

The modular choline-binding protein Pce, the phosphorylcholine esterase from S. pneumoniae, has been crystallized by the hanging-drop vapour-diffusion method. A SAD data set from a derivative with a gadolinium complex has been collected to 2.7 Å resolution.
Keywords: choline-binding proteins, phosphorylcholine esterases
Abstract
The pneumococcal phosphorylcholine esterase (Pce or CbpE) is a modular protein that hydrolyses the phosphorylcholine residues present in the teichoic and lipoteichoic acids of the pneumococcal cell wall. Pce has been crystallized using the hanging-drop vapour-diffusion method at 291 K. Diffraction-quality monoclinic crystals belong to space group C2, with unit-cell parameters a = 169.82, b = 57.26, c = 67.44 Å, β = 112.60°. A 2.7 Å resolution SAD data set from a non-isomorphous Gd-HPDO3A Pce derivative was collected at the gadolinium L III absorption edge using synchrotron radiation.
1. Introduction
Streptococcus pneumoniae is the major human Gram-positive pathogen. Many interactions with the host are mediated specifically by the phosphorylcholine residues present in the teichoic and lipoteichoic acids of the cell wall. These residues are recognized by components of the host response (Cundell et al., 1995 ▶; Pepis & Hirschfield, 2003 ▶) and serve as anchors for many surface-located proteins (Yother et al., 1992 ▶; Yother & White, 1994 ▶; López & García, 2004 ▶ and references therein), including proteins involved in cell adhesion and virulence (Yother et al., 1992 ▶; Hammerschmidt et al., 1997 ▶; Rosenow et al., 1997 ▶; Sánchez-Beato et al., 1998 ▶; Brooks-Walter et al., 1999 ▶). The biological role of choline during infection is a subject of current interest and it has been suggested that bacteria with a lower content of choline may escape the innate clearance mechanism in the bloodstream (Yother et al., 1998 ▶; Weiser & Kapoor, 1999 ▶). The pneumococcal pce gene encodes a teichoic acid phosphorylcholine esterase (Pce or CbpE) that removes phosphorylcholine residues from the cell-wall teichoic and lipoteichoic acids (de las Rivas et al., 2001 ▶; Vollmer & Tomasz, 2001 ▶). Therefore, Pce activity remodels the distribution of phosphorylcholine residues on the pneumococcal envelope, modulating the activity of choline-dependent enzymes and consequently the pathogen–host interactions. Pce carries an N-terminal signal sequence of 25 amino acids. The mature form of the protein (602 amino acids, 69 426 Da) contains a catalytic module localized in the N-terminal part of the protein and a choline-binding module at the C-terminal position that attaches the enzyme to the bacterial cell wall (de las Rivas et al., 2001 ▶; Vollmer & Tomasz, 2001 ▶). The sequence of the catalytic module reveals a low similarity with enzymes of the α/β-metallohydrolase family of the β-lactamase fold. The choline-binding module contains ten homologous repeating units of about 20 amino acids, similar to those found in other choline-binding proteins (Pfam PF01473), and a long C-terminal tail of 85 residues that does not present any known similarity with sequences in protein databases. Interestingly, a Pce mutant with significantly reduced colonization of the nasopharynx that was attributed to a decreased ability to adhere to human cells has been reported (Gosink et al., 2000 ▶) and Pce inactivation resulted in increased virulence in an intraperitoneal inoculation model (Vollmer & Tomasz, 2001 ▶). Therefore, it appears that solving the Pce structure may open new therapeutic prospects for treating pneumococcal diseases. In this communication, we present preliminary results obtained by X-ray crystallography on the complete Streptococcus pneumoniae phosphorylcholine esterase.
2. Experimental
2.1. Pce phosphorylcholinesterase production and purification
The pce truncated mutant (PceΔ55) was constructed by cloning into plasmid pT7-7 (Tabor & Richardson, 1985 ▶) the PCR fragment amplified with the oligonucleotide 5′-CCGAATTCAAGGAGATTAACATATGCAAGAAAGTTCAGGAAATAAAATCC-3′ (where the NdeI restriction site is underlined and the start codon is in bold), complementary to the 5′ end of the signal peptide of the pce gene (5′-CCGAATTCAAGGAGATTAACATATGCAAGAAAGTTCAGGAAATAAAATCC-3′) and with the oligonucleotide 5′-CGGGATCCTCATTATGTAGTTTTAATTGTAGCAGATTTCTC-3′ (where the BamHI restriction site is underlined and the complementary stop codon is in bold) complementary to the 3′ end of the deletion site (5′-CGGGATCCTCATTATGTAGTTTTAATTGTAGCAGATTTCTC-3′). The restriction sites were NdeI and BamHI. The recombinant strain culture was expressed using 50 µM isopropylthio-β-d-galactopyranoside as inducer in Escherichia coli strain BL21 (DE3). The DNA sequence of the pce mutant was confirmed with an automated Abi Prism 3700 DNA sequencer (Applied Biosystems). All primers were synthesized on a Beckman Oligo 1000M synthesizer. Restriction enzymes and other DNA-modifying enzymes were purchased from Amersham. Pce phosphorylcholine esterase and its truncated form were purified from cell extracts of E. coli BL21 (DE3) (pRGR12) or E. coli BL21 (DE3) (pAPM01), respectively, by affinity chromatography on DEAE cellulose equilibrated with phosphate/Zn buffer (20 mM sodium phosphate, 3 µM ZnCO3 pH 7.0) following the general procedure described by Sánchez-Puelles et al. (1990 ▶). The protein was specifically eluted using a linear gradient (0–70 mM choline in 2 h; elution rate 1 ml min−1) in phosphate/Zn buffer with 0.05 M NaCl. The homogeneity of the protein preparations was confirmed by SDS–PAGE and mass spectrometry. Pce activity was assayed at 298 K using p-nitrophenylphosphorycholine (NPPC) as substrate by measuring the absorbance of the product, p-nitrophenol, at 410 nm (de las Rivas et al., 2001 ▶). Fractions with high purity and activity were pooled and extensively dialyzed against Tris/Zn buffer (50 mM Tris–HCl, 3 µM ZnCO3 pH 8.0) in the absence and presence of 10 mM choline. The enzyme was then concentrated at 277 K with a 10 kDa cutoff protein concentrator (Amicon, YM-10) to approximately 11 mg ml−1. The final protein concentration was determined by spectrophotometry assuming a molar absorption coefficient of 194 020 M −1 cm−1 at 280 nm.
2.2. Mass spectrometry
MALDI–TOF measurements were performed using a Voyager DE-PRO mass spectrometer from Applied Biosystems equipped with a pulsed nitrogen laser (λ = 337 nm, 10 ns pulse width, 3 Hz frequency) and a delayed extraction ion source. Ions generated by the laser desorption were introduced into the flight tube (1.3 m flight path) with an acceleration voltage of 25 kV in the linear positive-ion mode. All mass spectra were collected by averaging the signals of 500 laser shots.
2.3. N-terminal sequencing
The N-terminal sequence of Pce and its proteolytic fragments was determined as described by Speicher (1994 ▶) using an automatic sequencer (model 477A from Applied Biosystems).
2.4. Crystallization
The initial crystallization conditions were established utilizing the sparse-matrix sampling technique (Jancarik & Kim, 1991 ▶) and the hanging-drop vapour-diffusion method at 291 K using Index Screens I and II from Hampton Research. Each hanging drop, made by mixing 1 µl protein solution and 1 µl well solution, was equilibrated against 500 µl well solution. With Pce that has been dialyzed in the presence of 10 mM choline, plate-shaped crystals were obtained in condition No. 45 of Index Screen I (0.1 M Tris pH 8.5, 25% PEG 3350). The crystals were systematically twinned thin plates that only diffracted to 3 Å resolution. In order to improve the crystallization process, the last 55 amino acids were deleted. The activity of the truncated form (PceΔ55) was similar to that of the wild-type protein. The initial conditions and all screens from Hampton Research were tested with the truncated form. In the absence of choline, PceΔ55 crystallized in the form of rods (Fig. 1 ▶ a) under condition No. 65 of Index Screen II (0.1 M bis-Tris pH 5.5, 0.1 M ammonium acetate, 17% PEG 10K) but the crystals diffracted very poorly. Of 72 detergents and additives tested with this condition (Detergent Screens 1, 2 and 3 and Additive Screens 1, 2 and 3 from Hampton Research), only n-dodecyl phosphocholine 12 (Detergent Screen 3) produced a new crystal form (Fig. 1 ▶ b). The initial drop composition with which the best crystals were obtained was 4 µl reservoir solution (17% PEG 10 000, 0.1 M bis-Tris pH 5.5 and 0.1 M ammonium acetate), 1 µl 1.5 mM n-dodecylphosphorylcholine and 4 µl protein solution. At 293 K, crystals reached maximum dimensions of 0.1 × 0.3 × 0.3 mm in 15–30 d.
Figure 1.

Effect of n-dodecylphosphocholine detergent on Pce crystals. (a) Crystals grown at 291 K in 0.1 M bis-Tris pH 5.5, 0.1 M ammonium acetate, 17% PEG 10K. (b) Crystals obtained under the same conditions of precipitant and buffer but with addition of 1.5 mM n-dodecylphosphocholine.
2.5. X-ray diffraction experiments
Diffraction data were collected from the native protein in-house on a MAR 345 image-plate detector with Cu Kα X-rays generated by an Enraf–Nonius rotating-anode generator equipped with a double-mirror focusing system and operated at 40 kV and 90 mA. Crystals were cryoprotected by a quick soak (10 s) in reservoir solution containing in addition 30%(v/v) glycerol.
A non-isomorphous Gd derivative was obtained by soaking the crystal for 1 min in mother liquor with 10 mM of the neutral gadolinium complex Gd-HPDO3A (Girard et al., 2003 ▶). The optimal wavelength for data collection was taken at the white line (λ = 1.7110 Å) of the L III absorption edge of Gd as determined from an X-ray fluorescence spectrum collected on the soaked crystal. A SAD data set was collected at 100 K using a CCD detector on beamline BM30A at the European Synchrotron Radiation Facility (ESRF). The crystal-to-detector distance was set to 110 mm. All data were processed and scaled using XDS (Kabsch, 1988 ▶) and SCALA from the CCP4 package (Collaborative Computational Project, Number 4, 1994 ▶). Data-collection statistics for the Gd-HPDO3A derivative of Pce are summarized in Table 1 ▶.
Table 1. Data-collection statistics for the Pce Gd-HPDO3A-derivative crystal.
Values in parentheses refer to the highest resolution shell.
| Crystal data | |
| Space group | C2 |
| Unit-cell parameters | |
| a (Å) | 169.82 |
| b (Å) | 57.26 |
| c (Å) | 67.44 |
| β (°) | 112.60 |
| Data collection | |
| Temperature (K) | 100 |
| Wavelength (Å) | 1.7110 |
| Resolution (Å) | 43.85–2.68 (2.82–2.68) |
| Total reflections | 58930 |
| Unique reflections | 16810 |
| Redundancy | 3.5 (2.2) |
| Completeness (%) | 97.8 (85.4) |
| I/σ(I) | 20.2 (5.0) |
| Rsym† | 0.04 (0.28) |
R
sym = 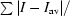
 , where the summation is over symmetry-equivalent reflections.
, where the summation is over symmetry-equivalent reflections.
3. Results and discussion
The first crystals of native Pce showed serious twinning problems. Careful analysis of protein stocks by SDS–PAGE and mass spectrometry showed the time-dependent appearance of two protein species corresponding to truncated forms of Pce (66.9 and 63.5 kDa) degraded at their C-terminal extreme, as confirmed by N-terminal sequence analysis. No further decrease in Pce molecular weight was found in protein stocks after several months. Analysis of Pce crystals by SDS–PAGE using silver staining showed that only the 63.5 kDa species, which lacked the last 55 amino acids, crystallized. The C-terminal region seems to be very flexible and no secondary-structure element is predicted for it. Therefore, in order to improve crystallization, the last 55 amino acids were deleted. The activity of the truncated form (PceΔ55) was similar to that of the wild-type protein. The best crystals of the truncated Pce form were obtained in the presence of a specific detergent (n-dodecylphosphorylcholine). They belong to the monoclinic space group C2, with unit-cell parameters a = 173.49, b = 58.12, c = 68.65 Å, β = 108.29°. Based on the Pce molecular weight and on the unit-cell volume, the Matthews coefficient of 4.0 Å3 Da−1 (Matthews, 1968 ▶) suggests the presence of a monomer in the asymmetric unit and a solvent content of 49%. Fast soaking in the presence of Gd-HPDO3A (Girard et al., 2003 ▶) led to derivative crystals that produced a high-quality diffraction pattern to 2.68 Å resolution (Fig. 2 ▶). A total of 58 930 measured reflections were merged into 16 810 unique reflections with an overall R sym of 0.04. An anomalous Patterson map was calculated using data in the entire resolution range. The resulting v = 0 and v = 1/2 Harker sections are shown in Fig. 3 ▶. Peaks corresponding to Gd-binding sites were clearly observed in anomalous Patterson maps and four sites were located using SHELX (Sheldrick, 1998 ▶). Initial phases were calculated at 2.68 Å resolution using the program SHARP (de La Fortelle &Bricogne, 1997 ▶). Model building of the complete modular Pce is currently in progress.
Figure 2.

X-ray diffraction pattern from a Pce crystal grown using the conditions described in Fig. 1 ▶(b) (oscillation range 1°). The edge of the plate corresponds to 1.9 Å resolution.
Figure 3.

(a) v = 0 Harker section and (b) v = 1/2 Harker section of the anomalous Patterson map for the Gd-HPDO3A derivative. Levels are contoured in 1σ steps starting at 2σ.
Acknowledgments
The authors thank Bracco Imaging (Milan) for kindly providing a sample of Gd-HPDO3A and the BM30A beamline at ESRF for access to synchrotron radiation and assistance in data collection. This work was supported by grants from the Spanish Ministry of Science and Technology (BIO2000-1307, BIO2002-02887, BIO2003-01952 and BMC2003-00074). LL holds a fellowship from the Spanish Ministry of Science and Technology.
References
- Brooks-Walter, A., Briles, D. E. & Hollingshead, S. K. (1999). Infect. Immun.67, 6533–6542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763. [Google Scholar]
- Cundell, D. R., Gerard, N. P., Gerard, C., Idanpaan-Heikkila, I. & Tuomanen, E. I. (1995). Nature (London), 377, 435–438. [DOI] [PubMed] [Google Scholar]
- Girard, E., Stelter, M., Vicat, J. & Kahn, R. (2003). Acta Cryst. D59, 1914–1922. [DOI] [PubMed] [Google Scholar]
- Gosink, K. K., Mann, E. R., Guglielmo, C., Tuomanen, E. I. & Masure, H. R. (2000). Infect. Immunol.68, 5690–5695. [DOI] [PMC free article] [PubMed]
- Hammerschmidt, S., Talay, S. R., Brandtzaeg, P. & Chhatwai, G. S. (1997). Mol. Microbiol.25, 1113–1124. [DOI] [PubMed] [Google Scholar]
- Jancarik, J. & Kim, S.-H. (1991). J. Appl. Cryst.24, 409–411. [Google Scholar]
- Kabsch, W. (1988). J. Appl. Cryst.21, 916–924. [Google Scholar]
- La Fortelle, E. de & Bricogne, G. (1997). Methods Enzymol.276, 472–494. [DOI] [PubMed]
- López, R. & García, E. (2004). FEMS Microbiol. Rev.28, 553–580. [DOI] [PubMed] [Google Scholar]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed] [Google Scholar]
- Pepis, M. B. & Hirschfield, G. M. (2003). J. Clin. Invest.111, 1808–1812.
- Rivas, B. de las, García, J. L., López, R. & García, P. (2001). Microb. Drug Resist.7, 213–222. [DOI] [PubMed] [Google Scholar]
- Rosenow, C., Ryan, P., Weiser, J. N., Johnson, S., Fontan, P., Ortqvist, A. & Masure, H. R. (1997). Mol. Microbiol.25, 819–829. [DOI] [PubMed] [Google Scholar]
- Sánchez-Beato, A. R., López, R. & García, J. L. (1998). FEMS Microbiol. Lett.164, 207–214. [DOI] [PubMed] [Google Scholar]
- Sánchez-Puelles, J. M., Sanz, J. M., García, J. L. & García, E. (1990). Gene, 89, 69–75. [DOI] [PubMed] [Google Scholar]
- Sheldrick, G. M. (1998). Direct Methods for Solving Macromolecular Structures, edited by S. Fortier, pp. 401–411. Dordrecht: Kluwer Academic Publishers.
- Speicher, D. W. (1994). Methods, 6, 262–273.
- Tabor, S. & Richardson, C. C. (1985). Proc. Natl Acad. Sci. USA, 82, 1074–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollmer, W. & Tomasz, A. (2001). Mol. Microbiol.39, 1610–1622. [DOI] [PubMed] [Google Scholar]
- Weiser, J. & Kapoor, M. (1999). Infect. Immun.67, 3690–3692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yother, J., Handsome, G. L. & Briles, D. E. (1992). J. Bacteriol.174, 610–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yother, J., Leopold, K., White, J. & Fischer, W. (1998). J. Bacteriol.180, 2093–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yother, J. & White, J. M. (1994). J. Bacteriol.176, 2976–2985. [DOI] [PMC free article] [PubMed] [Google Scholar]