin and the genes encoding the toxin-coregulated pilus (tcp) (21). In the absence of TcpH, TcpP is degraded by YaeL, the gene of which encodes the RseP homologue in V. cholerae (46). Upon degradation of TcpP, activation of ToxT is poor, which promotes a drastic reduction in expression of ctxAB and tcp (15, 63).
It is likely that proteases with modulatory activities similar to those of RseP are expressed by other bacterial species. In this study, we provide strong evidence that hurP, a gene that encodes a prospective S2P, is essential for heme-dependent expression of BhuR, the outer membrane heme receptor of B. bronchiseptica. Furthermore, using complementation experiments, we demonstrate that HurP, RseP, and YaeL exhibit highly conserved regulatory features.
MATERIALS AND METHODS
Culture media, strains, and growth conditions.
Bacterial strains and plasmids used in this study are listed in Table 1. Strains of B. bronchiseptica were maintained on brain heart infusion (BHI) agar or in BHI broth (Difco Laboratories, Detroit, MI). For Fe-replete growth conditions, BHI broth was supplemented with 36 μM FeSO4. Fe-limited and Fe-depleted conditions were achieved in BHI broth by supplementing the broth with ethylene-di-o-hydroxyphenylacetic acid (EDDHA) at 25 and 300 μM final concentrations, respectively. Strains of E. coli and V. cholerae were cultured on Luria-Bertani (LB) agar or in LB broth. Unless otherwise noted, antibiotics were used at the following concentrations: ampicillin (200 μg/ml), rifampin (25 μg/ml), streptomycin (200 μg/ml), tetracycline (10 μg/ml), kanamycin (50 μg/ml), and gentamicin (40 μg/ml). Antibiotics were obtained from Sigma Biochemicals (St. Louis, MO) and Amresco (Solon, OH). Biochemical reagents were purchased from Life Technologies, Inc. (Frederick, MD) and Sigma Biochemicals. Restriction enzymes and DNA-modifying enzymes were obtained from MBI Fermentas, Inc. (Hanover, MD). Deionized water with an electrical resistance of >18 MΩ was used to prepare all solutions.
TABLE 1.
Strains and plasmids
| Strain or plasmid | Relevant characteristic(s) | Source or reference |
|---|---|---|
| Strain | ||
| B. bronchiseptica | ||
| RB50 | Wild type | 18 |
| RB50-R1 | Spontaneous Rifr derivative of RB50 | This study |
| RB50hurP | RB50-R1 with hurP inactivated by pFUS2 integration | This study |
| RB50htpX | RB50-R1 with htpX inactivated by pFUS2 integration | This study |
| RB50mucD | RB50-R1 with mucD inactivated by pFUS2 integration | This study |
| RB50ctpA | RB50-R1 with ctpA inactivated by pFUS2 integration | This study |
| RB50degQ | RB50-R1 with degQ inactivated by pFUS2 integration | This study |
| V. cholerae | ||
| O395 | Wild type | 63 |
| O395ΔtcpH | O395 with internal tcpH deletion | 63 |
| O395ΔtcpHΔyaeL | O395ΔtcpH with internal yaeL deletion | 46 |
| E. coli | ||
| EC41 | Enterotoxigenic E. coli serotype O8:H36 | 28 |
| SA53 | Enterotoxigenic E. coli serotype O103:H36 | 28 |
| DH5αF′kan | ϕ80dlacZM15 Δ(lacZYA-argF)U169 deoR recA1 phoA hsdR17(rK− mK+) supE44λ−thi-1 gyrA96 relA1 [F′ proAB lacqZΔM15 Tn5(Kanr)] | Invitrogen (Carlsbad, CA) |
| DH5αF′tet | ϕ80dlacZM15 Δ(lacZYA-argF)U169 deoR recA1 phoA hsdR17(rK− mK+) supE44λ−thi-1 gyrA96 relA1 [F′ proAB lacqZΔM15 Tn10(Tetr)] | Invitrogen |
| HB101 | F− Δ(gpt-proA)62 leuB6 glnV44 ara-14 galK2 lacY1 Δ(mcr-mrr) rpsL20 xyl-5 mtl-1 recA13; Stpr | New England Biolabs (Beverly, MA) |
| Plasmid | ||
| pTOPO | Cloning vector; Ampr | Invitrogen |
| pKEL8 | hurP with pET21a-derived RBS in pTOPO | This study |
| pKEL10 | 630-bp hurP internal region in pTOPO | This study |
| pGEM-T | Cloning vector; Ampr | Promega (Madison, WI) |
| pNATX14b | rseP from E. coli EC41 with pET21a-derived RBS in pGEM-T | This study |
| pNATX14c | rseP from E. coli SA53 with pET21a-derived RBS in pGEM-T | This study |
| pNATX14 | rseP encoding E. coli K-12 RseP sequence derived from pNATX14b and pNATX14c in pGEM-T | This study |
| pMAN1 | 414-bp htpX internal region in pGEM-T | This study |
| pMAN2 | 513-bp mucD internal region in pGEM-T | This study |
| pMAN3 | 525-bp ctpA internal region in pGEM-T | This study |
| pMAN4 | 621-bp degQ internal region in pGEM-T | This study |
| pBAD18-Kan | Arabinose-inducible expression vector; Kanr | 29 |
| pBAD18-Kan-yaeL | V. cholerae yaeL in pBAD18-Kan | 46 |
| pNATX12.1 | hurP with pET21a-derived RBS in pBAD18-Kan | This study |
| pRK415 | Conjugative expression shuttle vector for B. bronchiseptica and E. coli; Plac; Tetr | 37 |
| pRK415Δ | α-Peptide gene in pRK415 frameshifted at the XhoI site | 39 |
| pKEL8.1 | hurP fused to a pET21a-derived ribosomal binding site in pRK415 | This study |
| pNATX14.1 | E. coli rseP with a pET21a-derived RBS in pRK415 | This study |
| pFUS2 | Suicide vector; Genr | 6 |
| pKEL10.1 | 630-bp hurP internal region in pFUS2 | This study |
| pMAN1.1 | 414-bp htpX internal region in pFUS2 | This study |
| pMAN2.1 | 513-bp mucD internal region in pFUS2 | This study |
| pMAN3.1 | 525-bp ctpA internal region in pFUS2 | This study |
| pMAN4.1 | 621-bp degQ internal region in pFUS2 | This study |
| pRK2013 | Conjugative helper plasmid; Kanr | 25 |
Cloning wild-type hurP.
The hurP open reading frame (ORF) was amplified from B. bronchiseptica RB50 by PCR using the upstream primer 5′-AAGCTTAaggagaTATACATATGCTTTTCACGCTGCTGGCC-3′, which contains a consensus ribosomal binding (RBS) site (lowercase sequence) located seven bases upstream from the hurP translational start codon (underlined), and the downstream primer 5′-GAGCTCGTAAGTGAACAGGCGCGCAAAATCATT- 3′, which contains the hurP translational stop sequence (underlined). The components for the PCR using RB50 genomic DNA as a template were the following: 1× EasyA buffer, 800 μM deoxynucleoside triphosphate [dNTP] mix, 200 nM each primer, 10% dimethylsulfoxide [DMSO], and 2.5 U of EasyA polymerase (Stratagene, La Jolla, CA). The PCR conditions were 30 cycles of 95°C for 45 s, 48°C for 45 s, and 72°C for 1.5 min. The amplified 1,361-bp DNA fragment was ligated into pTOPO (Invitrogen, Carlsbad, CA) to produce pKEL8. The insert of pKEL8 subsequently was confirmed by nucleotide sequencing. To engineer pNATX12.1, an EcoRI/SacI fragment from pKEL8 containing hurP and the RBS was directionally ligated into pBAD18-Kan. pKEL8.1 was engineered by directionally ligating a 1,345-bp HindIII/SacI DNA fragment from pKEL8 into pRK415, a broad-host-range mobilizable expression vector (37).
Cloning wild-type rseP.
A DNA fragment comprising the rseP ORF was amplified from E. coli strains EC41 (28) and SA53 (28) by PCR using the upstream primer 5′-AAGCTTAaggagaTATACATATGCTGAGTTTTCTCTGGGAT-3′, which contains a consensus RBS (lowercase sequence) located seven bases upstream from the rseP translational start codon (underlined), and downstream primer 5′-GAGCTCTCATAACCGAGAGAAATCATTGAAAAG-3′, which contains the translational stop sequence (underlined). The components for the PCR using EC41 and SA53 genomic DNA as a templates were the following: 1× EasyA buffer, 800 μM dNTP mix, 200 nM each primer, and 2.5 U of EasyA polymerase. The PCR conditions were 30 cycles of 95°C for 45 s, 50°C for 45 s, and 72°C for 1.5 min. The 1,379-bp DNA fragments obtained from EC41 and SA53 were ligated into pGEM-T (Promega, Madison, WI) to generate pNATX14b and pNATX14c, respectively. The nucleotide sequences of the inserts of pNATX14b and pNATX14c were compared to that of rseP from E. coli K-12 (8). PCR-derived mutations occurred in the inserts of pNATX14b and pNATX14c, i.e., a single-nucleotide substitution in pNATX14b encoded a methionine-to-threonine alteration at amino acid 78 (M78T); a single-nucleotide substitution in pNATX14c encoded a phenylalanine-to-leucine alteration at amino acid 120 (F120L). Several silent nucleotide substitutions also were found in both pNATX14b and pNATX14c. To engineer pNATX14, which encodes wild-type E. coli K-12 RseP, a 1,132-bp PauI/SacI fragment from pNATX14b (which contains the M78T alteration) was replaced with a 1,132-bp PauI/SacI fragment from pNATX14c. pNATX14.1 was engineered by directionally ligating a HindIII/SacI DNA fragment from pNATX14 into pRK415 (37).
Engineering pFUS2-derived mutants in B. bronchiseptica.
A 630-bp internal DNA fragment of hurP was amplified by PCR from B. bronchiseptica RB50 using primers 5′-GGCAAGCTTGCCGCTGTTCAATCTTTTTCTCG-3′ and 5′-ATGGTACCATCCCCGCCCAGCTGCACG-3′. The components for the PCR were the following: 1× Promega buffer, 800 μM dNTP mix, 200 nM each primer, 10% DMSO, and 2.5 U of Taq polymerase (Promega, Madison, WI). The PCR conditions were 30 cycles of 95°C for 45 s, 55°C for 45 s, and 72°C for 1 min. pKEL10 was engineered by ligating the amplified 630-bp DNA fragment into pTOPO. pKEL10.1 was engineering by directionally ligating a HindIII/KpnI fragment from pKEL10, containing the 630-bp hurP fragment, into the suicide vector pFUS2, which was designed for rapid gene inactivation by homologous recombination and generation of a transcriptional fusion between the interrupted gene and a promoterless lacZ (6). pKEL10.1 was introduced by conjugation into RB50-R1, a rifampin-resistant derivative of RB50 (50). Transconjugates containing Campbell-type insertions of pFUS2 into the chromosome were selected for resistance to gentamicin. The genotype of a prospective hurP mutant (RB50hurP), in which the 5′ end of hurP (encoding the first 305 amino acids of HurP) is in frame with the stop codon of the plasmid-carried groES (e.g., hurP:groES), was confirmed by colony PCR and Southern hybridization.
RB50htpX, RB50mucD, RB50ctpA, and RB50degQ were engineered using plasmid integration by employing the methods described above for constructing RB50hurP. The following primers were used to amplify internal DNA fragments of the respective target genes: 5′-GGGAAGCTTGAGGCAACCATGAAA TCC-3′ and 5′-GGGGGATCCCGTGTAGACGGCAATCTT-3′ to amplify a 414-bp fragment of htpX; 5′-GGGAAGCTTTGGATGCGGCGAAACAAC-3′ and 5′-GGGGGATCCGTAGATGTCGGTGGCATC-3′ to amplify a 513-bp fragment of mucD; 5′-GGGAAGCTTTGCATGAGCACTCGCAAG-3′ and 5′-GGGGGATCCCATGATCGTCAGCGTGAT-3′ to amplify a 525-bp fragment of ctpA; and 5′-GGGAAGCTTACAGTCAGTCTGGCCATT-3′ and 5′-GGGGGTACCGGTATTGATACCCAGGCC-3′ to amplify a 621-bp fragment of degQ. The genotypes of the prospective htpX, mucD, ctpA, and degQ mutants were confirmed by colony PCR.
Complementation of the V. cholerae yaeL mutant.
V. cholerae O395, O395ΔtcpH, and O395ΔtcpHΔyaeL (46) were each transformed by electroporation with pBAD18-Kan (29), pBAD18-Kan-yaeL (46), or pNATX12.1 (45). Overnight cultures of the transformants were subcultured in LB medium (pH 6.5) at 30°C to activate expression of virulence genes and subsequently were subcultured in the presence of 0.1% arabinose to induce expression of either yaeL or hurP from PBAD (46). Bacteria in 1 ml of mid-logarithmic-phase culture were pelleted by centrifugation and resuspended in solubilization buffer (31 mM Tris [pH 6.8], 2% sodium dodecyl sulfate [SDS], 2.5% 2-mercaptoethanol, 10% glycerol). Proteins were separated by SDS-polyacrylamide gel electrophoresis (PAGE) using 15% (wt/vol) polyacrylamide gels. Samples were boiled for 15 min prior to being loaded onto the gels. Loading volumes were adjusted to normalize for the culture optical density at 600 nm (OD600). Proteins were transferred to nitrocellulose membranes and then were probed with rabbit anti-TcpP antibodies (46), followed by being probed with goat anti-rabbit horseradish peroxidase-conjugated secondary antibody (Southern Biotech, Birmingham, AL). Immunoreactive proteins were visualized by using SuperSignal West Pico chemiluminescent substrate (Pierce, Rockford, IL).
Complementation of the B. bronchiseptica hurP mutant.
pKEL8.1 and pNATX14.1 were mobilized into B. bronchiseptica strains RB50-R1 and RB50hurP by conjugation using E. coli HB101(pRK2013) (25) as a helper strain. A transconjugant containing only pRK415Δ (39) was used as the negative control in all complementation experiments. Bacteria were cultured in BHI broth supplemented with 25 μM EDDHA for 8 h at 37°C to activate expression of Fe-repressed genes. For endpoint growth assays, Fe-limited cultures were diluted to an OD600 of 0.05 in BHI broth supplemented with 36 μM FeSO4, 300 μM EDDHA, or 300 μM EDDHA plus 5 μM hemin. Cells were cultured at 37°C for 18 h to stationary phase. All cultures were performed in triplicate.
To measure BhuR expression, bacteria were cultured at 37°C to stationary phase in BHI broth supplemented with 36 μM FeSO4, 25 μM EDDHA, or 25 μM EDDHA plus 1 μM hemin. Bacteria in 1 ml of culture were pelleted by centrifugation and resuspended in solubilization buffer (31 mM Tris [pH 6.8], 2% SDS, 2.5% 2-mercaptoethanol, 10% glycerol). Proteins were separated by SDS-PAGE using 7.5% (wt/vol) polyacrylamide gels. Samples were boiled for 15 min prior to being loaded onto the gels. Loading volumes were adjusted to normalize for the culture OD600. Proteins were transferred to polyvinylidene fluoride membranes and probed with rabbit anti-BhuR antibodies (J. C. Mocny and T. D. Connell, unpublished data) and with goat anti-rabbit horseradish peroxidase-conjugated secondary antibody. Immunoreactive proteins were visualized by using SuperSignal West Pico chemiluminescent substrate.
RT-PCR.
Total RNA was extracted from bacteria harvested after growth to mid-logarithmic phase (62). The following oligonucleotide primer sets were utilized in reverse transcription-PCRs (RT-PCRs) (One-Step RT-PCR; QIAGEN, Valencia, CA): 5′-TGTTCGACAACCGCTACCAGAACT-3′ and 5′-GCACGTTGATGGCTTCCCAGTATT-3′ to target a 513-nucleotide region of bhuR; 5′-AACGAGGTCAACGTCAAGACCGAT-3′ and 5′-ATGCCGTCCTTGGTGAAATACGGA-3′ to target a 264-nucleotide region of ORF BB2613; and 5′-GCACCAACTGCATGGTCATCTTCA-3′ and 5′-CGATGGCCATTTCCTTGTGCTCTT-3′ to target a 402-nucleotide region of the constitutively expressed recA (40).
Fifty nanograms of total RNA was employed in each reaction mixture. Reaction mixtures that lacked RNA template or reverse transcription enzyme were included as negative controls (data not shown). RT parameters included one 30-min cycle at 50°C followed by 15 min at 95°C to inactivate reverse transcriptase. Reverse transcribed DNA was amplified by PCR (25 cycles of 45 s at 95°C, 45 s at 50°C, and 1 min at 72°C). DNA within a volume of one fifth of that of each reaction mixture was resolved on a 2% agarose gel. Amplified DNA was visualized by staining the gel with ethidium bromide. The amounts of stained DNA were quantitated by densitometry. The expression of recA was used as an internal control. The results are presented from at least three independent RT-PCRs.
Statistics.
All experimental results were compared statistically using one- way analysis of variance (InStat version 3.00; GraphPad Software Inc., San Diego, CA).
RESULTS
hurP encodes a polypeptide with homology to S2Ps.
It is not uncommon for membrane proteins to be modified after transport. Thus, it was deemed plausible that posttranslational modifications were required to modulate heme-dependent expression of BhuR in B. bronchiseptica. A first supposition was that some modification of HurR likely was required for the membrane protein to accept BhuR-derived signals. S2Ps that modify cytoplasmic membrane proteins by regulated intramembrane proteolysis have been described for several bacteria (4, 10, 17, 35, 43, 46, 55). It has been suggested by Braun et al. that, for the E. coli FecI/FecR system, intramembrane proteolysis via RseP is involved in activating FecI (13). The FecI/FecR system of E. coli is analogous to the HurI/HurR system of B. bronchiseptica, in that it is an ECF sigma factor/sigma factor regulator involved in the substrate-dependent activation of an outer membrane receptor for an Fe-containing solute (12). Thus, an in silico analysis of the B. bronchiseptica genome (Sanger Institute Wellcome Trust Genome Campus, Cambridge, United Kingdom) (50) was performed to search for ORFs encoding polypeptides with structural similarities to RseP. These searches revealed an ORF (BB2612) encoding a prospective 444-amino-acid polypeptide that exhibited 56% similarity to RseP of E. coli (8), 57% similarity to YaeL of V. cholerae (32), 51% similarity to YluC of B. subtilis (55), and 48% similarity to MmpA of C. crescentus (17). The ORF subsequently was designated hurP. Orthologs to hurP, BP1426 and BPP1534, also were found in the genome sequences of Bordetella pertussis Tohama I and Bordetella parapertussis 12822, respectively (50).
Several conserved regions were revealed by a detailed amino acid sequence comparison of the predicted HurP polypeptide with RseP and YaeL (Fig. 1): (i) four putative transmembrane domains; (ii) HEXXH and LDG zinc-binding motifs required in RseP for proteolytic activity (19); and (iii) a periplasmic PDZ domain. While the PDZ domain has been shown for RseP to be dispensable for proteolytic activity, the domain temporally controls RseP activity with respect to the cleavage of RseA (9, 36). The conservation of these features in the prospective HurP polypeptide strongly suggested that hurP encoded an S2P.
FIG. 1.

Alignment of the amino acid sequences of RseP of E. coli (8), YaeL of V. cholerae (32), and HurP of B. bronchiseptica. Single-letter amino acid designations are employed. Conserved HEXXH and LDG motifs are highlighted in gray, transmembrane regions predicted by TMpred are boxed, and putative PDZ domains identified from the Pfam HMM database, available on the Sanger website (http://www.sanger.ac.uk/Software/Pfam/search.shtml), are indicated in boldface.
hurP complements a yaeL mutation.
To determine whether hurP encoded a polypeptide with S2P activity, the capacity of the gene to complement a mutation in an analogous S2P-modulated system was examined. In V. cholerae, TcpP is a membrane-bound transcriptional activator of virulence gene expression via toxT, a gene that encodes the direct activator of two critical virulence determinants: (i) cholera toxin (ctxAB), the extracellular enterotoxin (21), and (ii) the toxin-coregulated pilus (tcp) (21). Proteolytic cleavage of TcpP by YaeL inactivates TcpP. TcpH, a second membrane-bound protein expressed by V. cholerae, however protects TcpP from YaeL-dependent proteolysis. Expression of toxT occurs in the absence of cleavage of TcpP by YaeL (46). Functionally, this system can be observed by Western immunoblotting. Antibodies against TcpP bind to an ∼26-kDa polypeptide (Fig. 2, lanes 1 to 3) (46). This polypeptide is not detectable in an isogenic tcpH mutant (O395ΔtcpH) (Fig. 2, lanes 4 to 6) (46). A protein of ∼20 kDa, however, is observable in a tcpH yaeL double mutant (O395ΔtcpHΔyaeL) (Fig. 2, lane 7) (46). The truncated TcpP polypeptide is hypothesized to be a degradation product produced by an unidentified protease (46). When yaeL was expressed in trans in O395ΔtcpHΔyaeL, S2P activity was restored, i.e., the ∼20-kDa TcpP was no longer detectable (Fig. 2, lane 8) (46). To determine if HurP could substitute for YaeL in this system, pNATX12.1 (encoding hurP) was introduced into V. cholerae O395, O395ΔtcpH, and O395ΔtcpHΔyaeL (45). Transformants were cultured at 30°C in LB broth (pH 6.5) in the presence of 0.1% arabinose to induce expression of recombinant hurP (46). Cellular proteins from mid-logarithmic-phase cells were resolved by SDS-PAGE and analyzed by immunoblotting with anti-TcpP antibodies (46). In wild-type O395, TcpP is protected from degradation by TcpH, and in trans expression of yaeL or hurP in O395 had no detectible effect on TcpP, while in O395ΔtcpH, TcpP is completely degraded under all conditions examined due to the presence of endogenous YaeL. In O395ΔtcpHΔyaeL, however, a truncated TcpP is observed, and expression of either gene in O395ΔtcpHΔyaeL elicited degradation of truncated TcpP polypeptide (Fig. 2, lanes 8 and 9). These data indicated that HurP recognized TcpP as a substrate in a manner similar to that of YaeL, an established S2P (46). These data also indicated that the proteolytic activity and cleavage site recognition of YaeL was conserved in HurP, although the two proteins were expressed by two distantly related bacteria.
FIG. 2.
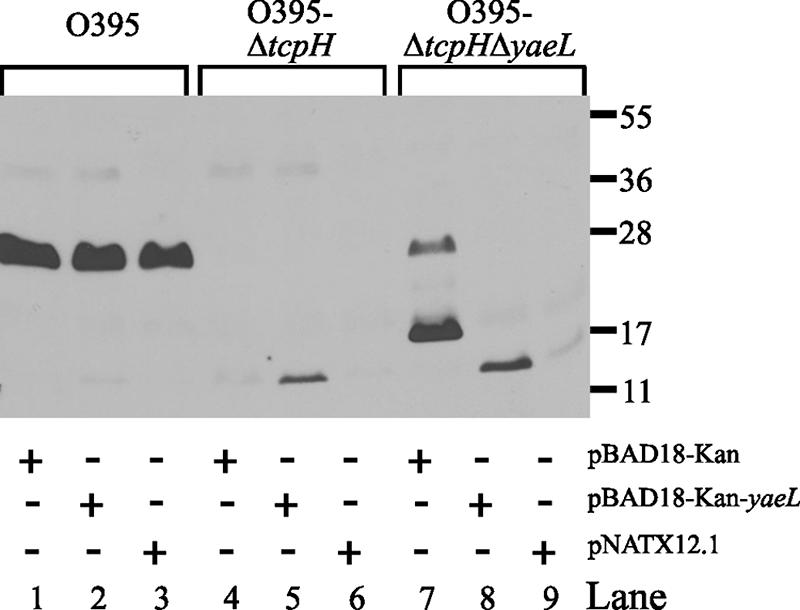
The capacity of a ΔtcpH ΔyaeL double mutant of V. cholerae to degrade TcpP is restored by introduction of hurP. V. cholerae O395, O395ΔtcpH, and O395ΔtcpHΔyaeL, each transformed with pBAD18-Kan, pBAD18-Kan-yaeL (yaeL), or pNATX12.1 (hurP), were cultured in LB broth (pH 6.5) at 30°C in the presence of 0.1% arabinose. Mid-logarithmic-phase cells were solubilized, the proteins were resolved by SDS-PAGE, and the resolved proteins were analyzed by immunoblotting with anti-TcpP antibodies. Protein standard molecular sizes are in kilodaltons. (Strains of V. cholerae, anti-TcpP antibodies, pBAD18-Kan, and pBAD18-Kan-yaeL were obtained from J. S. Matson and V. J. DiRita.)
hurP is essential for effective heme utilization by B. bronchiseptica.
RseP has an essential role in the activation of the extracytoplasmic stress response in E. coli and is required for viability (19). This requirement is alleviated if the cell is rendered defective in expression of rseA, which encodes the anti-sigma factor cleaved by RseP (35), or if expression of OmpA and OmpC, two outer membrane porins, the overexpression and subsequent accumulation of which in the periplasm is a signal for the extracytoplasmic stress response, is downregulated (22). In contrast, deletion of the RseP homologues in B. subtilis (55), V. cholerae (46), and C. crescentus (17) has no effect on the viability of these bacteria. To determine if deletion of hurP exerted a negative effect on growth and/or viability of B. bronchiseptica, a mutant with a defect in hurP was produced. RB50hurP was engineered by integrating pKEL10.1, a pFUS2-based suicide plasmid (6), into the hurP locus of RB50-R1, a rifampin-resistant derivative of RB50 (50). Insertional inactivation of hurP by pKEL10.1 had a small but statistically significant effect, as growth of RB50hurP(pRK415Δ) was slightly inhibited compared to that of RB50-R1(pRK415Δ) when both strains were cultured in Fe-replete BHI broth (Fig. 3). As expected, growth was arrested when RB50-R1(pRK415Δ) and RB50hurP(pRK415Δ) were cultured in Fe-depleted BHI broth. Growth of RB50-R1(pRK415Δ) was restored when the Fe-depleted BHI broth was supplemented with 5 μM hemin. In contrast, growth of RB50hurP(pRK415Δ) was severely inhibited when heme was the sole source of nutrient Fe (Fig. 3). These data demonstrated that, while hurP was not essential for viability of B. bronchiseptica, the capacity of the bacterium to utilize heme as a sole source of nutrient Fe required expression of hurP.
FIG. 3.
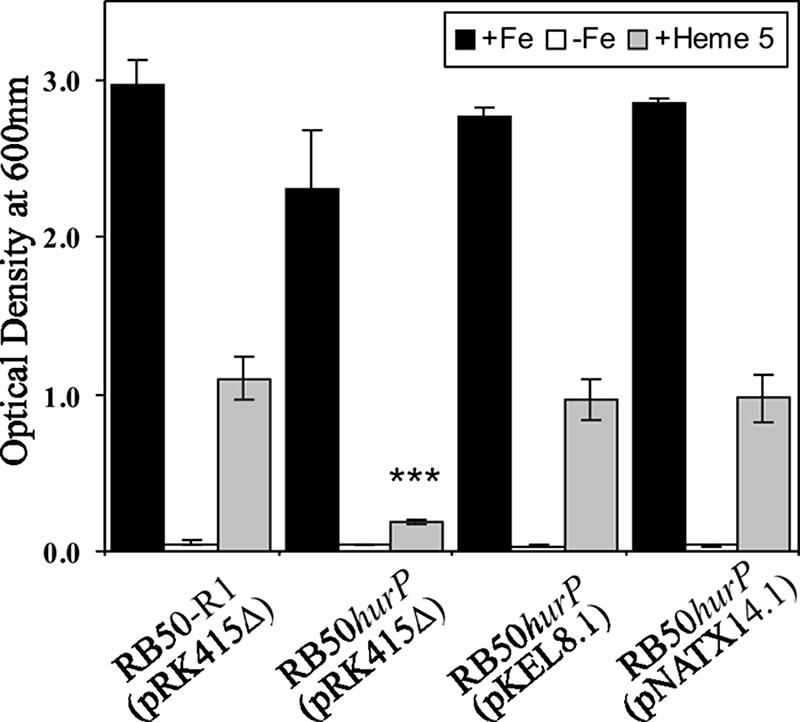
Expression of hurP is required by B. bronchiseptica for utilization of hemin as a source of nutrient Fe. B. bronchiseptica RB50-R1 and RB50hurP containing pRK415Δ, pKEL8.1 (hurP), or pNATX14.1 (rseP) were cultured at 37°C for 18 h in BHI broth supplemented with 36 μM FeSO4 (+Fe), 300 μM EDDHA (−Fe), or 300 μM EDDHA plus 5 μM hemin (+Heme 5). The density of triplicate cultures was determined spectrophotometrically at 600 nm. Error bars denote one standard deviation from the mean. The asterisks (***) denote statistical significance (P < 0.001) compared to the OD600 of RB50-R1 cultured in identical growth conditions.
hurP is essential for heme-dependent induction of BhuR.
Addition of hemin to Fe-depleted cultures of B. bronchiseptica induces high-level expression of the heme uptake locus bhuRSTUV (58). The inability of RB50hurP to utilize hemin as a source of nutrient Fe suggested that the mutant was defective in the expression of the bhuRSTUV operon. To evaluate this hypothesis, the expression of BhuR in RB50hurP was examined. B. bronchiseptica was cultured in BHI broth supplemented with 25 μM EDDHA to produce Fe-stressed conditions. Supplementation of the broth with 1 μM hemin is sufficient to induce maximal expression of bhuR; maximal growth of the bacterium is attained when the Fe-limited broth is supplemented with 5 μM hemin (57, 59; N. D. King-Lyons and T. D. Connell, unpublished data). RB50-R1 and RB50hurP, each conjugated with pRK415Δ, pKEL8.1, or pNATX14.1, were cultured in Fe-replete BHI broth, Fe-stressed BHI broth, and Fe-stressed BHI broth that had been supplemented with 1 μM hemin. Proteins from cells obtained from each culture condition were separated by SDS-PAGE, and the resolved proteins were analyzed by immunoblotting using anti-BhuR antibodies (J. C. Mocny and T. D. Connell, unpublished). As expected, BhuR was not expressed when the strains were cultured in Fe-replete conditions (Fig. 4, lanes 1 to 3). Under Fe-stressed conditions, however, BhuR was detected, albeit minimally, in all strains (Fig. 4, lanes 4 to 6). This low-level expression of BhuR in Fe-stressed conditions has been shown to be the result of read-through transcription from PhurI into the bhuRSTUV operon after Fur-dependent derepression of the promoter (59). Addition of 1 μM hemin to the Fe-stressed culture of RB50-R1(pRK415Δ) elicited significant induction of BhuR by the cells (Fig. 4, lanes 7 to 9). In contrast, hemin-dependent induction of BhuR was not detectable in RB50hurP(pRK415Δ) (Fig. 4, lanes 7 to 9). These data indicated that the defect in the capacity of RB50hurP to utilize hemin as a source of nutrient Fe likely was due to a paucity of BhuR in the outer membrane of the cell. Interestingly, BhuR expression also was absent in Fe-stressed cultures of RB50hurP(pRK415Δ) (Fig. 4, lane 4). This observation initially suggested that the hurP mutation also exerted an effect on Fe-dependent expression of BhuR. However, subsequent RT-PCR analysis demonstrated that Fe-dependent transcription of bhuR in RB50hurP was reduced only slightly compared to transcription of bhuR in RB50-R1 (Fig. 5). These data suggested that expression of BhuR in Fe-dependent cultures of RB50hurP(pRK415Δ) occur below the level of immunodetection. An alternative hypothesis was that the mutation in hurP exerted a negative effect on translation of the bhuR transcripts. Data from growth experiments, however, made this latter model less tenable. While RB50hurP(pRK415Δ) was severely inhibited for heme-dependent growth, the mutant retained a low but detectable capacity to proliferate in broth in which heme was the sole source of nutrient Fe (Fig. 3). These data indicated, therefore, that RB50hurP(pRK415Δ) expressed a small amount of BhuR in the outer membrane (and likely the other proteins involved in heme transport).
FIG. 4.

hurP is required for heme-dependent induction of BhuR. RB50-R1 and RB50hurP containing pRK415Δ, pKEL8.1, or pNATX14.1 were cultured at 37°C to stationary phase in BHI broth supplemented with 36 μM FeSO4 (+Fe), 25 μM EDDHA (low Fe), or 25 μM EDDHA plus 1 μM hemin (+Heme 1). Aliquots of cells were solubilized, the proteins were resolved by SDS-PAGE, and the resolved proteins were analyzed by immunoblotting with anti-BhuR antibodies (J. C. Mocny and T. D. Connell, unpublished). Protein standard molecular sizes are designated in kilodaltons.
FIG. 5.
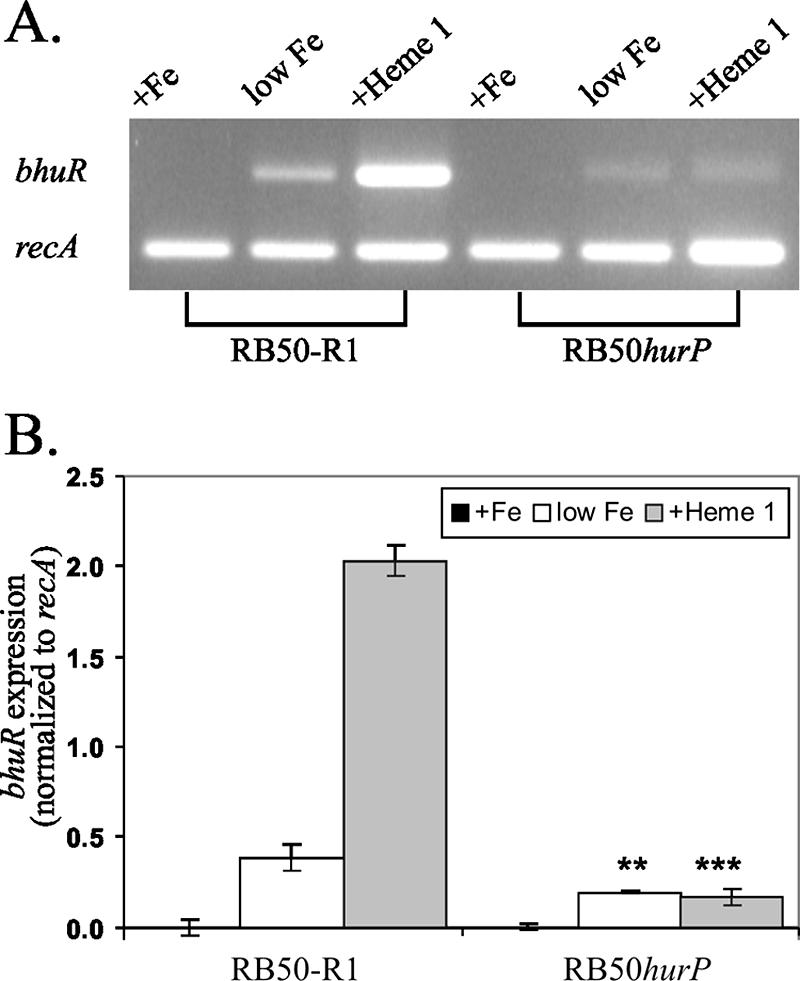
Transcription of bhuR. RB50-R1 and RB50hurP were cultured at 37°C in BHI broth supplemented with 36 μM FeSO4 (+Fe), 25 μM EDDHA (low Fe), or 25 μM EDDHA plus 1 μM hemin (+Heme 1). Total RNA was extracted from cells harvested after growth to mid-logarithmic phase (62). Primer sets that target a 513-bp region of bhuR and a 402-bp region of constitutively expressed recA (40) were employed in RT-PCRs (One-Step RT-PCR). (A) Amplified DNA in one-fifth of each RT-PCR was resolved on a 2% agarose gel and visualized by staining with ethidium bromide. (B) Data from panel A were quantitated by densitometry, and the extent of bhuR expression was determined by normalization with respect to recA expression. All RT-PCRs were performed in triplicate. Error bars denote one standard deviation from the mean. Statistically significant differences (**, P < 0.01; ***, P < 0.001) from RB50-R1 cultured in the identical growth conditions are indicated.
Complementation of RB50hurP.
RB50hurP was constructed by integration of pFUS2 into the hurP locus. To demonstrate that the defects in heme-dependent growth and heme-dependent induction of BhuR of RB50hurP were not caused by polar effects on downstream genes, complementation experiments were performed by introducing pKEL8.1 into the mutant. Introduction of plasmid-encoded wild-type hurP into RB50hurP restored Fe- and heme-dependent growth (Fig. 3). These data provided strong evidence that integration of the pFUS2-based suicide plasmid into the hurP locus in RB50hurP was unlikely to have exerted detrimental effects on downstream genes, at least with respect to Fe- and heme-dependent growth. Heterologous complementation experiments were performed by introducing rseP of E. coli (pNATX14.1) into RB50hurP. Introduction of pNATX14.1 also restored the growth phenotype of RB50hurP (Fig. 3). Thus, rseP exhibited the capacity to complement an hurP defect. Conclusive evidence that the insertion into hurP did not exert polar effects on downstream genes was provided by RT-PCR, which established that transcription of BB2613, the ORF located immediately downstream from hurP, was unaltered in RB50hurP (data not shown).
To evaluate whether complementation of the growth phenotype was correlated with increased levels of BhuR, expression of the heme receptor was evaluated in the complemented mutant by immunoblotting cells cultured under the various conditions of Fe stress and heme supplementation. Hemin-dependent expression of BhuR was restored in RB50hurP when either pKEL8.1 or pNATX14.1 was introduced into the mutant (Fig. 4, lanes 8 and 9).
Prospective S1Ps.
RseP and its homologues are classified as S2Ps (44). In the cases of RseP (4), TcpP (46), PodJ (17), and RsiW (55), the substrate for the respective S2P is produced after cleavage of the targeted polypeptide by a site 1 protease (S1P). To date, S1Ps have been identified in E. coli (4, 35), C. crescentus (16), and B. subtilis (23, 33). In E. coli, RseA initially must be cleaved by the membrane-anchored serine protease DegS to reveal the cleavage recognition site for RseP (4). In an attempt to identify a potential S1P involved in hemin-dependent expression of BhuR, the genomic and protein databases of B. bronchiseptica (50) were searched for genes or polypeptides with homology to degS or DegS, respectively (53). Searches also were conducted using several S1P motifs found in DegS (e.g., an N-terminal transmembrane anchor sequence, a protease signature, and the PDZ domain) as in silico probes. Fourteen ORFs were identified that contained at least one of these three motifs. Only 7 of those 14 ORFs, including hurP, contained at least two of the three motifs. BB4867 (degQ), which encodes the DegS homolog, and BB2112 (htpX), BB3749 (mucD), and BB0300 (ctpA) were chosen for further analysis. The other three ORFs encoded polypeptides that lacked a signal peptide. After pFUS2-based interruption of each of these ORFs, the mutants (RB50htpX, RB50mucD, RB50ctpA, and RB50degQ) were analyzed by immunoblotting for hemin-dependent expression of BhuR. Genetic interruption of the four genes had no effect on heme-induced expression of BhuR (data not shown). Whether heme-dependent induction of BhuR in B. bronchiseptica requires an S1P has yet to be determined.
DISCUSSION
While prior reports suggested that expression of HurI, HurR, and BhuR was sufficient for heme-dependent induction of BhuR, the experiments described herein indicated that at least one other ancillary factor is involved. Hemin-dependent expression of BhuR and heme-dependent growth of B. bronchiseptica required hurP (Fig. 4). Moreover, experiments demonstrating that hurP complemented a yaeL defect in V. cholerae and that rseP complemented an hurP defect in B. bronchiseptica indicated that the proteolytic activities and cleavage recognition capabilities of all three proteins are highly conserved in these taxonomically diverse bacteria. Thus, it might be hypothesized that the substrates for the three proteins should share amino acid homology at the cleavage site. However, RseA and TcpP, which are the natural substrates for RseP and YaeL, respectively, and HurR, which is the likely substrate for HurP (see below), exhibit little, if any, homology to each other. This observation suggests that the S2P cleavage sites recognized by HurP, RseP, and YaeL likely are not easily recognizable conserved primary amino acid sequences. RseP has been shown to cleave a broad range of model membrane proteins unrelated to RseA within the predicted transmembrane region, provided that the transmembrane region contains residues of low helical propensity (2). Perhaps the recognition sites in the substrates of HurP and YaeL also are located within a transmembrane region of low helical character in those proteins.
Heme-independent BhuR expression was diminished in the hurP mutant, albeit slightly. While a small amount of BhuR was detected by anti-BhuR antibodies in Fe-limited cultures of wild-type B. bronchiseptica, none was detected in similar cultures of the hurP-deficient mutant (Fig. 4). This result was unexpected given that, under Fe-limited conditions and in the absence of heme, bhuR is expressed from the Fur-dependent PhurI via read-through transcription (59). Involvement of HurP in bhuR expression from PhurI would preclude its direct role in the BhuR-HurR-HurI signal cascade. Data obtained from RT-PCR analysis, however, indicated that bhuR was expressed in an Fe-dependent manner in both RB50-R1 and RB50hurP (Fig. 5). Heme-independent bhuR expression occurred from PrhuI (by read-through transcription) and from PbhuR in an RhuI-dependent manner in Bordetella avium (38). It is conceivable that transcription of bhuR is controlled in a similar manner in B. bronchiseptica and that heme-independent expression of BhuR is diminished in the hurP-deficient strain due to the absence of HurI-dependent PbhuR activity. Additional experiments to transcriptionally map the hurIR bhuRSTUV locus are needed to evaluate that model.
hurP also had a slight effect on growth of Fe-replete cultures (Fig. 3), conditions in which hurP had no effect on BhuR expression (Fig. 4). The Fe-dependent growth defect of RB50hurP was rescued by in trans expression of hurP or rseP, (Fig. 3) indicating that the S2P is involved in a process or processes in addition to uptake and/or utilization of heme by B. bronchiseptica. For example, the B. bronchiseptica genome possesses hypothetical proteins homologous to those of E. coli σE (BB3752) and RseA (BB3751) (50), suggesting that hurP also is involved in regulating the stress response. The potential for hurP to have an effect on σE activity or the stress response, however, has not been investigated. Although hurP is not an essential gene in B. bronchiseptica, efficient utilization of heme as a sole source of Fe is negatively affected by a genetic defect in hurP (Fig. 3). This effect likely is due to lower-than-optimal amounts of BhuR occurring in the outer membrane that is the result of the disruption of the heme-dependent regulatory system that controls expression of BhuR (Fig. 4). High-level expression of BhuR depends on the heme response of a three-component signal transduction cascade composed of HurI (ECF sigma factor), HurR (sigma factor regulator), and BhuR (outer membrane heme receptor). Of the three proteins demonstrated to comprise the signal transduction complex, only HurR is located in the cytoplasmic membrane. Secondary-structure predictions of HurR using the TMpred algorithm, available online (http://www.ch.embnet.org), suggest that this protein contains a putative transmembrane region extending from residues 69 to 85, with the N-terminal portion of the protein in the cytoplasm and the C-terminal portion of the protein in the periplasm. The predicted structure of HurR is very similar to the structure of FecR (49), and it has been suggested that, in E. coli, FecR is a substrate for RseP (13). Since other S2Ps cleave cytoplasmic membrane-localized proteins, it is hypothesized, therefore, that HurI is activated, at least in part, by a process involving HurP-dependent proteolysis of HurR. Experiments to support this model by tracking HurP-dependent degradation of HurR are currently under way.
Regulated intermembrane proteolysis of membrane-spanning proteins is an important regulatory mechanism in many biological systems. Release of the membrane-bound protein generally occurs as the result of two sequential cleavage steps. In E. coli, RseA is degraded sequentially, initially by DegS, the S1P, and subsequently by RseP, the S2P (4). In V. cholerae, the substrate for YaeL is the truncated form of TcpP (46). These data suggested that TcpP initially is cleaved by an S1P to produce the truncated form of the protein. We surmised that the BhuR-HurR-HurI signal cascade that controls heme-dependent bhuR expression is activated by HurP and, therefore, may also be controlled by an S1P. Attempts to identify the S1P by systematic inactivation of genes encoding potential S1Ps based on their similarity to DegS, however, were unproductive, as were similar attempts made to identify the S1P for TcpP in V. cholerae (46). These results suggest that the putative S1Ps involved in bhuR expression in B. bronchiseptica and toxT repression in V. cholerae do not display similarities to known proteases. The uniqueness of the S1Ps may be the determinant of S2P specificity and is perhaps the reason why the S2Ps of B. bronchiseptica, V. cholerae, and E. coli are interchangeable.
In summary, these experiments established that hurP is a vitally important factor in the heme-dependent regulatory cascade of B. bronchiseptica for controlling expression of BhuR, the outer membrane receptor for heme. Investigations are ongoing to describe more precisely the activities of HurP and to identify additional factors encoded by B. bronchiseptica that are involved in the heme-dependent regulatory cascade. Genes encoding prospective HurP polypeptides also are found in the chromosomes of B. pertussis, B. parapertussis, and B. avium, a pattern that suggests that these three pathogenic species also require an hurP-encoded S2P for heme induction of their respective BhuR receptors. It should be noted that this report is the first in which an S2P has been shown to be involved in expression of a receptor controlling the uptake and metabolism of an essential micronutrient.
Acknowledgments
Funds to support this investigation were contributed to T.D.C. by The School of Medicine and Biomedical Sciences at The University at Buffalo. N.D.K.-L. was supported by a National Institutes of Health training grant (DE007034) awarded to The Department of Oral Biology at The University at Buffalo and a UB2020 Interdisciplinary Research Development Fund (35905-1-1054659), awarded to Matthew D. Disney and T.D.C.
We thank J. F. Miller for providing B. bronchiseptica strain RB50 and J. S. Matson and V. J. DiRita for providing the strains of V. cholerae, anti-TcpP antibodies, and the plasmids pBAD18-Kan and pBAD18-Kan-yaeL. We also thank Manish P. Shah for his assistance in the construction of the four prospective S1P mutants in B. bronchiseptica and in the immunoblotting experiments using those strains.
Footnotes
Published ahead of print on 22 June 2007.
REFERENCES
- 1.Agnoli, K., C. A. Lowe, K. L. Farmer, S. I. Husnain, and M. S. Thomas. 2006. The ornibactin biosynthesis and transport genes of Burkholderia cenocepacia are regulated by an extracytoplasmic function σ factor which is a part of the Fur regulon. J. Bacteriol. 188:3631-3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Akiyama, Y., K. Kanehara, and K. Ito. 2004. RseP (YaeL), an Escherichia coli RIP protease, cleaves transmembrane sequences. EMBO J. 23:4434-4442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alba, B. M., and C. A. Gross. 2004. Regulation of the Escherichia coli sigma-dependent envelope stress response. Mol. Microbiol. 52:613-619. [DOI] [PubMed] [Google Scholar]
- 4.Alba, B. M., J. A. Leeds, C. Onufryk, C. Z. Lu, and C. A. Gross. 2002. DegS and YaeL participate sequentially in the cleavage of RseA to activate the sigma(E)-dependent extracytoplasmic stress response. Genes Dev. 16:2156-2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Anderson, M. T., and S. K. Armstrong. 2004. The BfeR regulator mediates enterobactin-inducible expression of Bordetella enterobactin utilization genes. J. Bacteriol. 186:7302-7311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Antoine, R., S. Alonso, D. Raze, L. Coutte, S. Lesjean, E. Willery, C. Locht, and F. Jacob-Dubuisson. 2000. New virulence-activated and virulence-repressed genes identified by systematic gene inactivation and generation of transcriptional fusions in Bordetella pertussis. J. Bacteriol. 182:5902-5905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beck, N. A., E. S. Krukonis, and V. J. DiRita. 2004. TcpH influences virulence gene expression in Vibrio cholerae by inhibiting degradation of the transcription activator TcpP. J. Bacteriol. 186:8309-8316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blattner, F. R., G. Plunkett III, C. A. Bloch, N. T. Perna, V. Burland, M. Riley, J. Collado-Vides, J. D. Glasner, C. K. Rode, G. F. Mayhew, J. Gregor, N. W. Davis, H. A. Kirkpatrick, M. A. Goeden, D. J. Rose, B. Mau, and Y. Shao. 1997. The complete genome sequence of Escherichia coli K-12. Science 277:1453-1474. [DOI] [PubMed] [Google Scholar]
- 9.Bohn, C., J. Collier, and P. Bouloc. 2004. Dispensable PDZ domain of Escherichia coli YaeL essential protease. Mol. Microbiol. 52:427-435. [DOI] [PubMed] [Google Scholar]
- 10.Bramkamp, M., L. Weston, R. A. Daniel, and J. Errington. 2006. Regulated intramembrane proteolysis of FtsL protein and the control of cell division in Bacillus subtilis. Mol. Microbiol. 62:580-591. [DOI] [PubMed] [Google Scholar]
- 11.Braun, V., and H. Killmann. 1999. Bacterial solutions to the iron-supply problem. Trends Biochem. Sci. 24:104-109. [DOI] [PubMed] [Google Scholar]
- 12.Braun, V., and S. Mahren. 2005. Transmembrane transcriptional control (surface signalling) of the Escherichia coli Fec type. FEMS Microbiol. Rev. 29:673-684. [DOI] [PubMed] [Google Scholar]
- 13.Braun, V., S. Mahren, and A. Sauter. 2006. Gene regulation by transmembrane signaling. Biometals 19:103-113. [DOI] [PubMed] [Google Scholar]
- 14.Brown, M. S., J. Ye, R. B. Rawson, and J. L. Goldstein. 2000. Regulated intramembrane proteolysis: a control mechanism conserved from bacteria to humans. Cell 100:391-398. [DOI] [PubMed] [Google Scholar]
- 15.Carroll, P. A., K. T. Tashima, M. B. Rogers, V. J. DiRita, and S. B. Calderwood. 1997. Phase variation in tcpH modulates expression of the ToxR regulon in Vibrio cholerae. Mol. Microbiol. 25:1099-1111. [DOI] [PubMed] [Google Scholar]
- 16.Chen, J. C., A. K. Hottes, H. H. McAdams, P. T. McGrath, P. H. Viollier, and L. Shapiro. 2006. Cytokinesis signals truncation of PodJ polarity factor by a cell cycle-regulated protease. EMBO J. 25:377-386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen, J. C., P. H. Viollier, and L. Shapiro. 2005. A membrane metalloprotease participates in the sequential degradation of a Caulobacter polarity determinant. Mol. Microbiol. 55:1085-1103. [DOI] [PubMed] [Google Scholar]
- 18.Cotter, P. A., and J. F. Miller. 1994. BvgAS-mediated signal transduction: analysis of phase-locked regulatory mutants of Bordetella bronchiseptica in a rabbit model. Infect. Immun. 62:3381-3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dartigalongue, C., H. Loferer, and S. Raina. 2001. EcfE, a new essential inner membrane protease: its role in the regulation of heat shock response in Escherichia coli. EMBO J. 20:5908-5918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.De Las Peñas, A., L. Connolly, and C. A. Gross. 1997. σE is an essential sigma factor in Escherichia coli. J. Bacteriol. 179:6862-6864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.DiRita, V. J., C. Parsot, G. Jander, and J. J. Mekalanos. 1991. Regulatory cascade controls virulence in Vibrio cholerae. Proc. Natl. Acad. Sci. USA 88:5403-5407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Douchin, V., C. Bohn, and P. Bouloc. 2006. Down-regulation of porins by a small RNA bypasses the essentiality of the regulated intramembrane proteolysis protease RseP in Escherichia coli. J. Biol. Chem. 281:12253-12259. [DOI] [PubMed] [Google Scholar]
- 23.Ellermeier, C. D., and R. Losick. 2006. Evidence for a novel protease governing regulated intramembrane proteolysis and resistance to antimicrobial peptides in Bacillus subtilis. Genes Dev. 20:1911-1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fetherston, J. D., S. W. Bearden, and R. D. Perry. 1996. YbtA, an AraC-type regulator of the Yersinia pestis pesticin/yersiniabactin receptor. Mol. Microbiol. 22:315-325. [DOI] [PubMed] [Google Scholar]
- 25.Figurski, D. H., and D. R. Helinski. 1979. Replication of an origin-containing derivative of plasmid RK2 dependent on a plasmid function provided in trans. Proc. Natl. Acad. Sci. USA 76:1648-1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Flynn, J. M., I. Levchenko, R. T. Sauer, and T. A. Baker. 2004. Modulating substrate choice: the SspB adaptor delivers a regulator of the extracytoplasmic-stress response to the AAA+ protease ClpXP for degradation. Genes Dev. 18:2292-2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goldberg, M. B., S. A. Boyko, and S. B. Calderwood. 1991. Positive transcriptional regulation of an iron-regulated virulence gene in Vibrio cholerae. Proc. Natl. Acad. Sci. USA 88:1125-1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guth, B. E., C. L. Pickett, E. M. Twiddy, R. K. Holmes, T. A. Gomes, A. A. Lima, R. L. Guerrant, B. D. Franco, and L. R. Trabulsi. 1986. Production of type II heat-labile enterotoxin by Escherichia coli isolated from food and human feces. Infect. Immun. 54:587-589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guzman, L. M., D. Belin, M. J. Carson, and J. Beckwith. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 177:4121-4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Halliwell, B., and J. M. Gutteridge. 1984. Oxygen toxicity, oxygen radicals, transition metals and disease. Biochem. J. 219:1-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hantke, K. 1987. Selection procedure for deregulated iron transport mutants (fur) in Escherichia coli K12: fur not only affects iron metabolism. Mol. Gen. Genet. 210:135-139. [DOI] [PubMed] [Google Scholar]
- 32.Heidelberg, J. F., J. A. Eisen, W. C. Nelson, R. A. Clayton, M. L. Gwinn, R. J. Dodson, D. H. Haft, E. K. Hickey, J. D. Peterson, L. Umayam, S. R. Gill, K. E. Nelson, T. D. Read, H. Tettelin, D. Richardson, M. D. Ermolaeva, J. Vamathevan, S. Bass, H. Qin, I. Dragoi, P. Sellers, L. McDonald, T. Utterback, R. D. Fleishmann, W. C. Nierman, O. White, S. L. Salzberg, H. O. Smith, R. R. Colwell, J. J. Mekalanos, J. C. Venter, and C. M. Fraser. 2000. DNA sequence of both chromosomes of the cholera pathogen Vibrio cholerae. Nature 406:477-483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heinrich, J., and T. Wiegert. 2006. YpdC determines site-1 degradation in regulated intramembrane proteolysis of the RsiW anti-sigma factor of Bacillus subtilis. Mol. Microbiol. 62:566-579. [DOI] [PubMed] [Google Scholar]
- 34.Kanehara, K., Y. Akiyama, and K. Ito. 2001. Characterization of the yaeL gene product and its S2P-protease motifs in Escherichia coli. Gene 281: 71-79. [DOI] [PubMed] [Google Scholar]
- 35.Kanehara, K., K. Ito, and Y. Akiyama. 2002. YaeL (EcfE) activates the σE pathway of stress response through a site-2 cleavage of anti-σE, RseA. Genes Dev. 16:2147-2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kanehara, K., K. Ito, and Y. Akiyama. 2003. YaeL proteolysis of RseA is controlled by the PDZ domain of YaeL and a Gln-rich region of RseA. EMBO J. 22:6389-6398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Keen, N. T., S. Tamaki, D. Kobayashi, and D. Trollinger. 1988. Improved broad-host-range plasmids for DNA cloning in gram-negative bacteria. Gene 70:191-197. [DOI] [PubMed] [Google Scholar]
- 38.King, N. D., A. E. Kirby, and T. D. Connell. 2005. Transcriptional control of the rhuIR-bhuRSTUV heme acquisition locus in Bordetella avium. Infect. Immun. 73:1613-1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kirby, A. E., D. J. Metzger, E. R. Murphy, and T. D. Connell. 2001. Heme utilization in Bordetella avium is regulated by RhuI, a heme-responsive extracytoplasmic function sigma factor. Infect. Immun. 69:6951-6961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kuhl, S. A., R. P. McCreary, J. D. Bannan, and R. L. Friedman. 1990. Isolation and characterization of the recA gene of Bordetella pertussis. Mol. Microbiol. 4:1165-1172. [DOI] [PubMed] [Google Scholar]
- 41.Litwin, C. M., and J. Quackenbush. 2001. Characterization of a Vibrio vulnificus LysR homologue, HupR, which regulates expression of the haem uptake outer membrane protein, HupA. Microb. Pathog. 31:295-307. [DOI] [PubMed] [Google Scholar]
- 42.Lonetto, M. A., K. L. Brown, K. E. Rudd, and M. J. Buttner. 1994. Analysis of the Streptomyces coelicolor sigE gene reveals the existence of a subfamily of eubacterial RNA polymerase sigma factors involved in the regulation of extracytoplasmic functions. Proc. Natl. Acad. Sci. USA 91:7573-7577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lu, S., S. Cutting, and L. Kroos. 1995. Sporulation protein SpoIVFB from Bacillus subtilis enhances processing of the sigma factor precursor pro-σK in the absence of other sporulation gene products. J. Bacteriol. 177:1082-1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Makinoshima, H., and M. S. Glickman. 2006. Site-2 proteases in prokaryotes: regulated intramembrane proteolysis expands to microbial pathogenesis. Microbes Infect. 8:1882-1888. [DOI] [PubMed] [Google Scholar]
- 45.Marcus, H., J. M. Ketley, J. B. Kaper, and R. K. Holmes. 1990. Effects of DNase production, plasmid size, and restriction barriers on transformation of Vibrio cholerae by electroporation and osmotic shock. FEMS Microbiol. Lett. 56:149-154. [DOI] [PubMed] [Google Scholar]
- 46.Matson, J. S., and V. J. DiRita. 2005. Degradation of the membrane-localized virulence activator TcpP by the YaeL protease in Vibrio cholerae. Proc. Natl. Acad. Sci. USA 102:16403-16408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Maunsell, B., C. Adams, and F. O'Gara. 2006. Complex regulation of AprA metalloprotease in Pseudomonas fluorescens M114: evidence for the involvement of iron, the ECF sigma factor, PbrA and pseudobactin M114 siderophore. Microbiology 152:29-42. [DOI] [PubMed] [Google Scholar]
- 48.Michel, L., N. Gonzalez, S. Jagdeep, T. Nguyen-Ngoc, and C. Reimmann. 2005. PchR-box recognition by the AraC-type regulator PchR of Pseudomonas aeruginosa requires the siderophore pyochelin as an effector. Mol. Microbiol. 58:495-509. [DOI] [PubMed] [Google Scholar]
- 49.Ochs, M., S. Veitinger, I. Kim, D. Welz, A. Angerer, and V. Braun. 1995. Regulation of citrate-dependent iron transport of Escherichia coli: fecR is required for transcription activation by FecI. Mol. Microbiol. 15:119-132. [DOI] [PubMed] [Google Scholar]
- 50.Parkhill, J., M. Sebaihia, A. Preston, L. D. Murphy, N. Thomson, D. E. Harris, M. T. G. Holden, C. M. Churcher, S. D. Bentley, K. L. Mungall, A. M. Cerdeno-Tarraga, L. Temple, K. James, B. Harris, M. A. Quail, M. Achtman, R. Atkin, S. Baker, D. Basham, N. Bason, I. Cherevach, T. Chillingworth, M. Collins, A. Cronin, P. Davis, J. Doggett, T. Feltwell, A. Goble, N. Hamlin, H. Hauser, S. Holroyd, K. Jagels, S. Leather, S. Moule, H. Norberczak, S. O'Neil, D. Ormond, C. Price, E. Rabbinowitsch, S. Rutter, M. Sanders, D. Saunders, K. Seeger, S. Sharp, M. Simmonds, J. Skelton, R. Squares, S. Squares, K. Stevens, L. Unwin, S. Whitehead, B. G. Barrell, and D. J. Maskell. 2003. Comparative analysis of the genome sequences of Bordetella pertussis, Bordetella parapertussis and Bordetella bronchiseptica. Nat. Genet. 35:32-40. [DOI] [PubMed] [Google Scholar]
- 51.Pradel, E., N. Guiso, and C. Locht. 1998. Identification of AlcR, an AraC-type regulator of alcaligin siderophore synthesis in Bordetella bronchiseptica and Bordetella pertussis. J. Bacteriol. 180:871-880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Reyes-Ramirez, F., P. Dobbin, G. Sawers, and D. J. Richardson. 2003. Characterization of transcriptional regulation of Shewanella frigidimarina Fe(III)-induced flavocytochrome c reveals a novel iron-responsive gene regulation system. J. Bacteriol. 185:4564-4571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Riley, M., T. Abe, M. B. Arnaud, M. K. Berlyn, F. R. Blattner, R. R. Chaudhuri, J. D. Glasner, T. Horiuchi, I. M. Keseler, T. Kosuge, H. Mori, N. T. Perna, G. Plunkett III, K. E. Rudd, M. H. Serres, G. H. Thomas, N. R. Thomson, D. Wishart, and B. L. Wanner. 2006. Escherichia coli K-12: a cooperatively developed annotation snapshot—2005. Nucleic Acids Res. 34:1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rossi, M.-S., A. Paquelin, J. M. Ghigo, and C. Wandersman. 2003. Haemophore-mediated signal transduction across the bacterial cell envelope in Serratia marcescens: the inducer and the transported substrate are different molecules. Mol. Microbiol. 48:1467-1480. [DOI] [PubMed] [Google Scholar]
- 55.Schobel, S., S. Zellmeier, W. Schumann, and T. Wiegert. 2004. The Bacillus subtilis σW anti-sigma factor RsiW is degraded by intramembrane proteolysis through YluC. Mol. Microbiol. 52:1091-1105. [DOI] [PubMed] [Google Scholar]
- 56.Sexton, R., P. R. Gill, Jr., M. J. Callanan, D. J. O'Sullivan, D. N. Dowling, and F. O'Gara. 1995. Iron-responsive gene expression in 1943708M114: cloning and characterization of a transcription-activating factor, PbrA. Mol. Microbiol. 15:297-306. [DOI] [PubMed] [Google Scholar]
- 57.Vanderpool, C. K., and S. K. Armstrong. 2001. The Bordetella bhu locus is required for heme iron utilization. J. Bacteriol. 183:4278-4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vanderpool, C. K., and S. K. Armstrong. 2003. Heme-responsive transcriptional activation of Bordetella bhu genes. J. Bacteriol. 185:909-917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vanderpool, C. K., and S. K. Armstrong. 2004. Integration of environmental signals controls expression of Bordetella heme utilization genes. J. Bacteriol. 186:938-948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vasil, M. L., U. A. Ochsner, Z. Johnson, J. A. Colmer, and A. N. Hamood. 1998. The Fur-regulated gene encoding the alternative sigma factor PvdS is required for iron-dependent expression of the LysR-type regulator PtxR in Pseudomonas aeruginosa. J. Bacteriol. 180:6784-6788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wennerhold, J., A. Krug, and M. Bott. 2005. The AraC-type regulator RipA represses aconitase and other iron proteins from Corynebacterium under iron limitation and is itself repressed by DtxR. J. Biol. Chem. 280:40500-40508. [DOI] [PubMed] [Google Scholar]
- 62.Yang, J., I. Sangwan, and R. O'Brian. 2006. The Bradyrhizobium japonicum Fur protein is an iron-responsive regulator in vivo. Mol. Genet. Genomics 276:555-564. [DOI] [PubMed] [Google Scholar]
- 63.Yu, R. R., and V. J. DiRita. 1999. Analysis of an autoregulatory loop controlling ToxT, cholera toxin, and toxin-coregulated pilus production in Vibrio cholerae. J. Bacteriol. 181:2584-2592. [DOI] [PMC free article] [PubMed] [Google Scholar]