

Abstract
Genome sequencing projects on two relapsing fever spirochetes, Borrelia hermsii and Borrelia turicatae, revealed differences in genes involved in purine metabolism and salvage compared to those in the Lyme disease spirochete Borrelia burgdorferi. The relapsing fever spirochetes contained six open reading frames that are absent from the B. burgdorferi genome. These genes included those for hypoxanthine-guanine phosphoribosyltransferase (hpt), adenylosuccinate synthase (purA), adenylosuccinate lyase (purB), auxiliary protein (nrdI), the ribonucleotide-diphosphate reductase alpha subunit (nrdE), and the ribonucleotide-diphosphate reductase beta subunit (nrdF). Southern blot assays with multiple Borrelia species and isolates confirmed the presence of these genes in the relapsing fever group of spirochetes but not in B. burgdorferi and related species. TaqMan real-time reverse transcription-PCR demonstrated that the chromosomal genes (hpt, purA, and purB) were transcribed in vitro and in mice. Phosphoribosyltransferase assays revealed that, in general, B. hermsii exhibited significantly higher activity than did the B. burgdorferi cell lysate, and enzymatic activity was observed with adenine, hypoxanthine, and guanine as substrates. B. burgdorferi showed low but detectable phosphoribosyltransferase activity with hypoxanthine even though the genome lacks a discernible ortholog to the hpt gene in the relapsing fever spirochetes. B. hermsii incorporated radiolabeled hypoxanthine into RNA and DNA to a much greater extent than did B. burgdorferi. This complete pathway for purine salvage in the relapsing fever spirochetes may contribute, in part, to these spirochetes achieving high cell densities in blood.
The genus Borrelia consists of many species of spirochetes that are important human pathogens transmitted by ticks. The exception to the group is Borrelia recurrentis, which is transmitted by the human body louse (19). One phylogenetic subgroup includes those species that cause relapsing fever, an infection characterized by recurrent febrile episodes associated with high densities of spirochetes in the blood. In addition to the recurrent fever, the illness includes nonspecific symptoms such as headache, muscle and joint pain, chills, and vomiting (17). Borrelia hermsii and Borrelia turicatae are the primary agents of tick-borne relapsing fever in North America (17). These spirochetes are transmitted by the fast-feeding soft ticks Ornithodoros hermsi and Ornithodoros turicata, respectively (16).
A more prevalent tick-borne borreliosis in North America is Lyme disease caused by Borrelia burgdorferi (10, 47). Lyme disease spirochetes are transmitted by slow-feeding hard ticks, with Ixodes scapularis and Ixodes pacificus being the primary vectors (30). Early symptoms of Lyme disease include erythema migrans, low-grade fever, and a variety of nonspecific symptoms. Without antibiotic treatment, the disease may progress to more serious and persistent manifestations, including arthritis, carditis, and various neuropathies (46).
DNA-DNA hybridization studies with B. burgdorferi, B. hermsii, and B. turicatae revealed that these three spirochetes are closely related (25), despite major differences in pathogenicity. In mammals, the density of B. hermsii in the blood may reach 107 spirochetes per ml or more (14), whereas densities reported for B. burgdorferi in mouse plasma range from 1 × 103 to 4 × 105 spirochetes per ml (49). The long feeding time and large blood meal ingested by I. scapularis result in tick infection despite the relatively low number of spirochetes in the blood or skin of the host. The high concentration of relapsing fever spirochetes in the mammalian blood facilitates their efficient acquisition by Ornithodoros ticks, which feed in only 90 min or less. These differences suggest the coevolution of each tick and spirochete to maximize transmission in nature. Most of the mechanisms responsible for the differences in pathogenicity and vector specificity remain unknown.
DNA sequencing of B. hermsii DAH and B. turicatae 91E135 identified six open reading frames (ORFs) involved in purine metabolism in the relapsing fever spirochetes that are absent from the genome of B. burgdorferi (13, 20). Purine nucleotides can be generated via de novo synthesis or through the salvage of preformed purine bases (4, 48). Several pathways for purine salvage have been found in species of Spirochaeta, Treponema, and Leptospira (12, 26). Borrelia species apparently lack genes encoding enzymes required for the de novo synthesis of purines (20). Therefore, these spirochetes must utilize enzymes in the salvage pathway for the acquisition and incorporation of these bases into purine nucleotides. The goal of this study was to further characterize the genes involved in purine salvage and to identify the potential differences in purine metabolism between relapsing fever and Lyme disease spirochetes.
MATERIALS AND METHODS
Bacterial isolates and growth conditions.
The isolates of borreliae used in this study were cultured in modified Kelly's medium (2, 29) (complete BSK-H) (Sigma-Aldrich, St. Louis, MO) or BSK-II (2), both supplemented with rabbit serum to 12% (Table 1). Spirochetes were grown routinely at 34°C, but some experiments included cultures grown at 24°C.
TABLE 1.
Borrelia species and isolates used in this study
| Species | Isolate | Source |
|---|---|---|
| B. hermsii | DAH | Human |
| HS1 | Ornithodoros hermsi | |
| FRO | Human | |
| CON | Human | |
| BRO | Human | |
| YOR | Human | |
| RUM | Human | |
| CMC | Human | |
| HAN | Human | |
| B. turicatae | 91E135 | Ornithodoros turicata |
| RML | Ornithodoros turicata | |
| TCB-2 | Domestic dog | |
| B. parkeri | RML | Ornithodoros parkeri |
| B. crocidurae | CR2A | Ornithodoros erraticus |
| B. anserina | BA2 | Chicken |
| B. coriaceae | Co53 | Ornithodoros coriaceus |
| B. recurrentis | 132 | Human |
| B. miyamotoi | FR 64b | Apodemus argenteus |
| B. burgdorferi | B31 | Ixodes scapularis |
| B. afzelii | VS461 | Ixodes ricinis |
| B. bissettii | DN127 | Ixodes pacificus |
| B. valaisiana | VS116 | Ixodes ricinis |
| B. garinii | G2 | Human |
| B. japonica | HO14 | Ixodes ovatus |
DNA sequencing.
Genomic DNA samples of B. hermsii DAH and B. turicatae 91E135 were purified from 500-ml stationary-phase cultures as previously described (44). The library was constructed with DNA that had been sheared by nebulization as described in detail online at http://www.genome.ou.edu/protocol_book/protocol_partII.html. In brief, 25 μg of DNA was suspended in 500 μl Tris-EDTA and 25% glycerol. The mixture was forced by pressurized nitrogen gas through a plastic nebulizer (no. 4101; IPI Medical Products, Chicago, IL) to create DNA fragments of between 2 and 3 kb. The sheared DNA was gel purified, concentrated, and treated with Klenow and T4 DNA polymerase, and ligated into the vector pCR4Blunt-TOPO. The ligation mixture was transformed into Escherichia coli Top10 F′, and inserts were amplified by PCR and sequenced. DNA sequences were determined as described elsewhere (42).
Annotations.
Putative identifications of ORFs in the Borrelia genomes were made with the ERGO Genome Analysis and Discovery System (34). This bioinformatics suite is licensed from Integrated Genomics, Inc., Chicago, IL, for use at the Rocky Mountain Laboratories and incorporates public and proprietary algorithms with a minimum length cutoff of 40 amino acids when identifying ORFs. A detailed description and the greater predictive power of this package over publicly available annotation programs is presented elsewhere (34).
Southern blot assays.
Genomic DNA samples from borreliae were examined by Southern blot assays (45) as described previously (40). Briefly, EcoRI-digested DNA samples were separated by agarose gel electrophoresis. The DNA was visualized with ethidium bromide stain and UV transillumination. After depurination, denaturation, and neutralization, the DNA was transferred overnight by capillary action onto MagnaGraph nylon membranes (Micron Separations Inc., Westborough, MA). Hybridization probes were produced with the PCR digoxigenin probe synthesis kit (Roche Applied Science, Indianapolis, IN) as specified by the manufacturer. Genomic DNA of B. hermsii DAH was used as the template to produce probes using primers for hpt, purA, purB, nrdI, nrdE, nrdF, and ade (Tables 2 and 3). These primers amplified DNA fragments of 355, 452, 693, 343, 403, 406, and 320 bp, respectively. Genomic DNA of B. turicatae 91E135 was used as the template to produce a second ade probe that was 278 bp. The digoxigenin-labeled probes were denatured at 98°C for 10 min, added to 6 ml of fresh hybridization buffer with the membrane, and incubated at 55°C for 18 h. The membranes were washed with 2× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate)-0.1% sodium dodecyl sulfate for 10 min at room temperature and with 0.3× SSC-0.1% sodium dodecyl sulfate for 30 min at 65°C, which lowered the stringency suggested by the manufacturer. The blots were incubated with antidigoxigenin antibody conjugated to alkaline phosphatase and developed with the CDP-Star chemiluminescent substrate (both from Roche Applied Science). Hyperfilm ECL high-performance chemiluminescence film (Amersham Biosciences Inc., Piscataway, NJ) was exposed to membranes and developed to display the pattern of hybridization.
TABLE 2.
Primers and sequences for PCR amplification of hybridization probes
| Gene | Primer | Sequence (5′ to 3′) |
|---|---|---|
| hpt | hpt 5′ | CAAGAGAAATTGGATTAAACG |
| hpt 3′ | GCTACATTCTTTAAAGTTCTGTG | |
| purA | purA 5′ | ATCGGAACTACAAAACGAGG |
| purA 3′ | GGTCCTGAACCAACTCTTGATG | |
| purB | purB 5′ | GGCACATCTTTATGCTTATGC |
| purB 3′ | CCACCACTTGATTGTAGGCTC | |
| nrdI | nrdI 5′ | GCTTCAAAAACAGGAAATATAGAGC |
| nrdI 3′ | GTGCCTCATTTTTAATCCTTTCTAC | |
| nrdE | nrdE 5′ | GGATGGATTTTACAGATTAG |
| nrdE 3′ | TAAAAATGTAGGAGTTGCAG | |
| nrdF | nrdF 5′ | TTAGATGGTGAGGAGCGGGA |
| nrdF 3′ | CCATCTTACCTTGACCTGCA | |
| ade, intact | ade 5′ | CACAAAGTAAAACTATACCAAC |
| ade 3′ | GCACATAAGCCCCCTTTATTCTC | |
| ade, deletion | ade-del 5′ | GGTATTGATGGTATTAATTTCATGATAAACAACTCC |
| ade-del 3′ | GGTGCATGTCCGTCGACAAC |
TABLE 3.
Borrelia purine metabolism genes and ORF designations
| Gene product | Gene | ORF designation in:
|
||
|---|---|---|---|---|
| B. burgdorferi B31 | B. hermsii DAH | B. turicatae 91E135 | ||
| Hypoxanthine-guanine PRT | hpt | BH0421-A | BT0421-A | |
| Adenylosuccinate synthase | purA | BH0421-B | BT0421-B | |
| Adenylosuccinate lyase | purB | BH0421-C | BT0421-C | |
| Auxiliary (NrdI) protein | nrdI | BHP76-A | BTP76-A | |
| Ribonucleotide-diphosphate reductase alpha subunit | nrdE | BHP76-B | BTP76-B | |
| Ribonucleotide-diphosphate reductase beta subunit | nrdF | BHP76-C | BTP76-C | |
| Adenine PRT | apt | BB0777 | BH0777 | BT0777 |
| PRT | prt | BB0103 | BH0103 | BT0103 |
| GMP synthase | guaA | BBB18 | BHP-B18 | BTP-B18 |
| IMP dehydrogenase | guaB | BBB17 | BHP-B17 | BTP-B17 |
| Adenine deaminase | ade | BBK17 (BBH33) | Pseudogene | BTP-K17 |
| Guanine-hypoxanthine permease | pbuG | BBB22, BBB23 | BHP-B22 | BTP-B22 |
| Adenylate kinase | adk | BB0417 | BH0417 | BT0417 |
| Nucleoside-diphosphate kinase | ndk | BB0463 | BH0463 | BT0463 |
RNA isolation.
RNA was isolated from blood of two infected mice and spirochetes grown in BSK-H at 24°C and 34°C. The erythrocytes in the infected mouse blood were removed by centrifugation (100 × g for 5 min). Spirochetes in cultures and serum were harvested by centrifugation (14,000 × g for 15 min). The pellets were suspended in 100 μl Tris-EDTA containing 2 mg/ml lysozyme (Sigma-Aldrich) and incubated on ice for 3 min. Bacterial cell lysis and RNA isolation were performed with the RNeasy kit (QIAGEN, Valencia, CA) following the manufacturer's protocol. Contaminating DNA was removed with DNA-free (Ambion, Inc., Austin, TX). RNA quality was assessed by spectrophotometry and the RNA 6000 Nano assay kit (Agilent Technologies, Palo Alto, CA).
Real-time reverse transcription-PCR (RT-PCR).
Primers and probes were designed with Primer Express version 2.0 software (Applied Biosystems) and purchased from Applied Biosystems and MegaBases (Evanston, IL) (Table 4). TaqMan assays were performed as previously described (23). Assays were performed in triplicate with RNA samples isolated from at least two independent cultures of B. hermsii DAH, and standard curves were made with B. hermsii DAH genomic DNA. Triplicate assays using three independent RNAs confirmed that transcript levels of flaB were high and nearly identical at 24°C compared with 34°C (data not shown); therefore, 49 ng of flaB transcripts was used for each assay to normalize the data. TaqMan assays were also performed to determine transcription during infection in mice.
TABLE 4.
Primer and fluorescent probe sequences for real-time RT-PCR
| Gene | Primers and probe | Sequence (5′ to 3′) |
|---|---|---|
| hpt | Forward | AAGAATTAGCCCAAAAGATTAGAAACTACT |
| Probe | CACTTCTTAAAGGCTCTTTCATGTTT | |
| Reverse | CGTTTAATCCAATCTCTCTTGTAATATCTG | |
| purA | Forward | TCAAGAGTTGGTTCAGGACCTTTT |
| Probe | TCTTATCTCCAATAGGTTCTAGGATTTCAGTT | |
| Reverse | GCCATATTCCTGCCCCTTTT | |
| purB | Forward | GGCGGATTTATAGCTGCAACTC |
| Probe | TGCAAAAGACTAAACATTCCTCAAGCAT | |
| Reverse | TCAGGGCAGCATCAGTAGCTAAG | |
| flaB | Forward | AAGTCAGCTGCTCAAAATGTAAAAAC |
| Probe | TTTGCGGGTTGCATTCCAAGCTCTT | |
| Reverse | CAGCTAGTGATGCTGGTGTGTTAAT |
PRT activity assays.
Phosphoribosyltransferase (PRT) assays were performed as previously described (38). Briefly, borreliae were grown in 100 ml of BSK-H medium to a cell density of 1 × 108/ml as determined by microscopy. Spirochetes were pelleted by centrifugation at 14,300 × g for 20 min, suspended in phosphate-buffered saline, pelleted again, and suspended in cold assay buffer (47 mM Tris [pH 7.5], 2.1 mM dithiothreitol, 3.8 mM MgCl2, 0.21 mg/ml bovine serum albumin). The cells were sonicated on ice with a Branson Sonifier-cell disrupter 185 (VWR Scientific, San Francisco, CA). Cell fractions were centrifuged at 20,400 × g for 30 min to separate soluble and insoluble fractions. Each assay was done at 37°C in a 60-μl reaction mixture of the cold assay buffer supplemented with 2.1 mM 5-phosphoribosyl-pyrophosphate and various concentrations of borrelia-soluble lysates. The reaction was initiated with 12 or 24 μCi [3H]adenine, [3H]hypoxanthine, [3H]xanthine, or [3H]guanine (Moravek Biochemicals Inc., Brea, CA). At 0, 5, 10, 15, 30, and 45 min after initiation, 5 μl was transferred to a new tube on ice and the reaction was terminated with 1 μl 0.3 M EDTA. The purine substrate was separated from products using thin-layer chromatography on Baker-flex polyethyleneimine cellulose thin-layer chromatography sheets (J.T. Baker Inc., Phillipsburg, NJ) and visualized with UV light. Lanes were cut from the sheet, and activity was quantified with a Beckman Coulter LS6500 scintillation counter (Beckman Coulter, Fullerton, CA).
Nucleotide incorporation assays.
B. hermsii DAH and B. burgdorferi B31 cells were inoculated into BSK-II medium at a density of ∼5 × 105 cells/ml with 20 μCi (1 nmol) [3H]hypoxanthine (Moravek Biochemicals Inc.) and grown to a density of ∼8 × 107 cells/ml. Nucleic acids were purified from the 100-ml cultures using the QIAGEN RNA/DNA maxi kit following the manufacturer's protocol (QIAGEN). Purity was verified by agarose gel electrophoresis and an absorbance ratio at 260/280 nm. Incorporation of labeled nucleotides was quantified by counts per minute with a scintillation counter and normalized to the concentration of nucleic acids. To demonstrate the absence of RNA contamination, DNA samples were treated with DNase-free RNase A and purified again, and they showed no appreciable loss of radioactivity.
Nucleotide sequence accession numbers.
The 34 nucleotide sequences determined for B. hermsii and B. turicatae in this study have been deposited in the GenBank database under accession numbers DQ355750 to DQ355783.
RESULTS
DNA sequence analysis of Borrelia genes involved in purine metabolism.
Sequence analysis of the B. hermsii and B. turicatae genomes revealed three adjacent ORFs located between homologs of the B. burgdorferi chromosomal genes BB0421 and BB0422 (20) (Fig. 1A). These genes were homologous to genes in other organisms involved in purine salvage: hpt (hypoxanthine-guanine PRT), purA (adenylosuccinate synthase), and purB (adenylosuccinate lyase). Additionally, three plasmid-encoded genes involved in purine metabolism that are also absent in B. burgdorferi were identified in B. hermsii and B. turicatae: nrdI (auxiliary protein), nrdE (ribonucleotide-diphosphate reductase alpha subunit) and nrdF (ribonucleotide-diphosphate reductase beta subunit) (Fig. 1B). Additional genes involved in purine salvage that are present in B. burgdorferi were also found in B. hermsii and B. turicatae (Table 3) (13, 20, 21, 32). None of these spirochetes have genes encoding enzymes for the de novo synthesis of purines or pyrimidines; however, both B. hermsii and B. turicatae have a complete purine salvage pathway that is incomplete in B. burgdorferi.
FIG. 1.

Graphical representation of the ORFs in B. hermsii. (A) Chromosomal genes hpt, purA and purB; (B) plasmid-encoded genes nrdI, nrdE, and nrdF. In B. hermsii hpt, purA, and purB are located between orthologous genes BB0421 and BB0422 in B. burgdorferi. The ERGO bioinformatics suite (Integrated Genomics, Chicago, IL) (34) was used to produce these genetic maps. The scale bars indicate size in base pairs.
Southern blot analysis.
Genomic DNA samples from 17 borreliae were analyzed for the six genes that by sequence analysis were specific to B. hermsii and B. turicatae. DNA from the relapsing fever group bound all probes (Fig. 2 and data not shown). hpt, purA, and purB were located on the same-size EcoRI restriction fragment in B. hermsii DAH. Probes to these three genes also bound to restriction fragments of similar size in all relapsing fever isolates. In Borrelia coriaceae and Borrelia miyamotoi, the purB probe bound to a restriction fragment of a different size, indicating an additional EcoRI site between the sequences recognized by the purA and purB probes. The results were similar with probes to nrdI, nrdE, and nrdF (data not shown). These probes all hybridized to an EcoRI fragment of the same size in all species except Borrelia parkeri and B. turicatae, in which the nrdF probe bound to a restriction fragment of a different size. None of the DNA samples from the six isolates of B. burgdorferi sensu lato hybridized with these probes. Southern blot assays with undigested genomic DNAs from seven isolates of B. hermsii mapped the ribonucleotide-diphosphate reductase genes nrdI, nrdE, and nrdF to the largest linear plasmid of ∼200 kb (data not shown).
FIG. 2.

Southern blot analysis for the presence of purB and nrdF among Borrelia species. (A) Ethidium bromide-stained agarose gel of EcoRI-digested genomic DNA. (B) Hybridization pattern with the purB probe. (C) Hybridization pattern with the nrdF probe. Molecular size standards are shown on the left.
Quantitative RT-PCR analysis of hpt, purA, and purB transcripts.
Transcription of hpt, purA, and purB in B. hermsii DAH grown in vitro and in vivo was examined by TaqMan real-time RT-PCR. Specific RNAs were detected for each gene from spirochetes isolated from culture and mice, and there was a slight decrease in the amount of transcripts from spirochetes grown at the lower temperature (Table 5). However, the transcripts for the purine salvage genes were all much less abundant than the transcripts for flaB.
TABLE 5.
Transcription of B. hermsii purine salvage genes in vitro and in vivo
| Gene | Transcript amounts of hpt, purA and purB (ng, mean ± SD) compared to flaBa
|
Ratio (34°C/24°C) | ||
|---|---|---|---|---|
| 34°C culture | 24°C culture | In vivo | ||
| hpt | 12.54 ± 7.04 | 7.43 ± 2.64 | 2.64 ± 0.72 | 1.69 |
| purA | 4.88 ± 0.16 | 2.74 ± 0.20 | 0.53 ± 0.21 | 1.78 |
| purB | 3.57 ± 0.31 | 2.54 ± 1.13 | 1.38 ± 0.39 | 1.41 |
Relative amounts of mRNA from B. hermsii DAH grown at two culture temperatures or isolated from mouse blood were determined by real-time RT-PCR with flaB as the standard. flaB transcripts were normalized to 49 ng in each assay for comparison with the other genes.
We attempted to generate antibodies to the proteins encoded by these three genes to examine their presence in B. hermsii. Initial efforts to express the native genes in E. coli were unsuccessful. Examination of the codon usage revealed many rare codons, and such bias might explain the lack of expression in E. coli (data not shown). Therefore, we purchased a synthetic hpt gene (GenScript Corp., Piscataway, NJ) with codons optimized for expression in E. coli, as was done for B. burgdorferi BB0728 (9). We expressed this gene and purified the recombinant His-tagged Hpt. However, when rabbits were immunized with this material, the titer of the resulting antiserum was very poor, which suggested that Hpt was not immunogenic. Similar results were obtained when rabbits were immunized with synthetic Hpt peptides; therefore, the details of these efforts are not presented.
PRT activity.
PRT activity was examined with cell lysates of B. hermsii and B. burgdorferi (Fig. 3). The enzymatic activity of B. hermsii was generally greater than that of B. burgdorferi. Both species exhibited the most PRT activity with adenine, while no PRT activity was detected with xanthine. The B. hermsii lysate exhibited similar activities with hypoxanthine and guanine, while the B. burgdorferi lysate had no activity with guanine. However, B. burgdorferi had a small but reproducible PRT activity with hypoxanthine, in spite of the lack an hpt ortholog in this spirochete's genome (20).
FIG. 3.

PRT activities of B. burgdorferi and B. hermsii cell lysates with different purines as substrate. Activity was measured in femtomoles of purine base converted to ribonucleotide per milligram of lysate per minute. Error bars indicate standard deviations.
Nucleotide incorporation assays.
The results of the assays for PRT activity and the genomic differences in purine salvage pathways between B. hermsii and B. burgdorferi led us to examine the incorporation of 3H-labeled hypoxanthine into DNA and RNA by these spirochetes. Duplicate samples from three independent cultures of each species were examined. The results were consistent and therefore were combined for presentation (Fig. 4). B. hermsii incorporated labeled hypoxanthine equally into both DNA and RNA, with nearly ninefold-greater counts per minute than in B. burgdorferi. Unexpectedly, B. burgdorferi also incorporated a low but detectable level of hypoxanthine into DNA, suggesting that another, less efficient pathway was present. The labeled hypoxanthine used in these assays was 99.9% pure; therefore, the activity detected in B. burgdorferi was not due to the incorporation of other labeled bases.
FIG. 4.
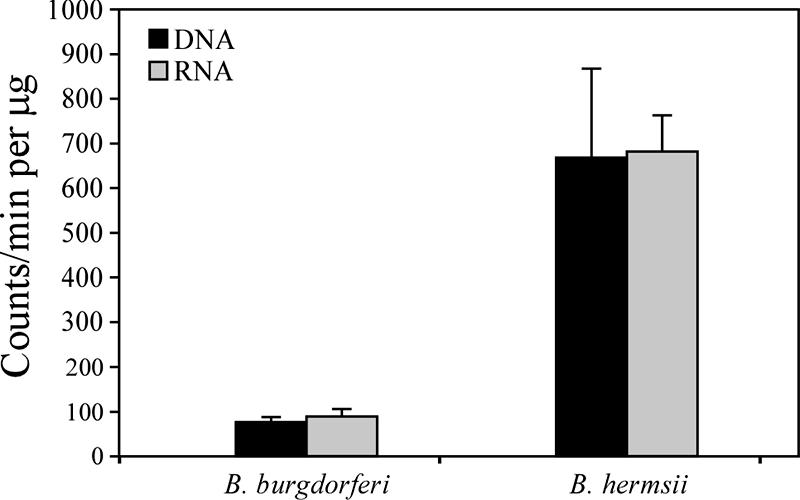
[3H]hypoxanthine incorporation into nucleic acids. The values are based on two measurements from each of three independent assays. The counts per minute per microgram of nucleic acid are represented by the mean and one standard deviation.
Examination of the ade locus.
Our sequencing efforts identified another ORF in the purine salvage pathway that coded for adenine deaminase (ade gene). B. turicatae 91E135 contained an intact ade gene, while the ortholog in B. hermsii DAH appeared to be a pseudogene with many premature stop codons and deletions. Therefore, we PCR amplified and sequenced this region in four additional isolates of B. hermsii to determine if the ade gene was also disrupted in these isolates. Multilocus sequence typing of 37 B. hermsii isolates recently identified two genomic groups in this species (35, 43). We sequenced the ade regions in two additional isolates from genomic group I (GGI) (HS1 and CON), two isolates from GGII (RUM and YOR), and two additional isolates of B. turicatae (RML and TCB-2). The DNA sequences of the two additional GGI isolates were identical to the DAH sequence and contained the same deletions and premature stop codons. However, the ade sequences in the two GGII isolates and the two additional isolates of B. turicatae contained an intact gene.
The ade pseudogene in the B. hermsii GGI isolates contained a 279-bp deletion that represented 17% of the coding sequence of the intact gene in the B. hermsii GGII isolates and B. turicatae. Therefore, two probes were made, one based on the region shared by all isolates and one based on the region present only in GGII isolates. Southern blot assays with four isolates in each genomic group confirmed the absence of an intact ade gene in the genomes of GGI isolates (Fig. 5).
FIG. 5.

Southern blot analysis for the adenine deaminase gene (ade) in B. hermsii. (A) Agarose gel with genomic DNA digested with EcoRI and stained with ethidium bromide. (B) Hybridization with the probe containing the sequence present in both the intact gene and pseudogene. (C) Hybridization with the probe with the sequence present only in the intact gene. Isolate designations are indicated above each lane. Molecular size standards are shown on the left.
DISCUSSION
Three chromosomal ORFs (hpt, purA, and purB) and three plasmid ORFs (nrdI, nrdE, and nrdF) that are involved in purine salvage and that are absent in the genomes of B. burgdorferi, Borrelia garinii, and Borrelia afzelii (20-22) were found in B. hermsii and B. turicatae. Another group has recently identified these loci in B. hermsii (3, 50). Southern blot assays confirmed that these genes (or highly related sequences) were present in all relapsing fever spirochetes examined but not in the Lyme disease group of bacteria. In general, the enzymes involved in purine salvage include PRTs, which catalyze the transfer of a phosphoribosyl group from phosphoribosyl-pyrophosphate to a purine base (e.g., adenine, hypoxanthine, xanthine, or guanine) that yields the corresponding nucleoside 5′-monophospate (e.g., AMP, IMP, XMP, or GMP) (Fig. 6). IMP is a central molecule in this process that can be used to generate both AMP and GMP via two pathways with adenylosuccinate synthase (PurA) and adenylosuccinate lyase (PurB) or with GMP synthase (GuaA) and IMP dehydrogenase (GuaB), respectively.
FIG. 6.

Schematic representation of purine metabolism pathways identified in B. burgdorferi, B. hermsii, and B. turicatae. Solid arrows represent enzymatic steps associated with their corresponding gene designation identified in Table 3. Genes present only in B. hermsii and B. turicatae are in a box, while those present in all three species are not enclosed. The dashed arrow represents a pathway with no gene yet identified. This figure was modified from reference 39.
Barbour and coworkers recently identified the hpt-purA-purB locus in B. hermsii HS1 and Borrelia miyamotoi and a partial sequence in Borrelia crocidurae (3). These investigators complemented an E. coli purA mutant with the B. hermsii purA gene, which restored the ability of the mutant to grow without adenine supplemented to the medium. They also performed a phylogenetic analysis with B. hermsii purA, purB, and orthologs from other organisms. The results suggested that these genes had a different evolutionary history from their orthologs in other spirochetes, and that the relapsing fever spirochetes likely acquired this locus via horizontal transfer from another bacterium after the division of the Lyme disease and relapsing fever groups of spirochetes. Zhong and coworkers recently identified the nrdIEF locus in B. hermsii HS1 and concluded that these genes were also likely acquired by horizontal transfer (50).
The B. burgdorferi genome lacks an obvious ribonucleotide-diphosphate reductase (20). These enzymes make deoxyribonucleotides from ribonucleotides, and their presence has been considered essential for all organisms to provide the deoxyribonucleotide pool required for DNA synthesis (27). Ribonucleotide-diphosphate reductases fall into three classes based, in part, on their cofactors and oxygen requirements (37). Class I ribonucleotide-diphosphate reductase (subgroups 1A and 1B) generate a tyrosyl radical with an iron-oxygen center and dioxygen. This process requires oxygen, and therefore class I ribonucleotide-diphosphate reductase can function only under aerobic or microaerophilic conditions (27).
Based on sequence comparisons, we agree with Zhong et al. (50) that the B. hermsii NrdE and NrdF proteins are likely class Ib ribonucleoside-diphosphate reductases. Zhong and coworkers (50) discussed the nrdIEF locus in B. hermsii only in the context of its role in the salvage of pyrimidines. However, these ribonucleoside-diphosphate reductases are also required to convert GDP and ADP to dGDP and dADP, respectively (33), and therefore play a critical role in the salvage of purines and the synthesis of purine nucleotides. Interestingly, the presence of a potential Fe-containing class 1b ribonucleoside-diphosphate reductase in the relapsing fever spirochetes may indicate a fundamental difference in metal utilization and/or metabolism compared to the Lyme disease spirochetes (36).
In general, B. hermsii exhibited greater PRT activity than did B. burgdorferi. We detected the highest PRT activity with adenine as the substrate but no activity with xanthine. One major difference between the two species was that B. hermsii exhibited similar activities with hypoxanthine and guanine whereas B. burgdorferi had no activity with guanine and only a small yet detectable activity with hypoxanthine. Also, B. hermsii incorporated much more labeled hypoxanthine than did B. burgdorferi. However, B. burgdorferi did incorporate some labeled hypoxanthine into DNA as well as into RNA. Since B. burgdorferi, B. garinii, and B. afzelii lack the hpt-purA-purB and nrdIEF loci (20-22), these findings were unexpected. The B. burgdorferi genome contains two genes encoding PRTs, apt (adenine PRT, BB0777) and prt (PRT, BB0103), with the latter the likely candidate responsible for the observed activity with hypoxanthine. Another possibility involves nucleoside deoxyribosyltransferases in B. burgdorferi (20), which our ERGO database assigns to BB0426 but which we have not found in relapsing fever spirochetes. These enzymes catalyze the transfer of the deoxyribosyl group from a nucleoside to another base (1, 28). What we interpreted as hypoxanthine PRT activity may have been the transfer of a deoxyribosyl group to hypoxanthine catalyzed by this enzyme. These results indicate some fundamental differences between the relapsing fever and the Lyme disease spirochetes in the salvage of purines. With the one exception noted below, the relapsing fever spirochetes have a complete purine salvage pathway while the Lyme disease spirochetes do not.
B. burgdorferi B31 has two copies of ade (adenine deaminase), each on a different plasmid: the full-length BBK17, consisting of 1,644 bp on lp36, and the severely truncated BBH33, which contains only 273 bp on lp28-3 (13, 20). An intact ade of 1,644 bp was also found in three isolates of B. turicatae and two isolates of B. hermsii in GGII. However, this gene was disrupted in the B. hermsii GGI isolates, with no evidence of another intact copy. Three of the GGI isolates examined came from clinically ill patients, and other clinical isolates in this genomic group are transmissible by ticks (35, 41). Thus, we conclude that spirochetes in GGI of B. hermsii lack one of the pathways for purine salvage, with no apparent loss of virulence or in their ability to persist in ticks. The inability to convert adenine to hypoxanthine in GGI isolates is probably compensated for by the ability of these spirochetes to take up hypoxanthine directly and convert it to IMP by hypoxanthine PRT.
The ability of the relapsing fever spirochetes to achieve such high cell densities in the blood is unique among the pathogenic spirochetes. Also, unlike the Lyme disease spirochetes, the relapsing fever spirochetes have a complete pathway for purine salvage. B. hermsii exhibited sevenfold more PRT activity with hypoxanthine than did B. burgdorferi, and B. hermsii incorporated ninefold more hypoxanthine into RNA and DNA than did B. burgdorferi. Hypoxanthine is the most abundant purine in human plasma (with hypoxanthine at 8.2 ± 1.3 μM, xanthine at 2.5 ± 0.6 μM, adenine at 0.3 ± 0.15 μM, and adenosine at 0.6 ± 0.2 μM) (24), is the primary product of purine catabolism within red blood cells (5, 8), is transported across the red blood cell membrane (31), and is effluxed to the outer surface of red blood cells (6). A common phenotype of relapsing fever spirochetes is their adherence to red blood cells during infection in humans and mice (11, 15, 23). We believe that this specific interaction of spirochetes with erythrocytes could provide a mechanism for the direct uptake of hypoxanthine, which is utilized for the synthesis of both RNA and DNA nucleotides. The human malarial parasite Plasmodium falciparum is also unable to synthesize purines de novo and relies on the salvage of intraerythrocytic hypoxanthine for its purine source to synthesize nucleotides (7, 18). The direct uptake of hypoxanthine by the relapsing fever spirochetes from the outer surface of red blood cells could account, in part, for the ability of these spirochetes to achieve such high cell densities in the blood, and efforts to test this hypothesis are under way.
Acknowledgments
We thank Anita Mora and Gary Hettrick for help with the figures, Greg Somerville for advice, Paul Policastro for animal work, Rebecca Byram for technical advice and reagents, and Mary Burtnick and Mark Fisher for reviewing the manuscript.
This research was supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases, NIH.
Editor: D. L. Burns
Footnotes
Published ahead of print on 14 May 2007.
REFERENCES
- 1.Anand, R., A. K. Kaminski, and S. E. Ealick. 2004. Structures of purine 2′-deoxyribosyltransferase, substrate complexes, and the ribosylated enzyme intermediate at 2.0 A resolution. Biochemistry 43:2384-2393. [DOI] [PubMed] [Google Scholar]
- 2.Barbour, A. G. 1984. Isolation and cultivation of Lyme disease spirochetes. Yale J. Biol. Med. 57:521-525. [PMC free article] [PubMed] [Google Scholar]
- 3.Barbour, A. G., A. D. Putteeet-Driver, and J. Bunikis. 2005. Horizontally acquired genes for purine salvage in Borrelia spp. causing relapsing fever. Infect. Immun. 73:6165-6168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Becerra, A., and A. Lazcano. 1998. The role of gene duplication in the evolution of purine nucleotide salvage pathways. Orig. Life Evol. Biosph. 28:539-553. [DOI] [PubMed] [Google Scholar]
- 5.Berman, P. A., D. A. Black, L. Human, and E. H. Harley. 1988. Oxypurine cycle in human erythrocytes regulated by pH, inorganic phosphate, and oxygen. J. Clin. Investig. 82:980-986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berman, P. A., and L. Human. 1990. Regulation of 5-phosphoribosyl 1-pyrophosphate and of hypoxanthine uptake and release in human erythrocytes by oxypurine cycling. J. Biol. Chem. 265:6562-6568. [PubMed] [Google Scholar]
- 7.Berman, P. A., L. Human, and J. A. Freese. 1991. Xanthine oxidase inhibits growth of Plasmodium falciparum in human erythrocytes in vitro. J. Clin. Investig. 88:1848-1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bontemps, F., G. Van den Berghe, and H. G. Hers. 1986. Pathways of adenine nucleotide catabolism in erythrocytes. J. Clin. Investig. 77:824-830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boylan, J. A., C. S. Hummel, S. Benoit, J. Garcia-Lara, J. Treglown-Downey, E. J. Crane III, and F. C. Gherardini. 2006. Borrelia burgdorferi bb0728 encodes a coenzyme A disulphide reductase whose function suggests a role in intracellular redox and the oxidative stress response. Mol. Microbiol. 59:475-486. [DOI] [PubMed] [Google Scholar]
- 10.Burgdorfer, W., A. G. Barbour, S. F. Hayes, J. L. Benach, E. Grunwaldt, and J. P. Davis. 1982. Lyme disease—a tick-borne spirochetosis? Science 216:1317-1319. [DOI] [PubMed] [Google Scholar]
- 11.Burman, N., A. Shamaei-Tousi, and S. Bergström. 1998. The spirochete Borrelia crocidurae causes erythrocyte rosetting during relapsing fever. Infect. Immun. 66:815-819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Canale-Parola, E., and G. W. Kidder. 1982. Enzymatic activities for interconversion of purines in spirochetes. J. Bacteriol. 152:1105-1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Casjens, S., N. Palmer, R. van Vugt, W. M. Huang, B. Stevenson, P. Rosa, R. Lathigra, G. Sutton, J. Peterson, R. J. Dodson, D. Haft, E. Hickey, M. Gwinn, O. White, and C. M. Fraser. 2000. A bacterial genome in flux: the twelve linear and nine circular extrachromosomal DNAs in an infectious isolate of the Lyme disease spirochete Borrelia burgdorferi. Mol. Microbiol. 35:490-516. [DOI] [PubMed] [Google Scholar]
- 14.Coffey, E. M., and W. C. Eveland. 1967. Experimental relapsing fever initiated by Borrelia hermsi. II. Sequential appearence of major serotypes in the rat. J. Infect. Dis. 117:29-34. [DOI] [PubMed] [Google Scholar]
- 15.Cook, A. R. 1904. Relapsing fever in Uganda. J. Trop. Med. Hyg. 7:24-26. [Google Scholar]
- 16.Davis, G. E. 1942. Species unity or plurality of the relapsing fever spirochetes, p. 41-47. In F. R. Moulton (ed.), A symposium of relapsing fever in the Americas. American Association for the Advancement of Science, Washington, DC.
- 17.Dworkin, M. S., T. G. Schwan, and D. E. Anderson. 2002. Tick-borne relapsing fever in North America. Med. Clin. North Am. 86:417-433. [DOI] [PubMed] [Google Scholar]
- 18.El Bissati, K., R. Zufferey, W. H. Witola, N. S. Carter, B. Ullman, and C. Ben Mamoun. 2006. The plasma membrane permease PfNT1 is essential for purine salvage in the human malaria parasite Plasmodium falciparum. Proc. Natl. Acad. Sci. USA 103:9286-9291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Felsenfeld, O. 1971. Borrelia. Strains, vectors, human and animal borreliosis. Warren H. Green, Inc., St. Louis, MO.
- 20.Fraser, C. M., S. Casjens, W. M. Huang, G. G. Sutton, R. Clayton, R. Lathigra, O. White, K. A. Ketchum, R. Dodson, E. K. Hickey, M. Gwinn, B. Dougherty, J.-F. Tomb, R. D. Fleischmann, D. Richardson, J. Peterson, A. R. Kerlavage, J. Quackenbush, S. Salzberg, M. Hanson, R. V. Vugt, N. Palmer, M. D. Adams, J. Gocayne, J. Weidman, T. Utterback, L. Watthey, L. McDonald, P. Artiach, C. Bowman, S. Garland, C. Fujii, M. D. Cotton, K. Horst, K. Roberts, B. Hatch, H. O. Smith, and J. C. Venter. 1997. Genomic sequence of a Lyme disease spirochaete, Borrelia burgdorferi. Nature 390:580-586. [DOI] [PubMed] [Google Scholar]
- 21.Glockner, G., R. Lehmann, A. Romualdi, S. Pradella, U. Schulte-Spechtel, M. Schilhabel, B. Wilske, J. Suhnel, and M. Platzer. 2004. Comparative analysis of the Borrelia garinii genome. Nucleic Acids Res. 32:6038-6046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Glockner, G., U. Schulte-Spechtel, M. Schilhabel, M. Felder, J. Suhnel, B. Wilske, and M. Platzer. 2006. Comparative genome analysis: selection pressure on the Borrelia vls cassettes is essential for infectivity. BMC Genomics 7:211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guyard, C., E. M. Chester, S. J. Raffel, M. E. Schrumpf, P. F. Policastro, S. F. Porcella, J. M. Leong, and T. G. Schwan. 2005. Relapsing fever spirochetes contain chromosomal genes with unique direct tandemly repeated sequences. Infect. Immun. 73:3025-3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hartwick, R. A., A. M. Krstulovic, and P. R. Brown. 1979. Identification and quantitation of nucleosides, bases and other UV-absorbing compounds in serum, using reversed-phase high performance liquid chromatography. II. Evaluation of human sera. J. Chromatogr. 186:659-676. [DOI] [PubMed] [Google Scholar]
- 25.Hyde, F. W., and R. C. Johnson. 1986. Genetic analysis of Borrelia. Zentbl. Bakteriol. Hyg. A 263:119-122. [DOI] [PubMed] [Google Scholar]
- 26.Johnson, R. C., and P. Rogers. 1967. Metabolism of leptospires. II. The action of 8-azaguanine. Can. J. Microbiol. 13:1621-1629. [DOI] [PubMed] [Google Scholar]
- 27.Jordan, A., and P. Reichard. 1998. Ribonucleotide reductases. Annu. Rev. Biochem. 67:71-98. [DOI] [PubMed] [Google Scholar]
- 28.Kaminski, P. A. 2002. Functional cloning, heterologous expression, and purification of two different N-deoxyribosyltransferases from Lactobacillus helveticus. J. Biol. Chem. 277:14400-14407. [DOI] [PubMed] [Google Scholar]
- 29.Kelly, R. 1971. Cultivation of Borrelia hermsi. Science 173:443-444. [DOI] [PubMed] [Google Scholar]
- 30.Lane, R. S., J. Piesman, and W. Burgdorfer. 1991. Lyme borreliosis: relation of its causative agent to its vectors and hosts in North America and Europe. Annu. Rev. Entomol. 36:587-609. [DOI] [PubMed] [Google Scholar]
- 31.Lassen, U. V. 1967. Hypoxanthine transport in human erythrocytes. Biochim. Biophys. Acta 135:146-154. [DOI] [PubMed] [Google Scholar]
- 32.Margolis, N., D. Hogan, K. Tilly, and P. A. Rosa. 1994. Plasmid location of Borrelia purine biosynthesis gene homologs. J. Bacteriol. 176:6427-6432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Neuhard, J., and P. Nygaard. 1987. Purines and pyrimidines, p. 445-473. In F. C. Neidhardt et al. (ed.), Escherichia coli and Salmonella typhimurium: cellular and molecular biology, vol. 1. American Society for Microbiology, Washington, DC. [Google Scholar]
- 34.Overbeek, R., N. Larsen, T. Walunas, M. D'Souza, G. Pusch, E. J. Selkov, K. Liolios, V. Joukov, D. Kaznadzey, I. Anderson, A. Bhattacharyya, H. Burd, W. Gardner, P. Hanke, V. Kapatral, N. Mikhailova, O. Vasieva, A. Osterman, V. Vonstein, M. Fonstein, N. Ivanova, and N. Kyrpides. 2003. The ERGO™ genome analysis and discovery system. Nucleic Acids Res. 31:164-171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Porcella, S. F., S. J. Raffel, D. E. Anderson Jr., S. D. Gilk, J. L. Bono, M. E. Schrumpf, and T. G. Schwan. 2005. Variable tick protein in two genomic groups of the relapsing fever spirochete Borrelia hermsii in western North America. Infect. Immun. 73:6647-6658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Posey, J. E., and F. C. Gherardini. 2000. Lack of a role for iron in the Lyme disease pathogen. Science 288:1651-1653. [DOI] [PubMed] [Google Scholar]
- 37.Reichard, P. 1993. From RNA to DNA, why so many ribonucleotide reductases? Science 260:1773-1777. [DOI] [PubMed] [Google Scholar]
- 38.Reyes, P., P. K. Rathod, D. J. Sanchez, J. E. Mrema, K. H. Rieckmann, and H. G. Heidrich. 1982. Enzymes of purine and pyrimidine metabolism from the human malaria parasite, Plasmodium falciparum. Mol. Biochem. Parasitol. 5:275-290. [DOI] [PubMed] [Google Scholar]
- 39.Saxild, H. H., K. Brunstedt, K. I. Nielsen, H. Jarmer, and P. Nygaard. 2001. Definition of the Bacillus subtilis PurR operator using genetic and bioinformatic tools and expansion of the PurR regulon with glyA, guaC, pbuG, xpt-pbuX, yqhZ-folD, and pbuO. J. Bacteriol. 183:6175-6183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schwan, T. G., J. M. Battisti, S. F. Porcella, S. J. Raffel, M. E. Schrumpf, E. R. Fischer, J. A. Carroll, P. E. Stewart, P. Rosa, and G. A. Somerville. 2003. Glycerol-3-phosphate acquisition in spirochetes: distribution and biological activity of glycerophosphodiester phosphodiesterase (GlpQ) among Borrelia spirochetes. J. Bacteriol. 185:1346-1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schwan, T. G., and B. J. Hinnebusch. 1998. Bloodstream- versus tick-associated variants of a relapsing fever bacterium. Science 280:1938-1940. [DOI] [PubMed] [Google Scholar]
- 42.Schwan, T. G., S. J. Raffel, M. E. Schrumpf, P. F. Policastro, J. A. Rawlings, R. S. Lane, E. B. Breitschwerdt, and S. F. Porcella. 2005. Phylogenetic analysis of the spirochetes Borrelia parkeri and Borrelia turicatae and the potential for tick-borne relasping fever in Florida. J. Clin. Microbiol. 43:3851-3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schwan, T. G., S. J. Raffel, M. E. Schrumpf, and S. F. Porcella. 2007. Diversity and distribution of Borrelia hermsii. Emerg. Infect. Dis. 13:436-442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Simpson, W. J., C. F. Garon, and T. G. Schwan. 1990. Analysis of supercoiled circular plasmids in infectious and non-infectious Borrelia burgdorferi. Microb. Pathog. 8:109-118. [DOI] [PubMed] [Google Scholar]
- 45.Southern, E. M. 1975. Detection of specific sequences among DNA fragments separated by gel electrophoresis. J. Mol. Biol. 98:503-517. [DOI] [PubMed] [Google Scholar]
- 46.Steere, A. C. 1989. Lyme disease. N. Engl. J. Med. 321:586-596. [DOI] [PubMed] [Google Scholar]
- 47.Steere, A. C., R. L. Grodzicki, A. N. Kornblatt, J. E. Craft, A. G. Barbour, W. Burgdorfer, G. P. Schmid, E. Johnson, and S. E. Malawista. 1983. The spirochetal etiology of Lyme disease. N. Engl. J. Med. 308:733-740. [DOI] [PubMed] [Google Scholar]
- 48.Switzer, R. L., H. Zalkin, and H. H. Saxild. 2002. Purine, pyrimidine, and pyridine nucleotide metabolism, p. 255-269. In A. L. Sonenshein, J. A. Hoch, and R. Losick (ed.), Bacillus subtilis and its closest relatives: from genes to cells. ASM Press, Washington, DC.
- 49.Wang, G., C. Ojaimi, R. Iyer, V. Saksenberg, S. A. McClain, G. P. Wormser, and I. Schwartz. 2001. Impact of genotype variation of Borrelia burgdorferi sensu stricto on kinetics of dissemination and severity of disease in C3H/HeJ mice. Infect. Immun. 69:4303-4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhong, J., S. Skouloubris, Q. Dai, H. Myllykallio, and A. G. Barbour. 2006. Function and evolution of plasmid-borne genes for pyrimidine biosynthesis in Borrelia spp. J. Bacteriol. 188:909-918. [DOI] [PMC free article] [PubMed] [Google Scholar]