

Abstract
The BAZF (BCL-6b) protein is highly similar to the BCL-6 transcriptional repressor. While BCL-6 has been characterized extensively, relatively little is known about the normal function of BAZF. In order to understand the physiological role of BAZF, we created BAZF-deficient mice. Unlike BCL-6-deficient mice, BAZF-deficient mice are healthy and normal in size. However, BAZF-deficient mice have a hematopoietic progenitor phenotype that is almost identical to that of BCL-6-deficient mice. Compared to wild-type mice, both BAZF-deficient and BCL-6-deficient mice have greatly reduced numbers of cycling hematopoietic progenitor cells (HPC) in the BM and greatly increased numbers of cycling HPC in the spleen. In contrast to HPC from wild-type mice, HPC from BAZF-deficient and BCL-6-deficient mice are resistant to chemokine-induced myelosuppression and do not show a synergistic growth response to granulocyte-macrophage colony-stimulating factor plus stem cell factor. Depletion of CD8 T cells in BAZF-deficient mice reverses several of the hematopoietic defects in these mice. Since both BAZF- and BCL-6-deficient mice have defects in CD8 T-cell differentiation, we hypothesize that both BCL-6 and BAZF regulate HPC homeostasis by an indirect pathway involving CD8 T cells.
The BCL-6 gene encodes a transcriptional repressor protein that is a critical regulator of lymphocyte and myeloid gene expression (19, 43, 50). Alterations in the BCL-6 gene are strongly associated with the diffuse large cell form of non-Hodgkin's B-cell lymphoma, and the BCL-6 gene is thought to act as an oncogene in this disease (15, 43). BAZF (also known as BCL-6b) is the protein most closely related to BCL-6 and, like BCL-6, has an N-terminal BTB/POZ domain, a unique middle region, and a C-terminal set of zinc finger modules (36). At the protein level, the BTB/POZ domain of BAZF is 65% similar to the BCL-6 BTB/POZ domain and the zinc finger region of BAZF is 94% similar to the BCL-6 zinc finger region, yet the middle region of BAZF is considerably shorter and divergent from the middle region of the BCL-6 protein (36). BAZF and BCL-6 bind an identical DNA motif, which is similar to a Stat factor motif (18, 23, 36). BAZF is a transcriptional repressor but requires functional BCL-6 for repressor activity (45). BAZF and BCL-6 proteins can bind to each other in cells that express both proteins (36). While BCL-6 is expressed ubiquitously, BAZF expression is restricted to heart, lung, and activated lymphocytes (36). While a number of target genes have been described for BCL-6 (41, 48), it is not yet clear if BAZF can also regulate these same targets.
The in vivo function of BCL-6 has been well characterized: BCL-6 controls the immune response by regulating the differentiation of B cells, T cells, and myeloid cells. More specifically, BCL-6 is required for germinal center formation and is also a critical inhibitor of Th2 responses and inflammation (18, 20, 49). BCL-6 also represses interleukin-6 (IL-6) production and regulates the IL-6 response by macrophages and myeloid progenitors (50). In contrast, the in vivo function of BAZF is poorly understood. A recent study found that BAZF augments CD4 T-cell proliferation, whereas BCL-6 has a negative effect on CD4 T-cell proliferation (44). This study suggests that BAZF and BCL-6 can play antagonistic roles in CD4 T cells. However, BCL-6 and BAZF may play similar roles in CD8 T cells, since recent studies showed that both BCL-6-deficient and BAZF-deficient mice develop faulty CD8 T-cell memory responses (25, 33).
Differentiation of hematopoietic progenitor cells (HPC) is tightly regulated by cytokines, chemokines, and the hematopoietic milieu. However, how specific transcription factors that are activated by these regulators of hematopoiesis control HPC proliferation, differentiation, and movement is poorly understood. Previously, we found that the transcription factors Stat4 and Stat6, which control T-helper-cell differentiation, can also control HPC by controlling the expression of the cytokine oncostatin M (3). We have also found that the Flt3 receptor, which plays a critical role in the proliferation and survival of hematopoietic cells, is dependent upon the transcription factor Stat5a for its function (51). A number of different transcriptional repressor proteins have been implicated in HPC survival and proliferation, such as Slug, FOG-1, Eed, Runx1, and SZF1 (16, 24, 31, 32, 37). The transcriptional repressor Slug is implicated in c-kit signaling, the transcriptional repressor FOG-1 is a repressor of eosinophil differentiation, the polycomb group repressor protein Eed is a negative regulator of hematopoietic proliferation, and the transcriptional repressor SZF1 may regulate the differentiation of CD34+ hematopoietic cells. Runx1 appears to control the early development of hematopoietic-lineage cells. Thus, transcriptional repressor proteins play a wide variety of key roles in hematopoiesis.
We therefore decided to investigate whether the transcriptional repressors BAZF and BCL-6 also regulate hematopoiesis. BCL-6 has been implicated in the development of erythroid lineage cells in neonatal mice; however, HPC were not analyzed (1). BAZF has been shown to be important for spermatogonial stem cell maintenance, suggesting that BAZF might also regulate HPC (35). Thus, we investigated the roles of BAZF and BCL-6 in HPC proliferation in bone marrow (BM) and spleen and differentiation in response to cytokines/chemokines. We found that disruption of BAZF or BCL-6 in the mouse germ line results in almost identical hematopoietic phenotypes. Both BAZF-deficient and BCL-6-deficient mice have decreased numbers and proliferation of CFU-granulocyte-macrophage (CFU-GM), burst-forming unit-erythroid (BFU-E), and multipotential (CFU-granulocyte/erythroid/megakaryocyte/macrophage [CFU-GEMM]) HPC in the BM and increased numbers and proliferation of these HPC in the spleen. HPC from both BAZF-deficient and BCL-6-deficient mice showed similar alterations in the response to stimulation/costimulation by growth factors and inhibition by suppressive chemokines. We found that the increased activity of HPC in the spleens of BAZF-deficient mice was blocked by deletion of CD8 lymphocytes. Taken together, these data show that BAZF and BCL-6 act through a common pathway to regulate hematopoiesis, possibly through their roles in generating CD8 T-cell memory.
MATERIALS AND METHODS
Mice.
BCL-6-deficient (BCL-6−/−) and BAZF-deficient (BAZF−/−) mice on a mixed C57BL/6 × 129/Sv strain background were bred in the animal facility at Indiana University School of Medicine. Animals were maintained under specific-pathogen-free conditions in animal facilities certified by the American Association of Laboratory Animal Care. BCL-6−/− mice have been described previously (18). Rag1-deficient mice were obtained from Jackson Labs (Bar Harbor, ME).
Disruption of the BAZF gene in mice.
Genomic fragments of the BAZF gene were produced by PCR using genomic DNA from embryonic stem (ES) cells. PCR primers (Table 1) were designed from the BAZF cDNA sequence (GenBank accession no. AB011665), and PCRs were performed with the Accutaq enzyme (Sigma). The 2.3-kb upstream fragment of the BAZF gene was amplified using primers BAF2 and BAR6. The 2.0-kb downstream fragment of the BAZF gene was amplified using primers BAF11 and BAR16. The PCR products were cloned into the pCRII-TOPO vector (Invitrogen, San Diego, CA) and then subsequently cloned into the pPNT vector (40) via EcoRI sites (upstream fragment) and XhoI/NotI sites (downstream fragment). The BAZF knockout construct was designed to delete all five of the BAZF gene zinc finger coding sequences and replace them with the neomycin resistance gene. The BAZF homologous recombination construct was electroporated into ES cells, neomycin-resistant clones were selected, and clones were screened for appropriate integration by PCR. Homologous recombination into the BAZF gene was verified by Southern blot analysis using EcoRI-digested genomic DNA. The 1-kb probe used for Southern blot analysis was prepared by PCR from ES DNA using the BAPF and BAPR primers (Table 1). Several homologous-recombination ES clones were identified, and these were injected into C57BL/6 blastocysts in order to produce chimeric mice. The offspring mice showing the greatest chimerism were then mated to C57BL/6 mice, and one clone was found to transmit via the germ line. The BAZF knockout allele was then carried on a mixed 129/Sv × C57BL/6 strain background. Litters were tested for the BAZF knockout allele via PCR with primers BAF10 and BAR10 (specific for BAZF) and 8969 (specific for the pgk promoter driving the neomycin gene). The sequences for BAF10, BAR10, 8969 are shown in Table 1.
TABLE 1.
DNA oligonucleotides used for the generation and analysis of BAZF-deficient mice
| Oligonucleotide | Sequence |
|---|---|
| BAF2 | GCTCTGGGCTACGTCCGAGAGTT |
| BAR6 | GCATTTATAGGGCTTGTCCTCGTC |
| BAF10 | CTACGCGCACACGTGCTCATCCA |
| BAR10 | TTTCACTGCCCCTAGGGAGCGAAGT |
| 8969 | GAGGCCACTTGTGTAGCGCCAAGT |
| BAF11 | CTGCGGCTGCATCTGCGTCAGAAA |
| BAR16 | GGCACAAAGAAGGGCTTCTTTATTC |
| BAPF | TTCTGGAAACTAATAACTTGGATTCTG |
| BAPR | TTTACATGGTCAAGATCCTTAACTTC |
RT-PCR analysis.
Reverse-transcription-PCR (RT-PCR) for BAZF mRNA expression was performed with BAF10 and BAR10 (Table 1). RT-PCR for β-tubulin mRNA was performed as described by Toney et al. (48).
Leukocyte subset analysis.
Complete white blood cell count analysis was done using a complete blood count instrument (Hemavet 850; CDC Technologies, Oxford, CT). The differences in neutrophil and monocyte percentages were confirmed by manual counting.
Analysis of HPC.
Total BM cells were plated at 5 × 104/ml, and total splenocytes were plated at 5 × 105/ml in 1% methylcellulose culture medium containing growth factors (30%, vol/vol, fetal bovine serum [HyClone, Logan, UT], 1 U/ml human erythropoietin [Epo; Amgen Biologicals, Thousand Oaks, CA], 50 ng/ml murine stem cell factor [SCF; R&D Systems, Minneapolis, MN], 5%, vol/vol, pokeweed mitogen mouse spleen cell-conditioned medium [PWMSCM], and 0.1 mM hemin [Eastman Kodak Co.]). Colonies derived from CFU-GM, BFU-E, and CFU-GEMM were scored after 7 days of incubation in a humidified environment at 5% CO2 and lowered (5%) O2 as previously described (14, 29). CFU and BFU are expressed as the total number of colonies per femur or spleen. Absolute numbers of progenitors per organ were calculated based on the number of viable, total nucleated cells per femur or spleen and on the number of colonies scored per number of cells plated. The percentage of progenitors in S phase was estimated by the high-specific-activity [3H]thymidine kill technique, which eliminates cells in cycle from dividing in culture to form colonies. Briefly, the cells are exposed to a 30-s pulse of 50 μCi/ml (20 Ci/mmol) tritiated thymidine (PerkinElmer, Wellesley, MA) followed by a cold thymidine chase, prior to plating (9, 34). Synergy assays with SCF and granulocyte-macrophage colony-stimulating factor (GM-CSF) and chemokine inhibition assays were performed as described previously (26). Statistics were performed using Student's t test. P values are indicated in the figure legends.
Depletion of CD4 T cells and CD8 T cells.
CD4 and CD8 T cells were depleted by antibody (Ab) injections as described previously (3). Briefly, mice received 200 μg of either anti-CD4 Ab (GK1.5; rat monoclonal immunoglobulin G2b [IgG2b]) or anti-CD8 Ab (2.43; rat monoclonal IgG2b) injected intraperitoneally four times over the course of 10 days. Control mice received injections of phosphate-buffered saline (PBS). Mice were sacrificed for hematopoietic analysis 1 day after the last Ab injection. Depletion of CD4 and CD8 T-cell subsets was verified by flow cytometry using different anti-CD4 and anti-CD8 Abs than the injected Abs. Depletion treatments resulted in the removal of 85 to 95% of the specific T-cell subset (CD4 or CD8).
RESULTS
Generation of BAZF-deficient mice.
In order to study the in vivo role of the BCL-6 homologue BAZF, we mutated BAZF in the mouse germ line using the technique of homologous recombination in ES cells. Our BAZF targeting construct was designed to mutate the complete zinc finger-coding region of the BAZF gene (about 460 bp; Fig. 1A). This was a gene targeting strategy similar to one that we used previously to mutate the BCL-6 gene in mice (18). Initial recombinant ES clones were identified by Southern blotting and RT-PCR methods. These clones were used to generate chimeric mice that could pass the BAZF mutation through the germ line. A Southern blot showing targeting of the BAZF gene in mice is shown in Fig. 1B. The wild-type BAZF gene is contained on an 8.7-kb EcoRI fragment. The mutated gene contains two extra EcoRI sites, and this mutated gene can be identified by Southern blotting as a 4.0-kb band in heterozygous and homozygous mutant animals. Figure 1C shows RT-PCR analysis of BAZF mRNA expression in the lung in wild-type and BAZF mutant mice. The oligonucleotides used for the RT-PCR correspond to the region of BAZF that is deleted in the BAZF mutant mice, and the results show that the zinc finger region of the BAZF gene is not expressed in BAZF mutant mice (hereafter called BAZF-deficient mice).
FIG. 1.

Scheme for mutating the BAZF gene in the mouse germ line. The mutation introduced by our gene-targeting construct deletes from the genome the region of the BAZF gene encoding 124 C-terminal amino acids of the 494-amino-acid BAZF protein. A region of BAZF encoding the most C-terminal 25 amino acids is not deleted by our targeting construct. The 124-amino-acid region deleted includes all of the zinc finger amino acids. The targeted BAZF gene is thus incapable of producing a DNA binding protein by alternative splicing. (A) Structure of the mouse BAZF gene locus, the targeting construct, and the recombined BAZF locus. ZF, zinc finger; E, EcoRI sites. Note that the four EcoRI sites in the targeting construct are not present in the endogenous gene. (B) Southern blot showing genomic DNA for wild-type (+/+) mice and mice heterozygous (+/−) and homozygous (−/−) for the BAZF mutation (BAZF-deficient mice). Genomic DNA was digested with EcoRI, and the blot was probed with the DNA fragment indicated in panel A. (C) RT-PCR for BAZF expression analyzing RNA from the lungs of wild-type (+/+) mice and BAZF-deficient (−/−) mice. The RT-PCR primers were designed to amplify the zinc finger region of BAZF that is deleted in BAZF-deficient mice. β-Tubulin is a control for the cDNA loading.
Comparison of BAZF-deficient- and BCL-6-deficient-mouse phenotypes.
BAZF-deficient mice are born at a normal Mendelian ratio and appear normal at birth. Unlike BCL-6-deficient mice (18, 49), which are severely growth retarded and frequently die before reaching maturity, BAZF-deficient mice are of normal size, are fertile, and appear to have a life span similar to that of wild-type mice. The majority of BCL-6-deficient mice spontaneously develop a severe Th2-type inflammation of the heart and lungs (18, 49). Careful histological analysis of the heart and lungs of multiple BAZF-deficient mice revealed no trace of inflammation (data not shown). We also assessed the ability of BAZF-deficient T cells to differentiate into Th2 cells, since BCL-6-deficient T cells have a strong bias towards Th2 differentiation (17, 30). We therefore activated naive CD4 T cells from BAZF-deficient mice under various costimulation conditions. However, in multiple experiments with over 10 different mice we were unable to detect significantly increased Th2 differentiation of the BAZF-deficient T cells compared to wild-type T cells (data not shown). The lack of bias towards Th2 differentiation in BAZF-deficient T cells is consistent with the lack of inflammation in these mice. Further, we mated BCL-6-deficient mice with BAZF-deficient mice to produce BCL-6/BAZF doubly deficient mice. BCL-6/BAZF doubly deficient mice had a phenotype very similar to that of BCL-6-deficient mice, displaying growth retardation and severe Th2-type inflammation (data not shown). These results indicate that loss of BCL-6 produces a dominant phenotype not significantly affected by loss of BAZF. As an additional test of the in vivo role for BAZF in controlling inflammation, we cultured macrophages from the BM of wild-type, BAZF-deficient mice, and BCL-6-deficient mice using macrophage colony-stimulating factor and then stimulated the macrophages and assessed production of proinflammatory genes known to be regulated by BCL-6 (48). Whereas the chemokines MCP-1, MCP-3, and MRP-1/C10 are expressed at greatly increased levels in BCL-6-deficient cells compared to wild-type cells, there is no increase seen with BAZF-deficient macrophages (data not shown). IL-6 mRNA expression is increased in BCL-6-deficient macrophages compared to wild-type macrophages, as reported previously (50), but BAZF-deficient macrophages showed levels of IL-6 gene expression similar to those of wild-type macrophages (data not shown). Thus, unlike BCL-6-deficient mice, BAZF-deficient mice do not develop increased Th2 responses, do not overproduce chemokines, and do not develop inflammatory disease of the heart and lung.
Hematopoiesis in BAZF-deficient and BCL-6-deficient mice.
When we performed complete white blood cell counts on the BM, spleen, and blood of BAZF-deficient mice, we observed essentially normal percentages for most cell types (Fig. 2). However, small but statistically significant differences were observed in the percentages of neutrophils and monocytes in the BM and peripheral blood. Furthermore, BAZF-deficient mice showed a significant increase in red blood cells per μl of blood. We therefore wondered if loss of BAZF might affect hematopoiesis and, specifically, the generation of HPC. The nucleated cellularity of spleen and BM of BAZF-deficient mice is normal (data not shown), indicating no gross defect in hematopoietic cell production. We first analyzed BM for numbers and cycling status of HPC (CFU-GM, BFU-E, and CFU-GEMM) using ex vivo cell colony-forming assays. We found that the numbers of HPC of all three types were strongly decreased in the BM of BAZF-deficient mice and that this difference was highly significant (Fig. 3A). Next we assayed the proportion of HPC that were actively proliferating in wild-type and BAZF-deficient mice. We found that, whereas 50% to 60% of the wild-type BM HPC were actively in the cell cycle, the BAZF-deficient BM HPC had a drastic and highly significant decrease in the percentage of cells that were actively proliferating, down to less than 10% (Fig. 3B). We next analyzed the spleens of BAZF-deficient mice for the number and the percentage of HPC in the cell cycle (Fig. 3C and D). In contrast to what was observed for HPC in the BM, all three types of HPC were strongly increased in the spleens of BAZF-deficient mice. Strikingly, whereas wild-type HPC in the spleen were almost completely in a quiescent state, 60% to 65% of the HPC in the spleens from BAZF-deficient mice were actively proliferating, and this difference was highly significant. Thus, BAZF plays a major role in regulating hematopoiesis at the level of HPC.
FIG. 2.

White blood cell and red blood cell counts of BAZF-deficient mice. Average percentages of lymphoid cells and myeloid subsets are shown for BM, peripheral blood leukocytes (PBL), and spleen (SP). Numbers of red blood cells (RBC) and platelets from PBL are shown. Results shown are averaged from four mice of each type. Error bars show standard deviations. P values were calculated with Student's t test. *, P < 0.05.
FIG. 3.

Decreased hematopoietic progenitor activity in the BM and increased hematopoietic progenitor activity in the spleens of BAZF-deficient mice. Results are the averages ± standard errors of the means from eight different wild-type and BAZF-deficient mice, assessed in two different experiments. P values were calculated with Student's t test. ***, P < 0.001; **, P < 0.01; *, P < 0.05. (A) BM cells were analyzed for HPC numbers per femur. (B) Cycling status of the BM hematopoietic progenitors, expressed as the percentage of cells in the cell cycle. (C) Spleen cells were analyzed for HPC numbers per spleen. (D) Cycling status of the splenic hematopoietic progenitors, expressed as the percentage of cells in the cell cycle.
In order to exclude the possibility that our germ line BAZF mutation led to the production of a truncated protein that could exert a dominant negative effect on hematopoiesis, we assessed whether BAZF-deficient heterozygote animals had alterations in hematopoiesis similar to what is seen in the BAZF-deficient homozygous mice. As shown in Fig. 4, BAZF-deficient heterozygote animals show an intermediate effect between wild-type and BAZF-deficient mice in terms of HPC numbers in the BM. A similar trend for BAZF-deficient heterozygote mice was observed with HPC numbers in the spleen, although, in contrast to the BM results, the results for BAZF-deficient heterozygote mice were not statistically significant (Fig. 4A versus B). When the cycling status of HPC was assayed, it was found that BAZF-deficient heterozygote mice had a phenotype identical to that of wild-type mice (Fig. 4), which was strikingly different from the phenotype of BAZF-deficient homozygous mice. These results indicate there may be a gene dosage effect for BAZF on the production of HPC but also indicate that the hematopoietic effects we observe in BAZF-deficient mice are not due to a dominant negative effect.
FIG. 4.

BAZF-heterozygous mice show different HPC numbers and cycling activity than BAZF-deficient mice. HPC numbers and cycling in the BM and spleen were calculated for wild type, BAZF+/−, and BAZF−/− mice. Results shown are averaged from four of each mouse type. Error bars show standard deviations. P values are relative to wild type and were calculated with Student's t test. ***, P < 0.0001; **, P < 0.001; *, P < 0.05. Only statistically significant comparisons are designated.
We next performed an analysis of HPC from BCL-6-deficient mice. The nucleated-cellularity levels of both spleens and BM of BCL-6-deficient mice were significantly decreased (BM, 45% decrease, P < 0.001; spleen, 26% decrease, P = 0.035) compared to wild-type mice; however, this decrease is consistent with the overall smaller size of BCL-6-deficient mice (18). We found that, similar to BAZF-deficient mice, BCL-6-deficient mice have greatly reduced numbers of all three types of hematopoietic progenitors in the BM (Fig. 5A) and that the HPC in the BM of BCL-6-deficient mice are very reduced in the proportion of cycling cells (Fig. 5B). Analysis of HPC from the spleens of BCL-6-deficient mice also revealed striking similarities with BAZF-deficient mice (Fig. 5C and D). Thus, the overall numbers of HPC were increased in the spleens of BCL-6-deficient mice, with increases in myeloid and multipotential progenitors showing the most significant difference with wild-type mice (Fig. 5C). In a pattern identical to that for the BAZF-deficient mice, HPC from the spleens of BCL-6-deficient mice were actively proliferating whereas the wild-type splenic HPC were almost completely quiescent (Fig. 5D). These results indicate that, at the level of HPC, the hematopoietic phenotype of BAZF-deficient mice is remarkably similar to that of BCL-6-deficient mice. These data also suggest that the increased Th2 differentiation, increased chemokine production by macrophages, and inflammatory disease are not the cause of the altered hematopoiesis in BCL-6-deficient mice, since BAZF-deficient mice have similarly altered hematopoiesis in the absence of other phenotypes observed in BCL-6-deficient mice.
FIG. 5.

Decreased hematopoietic progenitor activity in the BM and increased hematopoietic progenitor activity in the spleens of BCL-6-deficient mice. Results are the averages ± standard errors of the means from six different wild-type and BCL-6-deficient mice, assessed in two different experiments. P values were calculated with Student's t test. ***, P < 0.001; **, P < 0.01; N.S., not significant (P > 0.05). The reason the HPC numbers for the wild-type mice in this experiment are different from the HPC numbers for the wild-type mice in the experiment shown in Fig. 3 is that the experiment shown here was performed at a different time with different lots of serum and growth factors. (A) BM cells were analyzed for HPC numbers per femur. (B) Cycling status of the BM hematopoietic progenitors, expressed as the percentage of cells in the cell cycle. (C) Spleen cells were analyzed for HPC numbers per spleen. (D) Cycling status of the splenic hematopoietic progenitors, expressed as the percentage of cells in the cell cycle.
Lack of synergy with SCF in the absence of BCL-6 or BAZF.
SCF synergizes with other hematopoietic growth factors, such as GM-CSF, to induce a greatly enhanced proliferative state of HPC (42). We therefore tested if BAZF and BCL-6 affected the synergistic response of HPC to SCF plus GM-CSF. Total BM cells from wild-type mice were plated into agar-containing medium with SCF alone, GM-CSF alone, or SCF plus GM-CSF. CFU-GM-derived colonies were counted after 7 days of culture. As shown in Fig. 6, wild-type BM cells produced a strong synergistic response, where the number of colonies with SCF plus GM-CSF (105 ± 25) was greater than twice the number predicted if the effects of SCF alone (14 ± 3) and GM-CSF alone (31 ± 6) were additive (14 + 31 = 45). In contrast, the BAZF-deficient cells produced an additive response with SCF plus GM-CSF (Fig. 6). The BCL-6-deficient BM cells had a greater response to SCF alone than the BAZF-deficient cells, but otherwise the effect of SCF plus GM-CSF was additive for BCL-6-deficient BM cells (Fig. 6). Thus, both BAZF and BCL-6 control the synergistic response of HPC to SCF plus GM-CSF.
FIG. 6.
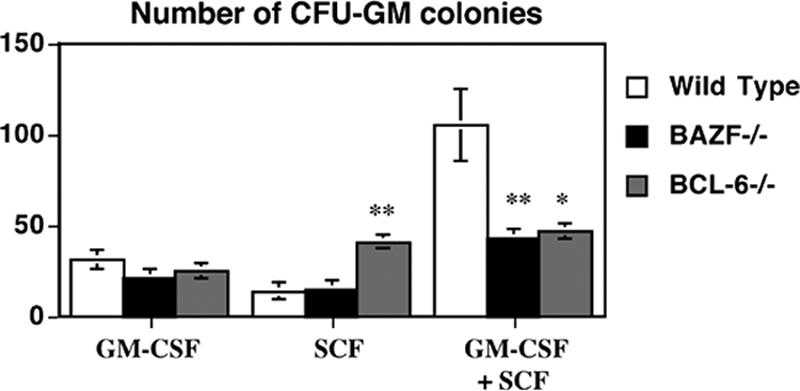
HPC from BAZF-deficient mice and BCL-6-deficient mice do not mount a synergistic response with SCF plus GM-CSF. Wild-type, BAZF-deficient, and BCL-6-deficient BM cells were stimulated in vitro with the indicated growth factors and then cultured in a methylcellulose colony assay. CFU-GM colony formation was determined after 7 days. Results shown are the averages for three different mice ± standard errors of the means for each type. P values were calculated with Student's t test. **, P < 0.01; *, P < 0.05 (compared to wild-type values).
Lack of chemokine-induced myelosuppression in both BAZF- and BCL-6-deficient mice.
Chemokines such as macrophage inflammatory protein 1α (MIP-1α; CCL2), IL-8 (CXCL8), and platelet factor 4 (PF-4) have suppressive effects on the proliferation of HPC (8). We therefore tested whether BAZF and BCL-6 might affect the ability of chemokines to suppress HPC development. While HPC from BAZF-deficient and BCL-6-deficient BM were insensitive to inhibition by MIP-1α, IL-8, and PF-4 (data not shown), these HPC are in a slow or noncycling state (Fig. 3B and 5B) and would not be expected to respond to these suppressive chemokines (8). Therefore, BM cells from wild-type, BAZF-deficient, and BCL-6-deficient mice were stimulated in vitro to proliferate with a cocktail of cytokines including Epo, SCF, and PWMSCM for 24 h, and then the cells were washed and plated in methylcellulose culture medium with Epo, SCF, PWMSCM, and either control medium, MIP-1α, IL-8, PF-4, or tumor necrosis factor alpha (TNF-α). BM HPC were prestimulated for 24 h with growth factors, such that after treatment there was an average cycling of 57% for wild-type cells and 56% for both BAZF-deficient and BCL-6-deficient cells. Colonies were scored 7 days later. We found that all four treatments, MIP-1α, IL-8, PF-4, and TNF-α, suppressed colony formation by at least 60% with wild-type cells (Fig. 7). Cells from both BAZF-deficient and BCL-6-deficient mice exhibited a dramatically different response to chemokines and TNF-α. The BM cells from both mutant mice were completely refractory to inhibition by chemokines MIP-1α, IL-8 and PF-4. However, BAZF-deficient and BCL-6-deficient cells were sensitive to inhibition by TNF-α, which signals through a different pathway than chemokines. These data show that lack of either BAZF or BCL-6 can lead to a striking shift in the ability of HPC to respond to chemokines. Stat4-deficient BM cells are also resistant to chemokine-induced HPC suppression (26). Both BCL-6-deficient and Stat4-deficient mice have an immunological bias towards Th2 differentiation, suggesting that increased Th2 activity may affect HPC sensitivity to chemokines. However, BAZF-deficient mice do not have increased Th2 differentiation, which indicates that another pathway is likely to be involved in the loss of chemokine suppression in these mice. Moreover, given the similarities in hematopoiesis in BAZF-deficient and BCL-6-deficient mice, it is likely that the loss of chemokine sensitivity in HPC in these mice occurs via a similar pathway.
FIG. 7.

HPC from BAZF-deficient mice and BCL-6-deficient mice do not respond to chemokine-mediated inhibition. Wild-type, BAZF-deficient, and BCL-6-deficient BM cells were stimulated in vitro to activate cycling and then, after a washing, cultured poststimulation in the presence of the indicated chemokines in a methylcellulose colony assay. Fifty-seven percent of wild-type BM cells were in cycle and 56% of both BAZF-deficient and BCL-6-deficient cells were in cycle following in vitro stimulation. CFU-GM colony formation was determined after 7 days. Results shown are the averages for three different mice ± standard errors of the means for each type. Results are plotted as percent inhibition of colony formation with chemokines and TNF-α, compared to colony formation without chemokine/TNF-α addition. P values were calculated with Student's t test. ***, P < 0.001; **, P < 0.01; *, P < 0.05 (compared to wild-type values).
A role for lymphocytes in controlling extramedullary hematopoiesis.
In order to understand if the increase in HPC in the spleens of BAZF-deficient mice was a direct effect of loss of BAZF on HPC or an indirect effect on HPC mediated by lymphoid cells, we mated BAZF-deficient mice to Rag1-deficient mice, which are unable to produce mature lymphocytes. When HPC were analyzed in the spleens of BAZF/Rag1 doubly deficient mice, we found that the deletion of mature lymphocytes led to a significant decrease in HPC numbers, down to wild-type levels (Fig. 8). Similar findings were seen when HPC cycling was analyzed in BAZF/Rag1 doubly deficient mice. These data therefore suggest that loss of BAZF function does not produce an intrinsic defect in HPC that leads to active extramedullary hematopoiesis, but rather that BAZF affects lymphoid cell function, and this alteration in turn affects HPC localization to the spleen. Like BAZF-deficient mice, Rag1-deficient mice have decreased numbers and cycling of HPC in the BM (54% decrease in numbers compared to wild-type mice, P < 0.001; 94% decrease in cycling compared to wild type mice, P < 0.001; data not shown). BAZF/Rag1 doubly deficient mice had a pattern of decreased numbers and cycling of HPC in the BM similar to those of BAZF-deficient mice and Rag1-deficient mice (data not shown).
FIG. 8.

Deletion of mature lymphocytes from BAZF-deficient mice ablates the increased development of splenic HPC. Spleen cells from the indicated mice were analyzed for the numbers of HPC (A) and the percentage of HPC in cycle (B). Error bars show standard errors. P values were calculated with Student's t test. ***, P < 0.0001; **, P < 0.001; *, P < 0.05. Only statistically significant comparisons are designated.
A role for CD8 T cells in controlling hematopoietic progenitor responses in BAZF-deficient mice.
Since we observed a role for lymphoid cells in the abnormal HPC phenotype of BAZF-deficient mice, we wondered if either CD4 or CD8 T cells might mediate the aberrant HPC responses. We tested this idea by using a strategy of using anti-CD4 and anti-CD8 Abs to deplete either CD4 or CD8 T cells in BAZF-deficient mice. We assessed HPC numbers and HPC cycling in the Ab-treated animals as well as in control PBS-injected animals. Depletion of CD4 T cells and CD8 T cells did not significantly increase the numbers of HPC grown from the BM under standard conditions, nor did anti-CD4 Ab or anti-CD8 Ab treatment decrease the numbers of HPC in the spleen in BAZF-deficient mice (data not shown). However, we found that depletion of either CD4 or CD8 T cells led to a significant increase in HPC cycling, approaching wild-type levels, in the BM of BAZF-deficient mice (Fig. 9A). Further, we found that depletion specifically of CD8 T cells led to a significant decrease in HPC cycling, approaching wild-type levels, in the spleens of BAZF-deficient mice (Fig. 9B). Next, we tested if CD4 or CD8 T-cell depletion affected the lack of SCF-plus-GM-CSF growth synergy that we saw with BAZF-deficient BM-derived HPC (Fig. 6). When BM cells from the Ab-treated mice were stimulated with specific amounts of SCF plus GM-CSF, we observed that depletion of CD8 T cells specifically led to a significant increase in CFU-GM colonies compared to control-treated and anti-CD4 Ab-treated mice (Fig. 10). The effect was consistent with a restoration of the normal synergistic growth response to SCF plus GM-CSF stimulation (compare with Fig. 6). Lastly, we tested if CD4 or CD8 T-cell depletion affected the lack of chemokine-mediated inhibition of CFU-GM colony formation that we observed with BAZF-deficient BM-derived HPC (Fig. 7). When BM cells from the Ab-treated mice were stimulated with growth factors in the presence of chemokines, we observed that CD8-T-cell-depleted BAZF-deficient animals had recovered the chemokine inhibition phenotype (Fig. 11). These data imply that BAZF-deficient CD8 T cells have a critical and aberrant effect on hematopoiesis and that removal of CD8 T cells from BAZF-deficient mice leads to the restoration of a more normal HPC growth response.
FIG. 9.

Depletion of T-lymphocyte subsets reverses abnormal HPC cycling activity in the BM and spleens of BAZF-deficient mice. Wild-type and BAZF-deficient (BAZF KO) mice were treated with Ab to CD4 or CD8 to deplete different subsets of T cells. BM cells from the indicated mice were analyzed for the numbers of HPC in (A) BM and (B) spleen. Results shown are averages for 10 mice per treatment. Error bars show standard errors. P values are in comparison to wild-type control mice and were calculated with Student's t test. *, P < 0.05 compared to wild-type “PBS control” mice; @, P < 0.05 compared to BAZF-deficient “PBS control” mice. Only statistically significant comparisons are designated.
FIG. 10.

Depletion of CD8 T lymphocytes promotes a normal synergistic response of BAZF-deficient (BAZF KO) BM progenitors to SCF plus GM-CSF stimulation. Wild-type and BAZF-deficient mice were treated with PBS alone, Ab to CD4, or Ab to CD8. BM cells from the indicated mice were analyzed for the numbers of HPC following in vitro stimulation with SCF and GM-CSF and culture in a methylcellulose colony assay. CFU-GM colony formation was determined after 7 days. Results shown are the averages of total CFU-GM per femur from four different mice ± standard errors of the means. P values are in comparison to wild-type control mice and were calculated with Student's t test. *, P < 0.05 compared to wild-type values.
FIG. 11.

Depletion of CD8 T lymphocytes restores the sensitivity of BAZF-deficient BM progenitors to chemokine-mediated inhibition. Wild-type and BAZF-deficient mice were treated with Ab to CD4 or CD8 to deplete different subsets of T cells. BM cells were stimulated in vitro to activate cycling and then, after a washing, cultured poststimulation in the presence of the indicated chemokines in a methylcellulose colony assay. CFU-GM colony formation was determined after 7 days. Results shown are the averages for four different mice ± standard errors of the means for each type. Results are plotted as percent inhibition of colony formation with chemokines and TNF-α, compared to colony formation without chemokine/TNF-α addition.
DISCUSSION
In this study we generated and characterized a novel strain of mice that are mutant for the gene encoding BAZF (also known as BCL-6b), a close homologue to the BCL-6 transcriptional repressor. Despite major differences in immunological function and general health between BAZF-deficient and BCL-6-deficient mice, we found that at the level of HPC numbers, cell cycling, and cytokine responses, the hematopoietic phenotypes of these two strains of mutant mice were almost identical. Further, we found that several of the abnormal hematopoietic phenotypes of BAZF-deficient mice are an indirect defect due to BAZF-deficient CD8 T cells.
There is a strong bias towards Th2 differentiation in BCL-6-deficient mice, and we have previously seen that T-helper-cell differentiation can alter hematopoiesis (3). Thus, one hypothesis is that the hematopoietic phenotype of BCL-6-deficient mice is due to increased Th2 differentiation. We think this is unlikely for two reasons. First, BAZF-deficient mice do not display increased Th2 differentiation, yet they have nearly the same hematopoietic phenotype as BCL-6-deficient mice. Second, Stat4-deficient mice also have a strong Th2 differentiation bias (27) but have a hematopoietic phenotype different from that of BCL-6-deficient mice. Specifically, Stat4-deficient mice have decreased numbers and cycling of HPC in the BM but do not have increased HPC in the spleen (3). We cannot rule out the possibility that some hematopoietic effects in BCL-6-deficient mice are due to increased Th2 responses, but it seems unlikely that the overall hematopoietic phenotype of BCL-6-deficient mice is due to increased Th2 responses.
The HPC phenotype shared by BCL-6-deficient mice and BAZF-deficient mice is unique, as the pattern of HPC responses we observe with these mice has not been seen in 20 other gene-targeted mice where HPC have been analyzed (2-7, 10-13, 21, 22, 28, 29, 34, 38, 39, 46, 47, 51). On the other hand, the hematopoietic phenotypes of both BCL-6-deficient and BAZF-deficient mice are similar to the effects on hematopoiesis of injecting lipopolysaccharide (LPS) into mice (21). Since BCL-6-deficient mice are immunocompromised and develop frequent bacterial infections (49), the effect of bacterially derived LPS on hematopoiesis could possibly explain the altered pattern of HPC activity in BCL-6-deficient mice. However, we think this is unlikely to be the case for BAZF-deficient mice. First, BAZF-deficient mice are completely healthy and have no signs of infections. Second, we assessed neutrophil function in BAZF-deficient mice, since neutrophils are the first line of defense against bacterial infections, and BAZF-deficient mice had normal neutrophil function (data not shown). Third, we investigated hematopoiesis in Rag1-deficient mice, which are highly immunocompromised due to a lack of mature B and T lymphocytes and are known to be more prone to microbial infections. We found that Rag1-deficient mice have similar numbers of HPC in the spleen as wild-type mice, and the splenic HPC are cycling at a similar rates in both types of mice (Fig. 8). Thus, the hematopoietic phenotypes of BCL-6-deficient mice and BAZF-deficient mice are not simply due to the mice being immunocompromised and having more infections.
Apart from alterations in HPC activity, the one other known common feature of BAZF-deficient and BCL-6-deficient mice is altered CD8 T-cell differentiation (25, 33). We found that the spleens of BAZF/Rag1 doubly deficient mice have greatly decreased HPC numbers compared to BAZF-deficient mice. Since BAZF/Rag1 doubly deficient mice do not have mature lymphoid cells, including CD8 T cells, this finding supports the idea that defective CD8 T cells in BAZF-deficient and BCL-6-deficient mice may drive at least some of the alterations in HPC we observe in these mice. This notion was supported further by experiments in which we depleted CD8 T cells from BAZF-deficient mice and saw a reversal of the cell cycle phenotype of the HPC, as well as a reversal of the loss of GM-CSF plus SCF synergy and the restoration of chemokine inhibition of HPC growth. Very little is known about a role for CD8 T cells in controlling HPC activity. Since the HPC colony assays are done as single-cell suspensions in agar-containing medium, we think it unlikely that the effect we observe involves cell-cell contact between CD8 T cells and HPC. Moreover, there are very few CD8 T cells in BM (about 1% of the nucleated-cell population) that could exert an effect in these cultures. Thus, we think it is most likely that BAZF-deficient CD8 T cells produce secreted factors that act at a distance to modulate HPC activity. While defective CD8 T cells may lead to increased viral infections and this may alter HPC activity in BAZF-deficient and BCL-6-deficient mice, our mice are kept in a specific-pathogen-free environment and are free of known pathogenic viruses.
A possible explanation for the decreased numbers and cycling of HPC in the BM of BCL-6-deficient and BAZF-deficient mice is the loss of synergy to SCF and GM-CSF that we observed with BM cells from these mice. We found that depletion of CD8 T cells reversed the loss of synergy to SCF and GM-CSF, and thus we can hypothesize that a factor made by CD8 T cells controls synergistic responses to growth factors in HPC. Further, since we found that HPC from both BAZF-deficient and BCL-6-deficient mice are refractory to chemokine-mediated suppression and that this effect was reversed by CD8 T-cell depletion, we can hypothesize that CD8-T-cell-derived factors can also affect chemokine responsiveness of HPC. The lack of growth factor synergy in the absence of BCL-6 or BAZF may also explain the enhanced numbers and cycling of HPC in the spleens of BAZF-deficient and BCL-6-deficient mice. The spleen, which contains abundant macrophages and T cells, may be a richer source of hematopoietic growth factors that can promote HPC proliferation and differentiation. Thus, the lack of growth factor synergy in BAZF-deficient and BCL-6-deficient HPC may drive these cells to proliferate where hematopoietic growth factors are more abundant. Our results with BAZF/Rag1 doubly deficient mice and depletion of T-cell subsets support the idea that T cells in the spleen are important for the increased proliferation of splenic HPC in BAZF-deficient mice. Specifically, CD8 T cells appear to promote the increased cycling of splenic HPC in BAZF-deficient mice.
A key issue arising from our studies is whether BAZF and BCL-6 affect hematopoiesis by the same indirect mechanism. We have evidence that BAZF acts upon CD8 T cells to regulate hematopoiesis by an HPC-extrinsic pathway. If BAZF and BCL-6 both regulate a specific key target gene in CD8 T cells, it would be interesting if normal expression of this target gene required repression by both BCL-6 and BAZF. This will be an area for future investigation since common target genes for BCL-6 and BAZF have not yet been described.
Our Ab depletion experiments showed a role for CD4 T cells as well as CD8 T cells in controlling the proliferation of BM HPC in BAZF-deficient mice (Fig. 9). However, only CD8 T cells appeared to control the proliferation of splenic HPC in BAZF-deficient mice (Fig. 9). This suggests that there are separate mechanisms controlling the cycling of BM versus splenic HPC. It also may indicate that BAZF plays a functional role in CD4 T cells. Future studies will be needed to clarify the relative role for BAZF in CD4 versus CD8 T cells.
In conclusion, our studies on BAZF-deficient mice have shown that BAZF regulates HPC activity via a highly novel pathway involving CD8 T cells and further that the BAZF homologue BCL-6 likely regulates hematopoiesis by a similar pathway.
Acknowledgments
This work was supported by National Institutes of Health grants AI46410 to A.L.D. and HL56416, HL67384, and DK53674 to H.E.B. A.L.D. also receives support from the Indiana Genomics Initiative (INGEN) of Indiana University. INGEN is supported in part by Lilly Endowment Inc.
We are very grateful to Mark Kaplan for the anti-Cd4 and anti-CD8 Abs. We also acknowledge Giao Hangoc for help with blood cell counts.
Footnotes
Published ahead of print on 25 May 2007.
REFERENCES
- 1.Asari, S., A. Sakamoto, S. Okada, Y. Ohkubo, M. Arima, M. Hatano, Y. Kuroda, and T. Tokuhisa. 2005. Abnormal erythroid differentiation in neonatal bcl-6-deficient mice. Exp. Hematol. 33:26-34. [DOI] [PubMed] [Google Scholar]
- 2.Boring, L., J. Gosling, S. W. Chensue, S. L. Kunkel, R. V. Farese, Jr., H. E. Broxmeyer, and I. F. Charo. 1997. Impaired monocyte migration and reduced type 1 (Th1) cytokine responses in C-C chemokine receptor 2 knockout mice. J. Clin. Investig. 100:2552-2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Broxmeyer, H. E., H. A. Bruns, S. Zhang, S. Cooper, G. Hangoc, A. N. McKenzie, A. L. Dent, U. Schindler, L. K. Naeger, T. Hoey, and M. H. Kaplan. 2002. Th1 cells regulate hematopoietic progenitor cell homeostasis by production of oncostatin M. Immunity 16:815-825. [DOI] [PubMed] [Google Scholar]
- 4.Broxmeyer, H. E., S. Cooper, G. Cacalano, N. L. Hague, E. Bailish, and M. W. Moore. 1996. Involvement of interleukin (IL) 8 receptor in negative regulation of myeloid progenitor cells in vivo: evidence from mice lacking the murine IL-8 receptor homologue. J. Exp. Med. 184:1825-1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Broxmeyer, H. E., S. Cooper, G. Hangoc, J. L. Gao, and P. M. Murphy. 1999. Dominant myelopoietic effector functions mediated by chemokine receptor CCR1. J. Exp. Med. 189:1987-1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Broxmeyer, H. E., S. Cooper, G. Hangoc, C. Mantel, and D. S. Franklin. 2000. Modulation of myeloid progenitor cell proliferation by cyclin-dependent kinase inhibitors as determined in p21cip1/waf1 p21cip1/waf1 p21cip1/waf1 p21cip1/waf1 gene knockout mice. Blood 96:539a. [Google Scholar]
- 7.Broxmeyer, H. E., S. Cooper, L. A. Lasky, and F. De Sauvage. 2005. Identification of a massive reserve of hematopoietic progenitors in mice. Stem Cells Dev. 14:105-110. [DOI] [PubMed] [Google Scholar]
- 8.Broxmeyer, H. E., and C. H. Kim. 1999. Regulation of hematopoiesis in a sea of chemokine family members with a plethora of redundant activities. Exp. Hematol. 27:1113-1123. [DOI] [PubMed] [Google Scholar]
- 9.Broxmeyer, H. E., D. E. Williams, S. Cooper, R. K. Shadduck, S. Gillis, A. Waheed, D. L. Urdal, and D. C. Bicknell. 1987. Comparative effects in vivo of recombinant murine interleukin 3, natural murine colony-stimulating factor-1, and recombinant murine granulocyte-macrophage colony-stimulating factor on myelopoiesis in mice. J. Clin. Investig. 79:721-730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carver-Moore, K., H. E. Broxmeyer, S. M. Luoh, S. Cooper, J. Peng, S. A. Burstein, M. W. Moore, and F. J. de Sauvage. 1996. Low levels of erythroid and myeloid progenitors in thrombopoietin- and c-mpl-deficient mice. Blood 88:803-808. [PubMed] [Google Scholar]
- 11.Cheng, J., S. Baumhueter, G. Cacalano, K. Carver-Moore, H. Thibodeaux, R. Thomas, H. E. Broxmeyer, S. Cooper, N. Hague, M. Moore, and L. A. Lasky. 1996. Hematopoietic defects in mice lacking the sialomucin CD34. Blood 87:479-490. [PubMed] [Google Scholar]
- 12.Christopherson, K. W., S. Cooper, G. Hangoc, and H. E. Broxmeyer. 2003. CD26 is essential for normal G-CSF-induced progenitor cell mobilization as determined by CD26−/− mice. Exp. Hematol. 31:1126-1134. [DOI] [PubMed] [Google Scholar]
- 13.Christopherson, K. W., II, S. Cooper, and H. E. Broxmeyer. 2003. Cell surface peptidase CD26/DPPIV mediates G-CSF mobilization of mouse progenitor cells. Blood 101:4680-4686. [DOI] [PubMed] [Google Scholar]
- 14.Cooper, S., and H. E. Broxmeyer. 1991. Clonogenic methods in vitro for the enumeration of granuloctye-macrophage progenitor cell (CFU-GM) in human bone marrow and mouse bone marrow and spleen. J. Tissue Cult. Methods 13:77-82. [Google Scholar]
- 15.Dalla-Favera, R., A. Migliazza, C. C. Chang, H. Niu, L. Pasqualucci, M. Butler, Q. Shen, and G. Cattoretti. 1999. Molecular pathogenesis of B cell malignancy: the role of BCL-6. Curr. Top. Microbiol. Immunol. 246:257-263. [DOI] [PubMed] [Google Scholar]
- 16.Deconinck, A. E., P. E. Mead, S. G. Tevosian, J. D. Crispino, S. G. Katz, L. I. Zon, and S. H. Orkin. 2000. FOG acts as a repressor of red blood cell development in Xenopus. Development 127:2031-2040. [DOI] [PubMed] [Google Scholar]
- 17.Dent, A. L., J. Hu-Li, W. E. Paul, and L. S. Staudt. 1998. T helper type 2 inflammatory disease in the absence of IL-4 and STAT6. Proc. Natl. Acad. Sci. USA 95:13823-13828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dent, A. L., A. L. Shaffer, X. Yu, D. Allman, and L. M. Staudt. 1997. Control of inflammation, cytokine expression, and germinal center formation by BCL-6. Science 276:589-592. [DOI] [PubMed] [Google Scholar]
- 19.Dent, A. L., F. H. Vasanwala, and L. M. Toney. 2002. Regulation of gene expression by the proto-oncogene BCL-6. Crit. Rev. Oncol. Hematol. 41:1-9. [DOI] [PubMed] [Google Scholar]
- 20.Fukuda, T., T. Yoshida, S. Okada, M. Hatano, T. Miki, K. Ishibashi, S. Okabe, H. Koseki, S. Hirosawa, M. Taniguchi, N. Miyasaka, and T. Tokuhisa. 1997. Disruption of the Bcl6 gene results in an impaired germinal center formation. J. Exp. Med. 186:439-448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gao, J. L., T. A. Wynn, Y. Chang, E. J. Lee, H. E. Broxmeyer, S. Cooper, H. L. Tiffany, H. Westphal, J. Kwon-Chung, and P. M. Murphy. 1997. Impaired host defense, hematopoiesis, granulomatous inflammation and type 1-type 2 cytokine balance in mice lacking CC chemokine receptor 1. J. Exp. Med. 185:1959-1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haneline, L. S., H. E. Broxmeyer, S. Cooper, G. Hangoc, M. Carreau, M. Buchwald, and D. W. Clapp. 1998. Multiple inhibitory cytokines induce deregulated progenitor growth and apoptosis in hematopoietic cells from Fac−/− mice. Blood 91:4092-4098. [PubMed] [Google Scholar]
- 23.Hartatik, T., S. Okada, S. Okabe, M. Arima, M. Hatano, and T. Tokuhisa. 2001. Binding of BAZF and Bc16 to STAT6-binding DNA sequences. Biochem. Biophys. Res. Commun. 284:26-32. [DOI] [PubMed] [Google Scholar]
- 24.Hirai, H., I. M. Samokhvalov, T. Fujimoto, S. Nishikawa, and J. Imanishi. 2005. Involvement of Runx1 in the down-regulation of fetal liver kinase-1 expression during transition of endothelial cells to hematopoietic cells. Blood 106:1948-1955. [DOI] [PubMed] [Google Scholar]
- 25.Ichii, H., A. Sakamoto, Y. Kuroda, and T. Tokuhisa. 2004. Bcl6 acts as an amplifier for the generation and proliferative capacity of central memory CD8+ T cells. J. Immunol. 173:883-891. [DOI] [PubMed] [Google Scholar]
- 26.Kaplan, M. H., H. C. Chang, S. Cooper, Y. Lee, and H. E. Broxmeyer. 2003. Distinct requirements for Stat4 and Stat6 in hematopoietic progenitor cell responses to growth factors and chemokines. J. Hematother. Stem Cell Res. 12:401-408. [DOI] [PubMed] [Google Scholar]
- 27.Kaplan, M. H., Y. L. Sun, T. Hoey, and M. J. Grusby. 1996. Impaired IL-12 responses and enhanced development of Th2 cells in Stat4-deficient mice. Nature 382:174-177. [DOI] [PubMed] [Google Scholar]
- 28.Kim, C. H., G. Hangoc, S. Cooper, C. D. Helgason, S. Yew, R. K. Humphries, G. Krystal, and H. E. Broxmeyer. 1999. Altered responsiveness to chemokines due to targeted disruption of SHIP. J. Clin. Investig. 104:1751-1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim, C. H., C. K. Qu, G. Hangoc, S. Cooper, N. Anzai, G. S. Feng, and H. E. Broxmeyer. 1999. Abnormal chemokine-induced responses of immature and mature hematopoietic cells from motheaten mice implicate the protein tyrosine phosphatase SHP-1 in chemokine responses. J. Exp. Med. 190:681-690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kusam, S., L. M. Toney, H. Sato, and A. L. Dent. 2003. Inhibition of Th2 differentiation and GATA-3 expression by BCL-6. J. Immunol. 170:2435-2441. [DOI] [PubMed] [Google Scholar]
- 31.Lessard, J., A. Schumacher, U. Thorsteinsdottir, M. van Lohuizen, T. Magnuson, and G. Sauvageau. 1999. Functional antagonism of the Polycomb-group genes eed and Bmi1 in hemopoietic cell proliferation. Genes Dev. 13:2691-2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu, C., M. Levenstein, J. Chen, E. Tsifrina, R. Yonescu, C. Griffin, C. I. Civin, and D. Small. 1999. SZF1: a novel KRAB-zinc finger gene expressed in CD34+ stem/progenitor cells. Exp. Hematol. 27:313-325. [DOI] [PubMed] [Google Scholar]
- 33.Manders, P. M., P. J. Hunter, A. I. Telaranta, J. M. Carr, J. L. Marshall, M. Carrasco, Y. Murakami, M. J. Palmowski, V. Cerundolo, S. M. Kaech, R. Ahmed, and D. T. Fearon. 2005. BCL6b mediates the enhanced magnitude of the secondary response of memory CD8+ T lymphocytes. Proc. Natl. Acad. Sci. USA 102:7418-7425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mantel, C., Y. J. Kim, S. Cooper, B. Kwon, and H. E. Broxmeyer. 1993. Polymerization of murine macrophage inflammatory protein 1 alpha inactivates its myelosuppressive effects in vitro: the active form is a monomer. Proc. Natl. Acad. Sci. USA 90:2232-2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oatley, J. M., M. R. Avarbock, A. I. Telaranta, D. T. Fearon, and R. L. Brinster. 2006. Identifying genes important for spermatogonial stem cell self-renewal and survival. Proc. Natl. Acad. Sci. USA 103:9524-9529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Okabe, S., T. Fukuda, K. Ishibashi, S. Kojima, S. Okada, M. Hatano, M. Ebara, H. Saisho, and T. Tokuhisa. 1998. BAZF, a novel Bcl6 homolog, functions as a transcriptional repressor. Mol. Cell. Biol. 18:4235-4244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Perez-Losada, J., M. Sanchez-Martin, A. Rodriguez-Garcia, M. L. Sanchez, A. Orfao, T. Flores, and I. Sanchez-Garcia. 2002. Zinc-finger transcription factor Slug contributes to the function of the stem cell factor c-kit signaling pathway. Blood 100:1274-1286. [PubMed] [Google Scholar]
- 38.Qu, C. K., W. M. Yu, B. Azzarelli, S. Cooper, H. E. Broxmeyer, and G. S. Feng. 1998. Biased suppression of hematopoiesis and multiple developmental defects in chimeric mice containing Shp-2 mutant cells. Mol. Cell. Biol. 18:6075-6082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reid, S., A. Ritchie, L. Boring, J. Gosling, S. Cooper, G. Hangoc, I. F. Charo, and H. E. Broxmeyer. 1999. Enhanced myeloid progenitor cell cycling and apoptosis in mice lacking the chemokine receptor, CCR2. Blood 93:1524-1533. [PubMed] [Google Scholar]
- 40.Schwartzberg, P. L., A. M. Stall, J. D. Hardin, K. S. Bowdish, T. Humaran, S. Boast, M. L. Harbison, E. J. Robertson, and S. P. Goff. 1991. Mice homozygous for the ablm1 mutation show poor viability and depletion of selected B and T cell populations. Cell 65:1165-1175. [DOI] [PubMed] [Google Scholar]
- 41.Shaffer, A. L., X. Yu, Y. He, J. Boldrick, E. P. Chan, and L. M. Staudt. 2000. BCL-6 represses genes that function in lymphocyte differentiation, inflammation, and cell cycle control. Immunity 13:199-212. [DOI] [PubMed] [Google Scholar]
- 42.Shaheen, M., and H. E. Broxmeyer. 2005. The humoral regulation of hematopoiesis, p. 223-265. In R. Hoffman, E. Benz, S. Shattil, B. Furie, H. Cohen, L. Silberstein, and P. MacGlave (ed.), Hematology: basic principles and practice, 4th ed. U.K. Elsevier Inc., Churchill Livingston, London, United Kingdom.
- 43.Staudt, L. M., A. L. Dent, A. L. Shaffer, and X. Yu. 1999. Regulation of lymphocyte cell fate decisions and lymphomagenesis by BCL-6. Intern. Rev. Immunol. 18:381. [DOI] [PubMed] [Google Scholar]
- 44.Takamori, M., M. Hatano, M. Arima, A. Sakamoto, L. Fujimura, T. Hartatik, T. Kuriyama, and T. Tokuhisa. 2004. BAZF is required for activation of naive CD4 T cells by TCR triggering. Int. Immunol. 16:1439-1449. [DOI] [PubMed] [Google Scholar]
- 45.Takenaga, M., M. Hatano, M. Takamori, Y. Yamashita, S. Okada, Y. Kuroda, and T. Tokuhisa. 2003. Bcl6-dependent transcriptional repression by BAZF. Biochem. Biophys. Res. Commun. 303:600-608. [DOI] [PubMed] [Google Scholar]
- 46.Taylor, G. A., E. Carballo, D. M. Lee, W. S. Lai, M. J. Thompson, D. D. Patel, D. I. Schenkman, G. S. Gilkeson, H. E. Broxmeyer, B. F. Haynes, and P. J. Blackshear. 1996. A pathogenetic role for TNF alpha in the syndrome of cachexia, arthritis, and autoimmunity resulting from tristetraprolin (TTP) deficiency. Immunity 4:445-454. [DOI] [PubMed] [Google Scholar]
- 47.Tian, M., H. E. Broxmeyer, Y. Fan, Z. Lai, S. Zhang, S. Aronica, S. Cooper, R. M. Bigsby, R. Steinmetz, S. J. Engle, A. Mestek, J. D. Pollock, M. N. Lehman, H. T. Jansen, M. Ying, P. J. Stambrook, J. A. Tischfield, and L. Yu. 1997. Altered hematopoiesis, behavior, and sexual function in mu opioid receptor-deficient mice. J. Exp. Med. 185:1517-1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Toney, L. M., G. Cattorretti, J. A. Graf, T. Merghoub, P. P. Pandolfi, R. Dalla-Favera, B. H. Ye, and A. L. Dent. 2000. BCL-6 regulates chemokine gene transcription in macrophages. Nat. Immunol. 1:214-220. [DOI] [PubMed] [Google Scholar]
- 49.Ye, B. H., G. Cattoretti, Q. Shen, J. Zhang, N. Hawe, R. de Waard, C. Leung, M. Nouri-Shirazi, A. Orazi, R. S. Chaganti, P. Rothman, A. M. Stall, P. P. Pandolfi, and R. Dalla-Favera. 1997. The BCL-6 proto-oncogene controls germinal-centre formation and Th2-type inflammation. Nat. Genet. 16:161-170. [DOI] [PubMed] [Google Scholar]
- 50.Yu, R. Y., X. Wang, F. J. Pixley, J. J. Yu, A. L. Dent, H. E. Broxmeyer, E. R. Stanley, and B. H. Ye. 2005. BCL-6 negatively regulates macrophage proliferation by suppressing autocrine IL-6 production. Blood 105:1777-1784. [DOI] [PubMed] [Google Scholar]
- 51.Zhang, S., S. Fukuda, Y. Lee, G. Hangoc, S. Cooper, R. Spolski, W. J. Leonard, and H. E. Broxmeyer. 2000. Essential role of signal transducer and activator of transcription (Stat)5a but not Stat5b for Flt3-dependent signaling. J. Exp. Med. 192:719-728. [DOI] [PMC free article] [PubMed] [Google Scholar]