

Abstract
Chimeric cowpea mosaic virus (CPMV) particles displaying foreign peptide antigens on the particle surface are suitable for development of peptide-based vaccines. However, commonly used PEG precipitation-based purification methods are not sufficient for production of high quality vaccine candidates because they do not allow for separation of chimeric particles from cleaved contaminating species. Moreover, the purified particles remain infectious to plants. To advance the CPMV technology further, it is necessary to develop efficient and scalable purification strategies and preferably eliminate the infectivity of chimeric viruses. CPMV was engineered to display a 25 amino acid peptide derived from the Bacillus anthracis protective antigen on the surface loop of the large coat protein subunit. The engineered virus was propagated in cowpea plants and assembled into chimeric virus particles displaying 60 copies of the peptide on the surface. An effective inactivation method was developed to produce non-infectious chimeric CPMV virus-like particles (VLPs). Uncleaved VLPs were separated from the contaminating cleaved forms by anion exchange chromatography. The yield of purified chimeric VLPs was 0.3 g·kg−1 of leaf tissue. The results demonstrate the ability to generate multi-gram quantities of non-infectious, chimeric CPMV VLPs in plants for use in the development of peptide-based vaccines.
Keywords: Vaccine, Plant, Chimeric virus-like particles, Inactivation, Purification
1. Introduction
Several vaccine candidates currently under development are based on short synthetic peptides of 10–40 amino acids derived from viral and bacterial pathogens, cancer cells, or protein allergens (Elsawa et al., 2004; Francis and Larche, 2005; Lazoura and Apostolopoulos, 2005). The immunogenicity of peptide-based vaccines have often been improved by formulating the peptides with liposomes (Ernst et al., 2006), chemically conjugating the peptides to a carrier (Fan et al., 2004), or alternatively, by genetically fusing the peptides to a carrier protein, such as heat shock protein (Peng et al., 2006) or viral coat protein (De Filette et al., 2005). Peptide-coat protein fusions can be produced in biological systems, such as E. coli or yeast, where they self-assemble into chimeric virus-like particles (VLPs) that display multiple copies of antigenic peptides on their surface. Cowpea mosaic virus (CPMV), produced in plants, is one example of a viral coat protein-peptide fusion system (Brennan et al., 2001).
CPMV, a plant virus, is a member of the family Comoviridae, genus Comovirus (Goldbach and Van Kammen, 1985). The genome of CPMV consists of two positive sense viral genomic RNAs. RNA1 (5889 nt) codes for proteins involved in virus replication and processing. RNA2 (3481 nt) encodes a 23.7 kDa small (S) coat protein, a 41.2 kDa large (L) coat protein, and a protein that facilitates virus movement through the plant (Goldbach and Van Kammen, 1985). Simultaneous infection of RNA1 and RNA2 is required for virus pathogenicity in plants (Goldbach and Van Kammen, 1985). CPMV RNAs are encapsidated separately into structurally identical icosahedral particles with an average diameter of 28.5 nm containing 60 copies each of L and S coat proteins per particle (Lomonossoff and Johnson, 1991). The three-dimensional structure of CPMV has been used to identify potential sites for insertion of heterologous peptides into the CPMV coat proteins (Lin et al., 1996). Peptides of up to 37 amino acids have been inserted between amino acids 23 and 24 in the S coat protein βB-βC surface loop (Porta et al., 1994) and between amino acids 98 and 99 in the L coat protein βE-αB surface loop (Brennan et al., 1999c). The engineered CPMV particles have been propagated in plants, purified by polyethylene glycol (PEG) precipitation (Dijstra and De Jager, 1998), and tested as vaccines in various animal models. High titers of antibodies specific for displayed peptides were reported after immunization of mice (Brennan et al., 1999a; Durrani et al., 1998; McLain et al., 1996; Nicholas et al., 2002; Nicholas et al., 2003). Furthermore, the engineered CPMV particles were shown to generate protective immunity from both viral (Dalsgaard et al., 1997; Langeveld et al., 2001) and bacterial (Brennan et al., 1999b; Gilleland et al., 2000) infections.
It has been reported that the insertion of epitopes into the CPMV coat proteins frequently leads to a cleavage between the two residues at the C-terminus of the inserted peptide (Taylor et al., 1999b). The epitope cleavage occurs during virus propagation in plants, but its mechanism is not understood (Taylor et al., 1999a). The cleavage may have undesirable effects on the integrity of the epitope and therefore on its immunogenic properties (Taylor et al., 1999a). Because the commonly used PEG-based CPMV purification method (Dijstra and De Jager, 1998) does not resolve the cleaved and uncleaved chimeric particles, the development of an improved method that separates these species would be desirable to ensure the highest quality and consistency of the CPMV-based vaccine candidates.
While no known plant viruses infect mammalian cells, the ability of chimeric CPMV to replicate in plants may lead to potential environmental concerns associated with the transport, distribution, and administration of CPMV-based vaccines. To render the chimeric virus non-infectious, Langeveld et al. (2001) developed an inactivation method based on UV irradiation. This treatment abolished the infectivity of CPMV in plants but special equipment was required for the inactivation. It would be useful to develop an alternative inactivation procedure that is simple, effective, and scalable.
The present study describes the development of a scalable method for purification of non-infectious, uncleaved, plant-derived chimeric CPMV virus-like particles (VLPs) that display 25 amino acid epitopes from the Bacillus anthracis protective antigen (PA).
2. Materials and methods
2.1 Cloning of CPMV constructs
CPMV RNA1 and RNA2 in vitro transcription plasmids were engineered by cloning the full length cDNAs downstream of the T7 RNA polymerase promoter and upstream of a unique restriction site (MluI for RNA1 and EcoRI for RNA2) into pUC19 and pUC18, respectively, as described previously (Vos et al., 1988). In vitro transcription plasmids containing the full-length infectious CPMV RNA1 and RNA2 were designated pCPMV-RNA1 and pCPMV-RNA2, respectively. Plasmid pCPMV-RNA2L for cloning the inserts into the βE-αB loop of the L coat protein was constructed as described previously (Brennan et al., 1999c). The PA1 peptide SNSRKKRSTSAGPTVPDRDNDGIPD, derived from Bacillus anthracis PA, was cloned into the L coat protein between amino acids 98 and 99. The PA1 oligonucleotide was synthesized by PCR amplification (Promega, Madison, WI) using overlapping primers 5′GGTTCCAATTCCAGGAAGAAGAGGTCCACTTCCGCTGGTCCTACT3′and 5′GTACTATACTTATCAGGAATACCATCATTATCCCTATCAGGCACAGTAGGACCAGCGGAAGTGGACCTCT3′ that served as templates for each other. The PCR product was cloned into pCPMV-RNA2L that was restricted with StuI and KpnI and blunted prior to ligation. The in vitro transcription plasmid for the chimeric RNA2 was designated pCPMV-PA1L. Each construct was verified by sequencing.
2.2 CPMV propagation in plants
Plasmid DNAs pCPMV-RNA1, pCPMV-RNA2, and pCPMV-PA1L were linearized with appropriate restriction enzymes prior to in vitro RNA transcription. The T7 mMessage mMachine (Ambion, Austin, TX) was used to generate run-off transcripts according to manufacturer’s instructions. RNA transcripts were capped during in vitro transcription using the RNA cap structure analog m7G(5′)ppp(5′)G (Ambion, Austin, TX). Cowpea plants (Vigna unguiculata), variety California Blackeye #5 (Ferry-Morse Seed, Fulton, KY), were grown in environmentally controlled growth chambers (Conviron, Winnipeg, Canada) at 25°C with 16/8 hr photoperiod. Primary leaves were inoculated mechanically at the two leaf stage, approximately 7 days after planting, by gently rubbing the RNA inoculum onto the leaf surface in the presence of an abrasive agent, carborundum powder (Fisher, Pittsburgh, PA). Each plant was inoculated with 2 μg of CPMV RNA1 and 2 μg of the appropriate CPMV RNA2 transcripts. Plants were grown for an additional 14–21 days. The leaf tissue was harvested and stored at −80°C until processing. Control plants were mock inoculated with water in the presence of the carborundum powder.
2.3 CPMV purification
For recovery of active virus particles, 40 g of frozen leaf tissue was blended in 150 ml of cold 30 mM Tris, pH 7.5 containing 0.2 mM phenylmethanesulfonyl fluoride (PMSF, Sigma-Aldrich, Saint Luis, MS) using a two-speed laboratory blender with a 1 liter stainless steel container (Warring, New Hartford, CT). The leaves were ground 2 times for 15 seconds at high speed. The plant slurry was centrifuged at 15,000xg for 30 min to remove plant cellular debris. For recovery of inactivated VLPs, 40 g of frozen leaf tissue was blended in 150 ml of cold 30 mM Tris, 0.5 M ammonium sulfate, 0.2 mM PMSF, pH 9.0 and the clarified extract was allowed to inactivate for 20 hr at 22°C. Cold 20% PEG 6000 (Sigma-Aldrich, Saint Luis, MS), 1 M NaCl solution was added to the clarified active or inactivated extract to a final concentration of 5% PEG 6000 and 0.25 M NaCl. After incubation for 1 hr at 4°C, the solution was centrifuged at 15,000xg for 30 min. The pellet was resuspended in 30 ml of 30 mM Tris, pH 7.5 and centrifuged at 15,000xg for 30 min. The supernatant was collected, diluted to 100 ml with ultra pure water, and filtered through a 0.45 μm filter (Pall Corporation, East Hills, NY) prior to chromatography. The diluted supernatant was split in half and run in two batches on a 16x100 mm column containing 20 ml of POROS 50 HQ strong anion exchange resin (Applied Biosystems, Foster City, CA) equilibrated with 30 mM Tris, pH 7.0. The chromatography was run at 600 cm·hr−1 at 4°C using an AKTA Explorer 100 FPLC system with a fraction collector Frac-950 and UNICORN software v5.0 (GE Healthcare, Piscataway, NJ). The column was washed with three column volumes (CVs) of 30 mM Tris, pH 7.0 to remove unbound proteins. VLPs were eluted with a gradient consisting of a one CV ramp from 0 to 150 mM NaCl, followed by a shallow one CV gradient to 186 mM NaCl, followed by a one CV ramp to 3 M NaCl. The chromatography run included an automated clean in place with a 0.5 M NaOH wash followed by an automated storage of the column and pumps into 20% EtOH. UV absorbance at 260 nm was used to monitor the virus as it eluted from the column. Fractions were collected, pooled, and buffer exchanged into phosphate buffered saline (PBS), pH 7.4 (Sigma-Aldrich, Saint Luis, MS) using a 100 kDa cutoff Amicon Ultra-15 Centrifugal Filter Device (Millipore, Billerica, MA). The final sample was filter sterilized using a 0.22 μm filter (Pall Corporation, East Hills, NY) and stored at −80°C.
2.4 Analytical work
Total protein concentration was determined by bicinchoninic acid (BCA) protein assay (Pierce Biotechnology, Rockford, IL) using 4 sample dilutions in triplicates. Encapsidated viral RNA was extracted using the RNaqueous kit (Ambion, Austin, TX) following manufacturer’s instructions. The isolated RNA was analyzed on a 1.2% agarose gel (Invitrogen, San Diego, CA) pre-stained with ethidium bromide (EtBr). The presence of the PA1L insert in the CPMV-PA1L constructs was verified by RT-PCR using the Access PCR system (Promega, Madison, WI) with the 5′CCTATTTTGGGACGGGTGC3′ and 5′GTGGTAGGAGATAAGGTCCATC3′ primers followed by sequencing of the PCR product with the 5′AGGTGACAGCTACCACCAAC3′ forward and 5′CGAGAAGTTCTTTTTGCACC3′ reverse primers. The VLP yield after each purification step was monitored by CPMV specific ELISA (Agdia, Elkhart, IN) using 4 sample dilutions in triplicates. Purified virus, quantified by BCA protein assay, was used as the quantitative ELISA standard. VLP samples were analyzed on a 10% SDS-PAGE gel (Invitrogen, San Diego, CA) stained with Simply Blue Safe Stain (Invitrogen, San Diego, CA). The samples were resuspended in the LDS sample buffer with reducing agent (Invitrogen, San Diego, CA) and boiled for 5 min prior to loading. The integrity of the purified VLPs was assayed by size exclusion chromatography (SEC HPLC) on an Agilent 1100 Series HPLC system (Agilent Technologies, Palo Alto, CA) using a TskGel G5000PWXL analytical column (Tosoh Biosciences, Tokyo, Japan). The mobile phase was 0.1 M sodium phosphate, pH 7.0. The flow rate was 0.5 ml·min−1 and the injection volume was 10 μl. UV absorbance at 215 nm was used to detect the virus as it eluted from the column.
2.5 Large scale CPMV inactivation testing
Cowpea plants were inoculated with 100 μl of active or inactivated wild type CPMV by gently rubbing the inoculum onto the leaf surface in the presence of the carborundum powder (Fisher, Pittsburgh, PA). Total RNA was extracted from leaf tissue 21 days post inoculation using the RNaqueous kit (Ambion, Austin, TX) and analyzed by RT-PCR using the Access RT-PCR system (Promega, Madison, WI). Primers 5′TGGCTGATTGTCAGAATTGG3′ and 5′TTGACCATCCCAATCTGCTC3′ were used for detection of the CPMV S coat protein gene. Primers 5′GCACAGTTTGGGTATATTG3′ and 5′GTAAAACTGGCAAAAATTAG3′ were used for detection of the cowpea house-keeping gene for the 40S ribosomal protein S15a (Simõnes-Arújo et al., 2002). PCR products were analyzed on 1.2% agarose gels (Invitrogen, San Diego, CA) pre-stained with EtBr. Total soluble protein was extracted from inoculated leaf tissue and used for Western dot blot analysis. Approximately 50 mg of leaf tissue per sample was disrupted manually in 500 μl of PBS, pH 7.4 buffer. The samples were centrifuged at 23,000xg for 10 min and the supernatant was used for subsequent analysis. 1 μl samples were dotted onto a wet nitrocellulose membrane (Bio-Rad, Hercules, CA) using a 1 μl transfer slot pin point tip transfer device (V&P Scientific, San Diego, CA). The membrane was dried for 20 min before blocking for 1.5 hr at room temperature in a solution of 3% BSA in 25 mM Tris, 0.15 M NaCl, pH 7.2 (TBS) buffer (Pierce, Rockford, IL). The membrane was then incubated for 1.5 hr in TBST (TBS with 0.05% Tween 20) containing the rabbit polyclonal anti-CPMV antibody J16 (Axis Genetics, Cambridge, UK), washed 3 times for 10 min with TBST, and incubated in TBST containing the secondary anti-rabbit antibody conjugated to alkaline phosphatase (Invitrogen, San Diego, CA). The membrane was washed 3 times for 10 min with TBST. CPMV specific signals were detected via the addition of the 5-bromo-4-chloro-3-indolyl phosphate and nitroblue tetrazolium (BCIP/NBT) substrate (Invitrogen, San Diego, CA). Positive signals became visible within 1–5 min as a purple color.
3. Results and discussion
3.1 Expression of CPMV VLPs in plants
In vitro RNA transcripts, resuspended in water, were used for inoculation of cowpea plants. The plants were co-inoculated with (1) the wild type CPMV RNA1 and RNA2 or (2) the wild type CPMV RNA1 and CPMV-PA1L containing the 25 amino acid PA1 insert derived from the Bacillus anthracis protective antigen in the L coat protein. Control plants were mock inoculated with water. Systemic mosaic symptoms characteristic of CPMV infection were observed within 7 days post inoculation on all plants inoculated with the wild type or chimeric CPMV (Fig. 1A). As expected, no symptoms developed on control plants mock-inoculated with water (Fig. 1B). The infected plant tissue was collected 2–3 weeks post inoculation. The presence of CPMV virus was verified by CPMV specific ELISA (Table 1). Though the entire plant was infected, the virus was at least 15 times more concentrated in the leaves than any other part of the plant (data not shown). Furthermore, the other plant structures, such as stems and roots, were too fibrous to be processed easily. Therefore, only the leaves were selected for processing.
Fig. 1.

Symptom development on cowpea plants inoculated with (A) wild type CPMV RNA1 and RNA2 in vitro RNA transcripts or (B) mock inoculated with water. The photographs were taken 30 days post inoculation.
Table 1.
CPMV-PA1L VLP purification
| Purification step | Total protein (g·kg−1)a | Yield (g·kg−1)b | Recovery (%) | Purity (%) |
|---|---|---|---|---|
| Tissue disruption | 26.9 | 1.35 | — | 5.0 |
| Inactivation | 24.5 | 1.28 | 95.0 | 5.2 |
| PEG precipitation | 1.66 | 1.06 | 78.8 | 64.2 |
| Anion exchange chromatography | 0.34 | 0.31 | 22.8 | 91.8 |
| Buffer exchange and sterile filtration | 0.31 | 0.30 | 22.0 | 94.6 |
Determined by BCA protein assay (Pierce Biotechnology, Rockford, IL).
Determined by CPMV specific ELISA (Agdia, Elkhart, IN).
3.2 Inactivation method development
The inactivation study was carried out on the clarified plant extract derived from leaves infected with wild type CPMV or CPMV-PA1L. Frozen leaf tissue was crushed, mixed, divided into equal 4 g samples, and blended in 15 ml of buffer using a high speed laboratory blender with a stainless steel mini-container (Warring, New Hartford, CT). Eight extraction buffers were tested that consisted of 30 mM Tris and 0.2 mM PMSF. The extraction buffers varied in pH (pH 7.0 or 9.0) and ammonium sulfate concentration (0, 0.1, 0.5, or 0.7M). The extracts were incubated at 22°C for 20 hr. The efficiency of virus inactivation was determined by monitoring the symptoms on cowpea plants inoculated with the treated plant extracts. The virus remained infectious after incubation at pH 7.0, regardless of the concentration of ammonium sulfate. The virus infectivity was also retained after incubation in 0 or 0.1 M ammonium sulfate at pH 9.0. No symptoms were detected on plants inoculated with extracts incubated in 0.5 M or 0.7 M ammonium sulfate at pH 9.0, thus suggesting that the virus was inactivated and rendered non-infectious.
The active and inactivated viruses were further purified by PEG precipitation and used for extraction of encapsidated viral RNAs. The isolated RNA was analyzed on a 1.2% agarose gel stained with EtBr. Intact viral genomic RNA1 and RNA2 were detected in the active wild type CPMV and CPMV-PA1L preparations (Fig. 2A and B). Smear but no full-length viral genomic RNAs were detected in the inactivated samples (Fig. 2A and B). The exact inactivation mechanism is unknown. We observed that pH 9.0 and at least 0.5 M ammonium sulfate was required for the inactivation. We speculate that the high pH and salt concentration caused the virus particles to become permeable and the encapsidated RNA was degraded by alkaline hydrolysis.
Fig. 2.
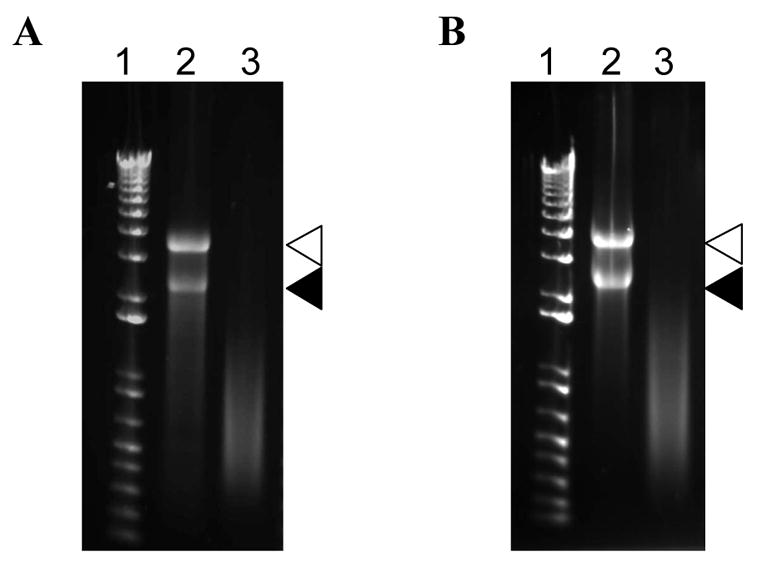
Analysis of encapsidated viral RNA. Encapsidated RNA was isolated from (A) the active or inactivated wild type CPMV and (B) the active or inactivated CPMV-PA1L VLPs. The RNA samples were run on 1.2% agarose gel pre-stained with EtBr (Invitrogen, San Diego, CA). Lane 1 is the 1 Kb Plus DNA Ladder (Invitrogen, San Diego, CA). Lanes 2 and 3 are the RNA samples isolated from the active and inactivated viruses, respectively. The CPMV RNA1 (open triangles) and RNA2 (closed triangles) are indicated.
3.3 Large scale inactivation study
To assure that the inactivation treatment resulted in non-infectious virus, large numbers of cowpea plants were inoculated with high concentrations of inactivated virus and monitored for symptom development. Wild type CPMV was chosen for these experiments, because it replicates most efficiently in plants and the infection can be achieved with a fraction of a microgram (Niblett and Semancik, 1970). Rae et al. (2005) reported that inoculation with 10 ng of CPMV was the minimum amount of virus required to observe symptoms in cowpea plants and inoculation with 1 ng of CPMV was sufficient to detect virus infection by RT-PCR. To prepare inactivated wild type CPMV, infected leaf tissue was blended in 30 mM Tris, 0.5 M ammonium sulfate, 0.2 mM PMSF, pH 9.0 and the clarified extract was incubated for 20 hr at 22°C. The inactivated virus was precipitated using PEG and resuspended in 30 mM Tris, pH 7.5. Fifty cowpea plants were inoculated with 100 μg of the inactivated virus per plant. This amount was 105 times higher than required for virus infectivity (Rae et al., 2005). Inoculation with 100 μg, 1 μg, 0.1 μg, and 0.01 μg of active wild type CPMV was used as a positive control. Inoculated plants were observed for CPMV symptoms over a period of 30 days. All plants inoculated with the active virus developed systemic symptoms 7 days post inoculation, but no symptoms were observed on plants inoculated with the inactivated CPMV (data not shown). The inoculated leaves were harvested 21 days post inoculation and used for extraction of total RNA and Western dot blot analysis. Total RNA was analyzed by RT-PCR designed to detect ~800 base pairs (bp) fragments derived from the CPMV S coat protein gene. A separate control RT-PCR reaction was set up to detect ~300 bp fragments derived from the cowpea housekeeping gene for the 40s ribosomal protein S15a (Simõnes-Arújo et al., 2002). While the cowpea housekeeping gene S15a was detected in all plants tested (Fig. 3A and B), the CPMV S coat protein gene was detected only in plants inoculated with the active CPMV (Fig. 3D). The absence of CPMV infection was verified by Western dot blot analysis. The CPMV signal was detected only in plants inoculated with the active CPMV (Fig. 3F). No signal was detected in extracts from plants inoculated with the inactivated virus (Fig. 3E). These results confirmed that 20 hr incubation in 30 mM Tris, 0.5 M ammonium sulfate, 0.2 mM PMSF, pH 9.0 at 22°C was sufficient to inactivate the virus. The described inactivation treatment is at least as effective as the UV inactivation method described by Langeveld et al. (2001). The method is scalable and can be seamlessly incorporated into the purification procedure as a part of the initial leaf processing.
Fig. 3.
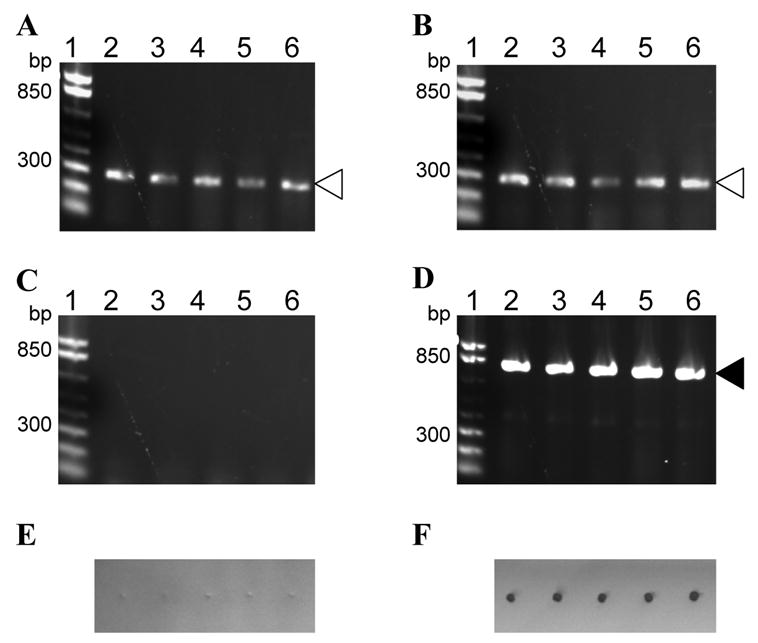
Large scale inactivation testing. Extracts from 5 plants inoculated with 100 μg of inactivated or active wild type CPMV, respectively, were analyzed by RT-PCR for the cowpea housekeeping gene S15a (A and B), RT-PCR for the CPMV S coat protein gene (C and D), and CPMV specific Western dot blot (E and F). The RT-PCR reactions (lanes 2–6) were run on 1.2% agarose gel pre-stained with EtBr (Invitrogen, San Diego, CA). Lane 1 is the 1 Kb Plus DNA Ladder (Invitrogen, San Diego, CA). PCR fragments derived from the cowpea housekeeping gene S15a and CPMV S coat protein gene are indicated by open and closed triangles, respectively.
3.4 Purification method development
The purification method development was carried out using frozen CPMV-PA1L infected leaf tissue as starting material. CPMV-PA1L yield, purity, and cumulative recovery efficiency were monitored by CPMV specific ELISA (Table 1) and SDS-PAGE (Fig. 4A). 40 g of frozen leaf tissue was blended in 150 ml of cold inactivation buffer. The clarified extract contained 1.35 g·kg−1 of CPMV-PA1L (Table 1). These yields were equivalent to those achieved with the wild type virus. After the inactivation, 95% of the CPMV-PA1L was detected in the extract by CPMV specific ELISA (Table 1). The inactivated VLPs were precipitated with PEG and analyzed by SDS-PAGE (Fig. 4A). The PEG preferentially precipitated CPMV-PA1L VLPs (Fig. 4A) with a cumulative recovery of 78.8% (Table 1).
Fig. 4.
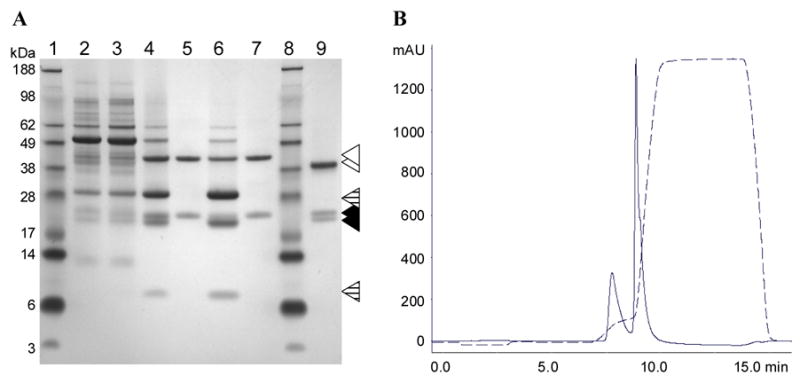
Purification of CPMV-PA1L VLPs. (A) SDS-PAGE analysis. The samples were run on 10% SDS-PAGE gel stained with Simply Blue Safe Stain (Invitrogen, San Diego, CA). Samples taken after the following purification steps were analyzed: Tissue disruption and clarification (lane 2), inactivation (lane 3), PEG precipitation (lane 4), anion exchange chromatography (lane 5 and 6), buffer exchange and sterile filtration (lane 7). Lanes 5 and 6 represent the first and second eluted peak, respectively. Lane 1 and 8 is the SeeBlue Plus2 molecular weight marker (Invitrogen, San Diego, CA). Lane 9 is the purified wild type CPMV control. The uncleaved CPMV-PA1L L coat protein and wild type CPMV L coat protein are indicated by open triangles. The full-length and trimmed S coat proteins are indicated by closed triangles. The CPMV-PA1L L coat protein cleavage products are indicated by dashed triangles. (B) CPMV-PA1L anion exchange chromatogram. Absorbance at 260 nm is indicated on the Y-axis and time in minutes is indicated on the X-axis. The conductivity trace is shown (dashed line).
The epitope cleavage that has been previously reported for chimeric CPMV particles (Brennan et al., 1999c; Lin et al., 1996; Taylor et al., 1999a; Taylor et al., 1999b) was detected in the PEG precipitated CPMV-PA1L VLP samples (Fig. 4A). In addition to the 44k Da uncleaved L coat protein, two L coat protein fragments were detected by SDS-PAGE (Fig. 4A). Interestingly, the cleaved and uncleaved VLPs could be separated by anion exchange chromatography using POROS 50 HQ strong anion exchange resin (Applied Biosystems, Foster City, California). The initial chromatography runs were performed using a 20 CV gradient from 0 to 1 M NaCl in 30 mM Tris, pH 7.0. The procedure produced two peaks that eluted at 110 and 230 mM NaCl, respectively. While the first chromatography peak consisted of VLPs containing uncleaved L coat proteins of predicted size (Fig. 4A), the second peak consisted of VLPs containing both the full-length and cleaved L coat proteins (Fig. 4A). We speculate that the VLPs eluting in the second chromatography peak were not a mixture of fully cleaved and uncleaved particles but rather contained anywhere from 1 to 60 cleaved L coat proteins per single particle.
In addition, the anion exchange chromatography resolved VLPs containing two forms of the S coat protein (Fig. 4A). The uncleaved CPMV-PA1L VLPs that eluted in the first chromatography peak contained full-length S coat proteins of the predicted size (Fig. 4A). The cleaved CPMV-PA1L VLPs that eluted in the second chromatography peak contained S coat proteins that migrated faster on the SDS-PAGE gel (Fig. 4A). Both the faster and slower migrating S coat proteins were also detected in the wild type CPMV control (Fig. 4A). We speculate that the faster migrating CPMV-PA1L S coat protein was degraded at the C-terminus. This phenomenon, called trimming, has been reported to occur naturally at the C-terminus of the wild type CPMV S coat protein during virus replication in plants (Lomonossoff and Johnson, 1991), and it has been shown to result in the loss of up to 24 amino acids from the C-terminus of the S coat protein (Taylor et al., 1999c). Our results indicate that the S coat protein trimming also occurs in the CPMV-PA1L chimeras and the trimmed and untrimmed VLPs can be resolved by anion exchange chromatography. The anion exchange chromatography also separated the trimmed and untrimmed wild type CPMV particles (data not shown).
The NaCl gradient was modified in subsequent chromatography runs to focus on the uncleaved CPMV-PA1L VLP target. The final gradient consisted of one CV ramp from 0 to 150 mM NaCl, followed by one CV shallow gradient to 186 mM NaCl, followed by one CV ramp to 3 M NaCl (Fig. 4B). The uncleaved CPMV-PA1L VLP fractions were pooled, and the VLP yield was determined by CPMV specific ELISA (Table 1). The cumulative recovery of uncleaved CPMV-PA1L VLPs was 22.8% (Table 1). The CPMV-PA1L VLPs were buffer exchanged into PBS, pH 7.4 using a 100 kDa cutoff Amicon Ultra-15 centrifugal filter device (Fig. 4A). The final samples were sterile filtered using the 0.22 μm filter and stored at −80°C. The final yield of uncleaved CPMV-PA1L VLPs was 0.3 g·kg−1 with an overall recovery of 22% (Table 2). The ratio of uncleaved to cleaved VLPs in the starting material was the most significant factor influencing the final CPMV-PA1L VLP yield.
The purified VLPs were analyzed by SEC HPLC. A single peak with retention time of 13.75 and 13.8 min was detected by SEC HPLC for the CPMV-PA1L VLPs and wild type CPMV particles, respectively (Fig. 5), thus indicating that the CPMV-PA1L VLPs are structurally intact and assembled into the predicted 120 coat protein unit particles of similar size and shape to wild type CPMV. To verify that the L coat protein contained the PA1L insert, the encapsidated viral RNA was isolated from the active CPMV-PA1L, purified in parallel with the inactivated virus, and used as a template for RT-PCR. Sequencing across the PA1L insert confirmed that there were no changes in the sequence (data not shown). This indicated that the PA1L insertion was stable and well tolerated during the virus replication in plants.
Fig. 5.
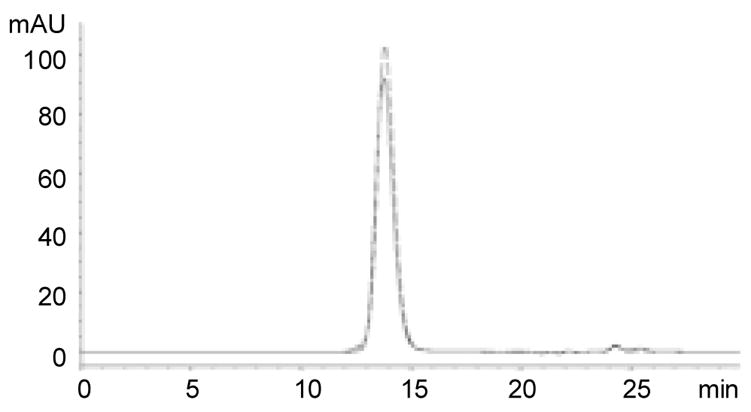
Analysis of purified VLPs. The CPMV-PA1L VLPs (solid line) and wild type CPMV (dashed line) were analyzed by SEC HPLC. Absorbance at 215 nm is indicated on the Y-axis and retention time in minutes is indicated on the X-axis.
4. Conclusion
The final purification scheme for production of non-infectious uncleaved plant CPMV VLPs engineered to display peptides derived from Bacillus anthracis PA consists of the following steps: (1) tissue disruption, (2) virus inactivation, (3) PEG precipitation, (4) anion exchange chromatography, (5) buffer exchange, and (6) sterile filtration. The inactivation step abolishes the infectivity of CPMV in plants and the anion exchange chromatography allows for the separation of uncleaved CPMV VLPs from the contaminating cleaved forms. This new purification method represents a very substantial improvement in comparison with existing methods and provides a good foundation for the development of a scaled up process. The recombinant CPMV-PA1L VLP candidates are currently being evaluated in non-human primates for their ability to generate immune responses.
Acknowledgments
We thank Carolyn Thomas, James Pack, Philip Dao, and Steve Maki for excellent technical assistance. We thank Gregory Mrachko and John E. Johnson for helpful discussions. We thank Kerr Anderson and Chuck Squires for critical review of the manuscript. This work has been supported by NIH Grant # 1U01AI054641-01.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Brennan FR, Bellaby T, Helliwell S, Jones TD, Kamstrup S, Dalsgaard K, Flock JI, Hamilton WDO. Chimeric plant virus particles administered nasally and orally induce systemic and mucosal immune responses in mice. J Virol. 1999a;73:930–938. doi: 10.1128/jvi.73.2.930-938.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan FR, Gilleland LB, Staczek J, Bendig MM, Hamilton WD, Gilleland HE. A chimaeric plant virus vaccine protects mice against a bacterial infection. Microbiology. 1999b;145:2061–2067. doi: 10.1099/13500872-145-8-2061. [DOI] [PubMed] [Google Scholar]
- Brennan FR, Jones TD, Gilleland LB, Bellaby T, Xu F, North PC, Thompson A, Gilleland LB, Staczek J, Lin T, Johnson JE, Hamilton WDO, Gilleland HE. Pseudomonas aeruginosa outer membrane protein F epitopes are highly immunogenic when expressed on a plant virus. Microbiology. 1999c;145:211–220. doi: 10.1099/13500872-145-1-211. [DOI] [PubMed] [Google Scholar]
- Brennan FR, Jones TD, Hamilton WDO. Cowpea mosaic virus as a vaccine carrier of heterologous antigens. Mol Biotech. 2001;17:15–26. doi: 10.1385/MB:17:1:15. [DOI] [PubMed] [Google Scholar]
- Dalsgaard K, Uttenthal A, Jones TD, Xu F, Merryweather AM, Hamilton WDO, Langeveld JP, Boshuizen RS, Kamstrup S, Lomonossoff GP, Porta C, Vela C, Casal JI, Meloen RH, Rodgers PB. Plant-derived vaccine protects target animals against a viral disease. Nat Biotechnol. 1997;15:248–252. doi: 10.1038/nbt0397-248. [DOI] [PubMed] [Google Scholar]
- De Filette M, Min Jou W, Birkett A, Lyons K, Schultz B, Tonkyro A, Resch S, Fiers W. Universal influenza A vaccine: optimization of M2-based constructs. Virology. 2005;337:149–161. doi: 10.1016/j.virol.2005.04.004. [DOI] [PubMed] [Google Scholar]
- Elsawa SF, Rodeberg DA, Celis E. T-cell epitope peptide vaccines. Expert Rev Vaccines. 2004;3:563–75. doi: 10.1586/14760584.3.5.563. [DOI] [PubMed] [Google Scholar]
- Dijstra J, De Jager CP. Practical plant virology: protocols and exercises. Springer; Berlin, Heidelberg, New York: 1998. [Google Scholar]
- Durrani Z, McInerney TL, McLain L, Jones T, Bellaby T, Brennan FR, Dimmock NJ. Intranasal immunization with a plant virus expressing a peptide from HIV-1 gp41 stimulates better mucosal and systemic HIV-1-specific IgA and IgG than oral immunization. J Immunol Methods. 1998;220:93–103. doi: 10.1016/s0022-1759(98)00145-8. [DOI] [PubMed] [Google Scholar]
- Ernst WA, Kim HJ, Tumpey TM, Jansen AD, Tai W, Cramer DV, Adler-Moore JP, Fujii G. Protection against H1, H5, H6 and H9 influenza A infection with liposomal matrix 2 epitope vaccines. Vaccine. 2006;24:5158–68. doi: 10.1016/j.vaccine.2006.04.008. [DOI] [PubMed] [Google Scholar]
- Fan J, Liang X, Horton MS, Perry HC, Citron MP, Heidecker GJ, Fu TM, Joyce J, Przysiecki CT, Keller PM, Garsky VM, Ionescu R, Rippeon Y, Shi L, Chastain MA, Condra JH, Davies ME, Liao J, Emini EA, Shiver JW. Preclinical study of influenza virus A M2 peptide conjugate vaccines in mice, ferrets, and rhesus monkeys. Vaccine. 2004;22:2993–3003. doi: 10.1016/j.vaccine.2004.02.021. [DOI] [PubMed] [Google Scholar]
- Francis JN, Larche M. Peptide-based vaccination: where do we stand? Curr Opin Allergy Clin Immunol. 2005;5:537–43. doi: 10.1097/01.all.0000191234.97760.88. [DOI] [PubMed] [Google Scholar]
- Gilleland HE, Gilleland LB, Staczek J, Harty RN, Garcia-Sastre A, Palese P, Brennan FR, Hamilton WD, Bendahmane M, Beachy RN. Chimeric animal and plant viruses expressing epitopes of outer membrane protein F as a combined vaccine against Pseudomonas aeruginosa lung infection. FEMS Immunol Med Microbiol. 2000;4:291–297. doi: 10.1111/j.1574-695X.2000.tb01442.x. [DOI] [PubMed] [Google Scholar]
- Goldbach R, Van Kammen A. Structure, replication and expression of the bipartitie genome of cowpea mosaic virus. In: Davies JW, editor. Molecular Plant Virology. CRC; Boca Raton: 1985. pp. 83–120. [Google Scholar]
- Langeveld JPM, Brennan FR, Martinez-Torrecuadrada JL, Jones TD, Boshuizen RS, Vela C, Casal JI, Kamstrup S, Dalsgaard K, Meloen RH, Bendig MM, Hamilton WDO. Inactivated recombinant plant virus protects dogs from a lethal challenge with canine parvovirus. Vaccine. 2001;19:3661–3670. doi: 10.1016/s0264-410x(01)00083-4. [DOI] [PubMed] [Google Scholar]
- Lazoura E, Apostolopoulos V. Rational peptide-based vaccine design for cancer immunotherapeutic applications. Curr Med Chem. 2005;12:629–639. doi: 10.2174/0929867053202188. [DOI] [PubMed] [Google Scholar]
- Lin T, Porta C, Lomonossoff G, Johnson JE. Structure-based design of peptide presentation on a viral surface: the crystal structure of a plant/animal virus chimera at 2.8 Å resolution. Fold Des. 1996;1:179–187. doi: 10.1016/s1359-0278(96)00030-2. [DOI] [PubMed] [Google Scholar]
- Lomonossoff GP, Johnson JE. The synthesis and structure of comovirus capsids. Prog Biophys Mol Biol. 1991;55:107–137. doi: 10.1016/0079-6107(91)90003-b. [DOI] [PubMed] [Google Scholar]
- McLain L, Durrani Z, Wisniewski LA, Porta C, Lomonossoff GP, Dimmock NJ. Stimulation of neutralizing antibodies to human immunodeficiency virus type 1 in three strains of mice immunized with a 22–mer amino acid peptide expressed on the surface of a plant virus. Vaccine. 1996;14:799–810. doi: 10.1016/0264-410x(95)00229-t. [DOI] [PubMed] [Google Scholar]
- Niblett CL, Semancik JS. The significance of the coat protein in infection by the electrophoretic forms of cowpea mosaic virus. Virology. 1970;41:201–207. doi: 10.1016/0042-6822(70)90072-3. [DOI] [PubMed] [Google Scholar]
- Nicholas BL, Brennan FR, Hamilton WD, Wakelin D. Effect of priming/booster immunisation protocols on immune response to canine parvovirus peptide induced by vaccination with a chimaeric plant virus construct. Vaccine. 2003;21:2441–2447. doi: 10.1016/s0264-410x(03)00054-9. [DOI] [PubMed] [Google Scholar]
- Nicholas BL, Brennan FR, Martinez-Torrecuadrada JL, Casal JI, Hamilton WD, Wakelin D. Characterization of the immune response to canine parvovirus induced by vaccination with chimaeric plant viruses. Vaccine. 2002;20:2727–2734. doi: 10.1016/s0264-410x(02)00200-1. [DOI] [PubMed] [Google Scholar]
- Peng M, Chen M, Ling N, Xu H, Qing Y, Ren H. Novel vaccines for the treatment of chronic HBV infection based on mycobacterial heat shock protein 70. Vaccine. 2006;24:887–896. doi: 10.1016/j.vaccine.2005.12.050. [DOI] [PubMed] [Google Scholar]
- Porta C, Spall VE, Loveland JE, Johnson JE, Barker PJ, Lomonosoff GP. Development of cowpea mosaic virus as a high-yielding system for the presentation of foreign peptides. Virology. 1994;202:949–955. doi: 10.1006/viro.1994.1417. [DOI] [PubMed] [Google Scholar]
- Rae CS, Khor IW, Wang Q, Destito G, Gonzalez MJ, Singh P, Thomas DM, Estrada MN, Powell E, Finn MG, Manchester M. Systemic trafficking of plant virus nanoparticles in mice via the oral route. Virology. 2005;343:224–235. doi: 10.1016/j.virol.2005.08.017. [DOI] [PubMed] [Google Scholar]
- Simõnes-Arújo JL, Rodrigues RL, Gerhardt LBA, Mondego JMC, Alves-Ferreira M, Rumjanek MG, Margis-Pinheiro M. Identification of differentially expressed genes by cDNA-AFLP technique during heat stress in cowpea nodules. FEBS Letters. 2002;515:44–50. doi: 10.1016/s0014-5793(02)02416-x. [DOI] [PubMed] [Google Scholar]
- Taylor KM, Lin T, Porta C, Mosser AG, Giesing HA, Lomonossoff GP, Johnson JE. Influence of three-dimensional structure on the immunogenicity of a peptide expressed on the surface of a plant virus. J Mol Recognit. 1999a;13:71–82. doi: 10.1002/(SICI)1099-1352(200003/04)13:2<71::AID-JMR489>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Taylor KM, Porta C, Lin T, Johnson JE, Barker PJ, Lomonossoff GP. Position-dependent processing of peptides presented on the surface of cowpea mosaic virus. Biol Chem. 1999b;380:387–392. doi: 10.1515/BC.1999.051. [DOI] [PubMed] [Google Scholar]
- Taylor KM, Spall VE, Butler PJG, Lomonossoff GP. The cleavable carboxyl-terminus of the small coat protein of cowpea mosaic virus is involved in RNA encapsidation. Virology. 1999c;255:129–137. doi: 10.1006/viro.1998.9567. [DOI] [PubMed] [Google Scholar]
- Vos P, Jaegle M, Wellink J, Verver J, Eggen R, Van Kammen A, Goldbach R. Infectious RNA transcripts derived from full-length DNA copies of the genomic RNAs of cowpea mosaic virus. Virology. 1988;165:33–41. doi: 10.1016/0042-6822(88)90655-1. [DOI] [PubMed] [Google Scholar]