

Abstract
For use of ribozymes in vivo, it is desirable to select functional ribozymes in the cellular environment (in the presence of inhibitory factors and limited concentrations of mandatory Mg2+ ions, etc.). As a first step toward this goal, we developed a new screening system for detection in vivo of an active ribozyme from pools of active and inactive ribozymes using the gene for dihydrofolate reductase (DHFR) as a selective marker. In our DHFR expression vector, the sequence encoding either the active or the inactive ribozyme was connected to the DHFR gene. The plasmid was designed such that, when the ribozyme was active, the rate of production of DHFR was high enough to endow resistance to trimethoprim (TMP). We demonstrated that the active ribozyme did indeed cleave the primary transcript in vivo, whereas the inactive ribozyme had no cleavage activity. Cells that harbored the active-ribozyme-coding plasmid grew faster in the presence of a fixed concentration of TMP than the corresponding cells that harbored the inactive-ribozyme-coding plasmid. Consequently, when cells were transformed by a mixture that consisted of active- and inactive-ribozyme-coding plasmids at a ratio of 1:1, (i) mainly those cells that harbored active ribozymes survived in the presence of TMP and (ii) both active- and inactive-ribozyme-harboring cells grew at an identical rate in the absence of TMP, a demonstration of a positive selection system in vivo. If the background “noise” can be removed completely in the future, the selection system might usefully complement existing selection systems in vitro.
Ribozyme and antisense technologies appear to have potential as methods for suppressing the expression of specific genes (1–9). Therefore, they could be powerful tools in gene therapy for some diseases caused by aberrant gene expression, including diseases caused by infectious agents such as HIV (7–10). There are several strategies for inhibition of the expression of specific genes during transcription and translation (11–15). The hammerhead ribozyme belongs to the class of molecules known as antisense RNAs (hereafter, the term ribozymes refers exclusively to hammerhead ribozymes unless otherwise noted). However, because of short extra sequences that form the so-called catalytic loop, this ribozyme can act as an enzyme. Since the substrate specificity of antisense and ribozyme molecules is high, antisense and ribozyme strategies seem likely to have some value for therapeutic purposes (7).
When the hammerhead ribozyme was engineered in such a way that it could cleave a specific RNA sequence “in trans” (16, 17), it was postulated that this ribozyme might be much more effective than simple antisense molecules in several respects (16–20). However, because of their instability and their lower-than-expected activities in vivo, ribozymes have not yet proven their superiority to antisense molecules. There seem to be several reasons for their low activity in vivo: (i) there may be many cellular proteins in vivo that inhibit the ribozymes’ catalytic activity (21, 22); (ii) the intracellular concentration of Mg2+ ions is much lower than that used in vitro for testing the ribozyme activity (23–25); and (iii) several cellular RNases contribute to the ribozymes’ instability (26–31). To overcome some of these problems, many approaches have been taken, including some attempts to select active ribozymes in vitro (32–37). The drawback to selection in vitro is that the activity in vitro does not always reflect the activity in vivo (38). Moreover, selection systems in vitro always involve reverse transcription. The activity of the ribozyme is associated with its specific structure, but reverse transcriptase activity is known to be inhibited by some secondary structures (39). Therefore, there is always a risk of missing the most effective ribozymes during selection in vitro.
Because of these limitations of screening systems in vitro, we need to design a screening system in vivo whereby selection can be made under the cellular conditions under which ribozymes must be active. As a first step toward the development of a screening system in vivo, we used the gene for dihydrofolate reductase (DHFR) as a selective marker in Escherichia coli. The addition by DHFR of a methyl group to deoxyuridylic acid to form thymidylic acid is an important reaction in DNA synthesis (40). Because DNA synthesis is required by all proliferating cells, inhibition of DNA synthesis is one of the most effective ways of controlling cell division. Several drugs, such as trimethoprim (TMP) and methotrexate, are potent inhibitors of DHFR, and consequently, they inhibit DNA synthesis and the multiplication of cells (40–43). When an inhibitor of DHFR, such as TMP, is present in the culture medium at a certain concentration, DHFR-producing clones, which have had already been transfected by a DHFR-expressing vector, are expected to survive and grow more rapidly than non-expressing clones (44). Therefore, if we can control the level of expression of the DHFR gene by a ribozyme, we should be able to determine the activities of ribozymes in terms of resistance to TMP. Namely, TMP resistance should be a function of ribozyme activity that can, in turn, be estimated from the concentration of TMP in the culture medium. We report here that clones that survived at a fixed concentration of TMP harbored mostly active ribozymes. Moreover, this selection system successfully identified a single base change in vivo and, therefore, to the best of our knowledge, this is the first report that suggests the possibility of positive selection in vivo of functional ribozymes.
MATERIALS AND METHODS
Bacterial Strains and Plasmids.
E. coli HB101 (recA13, supE44; Takara Shuzo, Kyoto) was used as a recipient for transformation. Several ribozyme expression vectors were constructed by modifying the DHFR expression vector pTZDHFR20 (45).
Synthesis of Oligonucleotides and Construction of Plasmids.
Oligodeoxynucleotides [active-ribozyme linkers (forward, 5′-AGC TTA ACT AAT TGA ATT CCT GAT GAG TCC CTA GGG ACG AAA CCA TGG ACT AAC TAA CTA AT-3′; and the corresponding reverse sequence), pseudo-ATG linkers (forward, 5′-CCG GAA AAG GAG GAA CTT CCA TGG TCG AAT TCA ACC TAT ATG ATC AGT CTG ATT GCG GCG-3′; and reverse), and 3′-terminator linkers (forward, 5′-TCG AGC GTC GTT AAA GCC CGC CTA ATG AGC GGG CTT TTT TTT TTA G-3′; and reverse)] were synthesized with a DNA synthesizer (model 392; Applied Biosystems) and purified by chromatography. Single base change (G5 → A or A14 → G) was introduced within the active-ribozyme catalytic core (see Fig. 1). These changes had already been shown to destroy cleavage activity (20, 46). Each linker was “tailed” with a recognition sequence for an appropriate restriction endonuclease. Each oligonucleotide linker was denatured at 95°C in a water bath and then gradually cooled to room temperature in TE buffer (10 mM Tris·HCl, pH 8.0/1 mM EDTA). After annealing, each linker set was then ligated to the digested vector pTZDHFR20 via its restriction sites and the tailed cohesive ends of the synthetic oligonucleotide linkers (Fig. 2).
Figure 1.

Secondary structure of an active ribozyme. A single point mutation (G5 to A, or A14 to G; circled) eliminates the ribozyme activity (20, 46). It is to be noted that the catalytic loop containing G5 and A14 captures Mg2+ ions, since a hammerhead ribozyme is a metalloenzyme (48–57).
Figure 2.

The ribozyme-connected DHFR expression vector. The plasmid vector has two ATG codons, one of which is a pseudo-initiation codon, located upstream of the authentic ATG, which is the initiation codon for the DHFR gene. If an active ribozyme is introduced upstream of the DHFR-coding region and if, upon transcription, the primary transcript is cleaved by this cis-acting ribozyme at the predetermined site between the two ATG codons, the excised mRNA can produce DHFR. Otherwise, the primary transcript starts translation at the pseudo-initiation codon, which is associated with a strong Shine–Dalgarno (SD) sequence and is out of frame with respect to the DHFR gene.
Composition of Culture Medium.
Luria–Bertani-modified plates, containing polypeptone, yeast extract, NaCl, and 16 mM MgSO4, were used for experiments to check the growth rate of individual clones. For the incubation of transformed E. coli cells on Luria–Bertani (LB)-modified plates, the medium contained ampicillin (100 μg/ml) and/or TMP (70 μg/ml).
Northern Blot Analysis.
Plasmid vector pTZDHFR harboring both a ribozyme and a DHFR gene was used to transform E. coli HB101. After overnight incubation at 37°C, total RNA was isolated with ISOGEN (Nippon Gene, Toyama, Japan) from 2 ml of cell culture in 2× YT (bacto-yeast extract and bacto-tryptone) medium. Ten micrograms of total RNA per sample were denatured in glyoxal/dimethyl sulfoxide, subjected to electrophoresis in a 1.8% Metaphor agarose (FMC) gel, and transferred to a Hybond-N nylon membrane (Amersham) (47). The membrane was probed with a synthetic oligonucleotide (5′-ATT CGC TGA ATA CCG ATT CCC AGT CAT CCG GCT CGT AAT C-3′; complementary to DHFR mRNA) that has been labeled with 32P using T4 Polynucleotide kinase (Takara Shuzo). Prehybridization and hybridization were performed in the same solution [5× standard saline phosphate/EDTA (SSPE; 0.15 M NaCl/10 mM phosphate, pH 7.4/1 mM EDTA)/50% formamide/5× Denhardt’s solution/0.5% SDS/150 μg/ml calf thymus DNA]. Final washing was performed in 0.1× SSPE/0.1% SDS at 70°C for 30 min.
Primer Extension Analysis.
An aliquot of a 0.2 pmol of 32P-labeled oligonucleotide primer (5′-GCC GAT AAC GCG ATC TAC-3′; complementary to DHFR mRNA) was allowed to hybridize to 5 μg of an RNA sample by heating at 65°C for 90 min and gradual cooling to room temperature in 15 μl of a solution of 10 mM Tris·HCl, pH 8.3/0.15 M KCl/1 mM EDTA. Then 15 μl of 2× reverse transcriptase reaction mixture containing 30 mM Tris·HCl (pH 8.3), 15 mM MgCl2, 8 mM DTT, 0.8 mM each dNTP, 6 units of human placental ribonuclease inhibitor, and 80 units of SuperScript RNaseH reverse transcriptase (GIBCO/BRL) was added. The reverse transcriptase reactions were carried out at 42°C for 60 min to avoid the influence of the secondary structure of the mRNA. After the reverse transcriptase reaction, 2 μl of stop solution, containing 95% formamide, 20 mM EDTA, 0.05% bromophenol blue, and 0.05% xylene cyanol, was mixed with 3 μl of the reaction mixture, and the resulting sample was fractionated on a 7 M urea/8% polyacrylamide gel. Four ddNTP sequencing reactions from the same 32P-labeled primer were fractionated together, creating sequencing ladders as markers.
RESULTS
Design and Construction of the Screening Vectors.
We designed a screening system in E. coli. In our screening vectors, either an active or an inactive ribozyme sequence (Fig. 1) was connected upstream of the E. coli DHFR gene (Fig. 2). The inactive ribozyme sequence differed from the active one by a single G5 → A (or A14 → G) mutation within catalytic core of the ribozyme. These mutations abolish ribozyme activity (20, 46). If the ribozyme were targeted to the DHFR gene itself, the growth of cells that had been transformed by active-ribozyme-coding plasmids should be slower in the presence of inhibitors of DHFR such as TMP and methotrexate. Then, clones surviving in the presence of TMP or methotrexate would turn out to have inactive-ribozyme-coding sequences, with resultant negative selection. Since it is desirable to select colonies that possess active ribozymes (positive selection), when we designed our vectors, the ribozyme was not targeted to the DHFR gene itself but to the region, designated the interspace, between two ATG codons (Fig. 2), of which one was the original initiation codon of the DHFR gene itself and the other was located upstream of the original initiation codon. The frame shift ATG was associated with a strong SD sequence and was out of frame relative to the DHFR gene. Therefore, because of the strong SD sequence associated with the upstream pseudo-ATG, the primary transcript would not produce a significant amount of DHFR. Only when the active ribozyme had cleaved the sequence between the two ATG codons would the control of translation by the pseudo-ATG with a strong SD sequence be abolished and the original ATG lead to production of DHFR.
To avoid any readthrough from the upstream regions, an “all stop codon” sequence (TAACTAACTAA) was introduced between the ribozyme and SD sequences. In this region, a stop codon would be encountered in all three possible frames. Furthermore, to facilitate the analysis of transcripts, a terminator sequence (58, 59) was introduced downstream of the DHFR gene. Then, if the active ribozyme were to attack the so-called interspace and cleave the primary transcript, which would consist of both the ribozyme and the DHFR genes connected in tandem, the resulting truncated (ribozyme-free) DHFR mRNA would potentially be detectable by Northern blot analysis.
Discrimination of Active Ribozymes from Inactive Ribozymes in the Presence of TMP.
Taking advantage of the direct relationship between the level of expression of DHFR and the strength of resistance to TMP (44), we constructed an active ribozyme-screening system. Among several concentrations of TMP tested, we found that at 70 μg TMP per ml of culture medium, E. coli clones that had been transformed with the active-ribozyme-expression vector grew more rapidly and made larger colonies compared with the clones with the inactive ribozymes. Fig. 3 shows the difference in growth rates between the active- and inactive-ribozyme-expressing colonies at 27°C and 37°C. Since the E. coli strain HB101 used in this study produces a low level of endogenous DHFR, formation of background colonies could not be avoided. Since the difference in growth rates between the active- and inactive-ribozyme-expressing colonies was greater at 27°C than at 37°C (Fig. 3B), selection of active ribozymes described below was made at 27°C in the presence of 70 μg of TMP and 100 μg of ampicillin per ml.
Figure 3.

(A) Colonies of E. coli HB101 cells that were transformed with the active- or inactive-ribozyme-expression plasmid. In the presence of 70 μg/ml TMP, colonies expressing active ribozymes (Left) grew faster than colonies expressing inactive ribozymes (Right). The difference in growth rates between the active- and inactive-ribozyme-expressing colonies was greater at 27°C (Upper) than at 37°C (Lower). (B) Distribution of colonies according to their colony size. About 4000 colonies that appeared in A were classified into 11 classes based on the diameter of colonies. The difference in growth rates between the active- and inactive-ribozyme-expressing colonies was greater at 27°C (Left) than at 37°C (Right). Since the E. coli strain HB101 used in this study produces a low level of endogenous DHFR, formation of background colonies could not be avoided.
Since active-ribozyme-expressing colonies grew more rapidly, as expected, than inactive-ribozyme-expressing colonies, we carried out a random screening assay according to the procedure outlined in Fig. 4. In this assay, equimolar amounts of active- and inactive-ribozyme-coding plasmids were mixed, and competent HB101 cells were transformed with the mixture. The transformed cells were divided into two portions, and each portion was plated either on a plate containing ampicillin (100 μg/ml) or on a plate containing ampicillin (100 μg/ml) and TMP (70 μg/ml). After incubation for 1 or more days, rapidly growing colonies were picked up at random from both plates. To check the reproducibility, we picked up only 10 colonies from each plate per day. Then, after minipreparation of plasmid DNA, sequences of the ribozyme regions of the selected clones were determined. Table 1 summarizes the sequencing results for the selected clones from more than seven independent experiments. Clones selected in the presence of TMP harbored mainly active ribozymes; in the case of the G5 and A5 mixture, only 1 out of 76 sequences turned out to be an inactive ribozyme sequence. In contrast, clones selected in the absence of TMP (in the presence of only ampicillin) yielded active and inactive sequences at a ratio of 1:1. Similar results were obtained in the case of the A14 and G14 mixture.
Figure 4.

Schematic diagram of the in vivo selection system. Competent cells are transformed with a mixture of equimolar amounts of active- and inactive-ribozyme-expression plasmids. In the absence of selection pressure (ampicillin plate), both active- and inactive-ribozyme-expressing colonies are expected to grow at the same rate. In contrast, active-ribozyme-expressing colonies are expected to grow faster on the ampicillin/TMP plate. Ampr, ampicillin resistance.
Table 1.
Numbers of selected colonies with active and inactive ribozymes on TMP-containing and/or ampicillin-containing plates
| Ribozyme | TMP plate | Ampicillin plate |
|---|---|---|
| G5 and A5 mixture | ||
| Active ribozyme | 75 | 29 |
| Inactive ribozyme | 1 | 28 |
| A14 and G14 mixture | ||
| Active ribozyme | 42 | 21 |
| Inactive ribozyme | 2 | 9 |
Plates were incubated at 27°C for 2–3 days, then larger colonies were picked up at random. TMP plates contained 70 μg of TMP and 100 μg of ampicillin per milliliter, and ampicillin plates contained 100 μg of ampicillin per milliliter without TMP.
To confirm that the phenotypic difference shown in Table 1 really originated from a single base change and not from any other mutations within the DHFR gene, we sequenced several clones in their entirety, including the DHFR region, and we further exchanged the HindIII–AccIII fragment (see Fig. 2) that contained the ribozyme sequence between the selected active and inactive clones. Since (i) no mutation was detected in the DHFR gene and (ii) the exchanged construct had the opposite phenotype, we could conclude that the phenotypic difference presented in Table 1 originated from a single base mutation. Therefore, we confirmed that the selection pressure of TMP was useful for identification of a single base change within the catalytic core of the ribozyme, which was correlated with ribozyme activity, which, in turn, was correlated with the level of expression of DHFR.
Detection of a Cleaved Fragment by Northern Blot Analysis.
To confirm that the phenotypic difference was associated with the cleavage activity of the ribozyme, Northern blot analysis was carried out with total RNA from E. coli HB101 cells that had been transfected with ribozyme expression vectors. Northern blot analysis is the most direct method for identifying cleavage activities of ribozymes in vivo. However, since cleaved fragments tend to undergo rapid degradation in vivo, Northern blot analysis failed in the past to detect cleaved fragments (60, 61). Our Northern blot analysis are shown in Fig. 5. As can be seen in Fig. 5 (lane 1), both the intact primary transcript and the cleaved fragment were detected in the analysis of total RNA extracted from cells that contained the active ribozyme vector. However, no cleavage activity was detected when we analyzed the total RNA extracted from cells that contained the inactive ribozyme vector (Fig. 5, lane 2). Although the inactive ribozyme lane (Fig. 5, lane 2) appears to show a weak signal at the size of the truncated fragment, this is not the cleavage product, as will be evidenced by the primer extension analysis (Fig. 6). The identification of the bands was based on mobilities of RNA size markers.
Figure 5.
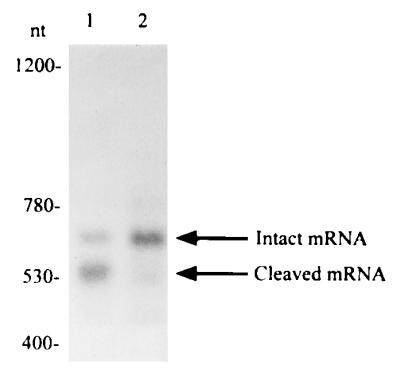
Northern blot analysis. Ten micrograms of total RNA from E. coli cells, transformed with the ribozyme-DHFR expression vector shown in Fig. 2, was subjected to electophoresis in 1.8% Metaphor agarose. After transfer to a membrane filter, the RNA was allowed to hybridize with the synthetic oligonucleotide probe (40-mer), which was complementary to part of the DHFR gene. Lane 1, active ribozyme, with G5 at the catalytic core; lane 2, inactive ribozyme, with A5 at the catalytic core. The active ribozyme expression vector produced the excised short fragment (lane 1), but there was no truncated fragment in lane 2, which originated from the inactive ribozyme expression vector. Lane 1 also shows the intact primary transcript. Fragment sizes were consistent with the expected lengths, estimated from a standard curve for mobilities of RNA size markers. The numbers indicate the length of fragments in nucleotides determined by use of size markers (not shown).
Figure 6.

Primer extension analysis. Five micrograms of total RNA was used as template for reverse transcription, with a 5′ end-labeled synthetic oligonucleotide primer. After transcription, the labeled transcribed product was subjected to electrophoresis on an 8% polyacrylamide gel. Lane 1, active ribozyme with the all stop codon was used as template; lane 2, inactive ribozyme with the all stop codon was used as template; lane 3, active ribozyme without the all stop codon; lane 4, inactive ribozyme without the all stop codon. Both lane 1 and lane 3 include cleaved fragments. On the other hand, no cleaved fragments are seen in lanes 2 and 4. The exact site of cleavage was determined by reference to the sequencing ladders.
Why did we detect the cleaved fragments when others have failed? In our case, the ribozyme target site was located upstream of the DHFR gene (Fig. 2). Therefore, the DHFR mRNA itself remained intact before and after the ribozyme-mediated cleavage. Thus, the protection from digestion by RNases “stored” within the sequence of DHFR mRNA did not change after the cleavage (protection by the binding of ribosomes, etc.).
Identification of the Cleavage Site by Primer Extension Analysis.
Although the sizes of intact and cleaved mRNAs (Fig. 5) were determined to be correct by reference to RNA size markers, the exact cleavage site was not determined by Northern blot analysis. To confirm that the cleaved fragment shown in Fig. 5 was really produced by the action of the ribozyme, primer extension analysis was carried out (Fig. 6). In these experiments, two different sets of constructs were used. In one case, the plasmids, shown in Fig. 2, that contained the all stop codon region and either the active (lane 1) or the G5 → A inactive (lane 2) ribozyme were used. In the second case, plasmids without the all stop codon region but with either an active (lane 3) or G5 → A inactive (lane 4) ribozyme were used.
As judged from the sequencing ladders on the left side, exactly the expected target sites were cleaved by the active ribozymes (lanes 1 and 3). In contrast, no cleavage products were detected with inactive ribozyme constructs (lanes 2 and 4), this strongly supports the conclusion for Fig. 5. A one-base-longer fragment was also observed for each transcript. These fragments can most probably be explained by the characteristics of reverse transcriptase, which has a “snap back” feature and incorporates one extra nucleotide independently of the template (62). It should be noted that, since the reaction mixture for the reverse transcriptase reaction contained Mg2+ ions, parts of the initial transcripts (intact mRNA) underwent ribozyme-mediated cleavage during reverse transcription. However, since there were no products other than the expected ones, we can safely conclude that the cleavage occurred specifically at the predetermined target site in vivo.
DISCUSSION
Successful selection in vitro of tailored RNA has been reported and is of considerable current interest (3, 32–37). However, to our knowledge, no such selection system exists in vivo. When ribozymes are to be used in vivo, we need to select the RNA that functions best in the cellular environment. Tsuchihashi and Herschlag reported that a protein derived from the p7 nucleocapsid protein of HIV type 1 can facilitate ribozyme cleavage (63, 64). Other proteins also probably facilitate ribozyme cleavage (65). In contrast, there are few reports of successful ribozyme-mediated gene inactivation in Saccharomyces cerevisiae (21, 22, 66–68). The difficulties in characterizing ribozyme action in vivo may hint at the existence of cellular inhibitory factors. Under such circumstances, it is desirable to be able to select functional ribozymes in the presence of such putative inhibitory factors in vivo. To this end, we first attempted to construct a positive selection system in vivo based on the general scheme shown in Fig. 7. When a toxin is expressed, cells harboring the gene for the toxin should be killed. If mRNA for the toxin can be successfully cleaved by the ribozyme that is coexpressed with the toxin mRNA, then cells harboring active ribozymes should survive and should form colonies. Consequently, all surviving colonies should hold information about active ribozyme sequences. In our first attempt, the toxin gene selected was the gene for RNase T1. However, despite some considerable effort, we failed to generate any plasmids that corresponded to the one shown in Fig. 7, when RNase T1 was used as a selective marker. No constructs with a gene for RNase T1 were rescued from transformed E. coli cells. Only frame-shifted constructs, with aborted production of RNase T1, could be rescued. In this first attempt, we could not control the extent of the toxicity of RNase T1.
Figure 7.

Schematic representation of a plasmid for the in vivo selection system. When the ribozyme is active, it can prevent expression of the toxin.
We, next, chose a potentially more controllable gene as a selective marker, namely, the gene for DHFR (44). As stated in the Introduction, DHFR is essential for DNA synthesis (40). Moreover, there exists a direct relationship between the level of expression of DHFR and the strength of resistance to TMP (44). As a first step toward constructing an in vivo screening system, we tested the feasibility of use of DHFR gene with a construct shown in Fig. 2. We initially examined two types of ribozyme, an active and an inactive ribozyme, in our initial test of the system.
At a fixed concentration of TMP of 70 μg/ml, E. coli cells harboring the active ribozyme expression vector grew faster than those harboring the inactive ribozyme expression vector (Fig. 3). Then we prepared a mixture of active and inactive ribozyme expression vectors in equimolar amounts and plated the transformed E. coli cells with the mixture on Luria–Bertani-modified plates that contained TMP at 70 μg/ml. After incubation at 27°C for 2–3 days, clones were harvested and their DNA sequences were examined to determine whether the clone contained the sequence of an active or an inactive ribozyme. In this way, we could judge whether there was any statistical significance to our method for selecting active ribozymes. Since for the most part, active ribozymes could be selected in the presence of TMP (Table 1), DHFR appeared more suitable as a selective marker than RNase T1. We also demonstrated, by Northern blot and primer extension analyses (Figs. 5 and 6), that the active ribozymes were fully functional in vivo, cleaving the primary mRNA of DHFR specifically at the predetermined site only. In both of these analyses (Figs. 5 and 6), the mutant ribozyme (G5 → A) did not have any cleavage activity. Another change, that eliminates ribozyme activity is a single base change at A14 (46). With this A14/G14 system, for the most part, active ribozymes could be selected in the presence of TMP (Table 1). Taking all these results into account, we can conclude that the difference in phenotypes of these clones originated from only a single-base mutation at the catalytic core of the hammerhead ribozyme (Fig. 1).
The examination of the construct shown in Fig. 2 revealed the possibility of selecting active ribozymes in vivo using DHFR as a selective marker. However, in its present form, the background noise could obscure selection of an active mutant from a large pool of inactive molecules (since the E. coli strain HB101 used in this study produces an endogenous DHFR, formation of background colonies could not be avoided); this is a preconstruction experiment and there was an escape of 1 inactive ribozyme among 76 clones selected in the G5/A5 system and two inactive ribozymes among 44 clones selected in the A14/G14 system (Table 1). We have not yet optimized this positive selection system in vivo. We know that the cleavage activity of the ribozyme depends strongly on the target site. Among several possible target sites, we arbitrarily chose, in this study, one target site close to the initiation codon. The ribozyme sequence was placed on the 5′ side of the DHFR gene, and no attempt has yet been made to compare the activity with that of ribozymes placed on the 3′ side (to avoid any reinitiation). Genes other than that for DHFR may also be more suitable as selective markers (the general positive selection system shown in Fig. 2 may be applicable to genes other than that for DHFR). We are, at present, trying to improve this system (trying to remove the noise) by several strategies, including the use of a DHFR-null strain. Nevertheless, as a first step toward the construction of an in vivo positive selection system, the present system allowed us successfully to identify a single base change that was associated with a change in ribozyme activity. While a bacterial cis-acting system is described in this report, it is clear that the approach might be adapted to a trans-acting eukaryotic system, which would be of value for the development of ribozyme gene therapies for human disease.
Footnotes
Abbreviations: DHFR, dihydrofolate reductase; TMP, trimethoprim; SD, Shine–Dalgarno.
References
- 1.Erickson R P, Izant J G, editors. Gene Regulation: Biology of Antisense RNA and DNA. New York: Raven; 1992. [Google Scholar]
- 2.Murray A H, editor. Antisene RNA & DNA: Antisense Techniques: An Overview. New York: Wiley–Liss; 1992. [Google Scholar]
- 3.Gray, M. W. & Cedergren, R. J., eds. (1993) FASEB J.7. [DOI] [PubMed]
- 4.Rossi J J. Curr Opin Biotechnol. 1992;3:3–7. doi: 10.1016/0958-1669(92)90117-2. [DOI] [PubMed] [Google Scholar]
- 5.Uhlmann E, Peyman A. Chem Rev (Washington, DC) 1990;90:544–584. [Google Scholar]
- 6.Cameron S H, Jennings P A. Proc Natl Acad Sci USA. 1989;86:9139–9143. doi: 10.1073/pnas.86.23.9139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sarver N, Cantin E M, Chang P S, Zaia J A, Ladne P A, Stephens D A, Rossi J J. Science. 1990;247:1222–1225. doi: 10.1126/science.2107573. [DOI] [PubMed] [Google Scholar]
- 8.Ojwang J O, Hampel A, Looney D J, Wong-Staal F, Rappaport J. Proc Natl Acad Sci USA. 1992;89:10802–10806. doi: 10.1073/pnas.89.22.10802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heidenreich O, Eckstein F. J Biol Chem. 1992;267:1904–1909. [PubMed] [Google Scholar]
- 10.Leavitt M C, Yu M, Yamada O, Kraus G, Looney D, Poeschla E, Wong-Staal F. Hum Gene Ther. 1995;5:1115–1120. doi: 10.1089/hum.1994.5.9-1115. [DOI] [PubMed] [Google Scholar]
- 11.Blume S W, Gee J E, Shrestha K, Miller D M. Nucleic Acids Res. 1992;20:1777–1784. doi: 10.1093/nar/20.7.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roy C. Nucleic Acids Res. 1993;21:2845–2852. doi: 10.1093/nar/21.12.2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mayfield C, Ebbinghaus S, Gee J, Jones D, Rodu B, Squibb M, Miller D. J Biol Chem. 1994;269:18232–18238. [PubMed] [Google Scholar]
- 14.Gee J E, Yen R L, Hung M C, Hogan M E. Gene. 1994;149:109–114. doi: 10.1016/0378-1119(94)90418-9. [DOI] [PubMed] [Google Scholar]
- 15.Choo Y, Sanchez-Garcia I, Klug A. Nature (London) 1994;372:642–645. doi: 10.1038/372642a0. [DOI] [PubMed] [Google Scholar]
- 16.Haseloff J, Gerlach W L. Nature (London) 1988;334:585–591. doi: 10.1038/334585a0. [DOI] [PubMed] [Google Scholar]
- 17.Uhlenbeck O C. Nature (London) 1987;328:596–600. doi: 10.1038/328596a0. [DOI] [PubMed] [Google Scholar]
- 18.Walbot V, Bruening G. Nature (London) 1988;334:196–197. [Google Scholar]
- 19.Maddox J. Nature (London) 1989;342:609–613. doi: 10.1038/342609a0. [DOI] [PubMed] [Google Scholar]
- 20.Inokuchi Y, Yuyama N, Hirashima A, Nishikawa S, Ohkawa J, Taira K. J Biol Chem. 1994;269:11361–11366. [PubMed] [Google Scholar]
- 21.Parker R, Muhlrad D, Deshler J O, Taylor N, Rossi J J. In: Gene Regulation: Biology of Antisense RNA and DNA. Erickson R P, Izant J G, editors. New York: Raven; 1992. pp. 183–195. [Google Scholar]
- 22.Taira K, Nishikawa S. In: Gene Regulation: Biology of Antisense RNA and DNA. Erickson R P, Izant J G, editors. New York: Raven; 1992. pp. 35–54. [Google Scholar]
- 23.Silver S, Clark D. J Biol Chem. 1971;246:569–576. [PubMed] [Google Scholar]
- 24.Flatman P W. J Membr Biol. 1984;80:1–14. doi: 10.1007/BF01868686. [DOI] [PubMed] [Google Scholar]
- 25.Romani A, Scarpa A. Arch Biochem Biophys. 1992;298:1–12. doi: 10.1016/0003-9861(92)90086-c. [DOI] [PubMed] [Google Scholar]
- 26.Pieken W A, Olsen D B, Benseler F, Aurup H, Eckstein F. Nature (London) 1991;253:314–317. doi: 10.1126/science.1857967. [DOI] [PubMed] [Google Scholar]
- 27.Olsen D B, Benseler F, Aurup H, Pieken W A, Eckstein F. Biochemistry. 1991;30:9735–9741. doi: 10.1021/bi00104a024. [DOI] [PubMed] [Google Scholar]
- 28.Heidenreich O, Eckstein F. J Biol Chem. 1992;267:1904–1909. [PubMed] [Google Scholar]
- 29.Taylor N R, Kaplan B E, Swiderski P, Li H, Rossi J J. Nucleic Acids Res. 1992;20:4559–4565. doi: 10.1093/nar/20.17.4559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Paolella G, Sproat B S, Lamond A I. EMBO J. 1992;11:1913–1919. doi: 10.1002/j.1460-2075.1992.tb05244.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shimayama T, Nishikawa F, Nishikawa S, Taira K. Nucleic Acids Res. 1993;21:2605–2611. doi: 10.1093/nar/21.11.2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pan T, Uhlenbeck O C. Biochemistry. 1992;31:3887–3895. doi: 10.1021/bi00131a001. [DOI] [PubMed] [Google Scholar]
- 33.Beaudry A A, Joyce G F. Science. 1992;257:635–641. doi: 10.1126/science.1496376. [DOI] [PubMed] [Google Scholar]
- 34.Lehman N, Joyce G F. Nature (London) 1993;361:182–185. doi: 10.1038/361182a0. [DOI] [PubMed] [Google Scholar]
- 35.Nakamaye K L, Eckstein F. Biochemistry. 1994;33:1271–1277. doi: 10.1021/bi00171a030. [DOI] [PubMed] [Google Scholar]
- 36.Cuenoud B, Szostak J W. Nature (London) 1995;375:611–614. doi: 10.1038/375611a0. [DOI] [PubMed] [Google Scholar]
- 37.Ishizaka M, Ohshima Y, Tani T. Biochem Biophys Res Commun. 1995;214:403–409. doi: 10.1006/bbrc.1995.2301. [DOI] [PubMed] [Google Scholar]
- 38.Denman R B, Smedman M, Ju W, Rubenstein R, Potempska A, Miller D L. Nucleic Acids Res. 1994;22:2375–2382. doi: 10.1093/nar/22.12.2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tuerk C, MacDougal S, Gold L. Proc Natl Acad Sci USA. 1992;89:6988–6992. doi: 10.1073/pnas.89.15.6988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blakley R L, Benkovic S J, editors. Folates and Pterins. Vol. 2. New York: Wiley Interscience; 1985. [Google Scholar]
- 41.Taira K, Benkovic S J. J Med Chem. 1988;31:129–137. doi: 10.1021/jm00396a019. [DOI] [PubMed] [Google Scholar]
- 42.Taira K, Fierke C A, Chen, Jin-Tann, Johnson K A, Benkovic S J. Trends Biochem Sci. 1987;12:275–278. [Google Scholar]
- 43.Iwakura M, Shimura Y, Tsuda K. J Biochem. 1982;91:1205–1212. doi: 10.1093/oxfordjournals.jbchem.a133804. [DOI] [PubMed] [Google Scholar]
- 44.Iwakura M, Shimura Y, Tsuda K. J Biochem. 1983;93:927–930. doi: 10.1093/jb/93.3.927. [DOI] [PubMed] [Google Scholar]
- 45.Iwakura M, Jones B E, Luo J, Matthews C R. J Biochem. 1995;117:480–488. doi: 10.1093/oxfordjournals.jbchem.a124733. [DOI] [PubMed] [Google Scholar]
- 46.Ruffner D E, Stormo G D, Uhlenbeck O C. Biochemistry. 1990;29:10695–10702. doi: 10.1021/bi00499a018. [DOI] [PubMed] [Google Scholar]
- 47.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd Ed. Vol. 1. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. pp. 7.40–7.42. [Google Scholar]
- 48.Pyle A M. Science. 1993;261:709–714. doi: 10.1126/science.7688142. [DOI] [PubMed] [Google Scholar]
- 49.Dahm S C, Derrick W B, Uhlenbeck O C. Biochemistry. 1993;32:13040–13045. doi: 10.1021/bi00211a013. [DOI] [PubMed] [Google Scholar]
- 50.Uebayasi M, Uchimaru T, Koguma T, Sawata S, Shimayama T, Taira K. J Org Chem. 1994;59:7414–7420. [Google Scholar]
- 51.Sawata S, Komiyama M, Taira K. J Am Chem Soc. 1995;117:2357–2358. [Google Scholar]
- 52.Kumar P K R, Zhou D-M, Yoshinari K, Taira K. In: Nucleic Acids and Molecular Biology. Eckstein F, Lilley D M J, editors. Vol. 10. Berlin: Springer; 1996. pp. 217–230. [Google Scholar]
- 53.Amontov S V, Taira K. J Am Chem Soc. 1996;118:1624–1628. [Google Scholar]
- 54.Zhou D-M, Usman N, Wincott F E, Matulic-Adamic J, Orita M, Zhang L-H, Komiyama M, Kumar P K R, Taira K. J Am Chem Soc. 1996;118:5862–5866. [Google Scholar]
- 55.Zhou D-M, Kumar P K R, Zhang L-H, Taira K. J Am Chem Soc. 1996;118:8969–8970. [Google Scholar]
- 56.Bassi G S, Mollegaard N-E, Murchie A I H, von Kitzing E, Lilley D M J. Nat Struct Biol. 1995;2:45–54. doi: 10.1038/nsb0195-45. [DOI] [PubMed] [Google Scholar]
- 57.Orita M, Vinayak R, Andrus A, Warashina M, Chiba A, Kaniwa H, Nishikawa F, Nishikawa S, Taira K. J Biol Chem. 1996;271:9447–9454. doi: 10.1074/jbc.271.16.9447. [DOI] [PubMed] [Google Scholar]
- 58.Yanofsky C. Nature (London) 1981;289:752–758. [Google Scholar]
- 59.Iwakura M, Tanaka T. J Biochem. 1992;111:31–36. doi: 10.1093/oxfordjournals.jbchem.a123714. [DOI] [PubMed] [Google Scholar]
- 60.Sioud M, Drlica K. Proc Natl Acad Sci USA. 1991;88:7303–7307. doi: 10.1073/pnas.88.16.7303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ferbeyre G, Bratty J, Chen H, Cedergren R. Gene. 1995;155:45–50. doi: 10.1016/0378-1119(94)00891-u. [DOI] [PubMed] [Google Scholar]
- 62.Frohman M A. In: in PCR Protocols: A Guide to Methods and Applications. Innis M A, Gelfand D H, Sninsky J J, White T J, editors. Orlando, FL: Academic; 1990. pp. 28–38. [Google Scholar]
- 63.Tsuchihashi Z, Khosla M, Herschlag D. Science. 1993;262:99–102. doi: 10.1126/science.7692597. [DOI] [PubMed] [Google Scholar]
- 64.Herschlag D, Khosla M, Tsuchihashi Z, Karpel R L. EMBO J. 1994;13:2913–2924. doi: 10.1002/j.1460-2075.1994.tb06586.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bertrand E L, Rossi J J. EMBO J. 1994;13:2904–2912. doi: 10.1002/j.1460-2075.1994.tb06585.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Egli C M, Braus G H. J Biol Chem. 1994;269:27378–27383. [PubMed] [Google Scholar]
- 67.Ferbeyre G, Bratty J, Chen H, Cedergren R. Gene. 1995;155:45–50. doi: 10.1016/0378-1119(94)00891-u. [DOI] [PubMed] [Google Scholar]
- 68.Ferbeyre G, Bratty J, Chen H, Cedergren R. J Biol Chem. 1996;271:19318–19323. doi: 10.1074/jbc.271.32.19318. [DOI] [PubMed] [Google Scholar]