

Abstract
Pocket proteins and cyclin-dependent kinase (CDK) inhibitors negatively regulate cell proliferation and can promote differentiation. However, which members of these gene families, which cell type they interact in, and what they do to promote differentiation in that cell type during mouse development are largely unknown. To identify the cell types in which p107 and p27 interact, we generated compound mutant mice. These mice were null for p107 and had a deletion in p27 that prevented its binding to cyclin-CDK complexes. Although a fraction of these animals survived into adulthood and looked similar to single p27 mutant mice, a larger number of animals died at birth or within a few weeks thereafter. These animals displayed defects in chondrocyte maturation and endochondral bone formation. Proliferation of chondrocytes was increased, and ectopic ossification was observed. Uncommitted mouse embryo fibroblasts could be induced into the chondrocytic lineage ex vivo, but these cells failed to mature normally. These results demonstrate that p27 carries out overlapping functions with p107 in controlling cell cycle exit during chondrocyte maturation. The phenotypic similarities between p107−/− p27D51/D51 and p107−/− p130−/− mice and the cells derived from them suggest that p27 and p130 act in an analogous pathway during chondrocyte maturation.
Differentiation of mammalian cells depends on the establishment of a specific transcriptional program that leads to a specialized function. Establishment of this program is generally coordinated with, or dependent upon, the cell exiting the cell cycle. Cell cycle exit is governed by the interactions of three families of proteins: the cyclin-dependent kinases (CDKs), the CDK inhibitors (CKI), and the pocket proteins (15, 65). CDKs promote proliferation in part by phosphorylating the pocket proteins (Rb, p107, and p130Rb2), leading to changes in their ability to interact with E2F transcription factors or decreasing their stability (6, 12). Opposing the activity of CDKs are the CKI. The Ink4 family proteins (p15, p16, p18, and p19) bind to cyclin D-CDK complexes or compete with cyclin D for binding to CDK subunits. Members of the Kip family (p21, p27, and p57) bind to and inhibit cyclin-CDK2 complexes (53).
The precise combinations of cyclins, CKI, and pocket proteins used to initiate or maintain cell cycle exit is cell type and signal specific. In some cases, a single protein is important, despite the fact that other family members are expressed. For example, p27-deficient oligodendrocyte progenitor cells and osteoblasts fail to exit the cell cycle in a timely manner, leading to a delay in differentiation (7, 8, 17). p21 and p57 are coexpressed in these cell types, respectively (8, 20, 62). In other cases, similar family members act in a redundant manner, and proliferative effects are more obvious when both are deleted. For example, p107 and p130 collaborate to control chondrocyte proliferation and maturation (13, 33). In yet other cases, deletion of members from different families is required to unmask proliferative phenotypes. For example, neuronal cells either fail to exit the cell cycle or reenter the cycle in p19 and p27 double knockout mice (72). Additionally, codeletion of p130 and p27 prevents cell cycle exit in angiogenic stem and progenitor cells, hindering their export from the bone marrow (67). Consequently, mouse knockouts are useful for identifying the specific cell type in which particular combinations of CKI and pocket proteins are required for proper development.
Cyclins, CKI, and pocket proteins can also affect the differentiation program independently of any role in cell cycle exit. p21 is required for the timely differentiation of oligodendrocyte progenitor cells, but not for growth arrest (70). In Xenopus laevis Muller glia, a mutant of p21 that fails to interact with cyclin D-CDK complexes can replace this differentiation-promoting function in p27Xic1 knockdown cells (44). Likewise, p27Xic1 plays a role in muscle and neuronal differentiation (63, 64). The carboxyl-half of p27, which does not bind cyclin-CDK complexes, plays a role in neuronal differentiation and cell migration (43). Finally, the interaction of Rb with Runx2/Cbfa1 regulates osteoblast differentiation (60). Additional possible targets have also emerged from protein interaction studies. For example, cyclin D1 can interact with a corepressor complex important in hormone receptor-dependent gene regulation (3, 46, 47, 73). Pocket proteins can interact with the Id transcription factors, chromatin remodeling enzymes, and other transcription factors in differentiating cells (22, 38). p21 can interact with Stat3, CBP/p300, and CEBPα, among a number of others (16, 45, 56). p57Kip2 interacts with B-myb (27).
In this work, we identified cell types in which a genetic interaction between p27 and p107 could be observed. We report that p107−/− p27D51/D51 mice display increased neonatal lethality and defects in chondrocyte maturation both in vivo and in vitro and that this is associated with a failure of cells to appropriately exit the cell cycle and initiate a differentiation program. The similarities between the phenotypes of p107−/− p130−/− and p107−/− p27D51/D51 mice suggest that p27 and p130 carry out qualitatively similar functions during chondrocyte maturation.
MATERIALS AND METHODS
Mice.
Generation of p130−/− (13), p107−/− (34), and p27D51/D51 (29) mice has been reported. Intercrossing p107−/− p27D51/+ animals generated p107−/− p27D51/D51 mice. p130−/− p27D51/D51 mice were generated by intercrossing p130−/− p27D51/+ animals. p107−/− p130−/− mice were generated by intercrossing p107+/− p130−/− mice. All these mice were in a mixed C57BL6/SvJ background.
Organ weights and thymocyte staining.
The weights of the brain, spleen, and kidney were determined as previously described (29). To determine the numbers of thymocytes at each developmental stage, the thymus was surgically removed, washed in phosphate-buffered saline (PBS), and mechanically disrupted. Pieces of tissue were removed by centrifugation and further filtered through a nylon mesh. Cell counts were performed in a hemocytometer. Single-cell suspensions were subsequently incubated with fluorescent antibodies against CD4 and CD8 (BD Pharmingen), and the number of cells that were CD4− CD8−, CD4+ CD8−, CD4− CD8+, and CD4+ CD8+ was determined by fluorescence-activated cell sorter analysis.
Morphometric analysis of long bone and sternal development.
Newborn animal cartilage and bone were stained with alcian blue and alizarin red as described elsewhere (25). Briefly, newborn animals were fixed overnight in 95% ethanol and then stained in alcian blue (1 mg per ml of 40% ethanol-60% glacial acetic acid) for 24 h at room temperature. These animals were then washed overnight in 100% ethanol and rehydrated through a series of ethanol dilutions (75%, 50%, and 25%; 2 h each), transferred to water, and then destained overnight in 0.5% KOH. The alcian blue-stained embryos were counterstained for 24 h in alizarin red (2 ml of a 0.1% solution of alizarin red in 8 ml 0.5% KOH) and then destained again in 0.5% KOH. Prior to imaging, the embryos were cleared in staged glycerol solutions (25% plus one drop of H2O2 to aid clearing, and then 50% and 75%, 24 h each) and stored in 80% glycerol.
Forelimb measurements were acquired with a high-resolution three-dimensional (3D) digitizer. This 3D measurement system consists of a three-axis Mitutoyo measuring microscope (MTI Corporation, Los Angeles, CA) fitted with high-precision linear scales in each x, y, and z axis, a noncontact measurement video-based zoom optical head (RAM Optical Instrumentation, Inc., Huntington Beach, CA) that included a through axis electronic crosshair, and a Hitachi color charge-coupled device camera integrated to a PC. Digital readout was transferred to 3D analytical measurement software (Measuregraph 1-2-3; RAM Optical Instrumentation, Inc.). All measurements were made with an accuracy of ±4 μm (0.004 mm) and recorded to the nearest 0.01 mm. The 3D capability of this instrument is essential for newborn mouse skeletal measurements at this accuracy, as it allows refocusing in the z axis to ensure correct positioning of the crosshairs, particularly on rounded surfaces. Each element was dissected free of its articulations and measured. Measurements in millimeters were taken as follows. The length of the humerus was taken as the maximum proximal-distal distance parallel with the anterior border (from the head to the trochlea), and the width was measured at midshaft (the 50% point) perpendicular to the length axis (from anterior to posterior, thus avoiding the deltoid tuberosity in the measurement). The length of the radius was taken as the maximum proximal-distal distance parallel with the interosseous border (from the head to the styloid process), and the width was measured at midshaft (the 50% point) perpendicular to the length axis. The length of the ulna was taken as the maximum proximal-distal distance parallel with the interosseous border (from the olecranon to the styloid process), and the width was measured at midshaft (the 50% point) perpendicular to the length axis.
In vivo BrdU staining.
Cell proliferation was assessed by incorporation of bromodeoxyuridine (BrdU; Sigma). Pregnant females were injected intraperitoneally with a 5-mg/ml BrdU solution in PBS (approximately 50 μg/g body weight). After 2 h, embryos were harvested and fixed in 4% paraformaldehyde, embedded in paraffin, and sectioned at 8 μm. Sections were stained with hematoxylin and eosin or immunostained with anti-BrdU antibody (Roche) as previously published (52). The number of BrdU-positive and the total number of proliferating chondrocytes in the fourth and fifth sternabrae were counted.
Serum starvation and BrdU incorporation in primary MEFs.
Mouse embryo fibroblasts (MEFs) were isolated and cultured as described elsewhere (66). All experiments were performed with MEFs from either passage 3 or 4 and plated at approximately 104 cells/cm2. For serum starvation, cells were cultured 24 h after plating and then washed twice with PBS and incubated for 60 h in medium containing 0.1% fetal bovine serum. For most experiments, a fraction of each harvested culture was pulsed with [3H]thymidine (40 μCi/ml) to verify growth arrest.
To measure the percentage of proliferating cells, BrdU incorporation was determined. In parallel to the cultures developed for protein and RNA extraction, cells were plated in two-well Permanox chamber slides (Nunc) and pulsed with BrdU (100 μM) for 4 h, washed once with PBS, and fixed with 70% ethanol for 30 min in the dark. After washing with PBS, the DNA was denatured by treatment with NaOH for 2 min. The cells were washed with 0.1 M Na2B4O7, pH 8.5, and then with PBS. The slides were then incubated for 30 min in the dark with 20 μl of fluorescein isothiocyanate-conjugated BrdU antibody (BD Pharmingen) diluted with 50 μl PBS containing 0.5% bovine serum albumin and 0.5% Tween 20. After several washes with PBS, the DNA was stained with 4′,6′-diamidino-2-phenylindole (5 μg/ml) for 5 to 10 min and washed with PBS again. Slides were mounted, and BrdU-positive cells were visualized with a fluorescence microscope. A total of at least 100 cells in five different fields were counted per sample.
Immunoblotting and immunokinase assay.
For the measurement of histone H1 kinase activity, cells were lysed at a concentration of 8 × 106 per 100 μl of NP-40-RIPA buffer as we previously described (30). The equivalent of 2 × 106 cells were immunoprecipitated with either cyclin E antiserum (30), cyclin A antibodies (H432; Santa Cruz), or rabbit anti-mouse antibodies (Zymed), and kinase assays were carried out as previously described (30).
To determine the amounts of CDK1 (C19), CDK2 (M2), CDK4 (C22), CDK6 (C21), cyclin D1 (72-13G), cyclin D3 (C16), cyclin E (M20), cyclin A2 (H432), p21 (F5), and p27 (C19), we extracted proteins in 20 mM HEPES-KOH pH 7.6, 5 mM KCl, 0.5 mM MgCl2, 100 mM NaCl containing 2 mM phenylmethylsulfonyl fluoride and 0.5 mM dithiothreitol as we previously described (57). To determine the amount of p130 (C20), p107 (C18), E2F4 (C20), and E2F5 (C20), proteins were extracted in a buffer containing 100 mM HEPES-KOH pH 7.5, 500 mM NaCl, 10 mM EDTA, 0.2% Triton X-100, 1 mM dithiothreitol, and 1 mM phenylmethylsulfonyl fluoride as described elsewhere (40). Ten to 80 μg of extract was resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and proteins were transferred to polyvinylidene difluoride membranes and blotted as we previously described (57). All antibodies were from Santa Cruz Biotechnology except for Rb (G3-245), which was purchased from Pharmingen.
Northern blotting.
Total RNA was isolated using the RNeasy kit (QIAGEN). Poly(A)+ RNA was selected with the Poly(A) Pure kit (Ambion) following the manufacturer's instructions. Eight μg of total RNA (for CDK1, cyclin A, and ARPP0) or 0.8 μg poly(A)+ RNA (for thymidylate synthase [TS], dihydrofolate reductase [DHFR], B-myb, E2F4, and p130) was denatured and resolved on agarose gels using the Northern Max-Gly kit (Ambion). RNA was transferred to a BrightStar-Plus nylon membrane (Ambion) overnight. Following UV cross-linking, the membrane was prehybridized with ULTRAhyb (Ambion) in a roller bottle for 30 min at 42°C. Full-length cDNA clones (ATCC) were excised from the vector, gel purified, and random-prime labeled with [α-32P]dCTP (Amersham) using the Prime-It II kit (Stratagene). Labeled probes were purified using Centrisep spin columns (Princeton Separations), heated at 90°C for 10 min, added to ULTRAhyb (106 cpm/ml), and incubated with membranes overnight at 42°C.
Chondrogenic differentiation and expression analysis.
High-density micromass cultures from mouse embryonic fibroblasts were prepared, and chondrogenic differentiation was induced by addition of BMP2 as previously described (21, 37). Briefly, 1 × 105 passage 2 primary mouse embryo fibroblasts were plated in 10-μl drops and cultured in the presence of recombinant human BMP2 (a kind gift from John Wozney, Wyeth Research, Cambridge, MA). Cells were either fixed with 2% formaldehyde for 10 min for alcian blue or alkaline phosphatase staining as described previously (21, 37) or harvested in TRIzol reagent (Invitrogen) for RNA preparation following the manufacturer's instructions. Protein extracts were prepared by sonicating cells in DIP buffer (48) as we previously described (57).
RNA was purified by RNase-free DNase I treatment, and cDNA was prepared from 1 μg of RNA using a First Strand synthesis kit (Invitrogen). Relative transcript levels were measured by quantitative reverse transcription-PCR (qRT-PCR) using SYBR Green 2× master mix (Applied Biosystems) in a 25-μl reaction volume. Primer sequences were as follows: collagen type 2a1 (5′CTGGAATGTCCTCTGGCGA and 5′TGAGGCAGTCTGGGTCTTCAC); sox 9 (5′GAGGCCACGGAAGAGACTCA and 5′CAGCGCCTTGAAGATAGCATT); Indian hedgehog (5′TCACCCCCAACTACAATCCC and 5′CCGTGTTCTCCTCGTCCTTG); collagen type 10a1 (5′CCTGCAGCAAAGGAAAACTC and 5′TGTGGTAGTGGTGGAGGACA); Runx2 (5′CGGCCCTCCCTGAACTCT and 5′TGCCTGCCTGGGATCTGTA); alkaline phosphatase (5′TTGTGCCAGAGAAAGAGAGAGA and 5′GTTTCAGGGCATTTTTCAAGGT); osteocalcin (5′CTGACAAAGCCTTCATGTCCAA and 5′GCGGGCGAGTCTGTTCACTA); histone H4 (5′CCAGCTGGTGTTTCAGATTACA3′ and 5′ACCCTTGCCTAGACCCTTTC3′); cyclin A2 (5′ACAGAGCTGGCCTGAGTGA3′ and 5′TTGACTGTTGGGCATGTTGT3′); CDK1 (5′GGCAGTTCATGGATTCTTCACTC3′ and 5′GCCAGTTTGATTGTTCCTTTGTC3′). Gene expression for each gene was normalized to glyceraldehyde 3-phosphate dehydrogenase levels as detected by primers from Applied Biosystems.
RESULTS
The phenotypes of adult p27D51/D51 and p107−/− p27D51/D51 mice are similar.
To determine whether p107 and p27 act in a redundant manner to control development of any particular cell lineage in mice, we intercrossed p107−/− p27+/D51 male mice with p107−/− p27+/D51 female mice. p107−/− mice had no significant phenotype in a mixed 129SvJ/C57BL6 background (13). On the other hand, p27D51/D51 mice show enhanced growth, multiorgan hyperplasia, female infertility, and the spontaneous development of pituitary intermediate lobe adenoma in this background (29). The D51 allele encodes a truncated protein that does not bind cyclin-CDK complexes, and mice expressing this allele have phenotypes similar to those described for p27-null animals (19, 41).
The phenotype of adult p107−/− p27D51/D51mice was generally similar to that observed in p27D51/D51 animals. The weight differences were more obvious in female mice than male mice (Fig. 1). p107−/− p27D51/D51mice generally weighed between the normal weight of wild-type (and p107−/−) mice and the increased weight of p27D51/D51 mice. However, a number of p107−/− p27D51/D51mice weighed the same or less than their siblings until approximately the third week of age, by which time they begin to overtake them. Additionally, females were infertile, and no litters were obtained from them despite their being able to mate.
FIG. 1.

Weights of mice. (A) The weights of two representative male and female mice of each genotype are shown over the first 8 to 10 weeks of age. (B) The average weight and standard deviation are indicated in the graphs for male and female mice of each genotype (n > 10 for each genotype).
The average wet weights of the kidney and spleen were significantly greater (P < 0.05) in the p27 knockout and the double mutant mice compared to wild-type or p107 single mutant mice. The brain weights were comparable (Fig. 2A). However, the weight of each individual organ relative to the total weight of the animal from which it was obtained was similar, indicating that combining p107 deficiency with the p27 mutation did not affect the proportional growth previously reported (19, 29, 41).
FIG. 2.
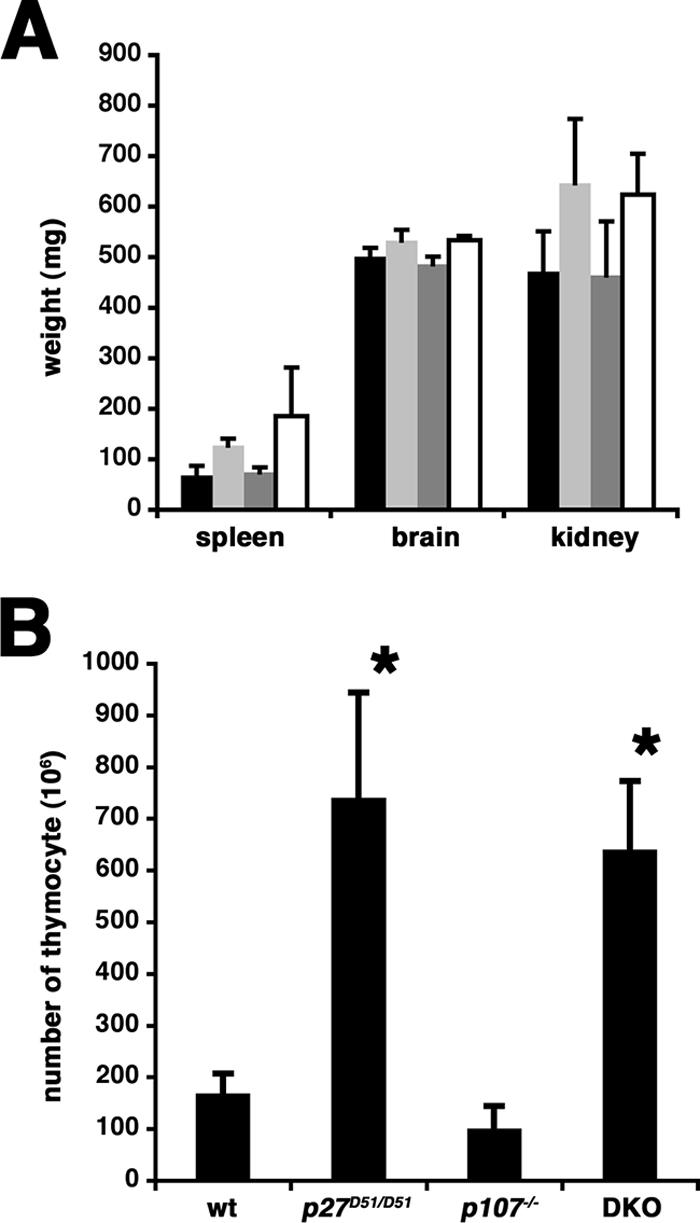
(A) Organ weights. The organs indicated were dissected from 23- to 41-week-old female mice, and the average weight and standard deviation were determined. Black, wild type, n = 5; light gray, p27D51/D51, n = 6; dark gray, p107−/−, n = 5; white, p107−/− p27D51/D51, n = 3. (B) Thymocytes from 8- to 10-week-old mice were collected, and the total cell number was determined. The mean number of thymocytes and the standard deviation are shown. Asterisks indicate those values significantly different (P < 0.01) from wild-type mice based on a two-tailed Student's t test.
The cellularity of the thymus, which was much greater and disproportionate to the body weight of the animal, was increased in female p107−/− p27D51/D51mice, almost to the same extent as that seen in p27D51/D51 mice (Fig. 2B). The number of thymocytes was nearly equivalent in wild-type and p107−/− mice (wild type, n = 5; p107−/−, n = 5). In p27D51/D51 mice (n = 4), consistent with what we reported before, we saw a roughly threefold increase in thymocyte number, and this was comparable to the increase in p107−/− p27D51/D51mice (n = 3). Furthermore, the genotype did not affect the distribution of thymocytes among the double-negative, double-positive, and single-positive CD4 or CD8 subsets (data not shown). Similar results were observed with 13- to 15-week-old female mice and in 8- to 10-week-old and 13- to 15-week-old male mice. These results suggest that there was limited genetic interplay between p27 and p107 in control of body mass and T-cell lymphogenesis.
Additionally, both male and female p107−/− p27D51/D51mice developed intermediate lobe pituitary adenomas reactive with antibodies against adrenocorticotropin but not growth hormone or prolactin. These tumors were histologically similar to the tumors arising in p27D51/D51 mice. In contrast to the p130−/− p27D51/D51 mice (67), there was no obvious change in the vascular architecture of the pituitary.
Nevertheless, although p107 deficiency did not exacerbate the phenotype of surviving p27D51/D51 mice, there was some interaction between these gene products, as the number of p107−/− p27D51/D51 animals identified at 3 weeks of age was significantly lower than expected (Table 1).
TABLE 1.
Effect of p27 dose on survival of embryonic and neonatal p107-deficient animals
| Age | No. of p107-deficient animals of genotype
|
No. of litters | χ2 test P valuea | ||
|---|---|---|---|---|---|
| p27+/+ | p27D51/+ | p27D51/D51 | |||
| Neonatal day 21 | 50 | 89 | 15 | 41 | <0.001 |
| Neonatal day 1 | 34 | 38 | 16 | 11 | 0.01 |
| Embryonic day 17.5 | 13 | 11 | 7 | 5 | 0.08 |
| Embryonic day 16.5 | 10 | 28 | 15 | 7 | 0.33 |
| Embryonic day 15.5 | 10 | 22 | 12 | 5 | 0.30 |
| Embryonic day 14.5 | 17 | 30 | 18 | 10 | 0.53 |
| Embryonic day 13.5 | 7 | 21 | 5 | 5 | 0.14 |
The χ2 test was based on an expected Mendelian inheritance pattern for the intercrossing of p107−/− p27D51/+ mice.
Neonatal lethality is associated with the loss of p107 and p27.
Because of the reduced frequency at which p107−/− p27D51/D51 animals were identified at weaning, we collected the dead and moribund newborns from 11 litters of the p107−/− p27+/D51 intercrosses. Of these pups, 95% were homozygous for the mutant p27 allele, and the remaining were heterozygous. Fostering newborn pups to wild-type mothers did not increase survival, suggesting that lethality was an intrinsic defect of the pups.
To determine when decreased viability first manifested, we determined the genotypes of embryos at various days postconception using timed pregnancies. Double knockout embryos were present at the expected Mendelian ratios from day 13.5 to day 16.5, with a slight, albeit statistically insignificant, decline at day 17.5 (Table 1). This phenotype was reminiscent of the one reported for p107−/− p130−/− mice (13), raising the possibility that the combined loss of p107 and p27 was qualitatively similar, but perhaps quantitatively weaker, than the loss of p107 and p130.
Reducing p130 exacerbates neonatal lethality in p107−/− p27D51/D51 mice.
The relationship between pocket proteins and CKI is complex, with each family of proteins affecting the expression levels of the other and compensating for each other to control CDK activity (10, 11, 35, 65). For example, p130 can be recruited into cyclin E-CDK2 complexes in Kip-deficient MEFs (11). Additionally, CDK-dependent ubiquitin-dependent pathways increase p130 turnover (4, 59).
Even though p107−/− p130+/− animals have diminished long-term viability and a modest reduction in growth, p107 and p130 act redundantly, and a single allele is sufficient to ensure neonatal viability (13). As we were interested in whether p27 and p130 compensated for each other, we asked if reducing the dose of p130 in p107−/− p27D51/D51 mice would exacerbate the viability phenotype. We did not recover any neonatal p107−/− p130+/− p27D51/D51 pups from nine crosses (n = 35) (Table 2), and the number of p107−/− p130+/− p27+/D51 mice recovered was also significantly lower than expected. In contrast, when we reduced p107 in p130−/− p27D51/D51animals, we recovered p107+/− p130−/− p27D51/D51 pups at the expected ratio. This suggests that p130 can compensate for the reduced amount of p27 in the p107−/− p27D51/D51 mice.
TABLE 2.
Effect of p130 dose on survival of neonatal p107−/− p27D51/D51 animals
| Basis of calculation | % of mice p130+/− and:
|
χ2 test P valueb | ||
|---|---|---|---|---|
| p107+/−p27D51/+ and p107+/−p27D51/D51 | p107−/−p27D51/+ | p107−/−p27D51/D51 | ||
| Mendeliana | 50 | 25 | 25 | |
| Observed, 3 wk | 93 | 7 | 0 | <10−11 |
The percentage of mice expected with Mendelian inheritance when intercrossing p107+/− p130−/− p27D51/D51 and p107−/− p130+/+ p27D51/+ mice.
The X2 test was based on a comparison between the expected and observed percentages of each genotype.
Loss of p107 and p27 affects morphology of the long bones.
The neonatal lethality in p107−/− p130−/− mice is related to defects in endochondral ossification (13). Endochondral ossification occurs in the long bones of the developing limbs and the sternum. During this process, chondrocytes in the cartilage of developing long bones proliferate, become quiescent, undergo a shift to hypertrophic growth, and are eventually replaced by bone. To determine if ossification was affected by the loss of p107 and p27, we generated wild-type, p107−/− p27D51/D51, p107−/− p130−/−, p27D51/D51, and p130−/− p27D51/D51 day 1 neonatal animals and stained whole-mount embryo preparations with alcian blue and alizarin red. Alcian blue stains nonmineralized cartilage, whereas alizarin red stains mineralized bone. Representative images of the forelimbs of p107−/−, p107−/− p130−/−, and p107−/− p27D51/D51mice are shown in Fig. 3A. Measurements of the humerus, radius, and ulna from all genotypes are compiled in Table 3.
FIG. 3.

Endochondral ossification in newborn mice. Neonatal mice were collected, and skeletal preparations were stained with alcian blue and alizarin red to differentiate the chondrocytic and bony regions. (A) Representative images of the forelimbs from p107−/−, p107−/− p27D51/D51, and p107−/− p130−/− mice are shown. Red, bone; blue, cartilage. The number of animals analyzed for this phenotype is indicated in Table 3. (B) Representative images of the sternal regions of mice. Note the presence of bone in the costal junctions in p107−/− p130−/− mice and the p107−/− p27D51/D51 deficient mice. The number of mice afflicted with this phenotype is reported in the text.
TABLE 3.
Forelimb measurements
| Genotype | n | Length (mm)
|
Width (mm)
|
||||
|---|---|---|---|---|---|---|---|
| Humerus | Radius | Ulna | Humerus | Radius | Ulna | ||
| Wild type | 4 | 4.68 ± 0.12 | 3.78 ± 0.15 | 4.93 ± 0.14 | 0.55 ± 0.06 | 0.40 ± 0.01 | 0.44 ± 0.03 |
| p27D51/D51 | 5 | 4.54 ± 0.33 | 3.57 ± 0.32 | 4.64 ± 0.42 | 0.53 ± 0.04 | 0.37 ± 0.03 | 0.43 ± 0.03 |
| p107−/−p27D51/D51 | 5 | 3.85 ± 0.31a | 2.94 ± 0.22a | 4.16 ± 0.18a | 0.70 ± 0.04a | 0.52 ± 0.03a | 0.59 ± 0.04a |
| p107−/−p130−/− | 5 | 2.79 ± 0.34a | 2.28 ± 0.19a | 3.26 ± 0.26a | 0.67 ± 0.05a | 0.44 ± 0.04 | 0.48 ± 0.02 |
| p130−/−p27D51/D51 | 3 | 4.41 ± 0.22 | 3.53 ± 0.21 | 4.54 ± 0.24 | 0.53 ± 0.02 | 0.35 ± 0.02 | 0.39 ± 0.06 |
Significantly different (P < 0.005) from the wild type (two-tailed Student's t test).
Looking at the forelimbs, we noted that there were significant changes in the length and width of the humerus, radius, and ulna in p107−/− p27D51/D51mice, consistent with a defect in endochondral ossification. This was reminiscent of the phenotype reported in p107−/− p130−/− mice and confirmed in this work (13). Nevertheless, whereas the length of the bones was more affected in p107−/− p130−/− animals, the width was more affected in the p107−/− p27D51/D51mice. While loss of p27 or p130 may be qualitatively similar, the latter finding may reflect potential quantitative differences in the mechanisms by which p130 and p27 control bone formation or remodeling.
We also noted phenotypic similarities between p107−/− p130−/− and p107−/− p27D51/D51 mice in the sternum (Fig. 3B). Wild-type, p130−/− p27D51/D51, and p27D51/D51 animals showed a typical pattern of bony elements (the sternabrae) separated by bands of cartilage (the sternal joints, points of fusion with the costal cartilage). In contrast, ossification of costal junctions was observed in one-third of the p107−/− animals (9/26; P = 0.02) and in over three-fourths of the p107−/− p27D51/D51 (15/18; P < 0.001) and p107−/− p130−/− (7/8; P < 0.001) animals. These findings are consistent with the idea that either p130 or p27 could compensate for the normal function of p107 during normal development of costal cartilage tissue.
Loss of p107 and p27 increases the S-phase fraction of proliferating chondrocytes.
p107 and p130 are required for growth arrest and maturation of chondrocytes in vivo (13). Thus, we asked whether p107 and p27 act redundantly to affect cell cycle exit in the developing sternum. To avoid potential problems associated with the developmental staging of different embryos, we only looked at littermates from a p107−/− p27+/D51 cross. The fraction of proliferating chondrocytes in the fourth and fifth sternal joints of day 16.5 embryos was increased in p107−/− p27D51/D51 mice (n = 2) compared to p107−/− mice (n = 3) (Fig. 4). Distinctions between the chondrocytes of the rib, the proliferating chondrocytes, the prehypertrophic chondrocytes, and the hypertrophic chondrocytes were observed (data not shown). Thus, p107 and p27 can cooperate to control proliferation in chondrocytes, suggesting that p27 and p130 might be on a common pathway controlling proliferation in these cells.
FIG. 4.

Cell proliferation in prehypertrophic chondrocytes in costal joints. Pregnant mice were injected with bromodeoxyuridine and sacrificed, and embryos were collected at day 16.5. BrdU incorporation was determined immunohistochemically, and the percentage of positive cells was plotted. Only littermates were used for comparisons; in one litter a single pair of p107−/− and p107−/− p27D51/D51 was obtained, and in another litter two p107−/− animals were paired with a single p107−/− p27D51/D51 sibling. Each tick mark represents the percentage of cells in one section that were BrdU positive. p107−/−, 3,5797 cells counted; p107−/− p27D51/D51, 2,7743 cells counted.
The combined loss of p107 and p27 does not affect cell proliferation, the cell cycle machinery, or p107/p130-regulated gene expression in mouse embryo fibroblasts.
To further explore the possibility that p27 deficiency and p130 deficiency were functionally equivalent, we began to look into cellular models in which the redundancy of p107 and p130 was already established. Specifically, we were interested in determining if there was a similar effect on cell behavior when p107 and p27 were both lost as when p107 and p130 were both lost. MEFs have a relatively neutral cellular phenotype that is not committed to a specific mesenchymal lineage. Combined loss of either p107 and p130, or of p107 and p27, did not affect the ability of MEFs to exit the cell cycle when deprived of serum (Fig. 5A). Furthermore, quiescent p107−/− p27D51/D51 MEFs entered S phase at a rate similar to wild-type cells and modestly slower than p107−/− p130−/− cells when serum was added back to the medium (Fig. 5B).
FIG. 5.

p130 function is not disrupted in p107−/− p27D51/D51 mouse embryo fibroblasts. (A) Cells deprived of serum exit the cell cycle. Passage 4 cells were serum starved for 60 h and pulsed with bromodeoxyuridine for 4 h. This experiment was repeated at least five times with different clones each time. (B) Serum-induced S-phase progression. Serum was added to serum-deprived quiescent cells, and their progress into S-phase was determined by incorporation of radiolabeled thymidine as described elsewhere (31). Two independent clones of each genotype were analyzed, as indicated in the figure. (C) Histone H1 kinase assay. Extracts were prepared from quiescent serum-starved cells or from quiescent cells that had been induced to reenter the cell cycle by serum addition 20 h previously, and the amount of immunoprecipitable cyclin E- and cyclin A-associated histone H1 kinase activity was measured. The genotypes are indicated above the lanes, with individual clone numbers noted. As a control for kinase activity, cyclin E-associated kinase activity was ascertained on wild-type clone 371 cells 20 h after serum addition to a quiescent culture. Cyclin A-associated kinase activity was ascertained on double mutant clone 342. Similar results were obtained when other clones with the indicated genotypes were used (wild type, n = 7; p107−/− p27D51/D51, n = 5). (D) Immunoblot assay. The protein noted on the left of the panel was determined by direct immunoblotting of whole-cell extracts. As a control for protein expression, extracts were also prepared from serum-stimulated wild-type clone 264 and double mutant clone 342. A nonspecific cross-reactive band is noted by the asterisk to the right of the p130Rb2 image. The positive sign to the right of the p27Kip1 image denotes the protein encoded by the D51 allele. (E) Northern blotting. In the top panel, the accumulation of CDK1 and cyclin A mRNA was determined in serum-starved and asynchronous cells (indicated by the − and + symbols below each lane). Genotypes and individual clone numbers are indicated above the lanes. Note that the accumulation of these mRNAs was much more pronounced in the serum-starved p107−/− p130−/− cells compared to the p107−/− p27D51/D51 or wild-type cells. ARPP0, a message that is not affected by serum starvation, was used as a loading control. In the bottom panel, we compared the expression of TS, DHFR, B-myb, and p130 in two additional clones, one wild type (372) and one p107−/− p27D51/D51 (345), and we found that they were similar.
CDK activity must surpass a threshold level to induce cell proliferation, and pocket protein binding to these complexes might inhibit activity to a subthreshold level in Kip-deficient cells without reaching the nadir established by Kip binding (65). Thus, we looked at whether CDK activity was higher in the quiescent p107−/− p27D51/D51 cells compared to wild-type cells (Fig. 5C). Cyclin A2-associated kinase activity was modestly increased in serum-starved p107−/− p27D51/D51 MEFs. Cyclin E-associated kinase activity was unaffected. This is consistent with the CDK-inhibitory binding of p107 to cyclin A2 in Kip-deficient cells (9).
To try and understand why cyclin A activity was slightly higher, we looked at the expression of cell cycle regulators in quiescent cells. Cyclin A and CDK1 proteins were modestly increased, consistent with the increased kinase activity, and there was no significant change in p21, the D-type cyclins, cyclin E, CDK2, CDK4, CDK6, or E2F4 and E2F5 (Fig. 5D). p130 accumulation was only occasionally affected (Fig. 5D). In three of the eight clones examined, p130 levels decreased by less than 50%. These data indicate that deficiency of both p107 and p27 caused only marginal differences in cyclin A2 and CDK1 accumulation and in cyclin A-associated kinase activity, the magnitudes of which were unable to affect quiescence in MEFs.
Although there was no significant effect on cell proliferation, we wanted to determine if the modest amount of cyclin A2 kinase activity could affect the p107/p130-controlled quiescence-associated gene expression program in MEFs. This program is associated with the repression of cyclin A2, CDK1, B-myb, TS, and DHFR mRNA (26). We found that cyclin A2 and CDK1 mRNA were expressed at comparable levels to those observed in wild-type cells and not at the increased levels seen in p107−/− p130−/− cells (Fig. 5E). ARPP0 served as a loading control. TS, DHFR, B-myb, and p130 mRNAs were not increased in quiescent p107−/− p27D51/D51 MEFs, either (Fig. 5E). Thus, there is no evidence that the loss of p27 and p107 function is sufficient to reduce the activity of p130 in MEFs, but it can modestly enhance cyclin A accumulation and activity in an apparently posttranscriptional manner.
Taken together, combined loss of p27 and p107 function does not have major effects on the levels, associations, and/or activities of any of the principal components of the cell cycle regulatory machinery in MEFs as measured in our studies, consistent with these cells retaining their normal dependence on growth factors for the stimulation of cell proliferation.
Defects in chondrogenic differentiation in p107−/− p27D51/D51 cells.
Chondrogenic differentiation in micromass cultures was previously reported to be disturbed in p107−/− p130−/− cells (33, 50). Thus, we wanted to examine chondrogenic differentiation of p107−/− p27D51/D51 cells ex vivo in micromass culture as well (37). To accomplish this, we induced the commitment of MEFs to the chondrogenic lineage by treating micromass-cultured early passage MEFs obtained from wild-type, p107−/− p27D51/D51, and p107−/− p130−/− embryos with BMP2. The extent and rate of differentiation were analyzed histochemically by alcian blue staining and alkaline phosphatase staining (a marker of hypertrophic chondrocytes). This revealed reduced deposition of extracellular matrix as well as delayed induction of hypertrophy in p107−/− p27D51/D51 and p107−/− p130−/− cells compared to wild type (Fig. 6A). qRT-PCR of chondrocyte-specific marker genes showed a normal induction pattern for the chondrocyte marker collagen type 2a1. Sox9, a marker of committed chondrogenesis, was also induced with similar kinetics in the three genotypes, but its continued presence was reduced in the knockout cells. The differentiation markers (Indian Hedgehog, collagen type 10a1, alkaline phosphatase, and osteocalcin) were not induced in p107−/− p27D51/D51 and p107−/− p130−/− cells (Fig. 6B). Runx2 mRNA was downregulated as expected in the wild-type cells (37). A similar pattern, but at a reduced level, was observed for p107−/− p130−/− mutant cells. In contrast, Runx2 levels in the p107−/− p27D51/D51 mutant cells showed a delayed profile as a result of higher proliferation, as suggested by higher expression of histone H4 (Fig. 6B). This suggests that p107−/− p27D51/D51 and p107−/− p130−/− MEFs committed to the chondrogenic lineage appropriately following treatment with BMP2, but their differentiation was inhibited. Commitment and differentiation of p107−/− cells was similar to that of wild-type cells (data not shown). These ex vivo findings are consistent with the phenotype observed in the developing sternabrae of p107−/− p27D51/D51 mice.
FIG. 6.

Ex vivo chondrogenic differentiation of mouse embryonic fibroblasts from wild-type, p107−/− p27D51/D51, and p107−/− p130−/− mice. (A) Chondrogenic differentiation was assessed by alcian blue and alkaline phosphatase staining of micromass cultures at day 9. (B) Quantitative real-time PCR analysis of chondrogenic marker genes during differentiation. Relative transcript levels were determined and plotted individually for the gene products indicated within each graph.
To address if the loss of p27 collaborated with p107 deficiency to affect p130 accumulation in these cells, we looked at the accumulation of p130 and other cell cycle regulators in pooled micromass cultures. We did not see any genotype-specific differences in either the accumulation of p130 or p27 or the reduction of hyperphosphorylated Rb, p21, or cyclin B1 that accompany chondrogenic commitment (Fig. 7A). There was a marginal increase in cyclin A2 and a more robust increase in CDK1 in the double knockout cells (Fig. 7A). Thus, a modest change in the amount of cyclin A2 and CDK1 was observed in both the p107−/− p130−/− and p107−/− p27D51/D51 cells; however, there was no significant change in p130 expression when we compared wild-type, p107−/−, and p107−/− p27D51/D51 cells, suggesting that the cellular phenotype in p107/p27 doubly deficient mice is not due to a direct loss of p130 protein.
FIG. 7.

Expression of cell cycle regulatory molecules during ex vivo chondrogenic differentiation of wild-type, p107−/−, p107−/− p27D51/D51, and p107−/− p130−/− MEFs. (A) Immunoblot assay. The protein noted on the left of the panel was determined by direct immunoblotting of whole-cell extracts. A 200-μg aliquot of extract was used to detect p130 and Rb, 20 μg was used for CDK1, and 80 μg was used for all others. The genotypes of the cells from which extracts were prepared are indicated above each lane and were confirmed by immunoblotting with p107 and p27 antibodies. BMP2 treatment was carried out for 24 h prior to harvest. (B) Northern blot assay. These results are arranged as for panel A, but the accumulation of CDK1 and cyclin A2 mRNA was determined. (C) Quantitative real-time PCR analysis of cyclin A2 and CDK1 in BMP2-treated micromass cultures was performed, and results are plotted as a percentage of the level in untreated micromass cultures of the same genotype. The mean and standard deviation from three independent experiments are plotted.
We could not recover enough protein from pooled micromass cultures to explore the interaction of p130 with other proteins in p107−/− p27D51/D51 cells; however, we reasoned that we could monitor cyclin A2 and CDK1 mRNA as surrogates of p107/p130-regulated gene expression, as we did previously in MEFs. We noted that CDK1 mRNA was higher in both double knockout cell types compared to either the wild-type or p107−/− control cells (Fig. 7B and C). Cyclin A2 mRNA was marginally higher in the double knockouts as well. ARPP0 served as a loading control (Fig. 7B). These results were confirmed by qRT-PCR (Fig. 7C). Thus, p27, like p130, can genetically interact with p107 to control differentiation and a gene expression program of chondrocytes. This suggests that p27 and p130 are on overlapping pathways that work with p107 to promote cell cycle exit and differentiation in the chondrogenic lineage. We suggest that p27 and p130 are in a single functional group, but they are unable to fully compensate for each other when one is absent.
DISCUSSION
In this report we provide evidence for the involvement of p27 in controlling chondrocyte differentiation and endochondral bone development in mice. We have generated and analyzed the phenotype of p107−/− p27D51/D51 mice. The combination of these deficiencies resulted in phenotypes similar to those previously reported for p107−/− p130−/− mice and recapitulated here, including neonatal lethality (Tables 1 and 2), aberrant or ectopic endochondral ossification with increased chondrocyte proliferation in cartilaginous regions of the limbs and sternum (Table 3; Fig. 3 and 4) (13), and chondrocyte maturation ex vivo (Fig. 6 and 7).
Endochondral ossification begins when mesenchymal cells are induced to enter a chondrocytic lineage. Many of the signaling molecules and the transcription factors involved in chondrocyte maturation have been identified (28, 32). A fetal cartilage mold is established and chondrocytes begin to produce PTHrP, which stimulates proliferation and further differentiation into postmitotic hypertrophic chondrocytes. Transforming growth factor β and Ihh both positively regulate chondrocytic PTHrP expression. On the other hand, fibroblast growth factor (FGF) signaling induces differentiation and the withdrawal of chondrocytes from the cell cycle. Ultimately, this process is controlled by a variety of transcription factors. Sox5 and -6 are essential for the induction of the chondrocytic lineage from mesenchymal cells (55). Sox9 is involved in the expansion of chondrocytic cells (5). Runx2/Cbfa1 is a determinant of the chondro-osteoprogenitor lineage in mesenchymal cells but must be downregulated for perichondrial progenitors to enter the chondrocyte lineage (24, 36). Runx2 is, however, required for hypertrophic chondrocyte maturation. Runx2 activation of vascular endothelial growth factor in hypertrophic chondrocytes is essential for progression of endochondral bone formation (69). Runx2 mediates PTH/PTHrP signaling in the hypertrophic zone (23) and FGF signaling in the perichondrium (24, 49). Thus, it is not surprising that skeletal malformations arise when the chondrocytic differentiation program is deregulated by either chronic activation or insufficient activation of Runx 2 (61, 68).
Skeletal manifestations can arise as a consequence of changes in chondrocyte proliferation. Skeletal defects in p107−/− p130−/− (13), p57−/− (71), and E2F1 transgenic mice (51) are associated with increased chondrocyte proliferation, without disruption of the normal morphological patterning of the growing cartilage column. We documented similar defects in bone development in p107−/− p27D51/D51 mice. The exaggeration of the p107 skeletal phenotype by either p130 or p27 insufficiency suggests that these proteins might share a function controlling proliferation and enforcing a p107-dependent cell cycle block.
How the cell cycle program is coordinated with chondrocyte maturation is becoming increasingly clear. Transforming growth factor β and PTHrP stimulate chondrocyte proliferation by increasing cyclin D1 (2) and decreasing p57Kip2 (39). Consistent with this, cyclin D1-deficient mice have reduced growth plate proliferation (54), and the proliferation of chondrocytes continues in p57−/− mice, leading to defects in endochondral ossification (71). However, the most compelling piece of evidence supporting a link between PTHrP signaling and p57 accumulation is the observation that skeletal abnormalities associated with PTHrP deficiency are not seen in a p57-deficient background (39). Interestingly, these skeletal abnormalities are also reversed in a p107−/− p130−/− background (36), suggesting that p57 and p107/p130 may act in a common pathway during PTHrP-regulated endochondral ossification.
Conversely, FGF induces chondrocyte growth arrest and maturation by inducing p107 and p130 (33). p107- and p130-deficient mice have defects in endochondral ossification associated with increased chondrocyte proliferation (13, 50). It has been suggested that p57 might act by inhibiting CDK activity, allowing p107 and p130 to accumulate and support chondrocyte differentiation (36). However, dephosphorylation of p107 is rapid, occurring within 1 hour of FGF treatment, and is not dependent on gene transcription or protein translation (30). Consequently, it is more likely that changes in the pocket proteins precede cell cycle exit induced by p57.
The regulation of p27 levels in relation to p57 might account for the differences in the penetrance of the skeletal phenotype that arise from compound mutations in the p27 and p107 loci (17, 20, 62). In osteoblasts, the level of p27 is correlated with Runx2 and inversely related to proliferation. p57 levels are increased as chondrocytes exit the cell cycle with decreased Runx2, whereas p27 levels only increase in conjunction with the reactivation of Runx2 in hypertrophic chondrocytes. There are a number of reports that show that p27 accumulates in hypertrophic chondrocytes (1, 33, 42, 58) However, as there is little change in the spatial or temporal regulation of chondrocyte proliferation in p27-deficient mice, it was suggested that its contribution might be minimal (18). Our crossing the p27 deficiency into the sensitized background of p107-deficient mice clearly unmasks a significant contributory function for p27. In the chondrogenic lineage, differences in the temporal regulation of these two CDK inhibitors may cause either protein to become genetically indispensable for normal cartilage.
Thus, there are four important cell cycle regulators implicated in chondrocyte maturation: p57, p27, p107, and p130. To understand the relationships between these proteins, we find it helpful to think of the impact of the cell cycle on chondrocytic maturation in two stages (14). First, because the loss of either p57 or p107 alone had the greatest impact on chondrocyte maturation in both mice and primary cultures, we think these are at the top of the heirarchy. Additionally, we suspect that p107 is the major input through which signaling affects maturation, because p107 accumulates before p130 and before cell cycle exit occurs in FGF-treated RCS cells (14). In the absence of p107, compensatory upregulation of p130 might substitute for the initial decision to exit the cycle, as suggested previously (13), but it is also plausible that a parallel pathway controlled by increasing p57 as cells enter the G1 phase would operate a bit more inefficiently to induce the initial growth arrest. Indeed, it is clear that p107 single mutant mice do have a subtle phenotype, albeit subclinical in regards to viability. So what do p27 and p130 do? Because any role for p27 and p130 is only unmasked in p107-deficient cells, p27 accumulates during the hypertrophic transition in vivo and in vitro, and p130 accumulates in RCS cells once they exit the cell cycle, we suspect that these proteins normally accumulate only when the cells have exited the cell cycle. Thus, they might serve to buffer the cell against continual mitogenic signaling which could otherwise induce promiscuous proliferation. Thus, p130 and p27 may simply reinforce the cell cycle exit decision made by p107 and p57. Ultimately, collaboration between the molecules that induce cell cycle exit and those that maintain quiescence might be required to permit the elaboration of appropriate transcriptional program leading to the hypertrophic fate.
Acknowledgments
We thank David Cobrinik (Cornell University) and Antonio Giordano (Temple University) for stimulating discussions as the work progressed and Andrea Brendolan (Cornell University), Elizabeth Poynor (MSKCC), Sukhee Lee (University of Massachusetts Medical Center), Scott Weatherbee (MSKCC), and Laura Meltzer (MSKCC) for experimental advice and assistance during the course of the work.
This work was funded by grants from the NCI (CA89563) and the Golfers Against Cancer Foundation to Andrew Koff and by Caja Madrid and Cultek Foundations to Andrew Koff, Anxo Vidal, and Timothy G. Bromage.
Footnotes
Published ahead of print on 14 May 2007.
REFERENCES
- 1.Ballock, R. T., X. Zhou, L. M. Mink, D. H. Chen, B. C. Mita, and M. C. Stewart. 2000. Expression of cyclin-dependent kinase inhibitors in epiphyseal chondrocytes induced to terminally differentiate with thyroid hormone. Endocrinology 141:4552-4557. [DOI] [PubMed] [Google Scholar]
- 2.Beier, F., Z. Ali, D. Mok, A. C. Taylor, T. Leask, C. Albanese, R. G. Pestell, and P. LuValle. 2001. TGFβ and PTHrP control chondrocyte proliferation by activating cyclin D1 expression. Mol. Biol. Cell 12:3852-3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bernards, R. 1999. CDK-independent activities of D type cyclins. Biochim. Biophys. Acta 1424:M17-22. [DOI] [PubMed] [Google Scholar]
- 4.Bhattacharya, S., J. Garriga, J. Calbo, T. Yong, D. S. Haines, and X. Grana. 2003. SKP2 associates with p130 and accelerates p130 ubiquitylation and degradation in human cells. Oncogene 22:2443-2451. [DOI] [PubMed] [Google Scholar]
- 5.Bi, W., J. M. Deng, Z. Zhang, R. R. Behringer, and B. de Crombrugghe. 1999. Sox9 is required for cartilage formation. Nat. Genet. 22:85-89. [DOI] [PubMed] [Google Scholar]
- 6.Cam, H., and B. D. Dynlacht. 2003. Emerging roles for E2F: beyond the G1/S transition and DNA replication. Cancer Cell 3:311-316. [DOI] [PubMed] [Google Scholar]
- 7.Casaccia-Bonnefil, P., R. J. Hardy, K. K. Teng, J. M. Levine, A. Koff, and M. V. Chao. 1999. Loss of p27Kip1 function results in increased proliferative capacity of oligodendrocyte progenitors but unaltered timing of differentiation. Development 126:4027-4037. [DOI] [PubMed] [Google Scholar]
- 8.Casaccia-Bonnefil, P., R. Tikoo, H. Kiyokawa, V. Friedrich, Jr., M. V. Chao, and A. Koff. 1997. Oligodendrocyte precursor differentiation is perturbed in the absence of the cyclin-dependent kinase inhibitor p27Kip1. Genes Dev. 11:2335-2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chibazakura, T., S. G. McGrew, J. A. Cooper, H. Yoshikawa, and J. M. Roberts. 2004. Regulation of cyclin-dependent kinase activity during mitotic exit and maintenance of genome stability by p21, p27, and p107. Proc. Natl. Acad. Sci. USA 101:4465-4470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Classon, M., and E. Harlow. 2002. The retinoblastoma tumour suppressor in development and cancer. Nat. Rev. Cancer 2:910-917. [DOI] [PubMed] [Google Scholar]
- 11.Coats, S., P. Whyte, M. L. Fero, S. Lacy, G. Chung, E. Randel, E. Firpo, and J. M. Roberts. 1999. A new pathway for mitogen-dependent cdk2 regulation uncovered in p27Kip1-deficient cells. Curr. Biol. 9:163-173. [DOI] [PubMed] [Google Scholar]
- 12.Cobrinik, D. 2005. Pocket proteins and cell cycle control. Oncogene 24:2796-2809. [DOI] [PubMed] [Google Scholar]
- 13.Cobrinik, D., M. H. Lee, G. Hannon, G. Mulligan, R. T. Bronson, N. Dyson, E. Harlow, D. Beach, R. A. Weinberg, and T. Jacks. 1996. Shared role of the pRB-related p130 and p107 proteins in limb development. Genes Dev. 10:1633-1644. [DOI] [PubMed] [Google Scholar]
- 14.Dailey, L., E. Laplantine, R. Priore, and C. Basilico. 2003. A network of transcriptional and signaling events is activated by FGF to induce chondrocyte growth arrest and differentiation. J. Cell Biol. 161:1053-1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.De Clercq, A., and D. Inze. 2006. Cyclin-dependent kinase inhibitors in yeast, animals, and plants: a functional comparison. Crit. Rev. Biochem. Mol. Biol. 41:293-313. [DOI] [PubMed] [Google Scholar]
- 16.Dotto, G. P. 2000. p21WAF1/Cip1: more than a break to the cell cycle? Biochim. Biophys. Acta 1471:M43-M56. [DOI] [PubMed] [Google Scholar]
- 17.Drissi, H., D. Hushka, F. Aslam, Q. Nguyen, E. Buffone, A. Koff, A. van Wijnen, J. B. Lian, J. L. Stein, and G. S. Stein. 1999. The cell cycle regulator p27kip1 contributes to growth and differentiation of osteoblasts. Cancer Res. 59:3705-3711. [PubMed] [Google Scholar]
- 18.Emons, J. A., R. Marino, O. Nilsson, K. M. Barnes, N. Even-Zohar, A. C. Andrade, N. A. Chatterjee, J. M. Wit, M. Karperien, and J. Baron. 2006. The role of p27Kip1 in the regulation of growth plate chondrocyte proliferation in mice. Pediatr. Res. 60:288-293. [DOI] [PubMed] [Google Scholar]
- 19.Fero, M. L., M. Rivkin, M. Tasch, P. Porter, C. E. Carow, E. Firpo, K. Polyak, L. H. Tsai, V. Broudy, R. M. Perlmutter, K. Kaushansky, and J. M. Roberts. 1996. A syndrome of multiorgan hyperplasia with features of gigantism, tumorigenesis, and female sterility in p27Kip1-deficient mice. Cell 85:733-744. [DOI] [PubMed] [Google Scholar]
- 20.Galindo, M., J. Pratap, D. W. Young, H. Hovhannisyan, H. J. Im, J. Y. Choi, J. B. Lian, J. L. Stein, G. S. Stein, and A. J. van Wijnen. 2005. The bone-specific expression of Runx2 oscillates during the cell cycle to support a G1-related antiproliferative function in osteoblasts. J. Biol. Chem. 280:20274-20285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gaur, T., L. Rich, C. J. Lengner, S. Hussain, B. Trevant, D. Ayers, J. L. Stein, P. V. Bodine, B. S. Komm, G. S. Stein, and J. B. Lian. 2006. Secreted frizzled related protein 1 regulates Wnt signaling for BMP2 induced chondrocyte differentiation. J. Cell Physiol. 208:87-96. [DOI] [PubMed] [Google Scholar]
- 22.Goodrich, D. W. 2006. The retinoblastoma tumor-suppressor gene, the exception that proves the rule. Oncogene 25:5233-5243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guo, J., U. I. Chung, D. Yang, G. Karsenty, F. R. Bringhurst, and H. M. Kronenberg. 2006. PTH/PTHrP receptor delays chondrocyte hypertrophy via both Runx2-dependent and -independent pathways. Dev. Biol. 292:116-128. [DOI] [PubMed] [Google Scholar]
- 24.Hinoi, E., P. Bialek, Y. T. Chen, M. T. Rached, Y. Groner, R. R. Behringer, D. M. Ornitz, and G. Karsenty. 2006. Runx2 inhibits chondrocyte proliferation and hypertrophy through its expression in the perichondrium. Genes Dev. 20:2937-2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Horton, C., and M. Maden. 1995. Endogenous distribution of retinoids during normal development and teratogenesis in the mouse embryo. Dev. Dyn. 202:312-323. [DOI] [PubMed] [Google Scholar]
- 26.Hurford, R. K., Jr., D. Cobrinik, M. H. Lee, and N. Dyson. 1997. pRB and p107/p130 are required for the regulated expression of different sets of E2F responsive genes. Genes Dev. 11:1447-1463.9192872 [Google Scholar]
- 27.Joaquin, M., and R. J. Watson. 2003. The cell cycle-regulated B-Myb transcription factor overcomes cyclin-dependent kinase inhibitory activity of p57KIP2 by interacting with its cyclin-binding domain. J. Biol. Chem. 278:44255-44264. [DOI] [PubMed] [Google Scholar]
- 28.Karsenty, G., and E. F. Wagner. 2002. Reaching a genetic and molecular understanding of skeletal development. Dev. Cell 2:389-406. [DOI] [PubMed] [Google Scholar]
- 29.Kiyokawa, H., R. D. Kineman, K. O. Manova-Todorova, V. C. Soares, E. S. Hoffman, M. Ono, D. Khanam, A. C. Hayday, L. A. Frohman, and A. Koff. 1996. Enhanced growth of mice lacking the cyclin-dependent kinase inhibitor function of p27Kip1. Cell 85:721-732. [DOI] [PubMed] [Google Scholar]
- 30.Koff, A., F. Cross, A. Fisher, J. Schumacher, K. Leguellec, M. Philippe, and J. M. Roberts. 1991. Human cyclin E, a new cyclin that interacts with two members of the CDC2 gene family. Cell 66:1217-1228. [DOI] [PubMed] [Google Scholar]
- 31.Koff, A., M. Ohtsuki, K. Polyak, J. M. Roberts, and J. Massague. 1993. Negative regulation of G1 in mammalian cells: inhibition of cyclin E-dependent kinase by TGF-beta. Science 260:536-539. [DOI] [PubMed] [Google Scholar]
- 32.Kronenberg, H. M. 2006. PTHrP and skeletal development. Ann. N. Y. Acad. Sci. 1068:1-13. [DOI] [PubMed] [Google Scholar]
- 33.Laplantine, E., F. Rossi, M. Sahni, C. Basilico, and D. Cobrinik. 2002. FGF signaling targets the pRb-related p107 and p130 proteins to induce chondrocyte growth arrest. J. Cell Biol. 158:741-750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee, M. H., B. O. Williams, G. Mulligan, S. Mukai, R. T. Bronson, N. Dyson, E. Harlow, and T. Jacks. 1996. Targeted disruption of p107: functional overlap between p107 and Rb. Genes Dev. 10:1621-1632. [DOI] [PubMed] [Google Scholar]
- 35.Lees, E., B. Faha, V. Dulic, S. I. Reed, and E. Harlow. 1992. Cyclin E/cdk2 and cyclin A/cdk2 kinases associate with p107 and E2F in a temporally distinct manner. Genes Dev. 6:1874-1885. [DOI] [PubMed] [Google Scholar]
- 36.Lengner, C. J., M. Q. Hassan, R. W. Serra, C. Lepper, A. J. van Wijnen, J. L. Stein, J. B. Lian, and G. S. Stein. 2005. Nkx3.2-mediated repression of Runx2 promotes chondrogenic differentiation. J. Biol. Chem. 280:15872-15879. [DOI] [PubMed] [Google Scholar]
- 37.Lengner, C. J., C. Lepper, A. J. van Wijnen, J. L. Stein, G. S. Stein, and J. B. Lian. 2004. Primary mouse embryonic fibroblasts: a model of mesenchymal cartilage formation. J. Cell Physiol. 200:327-333. [DOI] [PubMed] [Google Scholar]
- 38.Macaluso, M., M. Montanari, and A. Giordano. 2006. Rb family proteins as modulators of gene expression and new aspects regarding the interaction with chromatin remodeling enzymes. Oncogene 25:5263-5267. [DOI] [PubMed] [Google Scholar]
- 39.MacLean, H. E., J. Guo, M. C. Knight, P. Zhang, D. Cobrinik, and H. M. Kronenberg. 2004. The cyclin-dependent kinase inhibitor p57Kip2 mediates proliferative actions of PTHrP in chondrocytes. J. Clin. Investig. 113:1334-1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mittnacht, S., and R. A. Weinberg. 1991. G1/S phosphorylation of the retinoblastoma protein is associated with an altered affinity for the nuclear compartment. Cell 65:381-393. [DOI] [PubMed] [Google Scholar]
- 41.Nakayama, K., N. Ishida, M. Shirane, A. Inomata, T. Inoue, N. Shishido, I. Horii, D. Y. Loh, and K. Nakayama. 1996. Mice lacking p27Kip1 display increased body size, multiple organ hyperplasia, retinal dysplasia, and pituitary tumors. Cell 85:707-720. [DOI] [PubMed] [Google Scholar]
- 42.Negishi, Y., N. Ui, M. Nakajima, K. Kawashima, K. Maruyama, T. Takizawa, and H. Endo. 2001. p21Cip-1/SDI-1/WAF-1 gene is involved in chondrogenic differentiation of ATDC5 cells in vitro. J. Biol. Chem. 276:33249-33256. [DOI] [PubMed] [Google Scholar]
- 43.Nguyen, L., A. Besson, J. I. Heng, C. Schuurmans, L. Teboul, C. Parras, A. Philpott, J. M. Roberts, and F. Guillemot. 2006. p27kip1 independently promotes neuronal differentiation and migration in the cerebral cortex. Genes Dev. 20:1511-1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ohnuma, S., A. Philpott, K. Wang, C. E. Holt, and W. A. Harris. 1999. p27Xic1, a Cdk inhibitor, promotes the determination of glial cells in Xenopus retina. Cell 99:499-510. [DOI] [PubMed] [Google Scholar]
- 45.Perkins, N. D. 2002. Not just a CDK inhibitor: regulation of transcription by p21WAF1/CIP1/SDI1. Cell Cycle 1:39-41. [PubMed] [Google Scholar]
- 46.Petre, C. E., Y. B. Wetherill, M. Danielsen, and K. E. Knudsen. 2002. Cyclin D1: mechanism and consequence of androgen receptor co-repressor activity. J. Biol. Chem. 277:2207-2215. [DOI] [PubMed] [Google Scholar]
- 47.Petre-Draviam, C. E., E. B. Williams, C. J. Burd, A. Gladden, H. Moghadam, J. Meller, J. A. Diehl, and K. E. Knudsen. 2005. A central domain of cyclin D1 mediates nuclear receptor corepressor activity. Oncogene 24:431-444. [DOI] [PubMed] [Google Scholar]
- 48.Reed, M. F., V. F. Liu, M. H. Ladha, K. Ando, J. D. Griffin, D. T. Weaver, and M. E. Ewen. 1998. Enforced CDK4 expression in a hematopoietic cell line confers resistance to the G1 arrest induced by ionizing radiation. Oncogene 17:2961-2971. [DOI] [PubMed] [Google Scholar]
- 49.Reinhold, M. I., and M. C. Naski. 2006. Direct interactions of Runx2 and canonical Wnt signaling induce FGF18. J. Biol. Chem. 282:3653-3663. [DOI] [PubMed] [Google Scholar]
- 50.Rossi, F., H. E. MacLean, W. Yuan, R. O. Francis, E. Semenova, C. S. Lin, H. M. Kronenberg, and D. Cobrinik. 2002. p107 and p130 coordinately regulate proliferation, Cbfa1 expression, and hypertrophic differentiation during endochondral bone development. Dev. Biol. 247:271-285. [DOI] [PubMed] [Google Scholar]
- 51.Scheijen, B., M. Bronk, T. van der Meer, and R. Bernards. 2003. Constitutive E2F1 overexpression delays endochondral bone formation by inhibiting chondrocyte differentiation. Mol. Cell. Biol. 23:3656-3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shaffer, D. R., A. Viale, R. Ishiwata, M. Leversha, S. Olgac, K. Manova, J. Satagopan, H. Scher, and A. Koff. 2005. Evidence for a p27 tumor suppressive function independent of its role regulating cell proliferation in the prostate. Proc. Natl. Acad. Sci. USA 102:210-215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sherr, C. J., and J. M. Roberts. 1999. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 13:1501-1512. [DOI] [PubMed] [Google Scholar]
- 54.Sicinski, P., J. L. Donaher, S. B. Parker, T. Li, A. Fazeli, H. Gardner, S. Z. Haslam, R. T. Bronson, S. J. Elledge, and R. A. Weinberg. 1995. Cyclin D1 provides a link between development and oncogenesis in the retina and breast. Cell 82:621-630. [DOI] [PubMed] [Google Scholar]
- 55.Smits, P., P. Li, J. Mandel, Z. Zhang, J. M. Deng, R. R. Behringer, B. de Crombrugghe, and V. Lefebvre. 2001. The transcription factors L-Sox5 and Sox6 are essential for cartilage formation. Dev. Cell 1:277-290. [DOI] [PubMed] [Google Scholar]
- 56.Snowden, A. W., L. A. Anderson, G. A. Webster, and N. D. Perkins. 2000. A novel transcriptional repression domain mediates p21WAF1/CIP1 induction of p300 transactivation. Mol. Cell. Biol. 20:2676-2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Soos, T. J., H. Kiyokawa, J. S. Yan, M. S. Rubin, A. Giordano, A. DeBlasio, S. Bottega, B. Wong, J. Mendelsohn, and A. Koff. 1996. Formation of p27-CDK complexes during the human mitotic cell cycle. Cell Growth Differ. 7:135-146. [PubMed] [Google Scholar]
- 58.Stewart, M. C., R. M. Kadlcek, P. D. Robbins, J. N. MacLeod, and R. T. Ballock. 2004. Expression and activity of the CDK inhibitor p57Kip2 in chondrocytes undergoing hypertrophic differentiation. J. Bone Miner. Res. 19:123-132. [DOI] [PubMed] [Google Scholar]
- 59.Tedesco, D., J. Lukas, and S. I. Reed. 2002. The pRb-related protein p130 is regulated by phosphorylation-dependent proteolysis via the protein-ubiquitin ligase SCF(Skp2). Genes Dev. 16:2946-2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Thomas, D. M., S. A. Carty, D. M. Piscopo, J. S. Lee, W. F. Wang, W. C. Forrester, and P. W. Hinds. 2001. The retinoblastoma protein acts as a transcriptional coactivator required for osteogenic differentiation. Mol. Cell 8:303-316. [DOI] [PubMed] [Google Scholar]
- 61.Ueta, C., M. Iwamoto, N. Kanatani, C. Yoshida, Y. Liu, M. Enomoto-Iwamoto, T. Ohmori, H. Enomoto, K. Nakata, K. Takada, K. Kurisu, and T. Komori. 2001. Skeletal malformations caused by overexpression of Cbfa1 or its dominant negative form in chondrocytes. J. Cell Biol. 153:87-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Urano, T., H. Yashiroda, M. Muraoka, K. Tanaka, T. Hosoi, S. Inoue, Y. Ouchi, and H. Toyoshima. 1999. p57Kip2 is degraded through the proteasome in osteoblasts stimulated to proliferation by transforming growth factor beta1. J. Biol. Chem. 274:12197-12200. [DOI] [PubMed] [Google Scholar]
- 63.Vernon, A. E., C. Devine, and A. Philpott. 2003. The cdk inhibitor p27Xic1 is required for differentiation of primary neurones in Xenopus. Development 130:85-92. [DOI] [PubMed] [Google Scholar]
- 64.Vernon, A. E., and A. Philpott. 2003. A single cdk inhibitor, p27Xic1, functions beyond cell cycle regulation to promote muscle differentiation in Xenopus. Development 130:71-83. [DOI] [PubMed] [Google Scholar]
- 65.Vidal, A., and A. Koff. 2000. Cell-cycle inhibitors: three families united by a common cause. Gene 247:1-15. [DOI] [PubMed] [Google Scholar]
- 66.Vidal, A., S. S. Millard, J. P. Miller, and A. Koff. 2002. Rho activity can alter the translation of p27 mRNA and is important for RasV12-induced transformation in a manner dependent on p27 status. J. Biol. Chem. 277:16433-16440. [DOI] [PubMed] [Google Scholar]
- 67.Vidal, A., S. Zacharoulis, W. Guo, D. Shaffer, F. Giancotti, A. H. Bramley, C. de la Hoz, K. K. Jensen, D. Kato, D. D. Macdonald, J. Knowles, N. Yeh, L. A. Frohman, S. Rafii, D. Lyden, and A. Koff. 2005. p130Rb2 and p27kip1 cooperate to control mobilization of angiogenic progenitors from the bone marrow. Proc. Natl. Acad. Sci. USA 102:6890-6895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yoshida, C. A., and T. Komori. 2005. Role of Runx proteins in chondrogenesis. Crit. Rev. Eukaryot. Gene Expr. 15:243-254. [DOI] [PubMed] [Google Scholar]
- 69.Zelzer, E., D. J. Glotzer, C. Hartmann, D. Thomas, N. Fukai, S. Soker, and B. R. Olsen. 2001. Tissue specific regulation of VEGF expression during bone development requires Cbfa1/Runx2. Mech. Dev. 106:97-106. [DOI] [PubMed] [Google Scholar]
- 70.Zezula, J., P. Casaccia-Bonnefil, S. A. Ezhevsky, D. J. Osterhout, J. M. Levine, S. F. Dowdy, M. V. Chao, and A. Koff. 2001. p21cip1 is required for the differentiation of oligodendrocytes independently of cell cycle withdrawal. EMBO Rep. 2:27-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang, P., N. J. Liegeois, C. Wong, M. Finegold, H. Hou, J. C. Thompson, A. Silverman, J. W. Harper, R. A. DePinho, and S. J. Elledge. 1997. Altered cell differentiation and proliferation in mice lacking p57KIP2 indicates a role in Beckwith-Wiedemann syndrome. Nature 387:151-158. [DOI] [PubMed] [Google Scholar]
- 72.Zindy, F., J. J. Cunningham, C. J. Sherr, S. Jogal, R. J. Smeyne, and M. F. Roussel. 1999. Postnatal neuronal proliferation in mice lacking Ink4d and Kip1 inhibitors of cyclin-dependent kinases. Proc. Natl. Acad. Sci. USA 96:13462-13467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zwijsen, R. M., E. Wientjens, R. Klompmaker, J. van der Sman, R. Bernards, and R. J. Michalides. 1997. CDK-independent activation of estrogen receptor by cyclin D1. Cell 88:405-415. [DOI] [PubMed] [Google Scholar]